DOI:
10.1039/D5NA00224A
(Paper)
Nanoscale Adv., 2025,
7, 4691-4709
Periodic mesoporous organosilica functionalized with p-aminobenzenesulfonic acid: an efficient solid acid catalyst for one-pot synthesis of 5-substituted 1H-tetrazoles under eco-friendly conditions†
Received
5th March 2025
, Accepted 17th May 2025
First published on 19th May 2025
Abstract
In this study, a new periodic mesoporous organosilica (PMO) functionalized with p-aminobenzenesulfonic acid (PABSA) was prepared via a novel co-condensation and sequential integration strategy. The obtained periodic mesoporous organosilica (PABSA-Pr-PMO) nanomaterial was characterized using various techniques, including Fourier-transform infrared (FT-IR) spectroscopy, field emission scanning electron microscopy (FESEM), thermogravimetric/differential thermogravimetry analysis (TGA/DTA), the Brunauer–Emmett–Teller (BET) analysis, energy-dispersive X-ray spectroscopy (EDS), EDS elemental mapping, and X-ray diffraction (XRD). The thermally stable PABSA-Pr-PMO nanomaterial demonstrates high surface area, uniform particle size and well-defined active site as a solid acid. This material was successfully employed as an efficient catalyst for the synthesis of pharmaceutically relevant tetrazole derivatives via a cascade condensation and concerted 1,3-cycloaddition reaction. The desired 5-substituted 1H-tetrazole derivatives were efficiently synthesized through multicomponent reactions between aromatic aldehydes, malononitrile and sodium azide in the presence of PABSA-Pr-PMO in EtOH under reflux conditions. The protocol exhibited excellent yields and high purity. Low catalyst loading, short reaction times, and the use of environmentally friendly solvents such as EtOH and water, which replace the hazardous dimethylformamide (DMF) solvent, are significant advantages of this novel protocol. Additionally, the straightforward separation and recyclability of the catalyst, which maintains its activity for at least five consecutive runs without significant loss, contribute to its robustness and sustainability. This protocol can be distinguished by its efficiency, environmental friendliness, and sustained catalytic activity, highlighting it as an ideal method for the synthesis of 5-substituted 1H-tetrazole derivatives.
1 Introduction
Nanocatalysis has emerged as a prominent research area in recent years. Functionalized materials at the nano- or submicro-scale exhibit significantly enhanced catalytic activity compared to conventional catalysts in organic reactions, primarily due to their increased surface area and abundant catalytic sites.1–3 This has encouraged materials scientists to actively pursue the synthesis of new nanomaterials having simultaneously higher activity and lower toxicity,4 seeking to leverage their distinctive properties across diverse fields, including chemistry, medicine, biology, and physics.5–10 Catalysis is pivotal in numerous chemical processes, supporting many synthetic advancements in both academic and industrial scales. A core principle of green chemistry is the design of chemical products and processes that minimize or eliminate the use and generation of hazardous substances.11–13 Nano-ordered catalytic systems offer a powerful approach to significantly enhance reaction yields and selectivity while facilitating precise control over reaction conditions. Novel catalytic systems not only contribute to environmental sustainability and economic efficiency but also accelerate reaction rates and suppress the formation of undesirable side products. Heterogeneous nano-ordered catalytic systems, particularly, have garnered significant attention due to their recyclability, reusability, and reduced environmental impact.14–20
In recent years, the field of nanotechnology has inspired researchers to investigate a variety of nanostructured materials for broad applications. A significant category within these materials is porous solids. Mesoporous silica nanomaterials (MSNs) have emerged as a class of versatile materials with unique properties and wide-ranging applications.21,22 Compared to traditional silica nanoparticles,23 MSNs offer superior characteristics such as high surface area, tunable pore size, high pore volume and excellent biocompatibility. Periodic mesoporous organosilicas (PMOs), a class of hybrid nanomaterials, are synthesized through sol–gel processes involving organo-bridged alkoxysilanes and surfactants.23–35 PMOs have received significant attention due to their exceptional properties, including high thermal and chemical stability, low toxicity, excellent biocompatibility, high surface area, large pore volume, and uniform pore size distribution. These attributes make PMOs ideal candidates for a wide range of applications, such as sensing,36 gas adsorption,37 catalysis,38 drug delivery,39 gene therapy,40 and electronics.41–47
The scalability of PMOs enables large-scale production, making them a promising material for various applications.45 PMOs offer the special advantage of uniform incorporating the desired organic structures directly into the silica structure, which is completely different from the structure of post-modified porous materials such as MCM (mobile composite material) and SBA (Santa Barbara amorphous) materials. In PMOs, organic groups inside the channel walls act as bridges between Si centers ((R′O)3Si-R-Si(OR′)3)(R′ = methyl or ethyl, R = bridge organic groups are placed). Furthermore, in the majority of instances pertaining to large-pore PMOs, the macroscopic morphology of the resultant products has not been subject to precise control.48–52 However, there is a promising solution to these limitations. For instance, pendant-type organotrialkoxysilanes can be utilized alongside traditional bridged organoatrilkoxysilanes [RSi(OEt)3, R = organic groups] as precursors.52,53 The pendant-type structure allows for the exposure of functional groups to the pore surface, potentially enhancing interactions with large biomolecules during adsorption or immobilization. In biodegradable PMOs (BPMOs) synthesized by combining bridged and pendant-type organoalkoxysilanes, the bridged organic groups negatively influence the physical properties of the PMO framework, while the pendant organic groups within the pores contribute to chemical reactions.54 Pendant organic groups typically exhibit higher reactivity compared to wall groups. Nevertheless, BPMO structures that incorporate more than 30 mol% of pendant organic silane tend to experience structural collapse.52 The introduction of a diverse set of high-density pendant groups offers a promising avenue for developing materials with superior performance characteristics in catalytic, adsorptive, and separation-based processes. All PMOs, as organic–inorganic hybrid materials can be synthesized by hydrolysis reactions and condensation of bridged organic silica precursors through the self-assembly process of a structure-directing agent, which is related to a similar process for the preparation of mesoporous silica materials. Indeed, incorporation of well-defined organic groups into the PMO structure increases the hydrophobicity and thermostability of the material, while leaching issues are usually minimized by external cross-linking functionalities.52
On the other hand, multicomponent reactions (MCRs), as a method for organicl synthesis performed through one-pot, tandem, cascade, or domino reactions, have received much attention.55,56 During MCRs, more than two substrates are combined that form a single product with a high bond-formation index (BFI) and atom efficiency.57 Heterocyclic compounds that are synthesized by the MCR strategy are very important and widely used because of their presence in most medicinal and pharmaceutically compounds. Among them, tetrazoles are a group of nitrogen-containing heterocyclic scaffolds that are found in the structure of several commercial drugs such as cefamandole, ceftezole, irbesartan, losartan and valsartan (Fig. 1).58
 |
| | Fig. 1 Active pharmaceutical ingredients having the tetrazole moiety: losartan, forasartan, irbesartan. | |
Tetrazole heterocycles, characterized by a five-membered ring incorporating four nitrogen atoms and one carbon atom, demonstrate exceptional stability toward thermal, microwave, and chemical stress, even with their high nitrogen content.59 Tetrazoles exist in two tautomeric forms: the 1H-tetrazole and 2H-tetrazole tautomers. The 1H-tetrazole tautomer is generally more stable in the solution phase, and the 2H-tetrazole tautomer exhibits greater stability in the gas phase.14,60 These compounds are very important and have found effective applications in various fields including medicine, biochemistry, pharmacology and various industries.61,62 Drugs containing tetrazole compounds have shown a wide range of useful properties in various therapeutic areas, including antibacterial,63 antifungal,64 anticancer,65 antituberculosis,65 peptide inhibitors,60 and antimalarial therapies. Tetrazoles serve as valuable building blocks in organic synthesis and have also found applications in high-energy materials, such as explosives and rocket propellants, as well as in specialized fields such as photography, chemical imaging, and military technology.59 Tetrazole is a crucial scaffold in drug design because it effectively mimics carboxylic acids (a concept called bioisosterism), often leading to improved drug properties such as absorption, stability, and potency.66–68 The classic synthesis of 5-substituted 1H-tetrazoles relies on a [3 + 2] cycloaddition of an azide and a nitrile.69 However, driven by sustainability concerns, the development of novel, greener synthetic routes for tetrazoles has gained significant attention recently. These efforts include the discovery of new green and environmentally friendly catalysts as well as the use of green solvents or environmentally friendly conditions.70 Future research in tetrazole chemistry may focus on the discovery of novel synthetic methodologies, the development of multifunctional tetrazole-based compounds, and the exploration of their potential applications in emerging fields such as click chemistry and materials science. Due to their considerable importance, many studies have been reported for the synthesis of 5-substituted 1H-tetrazoles through the multicomponent reaction of malononitrile (2), sodium azide (3) and various aldehydes (4) using different types of catalysts such as Fe3O4–PTh–SO3H,71 nano-NiO,72 Cu@APS-TDU-PMO,73 Fe3O4@PMO–ICS–ZnO,74 ZrP2O7 NPs,75 Pd/AlO(OH) NPs,15 the CFHT nanocomposite,76 and the CoFe2O4/Ni-BDC hybrid composite.77
According to many reports on the synthesis of tetrazole derivatives, most of these protocols have serious disadvantages such as the use of hazardous or precious metals, the inclusion of strong Lewis acids, harsh reaction conditions, low to moderate product yield, prolonged reaction time, and the use of highly toxic organic solvents, or expensive reagents. To address these challenges, a protocol based on a PMO functionalized with PABSA (PABSA-Pr-PMO nanocatalyst, 1) has been developed, which provides a novel, highly efficient, cost-effective, inexpensive, robust, and environmentally friendly approach for the synthesis of a series of 5-substituted 1H-tetrazoles. This solid acidic catalyst (1) exhibits high recyclability and promotes the facile synthesis of diverse tetrazole derivatives from aldehydes, malononitrile, and sodium azide via a multicomponent reaction route.67,78–80
2 Experimental
2.1. Materials and instrumentation
All chemicals and reagents were purchased with high purity from Merck or Aldrich and used as received, except for liquid aldehydes which were distilled before their use. The progress of reactions and the purity of the obtained products were monitored by thin layer chromatography (TLC) using Merck aluminum plates coated with 0.2 mm silica gel F254. Melting points were measured using an electrothermal 9100 device and are uncorrected. FTIR characterization of the PABSA-Pr-PMO nanocatalyst (1), as well as identification of products, was performed using KBr discs on a 1720-X PerkinElmer spectrometer. A Bruker Avance III 300 MHz and Varian Inova 500 MHz NMR system spectrometer were used for the recording of 1H NMR spectra of products in DMSO-d6 at ambient temperature. Thermal gravimetric analysis data were gathered by using a Bahr company STA 504 instrument. The X-ray diffraction pattern was obtained using STOE apparatus with a Cu Kα radiation source. Field emission scanning electron microscopy images were recorded by using a TESCAN-MIRA II device. All the products are known compounds and were identified by comparison of their physical and spectral data with those of authentic samples.
2.2. General procedure for the preparation of the PABSA-(3-chloropropyl)trimethoxysilane precursor
In this step, (3-chloropropyl)trimethoxysilane (CPTMS, 5.7 mmol, d = 1.09 g ml−1, 1.04 ml) was added dropwise into p-aminobenzenesulfonic acid (PABSA, 5.7 mmol, 1.00 g) dissolved in dry toluene (30 ml) under reflux conditions. Then, dry trimethylamine was added to the reaction mixture with a molar ratio of 1.2 equivalent (d = 0.726 g ml−1, 0.95 ml). The reaction was carried out under a nitrogen atmosphere for 24 h to afford a white gel. The solvent was then removed on a rotary evaporator and the residue was dried. The obtained mixture was washed sequentially with hexane, diethyl ether, and absolute EtOH twice (5.0 ml), respectively, and then dried for 24 h. A white powder, the PABSA-silyl precursor (I), was obtained in 95% yield.58 The PABSA-silyl precursor (I) was characterized through 1H and 13C NMR spectroscopy.
FT-IR (KBr, ν, cm−1) ν: 3420 (O–H), 3335 (N–H), 1400–1600 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1389, 1154, 1111, 697 cm−1; 1H NMR (500 MHz, DMSO-d6): δ (ppm) 9.19 (s, 1H, OH), 7.20 (d, J = 8.2 Hz, 2H), 6.46 (d, J = 8.2 Hz, 2H), 5.30 (s, 1H, NH), 3.61 (s, 9H, –OMe), 3.03 (q, J = 7.2 Hz, 2H, –NH–CH2−), 1.13 (m, 2H, –CH2−), 0.84 (t, J = 7.2 Hz, 2H, Si–CH2−). 13C NMR (125 MHz, DMSO-d6) δ (ppm) 149.18, 135.32, 126.65, 112.32, 46.68, 42.66, 34.87, 8.55.
C), 1389, 1154, 1111, 697 cm−1; 1H NMR (500 MHz, DMSO-d6): δ (ppm) 9.19 (s, 1H, OH), 7.20 (d, J = 8.2 Hz, 2H), 6.46 (d, J = 8.2 Hz, 2H), 5.30 (s, 1H, NH), 3.61 (s, 9H, –OMe), 3.03 (q, J = 7.2 Hz, 2H, –NH–CH2−), 1.13 (m, 2H, –CH2−), 0.84 (t, J = 7.2 Hz, 2H, Si–CH2−). 13C NMR (125 MHz, DMSO-d6) δ (ppm) 149.18, 135.32, 126.65, 112.32, 46.68, 42.66, 34.87, 8.55.
2.3. General procedure for the preparation of PABSA-Pr-PMO (1)
PABSA-Pr-PMO (1) was prepared by the co-condensation reaction of tetraethyl orthosilicate (TEOS) as a silica source, and PABSA-silyl precursor (I) was used in the presence of Pluronic P123 as a soft-template under basic conditions. 1.2 g of Pluronic P123 was dissolved in a mixture of 43 ml of 2.0 M HCl and 10 ml of distilled water and stirred for 4 h at room temperature. PABSA-silyl precursor (I, 0.78 g) was added to the mixture and stirred for 30 min. Subsequently, TEOS (d = 0.933 g ml−1, 2.86 ml) was added dropwise and the mixture was stirred at 40 °C for 24 h. The temperature was then increased to 100 °C and the mixture was aged statically at this temperature for 48 h. The solid product was then subjected to Soxhlet extraction by using an EtOH-hydrochloric acid solution (60 ml of 96% EtOH and 2 ml of concentrated hydrochloric acid) for 72 h to remove the polymer template (P123 surfactant). The resulting white powder was dried at 100 °C for 12 h (Scheme 1).52,54,58,81–83
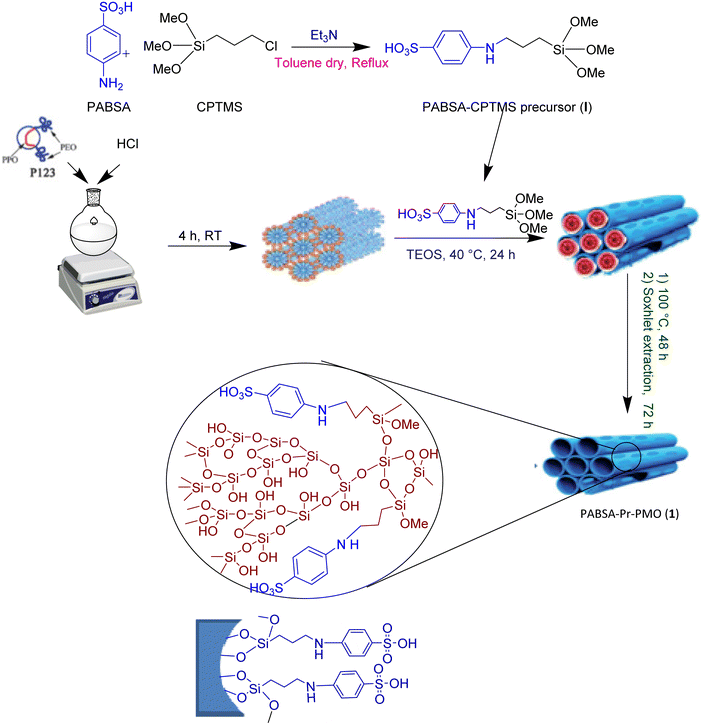 |
| | Scheme 1 Schematic description for the synthesis of the PABSA-Pr-PMO nanocatalyst (1). | |
2.4. General procedure for the synthesis of 5-substituted 1H-tetrazole derivatives 5a–s catalyzed by the PABSA-Pr-PMO nanocatalyst (1)
A mixture of malononitrile (2, 1.0 mmol), sodium azide (3, 1.2 mmol) and aldehyde (4, 1.0 mmol) in 2.0 ml EtOH was heated in the presence of PABSA-Pr-PMO nanocatalyst (1, 5.0 mg) in a 10 ml single necked round bottom flask equipped with a condenser under reflux conditions for the time shown in Table 3. The reaction progress was monitored by TLC (Eluent: EtOAc/n-hexane, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). After the reaction was completed, the obtained mixture was cooled to room temperature. The PABSA-Pr-PMO nanocatalyst (1) was easily separated from the reaction mixture using filter paper. The solvent was evaporated under reduced pressure on a rotary evaporator and the residue was diluted with distilled water and treated with HCl (2.0 N, 5.0 ml) under vigorous stirring. The solid product formed was filtered from the aqueous solution and collected as the desired corresponding tetrazole. The product was then dried at 70 °C for 2 h. The structure of products 5a–s was confirmed by using the melting point as well as FT-IR,1H NMR and 13C NMR spectroscopy.
:3). After the reaction was completed, the obtained mixture was cooled to room temperature. The PABSA-Pr-PMO nanocatalyst (1) was easily separated from the reaction mixture using filter paper. The solvent was evaporated under reduced pressure on a rotary evaporator and the residue was diluted with distilled water and treated with HCl (2.0 N, 5.0 ml) under vigorous stirring. The solid product formed was filtered from the aqueous solution and collected as the desired corresponding tetrazole. The product was then dried at 70 °C for 2 h. The structure of products 5a–s was confirmed by using the melting point as well as FT-IR,1H NMR and 13C NMR spectroscopy.
2.5. Spectral data of the selected products
2.5.1 (E)-3-(4-Chlorophenyl)-2-(1H-tetrazole-5-yl) acrylonitrile (5a).
Pale yellow powder; M.p: 156–159 °C; FT-IR (KBr, ν, cm−1): 3375 (NH), 2204 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1603 (CN), 1567 (CCaliph.), 1521 (CCarom.), 1351, 1287, 1111, 857, 609 cm−1; 1H NMR (300 MHz, DMSO-d6): δ (ppm) 8.49 (s, J = 8.0 Hz, 1H), 7.82 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 3.35 (s, br, NH overlap with solvent); 13C NMR (75 MHz, DMSO-d6) δ (ppm) 159.27, 139.31, 133.97, 131.37, 127.53, 118.64, 116.33, 100.25.
N), 1603 (CN), 1567 (CCaliph.), 1521 (CCarom.), 1351, 1287, 1111, 857, 609 cm−1; 1H NMR (300 MHz, DMSO-d6): δ (ppm) 8.49 (s, J = 8.0 Hz, 1H), 7.82 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 3.35 (s, br, NH overlap with solvent); 13C NMR (75 MHz, DMSO-d6) δ (ppm) 159.27, 139.31, 133.97, 131.37, 127.53, 118.64, 116.33, 100.25.
2.5.2 (E)-3-(4-Hydroxyphenyl)-2-(1H-tetrazol-5-yl) acrylonitrile (5k).
Cream solid powder; M.p: 162–165 °C; FT-IR (KBr, ν, cm−1): 3505 (OH), 3368 (NH), 2204 (CN), 1603 (CN), 1568 (CCaliph.), 1519 (CCarom.), 1470, 1350, 857, 637 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 10.65 (br.s, OH, 1H), 8.23 (s, 1H), 7.96 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.7 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ (ppm) 161.61, 155.17, 147.72, 133.52, 123.25, 116.46, 116.24, 92.56.
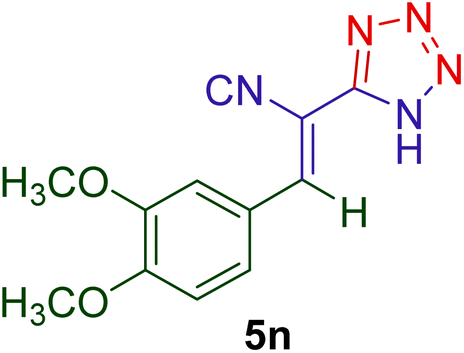
2.5.3 (E)-3-(3,4-Dimethoxyphenyl)-2-(1H-tetrazol-5-yl) acrylonitrile (5n).
Yellow crystals; M.p: 189–191 °C; FT-IR (KBr, ν, cm−1): 3370 (NH), 2223 (CN), 1600 (CN), 1568 (CCaliph.), 1510 (CCarom.), 1445, 1277 (C–Oarom.), 1018 (C–Oaliph.), 932, 850, 734, 630 cm−1; 1H NMR (300 MHz, DMSO-d6) δ (ppm) 8.35 (s, 1H), 7.63 (s, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.20 (d, J = 8.5 Hz, 1H), 3.88 (s, 3H), 3.83 (s, 3H), 3.37 (br.s, 1H, NH, exchangeable); 13C NMR (75 MHz, DMSO-d6) δ (ppm) 155.27, 154.31, 147.97, 146.37, 127.53, 125.25, 116.64, 112.23, 94.16, 56.67, 55.77.
2.5.4 (E)-3-(4-Methoxyphenyl)-2-(1H-tetrazole-5-yl) acrylonitrile (5l).
Pale yellow crystal; M.p: 157–158 °C, FT-IR (KBr, ν, cm−1): 3150 (NH), 2224 (CN), 1696 (CN), 1542 (CCaliph.), 1494 (CCarom.), 1090, 822 cm−1; 1H NMR (300 MHz, DMSO-d6): δ (ppm), 13.80 (br.s, NH), 8.51 (s, 1H), 7.87–7.91 (d, J = 8.4 Hz, 1H), 7.31–7.19 (d, J = 8.3 Hz, 1H), 3.78 (s, 3H); 13C NMR (75 MHz, DMSO-d6): δ (ppm), 164.61, 156.15, 147.77, 131.62, 125.25, 116.44, 115.35, 94.76, 56.27.
3 Results and discussion
3.1. Characterization of the periodic mesoporous organosilica functionalized with the p-aminobenzenesulfonic acid (PABSA-Pr-PMO) nanomaterial (1)
The structural, morphological, and textural properties of the synthesized PABSA-Pr-PMO nanomaterial (1) were characterized using Fourier-transform infrared (FT-IR) spectroscopy, field emission scanning electron microscopy (FESEM), Brunauer–Emmett–Teller (BET) analysis, thermogravimetric/differential thermogravimetry analysis (TGA/DTA), energy-dispersive X-ray spectroscopy (EDS), elemental mapping, and X-ray diffraction (XRD).
3.1.1. Fourier transform infrared (FTIR) analysis.
The FT-IR spectrum of the CPTMS-PABSA precursor (l, Fig. 2a) shows asymmetric and symmetric stretching modes of aliphatic C–H at 2987 and 2870 cm−1, respectively, and N–H stretching absorption bands at about 3335 cm−1 indicate type II amine groups. Also, the spectral band observed at 3420 cm−1 corresponds to the acidic O–H.84 On the other hand, the signals observed at 1400 cm−1 to 1600 cm−1 are related to the aromatic stretching bonds of the CC ring. The absorption band at 1114 cm−1 shows asymmetric stretching of C–N–C. In PABSA-Pr-PMO (1), the absorption band at 1070 cm−1 is attributed to Si–O–Si symmetric stretching vibration. The absorption of the Si–OH stretching vibration is visible at around 954 cm−1. Furthermore, signals at 1154 (OSO symmetric stretching vibration overlapped with Si–O–Si asymmetric stretching vibration), and 1399 cm−1 (asymmetric OSO stretching vibration) can be related to the SO3H groups.85 Also, the signal appearing at 695 cm−1 related to the S–O band is observed. In the FT-IR spectrum of PABSA-Pr-PMO (1, Fig. 2b), overlap of the acidic O–H stretching vibration of PABSA with the free O–H stretching mode of silanol groups on the PMO surface appears at 3435 cm−1. All these data confirmed the successful synthesis of PABSA-Pr-PMO (1).83,86
 |
| | Fig. 2 FT-IR spectra of the (a) CPTMS-PABSA and (b) PABSA-Pr-PMO nanocatalyst (1). | |
3.1.2. Field emission scanning electron microscopy (FESEM) analysis.
The morphology and particle size distribution of PABSA-Pr-PMO nanoparticles (1) were analyzed using field emission scanning electron microscopy (FESEM) (Fig. 3). Nanoparticles showed a uniform and regular tubular morphology with a narrow particle size distribution and almost undeviating spreading of the PABSA moiety over the PABSA-Pr-PMO (1) structure. The average particle size was determined to be between 35 and 63 nm, which indicates uniform distribution of particles.
 |
| | Fig. 3 FESEM images of the PABSA-Pr-PMO nanocatalyst (1) with 1.0 μm (a), 500 nm (b and c) and 200 nm (d) scales. | |
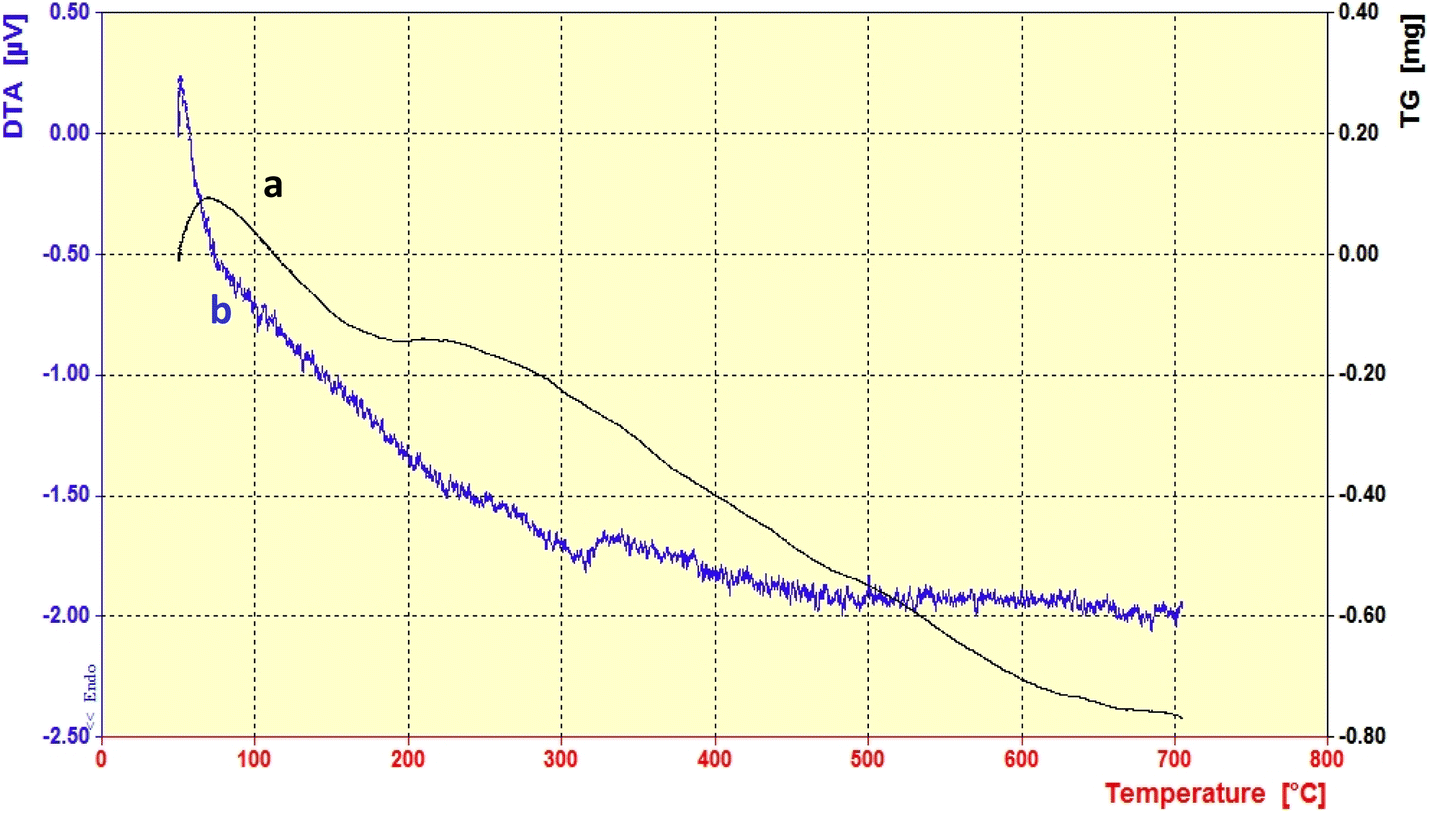
3.1.3. Thermogravimetric analysis (TGA).
Thermogravimetric analysis (TGA) was performed to investigate the thermal stability of the PABSA-Pr-PMO nanocatalyst (1) from 50 °C to 800 °C. An initial weight gain up to 115 °C can be caused by several factors especially buoyancy effects and the way the sample is packed. The first weight loss between 115 and 200 °C can be attributed to the removal of adsorbed water or EtOH solvents as well as decomposition of the residual P123 surfactant.87 The second weight loss, occurring between 200 °C and 300 °C, was associated with the decomposition of the remaining organic structure and the sulfonic acid functional group of PABSA. The final weight loss observed between 300 °C and 500 °C was attributed to the condensation of silanol groups siloxane ones. All these results confirm the successful synthesis of the PABSA-Pr-PMO nanocatalyst (1, Fig. 4a).86,88 It is important to note that the differential thermal analysis (DTA) curve demonstrates good agreement with the thermal gravimetric analysis (TGA) results. The DTA curve indicates that the removal of water and PABSA is an endothermic process. Conversely, the decomposition of the remaining organic structure is exothermic. These observations further validate the thermal stability and the structural integrity of the synthesized PABSA-Pr-PMO nanocatalyst (1, Fig. 4b).
 |
| | Fig. 4 (a) TGA and (b) DTA analysis of the PABSA-Pr-PMO nanocatalyst (1). | |
3.1.4. Energy dispersive X-ray (EDX) spectroscopy analysis.
Energy-dispersive X-ray (EDX) spectroscopy analysis was employed to determine the elemental composition of the PABSA-Pr-PMO nanocatalyst (1). The EDS spectrum revealed well-defined peaks corresponding to oxygen (O, 50.15%), sulfur (S, 4.80%), carbon (C, 25.69%), silicon (Si, 12.74%), and nitrogen (N, 6.62%), confirming the presence of these elements in the structure of the nanocatalyst (1, Fig. 5).
 |
| | Fig. 5 The EDX spectrum of the PABSA-Pr-PMO nanocatalyst (1). | |
3.1.5. Elemental mapping analysis.
Another valuable technique for identifying the distribution of elements within the structure of the nanomaterial is elemental mapping. As illustrated in Fig. 6, the elements of silicon (Si), nitrogen (N), sulfur (S), carbon (C), and oxygen (O) are uniformly dispersed throughout the selected area of the energy dispersive X-ray (EDX) image. This property is particularly important in heterogeneous catalytic systems, as it allows for the evaluation of catalyst uniformity and identification of potential defects or impurities that may impact catalytic activity and performance.
 |
| | Fig. 6 Elemental mapping of the PABSA-Pr-PMO nanocatalyst (1). | |
3.1.6. X-ray diffraction (XRD) analysis.
The low-angle XRD pattern of PABSA-Pr-PMO (1) exhibits a sharp peak at 2θ = 1.52°, indicative of a well-ordered mesoporous organosilica structure (Fig. 7a).81,82,89 The wide-angle XRD patterns shown in Fig. 7b demonstrate the crystallinity and arrangement of the PABSA-Pr-PMO nanocatalyst (1) as well as in full compliance with silicon oxide (JCPDS card no: 00-046-1045) and PABSA (JCPDS card no: 96–201-2658).
 |
| | Fig. 7 (a) Low and (b) wide angle X-ray powder diffraction (XRD) patterns of the PABSA-Pr-PMO nanomaterial (1). | |
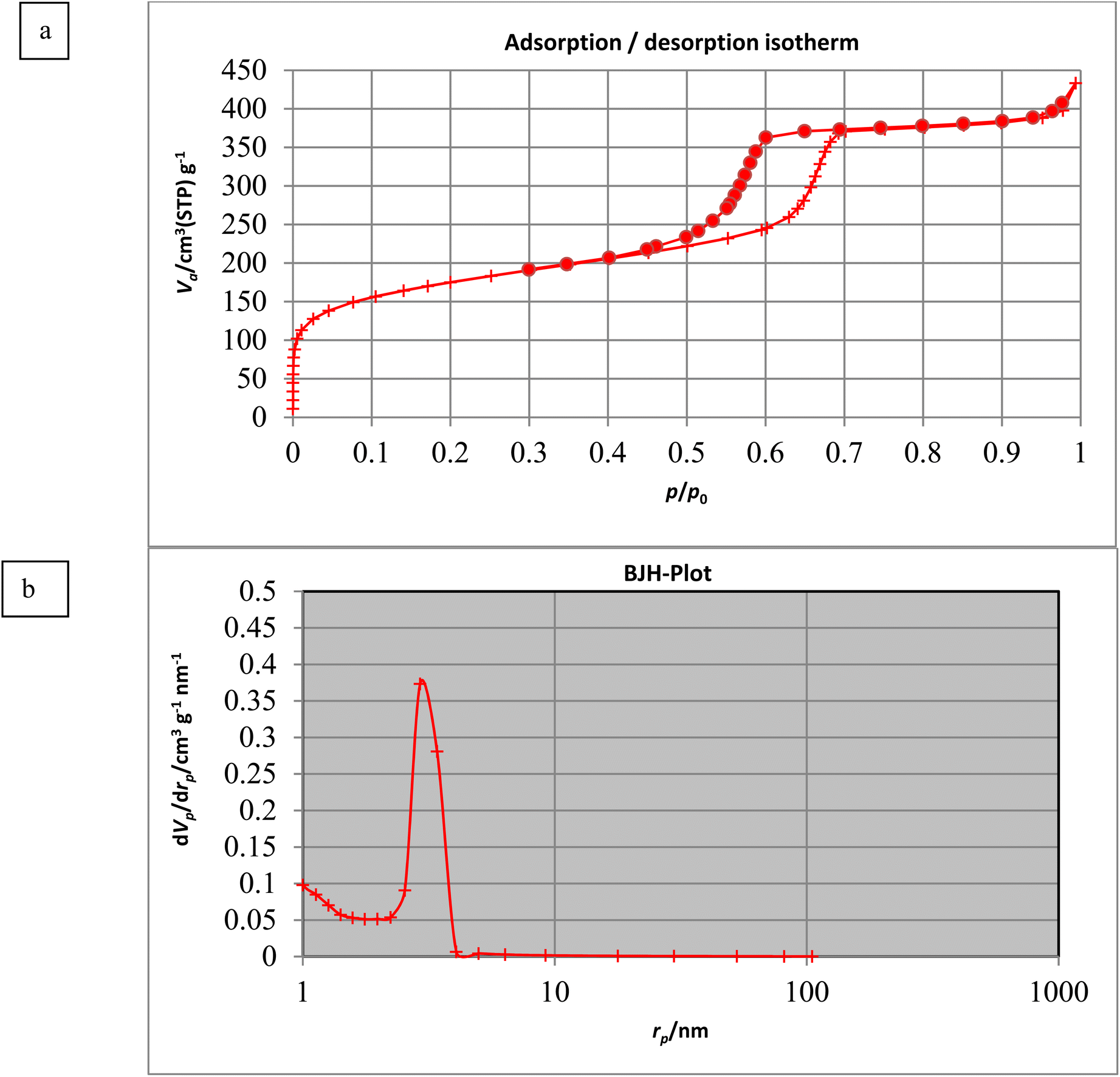
3.1.7. Brunauer–Emmett–Teller (BET) analysis.
The textural properties of PABSA-Pr-PMO nanomaterial (1) were investigated by N2 adsorption–desorption analysis and its surface area was measured using the Brunauer–Emmet–Teller (BET) method.87,90 It showed type IV isotherms with a H1 hysteresis loop, characteristic of mesoporous materials; this analysis showed that the specific BET surface area of the PABSA-Pr-PMO nanocatalyst (1) is about 617.44 m2 g−1 (Fig. 8a). The pore size distribution of the catalyst (1) was estimated by using the Barrett–Joyner–Halenda (BJH) method.83,86 The average total pore diameter and total pore volume were measured to be 4.2606 nm and 0.6274 cm3 g−1, respectively (Fig. 8b).
 |
| | Fig. 8 (a) Nitrogen adsorption–desorption and (b) pore size distribution isotherms of the PABSA-Pr-PMO nanocatalyst (1). | |
3.1.8. Determination of the acidity of the PABSA-Pr-PMO nanocatalyst (1).
Back-titration analysis verified the presence of acidic sites within the PMO-PABSA nanocatalyst (1), a technique that involves adding an excess of a standard solution to a sample and then titrating the remaining excess with another standard solution. In this procedure, 0.1 g of PMO-PABSA (1), 0.1 g of NaCl and 2 ml of NaOH (0.1 M) were introduced to 7 ml of distilled water. The resulting mixture was then subjected to stirring at room temperature for a duration of 24 h. Throughout this timeframe, the acidic protons released from the PABSA-Pr-PMO nanocatalyst (1) were fully neutralized by the hydroxide ions present in the NaOH solution. Subsequently, two drops of phenolphthalein indicator solution were added to the mixture, resulting in a pink coloration of the solution. Finally, the excess hydroxide ions were neutralized through titration with 1.11 ml of a HCl solution (0.1 M). This titration process continued until the pink coloration of the mixture, indicative of the presence of excess hydroxide ions, was completely disappeared. This neutralization point signifies a pH = 7, indicating an acidity of 2.48 mmol.g−1 of the PABSA-Pr-PMO nanocatalyst (1).
3.2. Optimization of conditions for the synthesis of tetrazole derivatives in the presence of the PABSA-Pr-PMO nanocatalyst (1)
After characterizing the PABSA-Pr-PMO nanocatalyst (1), its efficiency in the synthesis of 1H-tetrazole derivatives was thoroughly investigated. To determine the optimal conditions, a systematic study was conducted, varying the catalyst loading, solvent, and temperature. The effects on reaction yield and time were systematically examined, ultimately leading to the determination of the most efficient conditions for the synthesis of 1H-tetrazole derivatives. Thus, the reaction between malononitrile (2, 1.0 mmol), sodium azide (3, 1.2 mmol), and 4-chlorobenzaldehyde (4a, 1.0 mmol) in 2.0 ml of EtOH was chosen as the model reaction to evaluate the efficiency of the PABSA-Pr-PMO nanocatalyst (1). The results, summarized in Table 1, clearly demonstrate the significance of catalyst use and temperature in the model reaction. When the reaction was conducted in the absence of any catalyst under different conditions such as in EtOH at room temperature or under reflux conditions, the yields of the desired product, (E)-3-(4-chlorophenyl)-2-(1H-tetrazol-5-yl) acrylonitrile, were very low. This underscores the necessity of employing a catalyst to enhance the reaction efficiency. When the model reaction was performed under solvent-free conditions or under reflux conditions, the yields were extremely low and the reaction required prolonged reaction times (entries 1–4, Table 1). Notably, a substantial improvement in the yield of the desired product 5a was observed when the PABSA-Pr-PMO nanocatalyst (1) was employed even at remarkably short reaction times. Then, the catalyst was tested at room temperature with EtOH and water solvents. The yields obtained were low and the reaction required a long time (entries 5–6, Table 1). These findings demonstrate high catalytic activity of the nanocatalyst for the synthesis of the desired product 5a, which originates from very good dispersion of catalytically active sites and appropriate surface area of the PABSA-Pr-PMO nanocatalyst (1). The effect of different solvents including water, EtOH, their mixtures, toluene, dichloromethane, and DMF on the reaction rate was investigated. Solvents such as dichloromethane, H2O/EtOH and toluene increased the time and decreased the yield percentage. The yield was low in DMF solvents, which can be attributed to the reaction of DMF with azides at high temperatures (entries 7–14, Table 1). Both water and EtOH proved to be effective solvents, yielding excellent product 5a in comparable reaction times (entries 7, 13, Table 1). In addition, experiments were also conducted in the H2O/EtOH (1:1) and water solvents with a higher amount of catalyst loading. The obtained results results did not show any significant changes in yield and reaction time (entries 15–16, Table 1). Also, to determine the optimal stoichiometry of sodium azide (3), different amounts were compared and the best stoichiometry was selected as 1.2 mmol (entries 17–18, Table 1). Interestingly, the separation of catalyst 1 was found to be significantly more efficient in EtOH compared to water, primarily due to the higher solubility of the desired 1H-tetrazole products at elevated temperatures. This facilitated simple separation of the heterogeneous catalyst 1. Consequently, the optimal reaction conditions were identified as 5.0 mg of PABSA-PrPMO (1) loading in EtOH under reflux conditions, which were employed in subsequent experiments.
Table 1 Optimization of conditions in the model reaction of malononitrile (2), NaN3 (3) and 4-chlorobenzaldehyde (4a) under different conditions for the synthesis of 1H-tetrazole 5aa
In the next step of our research, the optimized conditions were extended to a variety of other aromatic aldehydes 4b–s to explore the scope of the reaction for the synthesis of diverse 1H-tetrazole derivatives using the PABSA-Pr-PMO nanocatalyst (1). A variety of aromatic carbocyclic and heterocyclic aldehydes were employed under optimized conditions, affording high to excellent yields of the desired products 5a–s within short reaction times. Notably, aromatic aldehydes with carbocyclic rings bearing electron-withdrawing groups 4a–e and 4g, and electron-deficient heterocycle 4i reacted efficiently with high yields to produce the corresponding 1H-tetrazole derivatives (entries 1–6, 8 and 9, Table 2). Furthermore, aromatic aldehydes with electron-donating groups with a carbocyclic ring 4f, 4k–o, and electron-rich heterocycles 4p–r as well as cinnamaldehyde (4s), as an α,β-unsaturated compound, were also examined. Interestingly, no polymerization product was detected for these acid-sensitive substrates.91 Under the optimized conditions, they successfully afforded the corresponding 1H-tetrazole derivatives 5f and 5j–s with high efficiency (entries 6 and 10–19, Table 2). The reaction rate of the aforementioned aldehydes, particularly those bearing electron-donating groups, is typically higher compared to that of aldehydes with perfect electron-withdrawing groups, such as –NO2. The presence of electron-donating groupsincreases the electron density in the intermediates A to take part in the 1,3-dipolar cycloaddition, thus enhancing the formation rate of the corresponding 1H-tetrazole derivatives (Scheme 2). In general, the PABSA-Pr-PMO nanocatalyst (1) demonstrates its effectiveness in the synthesis of 1H-tetrazole derivatives by providing high to excellent yields (80–97%) in a relatively fast reaction time from 4 to 40 min.
Table 2 Synthesis of 5-substituted 1H-tetrazole derivatives 5a–s catalyzed by the PABSA-Pr-PMO nanomaterial (1)a
 |
| | Scheme 2 The proposed mechanism for the synthesis of tetrazole derivatives 5a–s catalyzed by the heterogeneous PABSA-Pr-PMO nanocatalyst (1). | |
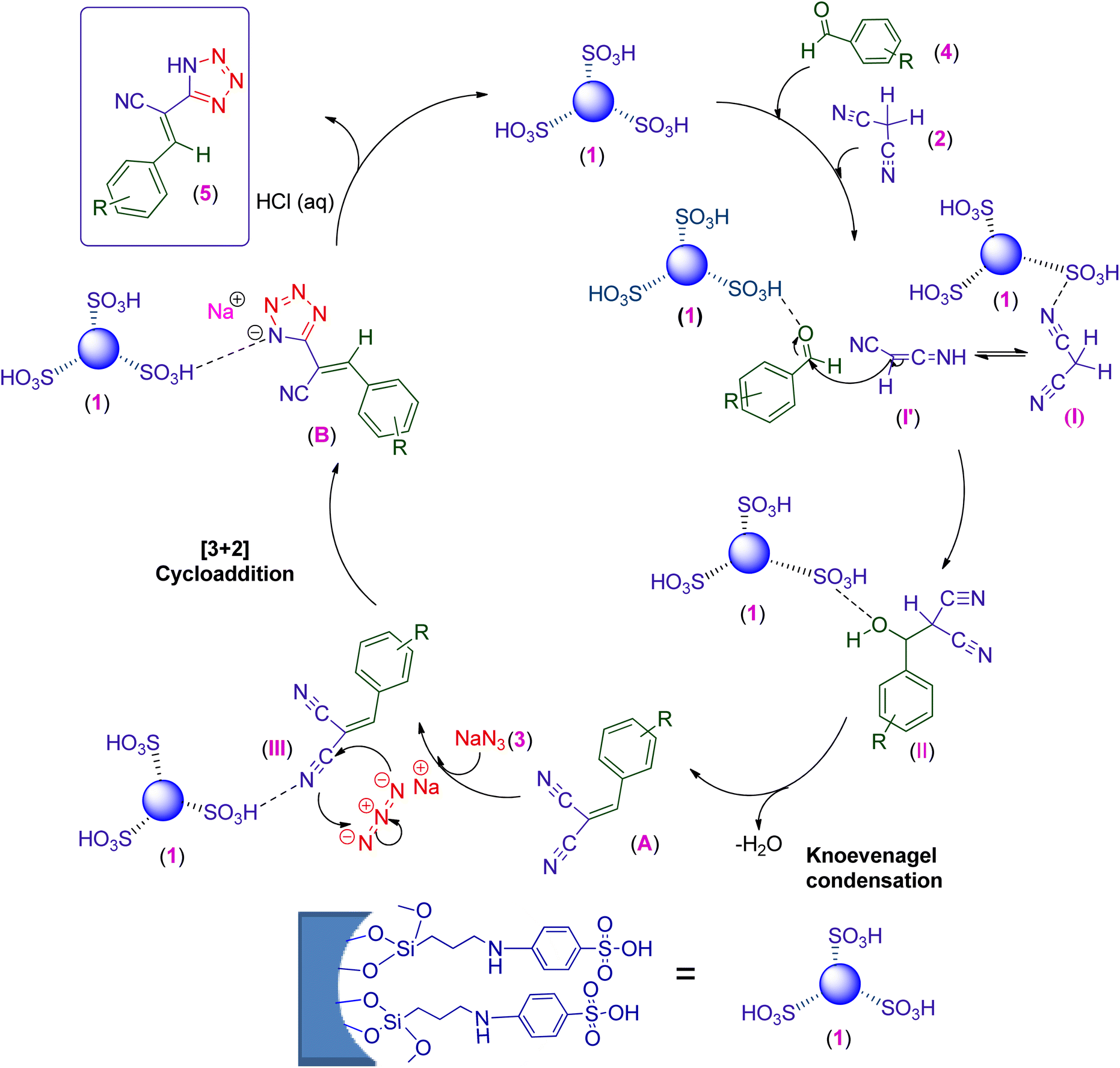
3.3. Proposed mechanism for the preparation of 5-substituted 1H-tetrazole derivatives catalyzed by the PABSA-Pr-PMO nanocatalyst (1)
A plausible mechanism for the synthesis of tetrazole derivatives catalyzed by PABSA-Pr-PMO (1) is shown in Scheme 2. Initially, a Knoevenagel condensation occurs, involving the nucleophilic addition of the malononitrile C=H acid (2) through intermediate (I′) to the carbonyl group of aromatic aldehydes 4 to afford intermediate (A) after dehydration of intermediate (II), all activated by the acidic PABSA-Pr-PMO nanocatalyst (1). The nitrogen atom of the nitrile group in intermediate A is also activated by the catalyst (1) for the concerted 1,3-dipolar cycloaddition with the azide ions. The click reaction of the intermediate A and the azide ion leads to the formation of 5-substituted 1H-tetrazole derivative sodium salt B. Finally, the addition of HCl (aq) produces the formation of the desired tetrazole derivatives.96,97,103
3.4. Comparison of the catalytic activity of PABSA-Pr-PMO (1) in the synthesis of tetrazole derivatives with other catalytic systems
The current protocol for synthesizing tetrazole 5a offers notable advantages compared to traditional methods, including high yields, avoidance of toxic solvents such as DMF,104–106 and a shorter reaction time as they have been compared and shown in Table 3. Additionally, the ease of catalyst separation simplifies purification, enhancing process efficiency and sustainability. These features make the current method superior due to its alignment with green chemistry principles, offering significant improvements in both environmental impact and practical application.
Table 3 Comparison of the catalytic efficiency of PABSA-Pr-PMO (1) with other catalytic systems
| Entry |
Catalyst |
Catalyst loading |
Reaction conditions |
Time (min) |
Yield (%) |
Reference |
| 1 |
Cu@APS-TDU-PMO |
30 mg |
Solvent-free/110 °C |
30–60 |
89–97 |
73
|
| 2 |
Fe3O4@fibroin-SO3H |
10 mol% |
Solvent-free/100 °C |
2–3 h |
76–86 |
103
|
| 3 |
Fe3O4@PMO–ICS–ZnO |
10 mg |
EtOH/reflux |
5–30 |
78–98 |
74
|
| 4 |
Pd/AlO(OH) NPs |
30 mg |
EtOH/95–100 °C |
3–5 h |
75–95 |
15
|
| 5 |
Fe3O4@BNPs-CPTMS-Chitosan-Pd(0) |
20 mg |
Solvent-free/70 °C |
30–65 |
93–97 |
98
|
| 6 |
Nano-Fe3O4 |
20 mol% |
Solvent-free/microwave |
35 |
82–92 |
93
|
| 7 |
Mg–Al LDH-CPTMS-Guanidine-Cu(II) |
30 mg |
DMF/110 °C |
15–23 |
82–97 |
104
|
| 8 |
NH–Cu(II)@MNP |
20 mg |
EtOH/reflux |
5–7 h |
0–92 |
107
|
| 9 |
Silica molybdic acid |
0.1 mol% |
H2O/MW/70 °C |
15 |
89–91 |
97
|
| 10 |
Cu-MCM-41 |
500 mg |
DMF/130 °C |
720 |
72–95 |
105
|
| 11 |
BaWO4 |
0.26 mmol |
DMF/120 °C |
24 h |
5–93 |
106
|
|
12
|
PABSA-Pr-PMO (1)
|
5.0 mg
|
EtOH/80 °C
|
4–40
|
80–97
|
This work |
3.5 Examining the recyclability of the PABSA-Pr-PMO nanocatalyst (1) for the synthesis of 5-substituted 1H-tetrazole derivative 5a
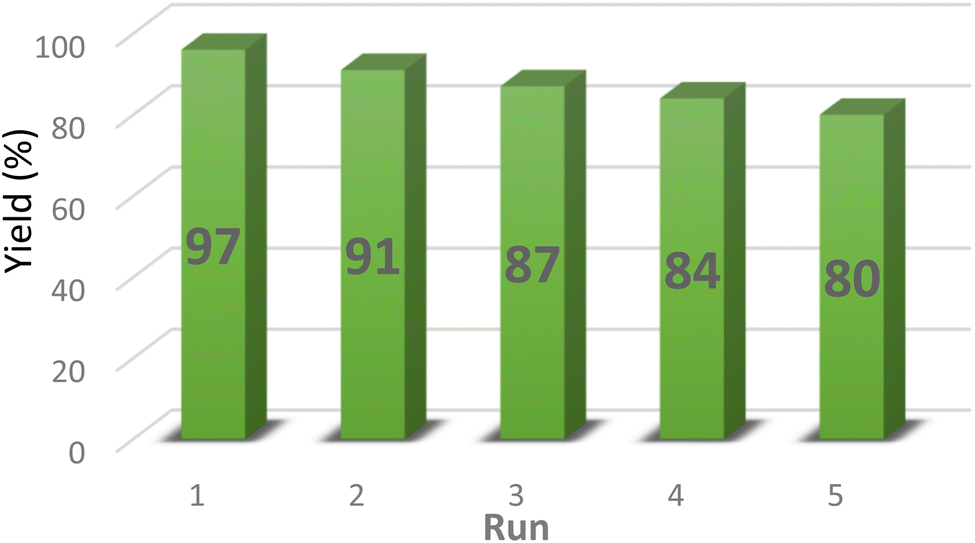
In this part of our study, we explored the reusability of the PABSA-functionalized periodic mesoporous organosilica catalyst (PABSA-Pr-PMO, 1) in the synthesis of (E)-3-(4-chlorophenyl)-2-(1H-tetrazol-5-yl) acrylonitrile 5a under optimal conditions in consecutive runs. The nanocatalyst (1) was simply separated from the reaction mixture using filtration. To ensure its readiness for reuse, the catalyst was sequentially dispersed in EtOH and water for 15 min three times, followed by drying at 60 °C for 30 min. As depicted in Fig. 9, the results indicate that over five consecutive cycles, there was only a slight decrease in reaction yield with no significant change in reaction time. These findings highlight the remarkable structural and functional stability of PABSA-Pr-PMO (1) in the synthesis of 5-substituted 1H-tetrazole derivatives, illustrating its significant potential for repeated use without substantial loss of catalytic efficiency. This stability not only underscores the robustness of the catalyst under various reaction conditions but also supports its practical application in scalable and sustainable chemical processes. Also, the FT-IR spectra of the fresh and recycled PABSA-Pr-PMO nanocatalyst (1) after the fifth cycle were compared. The results are summarized in Fig. 10. The obtained data indicate that the catalyst remained quite stable without significant changes in its structure (Fig. 10). Indeed, the catalytic activity of PABSA-Pr-PMO (1) decreases slightly after each run due to factors such as changes in the structure of the pores over time that can affect the diffusion of reactants into the active sites and the diffusion of products away from them.
 |
| | Fig. 9 Reusability data for the PABSA-Pr-PMO nanocatalyst (1) in the synthesis of 5a. | |
 |
| | Fig. 10 FT-IR spectra of the (a) fresh and (b) recovered PABSA-Pr-PMO nanocatalyst (1) after the fifth run. | |
4 Conclusions
In brief, a new periodic mesoporous organosilica functionalized with p-aminobenzenesulfonic acid (PABSA-Pr-PMO) was prepared through a convenient and environmentally friendly procedure. The PABSA-Pr-PMO nanomaterial shows good properties including high surface area, high pore volume, uniform pore size distribution, and excellent thermal stability. These properties are essential for showing appropriate Brønsted acid catalytic activity. The catalytic activity of PABSA-Pr-PMO solid acid was confirmed in the one-pot synthesis of tetrazole derivatives via a cascade condensation and concerted 1,3-cycloaddition reaction according to the principles of green chemistry. The PABSA-Pr-PMO catalyst facilitated efficient conversion of various aromatic and α,β-unsaturated aldehydes, malononitrile and sodium azide under low loading and provided the corresponding 5-substituted 1H-tetrazole derivatives in a short reaction time and excellent purity. Key advantages of this protocol include low catalyst loading, the use of green solvents such as EtOH or water instead of carcinogenic alternatives such as DMF, and short reaction times. Additionally, the easy separation and recyclability of the catalyst for at least five consecutive runs without significant loss of activity underscore its practical effectiveness. These attributes mutually highlight our method as a superior alternative to recently introduced procedures, offering a sustainable and efficient approach for the synthesis of tetrazole derivatives.
Data availability
The data supporting this article have been included as part of the ESI.†
Note added after first publication
This article replaces the version published on 19th May 2025, which due to a production error contained an instruction from the proofing process. This sentence has been removed, and all other text remains unchanged.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the financial support from The Research Council of Iran University of Science and Technology (IUST), Tehran, Iran (Grant No. 160/24293). We would also like to acknowledge the support of the Iran Nanotechnology Initiative Council (INIC), Iran.
References
-
P. V. Tawade and K. L. Wasewar, in Environmental Applications of Microbial Nanotechnology, ed. P. Singh, V. Kumar, M. Bakshi, C. M. Hussain and M. Sillanpää, Elsevier, 2023, pp. 43–64, DOI:10.1016/B978-0-323-91744-5.00015-1, .
- N. Shahcheraghi, H. Golchin, Z. Sadri, Y. Tabari, F. Borhanifar and S. Makani, 3 biotech, 2022, 12, 65 CrossRef PubMed.
-
V. Chaudhary, S. Vasistha and M. P. Rai, in Emerging Sustainable Nanomaterials for Biomedical Applications, ed. S. Garg, A. Chandra and S. Sagadevan, Springer Nature Switzerland, Cham, 2024, DOI:10.1007/978-3-031-63961-6_10, pp. 243–268.
- R. Holghoomi and A. Hosseinzadeh Colagar, Inorg. Chem. Commun., 2024, 167, 112753 CrossRef CAS.
-
R. Paliwal and S. R. Paliwal, Nanomedicine, Nanotheranostics and Nanobiotechnology: Fundamentals and Applications, CRC Press, 2024 Search PubMed.
- J. Wang, W. Zhao, Z. Zhang, X. Liu, T. Xie, L. Wang, Y. Xue and Y. Zhang, Adv. Mater., 2024, 36, 2308915 CrossRef CAS PubMed.
- Y. Khan, H. Sadia, S. Z. Ali Shah, M. N. Khan, A. A. Shah, N. Ullah, M. F. Ullah, H. Bibi, O. T. Bafakeeh, N. B. Khedher, S. M. Eldin, B. M. Fadhl and M. I. Khan, Catalysts, 2022, 12, 1386 CrossRef CAS.
- R. Mo, T. Jiang, J. Di, W. Tai and Z. Gu, Chem. Soc. Rev., 2014, 43, 3595–3629 RSC.
- J. Kim and E. Franco, Curr. Opin. Biotechnol., 2020, 63, 135–141 CrossRef CAS PubMed.
- R. Athavale, S. Gardi, F. Choudhary, D. Patil, N. Chandan and P. More, Appl. Catal., A, 2024, 669, 119505 CrossRef CAS.
- M. D. Argyle and C. H. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef CAS.
- B. Changmai, C. Vanlalveni, A. P. Ingle, R. Bhagat and S. L. Rokhum, RSC Adv., 2020, 10, 41625–41679 RSC.
- P. Patil, S. Kadam, D. Patil and P. More, Catal. Commun., 2022, 170, 106500 CrossRef CAS.
- A. Maurya, U. K. Patel, S. Kumar and A. Agarwal, RSC Adv., 2024, 14, 29505–29517 RSC.
- E. Aydinli, Z. A. Hameed, H. Goksu and S. Adem, J. Chem. Sci., 2024, 136, 22 CrossRef CAS.
- H. FaniMoghadam, M. G. Dekamin and N. Rostami, Res. Chem. Intermed., 2022, 48, 3061–3089 CrossRef CAS.
- B. A. D. Neto, R. O. Rocha and M. O. Rodrigues, Molecules, 2022, 27, 132 CrossRef CAS PubMed.
- T. Zarganes-Tzitzikas, A. L. Chandgude and A. Dömling, Chem. Rec., 2015, 15, 981–996 CrossRef CAS PubMed.
- H. A. Younus, M. Al-Rashida, A. Hameed, M. Uroos, U. Salar, S. Rana and K. M. Khan, Expert Opin. Ther. Pat., 2021, 31, 267–289 CrossRef CAS PubMed.
- S. Arade, S. Balgude, J. Kounsalye, D. Hingane and P. More, J. Mater. Sci.: Mater. Electron., 2024, 35, 1636 CrossRef CAS.
- S.-H. Wu, C.-Y. Mou and H.-P. Lin, Chem. Soc. Rev., 2013, 42, 3862–3875 RSC.
- R. K. Kankala, Y. H. Han, J. Na, C. H. Lee, Z. Sun, S. B. Wang, T. Kimura, Y. S. Ok, Y. Yamauchi and A. Z. Chen, Adv. Mater., 2020, 32, 1907035 CrossRef CAS PubMed.
- H. Li, X. Chen, D. Shen, F. Wu, R. Pleixats and J. Pan, Nanoscale, 2021, 13, 15998–16016 RSC.
- P. Van Der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- K. B. Baharudin, Y. H. Taufiq-Yap, J. Hunns, M. Isaacs, K. Wilson and D. Derawi, Microporous Mesoporous Mater., 2019, 276, 13–22 CrossRef CAS.
- C. Pirez, J.-M. Caderon, J.-P. Dacquin, A. F. Lee and K. Wilson, ACS Catal., 2012, 2, 1607–1614 CrossRef CAS.
- J. Dhainaut, J.-P. Dacquin, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 296–303 RSC.
- V. Poscher and Y. Salinas, Materials, 2020, 13, 3668 CrossRef CAS PubMed.
- R. Narayan, U. Y. Nayak, A. M. Raichur and S. Garg, Pharmaceutics, 2018, 10, 118 CrossRef CAS PubMed.
- J. G. Croissant, Y. Fatieiev, A. Almalik and N. M. Khashab, Adv. Healthc. Mater., 2018, 7, 1700831 CrossRef PubMed.
- S. Chinnathambi and F. Tamanoi, Pharmaceutics, 2020, 12, 890 CrossRef CAS PubMed.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Int. J. Nanosci. Nanotechnol., 2006, 6, 265–288 CrossRef CAS PubMed.
- W. J. Hunks and G. A. Ozin, J. Mater. Chem., 2005, 15, 3716–3724 RSC.
- A. Zebardasti, M. G. Dekamin, E. Doustkhah and M. H. N. Assadi, Inorg. Chem., 2020, 59, 11223–11227 CrossRef CAS PubMed.
- M. G. Dekamin, E. Arefi and A. Yaghoubi, RSC Adv., 2016, 6, 86982–86988 RSC.
- D. M. Fernandes, M. Nunes, B. Bachiller-Baeza, I. Rodríguez-Ramos, A. Guerrero-Ruiz, C. Delerue-Matos and C. Freire, J. Solid State Electrochem., 2017, 21, 1059–1068 CrossRef CAS.
- M. A. Lourenço, P. Ferreira and J. R. Gomes, Phys. Chem. Chem. Phys., 2018, 20, 16686–16694 RSC.
- Q. Yang, J. Liu, L. Zhang and C. Li, J. Mater. Chem., 2009, 19, 1945–1955 RSC.
- E. Romaní-Cubells, S. Martínez-Erro, V. Morales, A. Chocarro-Calvo, J. M. García-Martínez, R. Sanz, C. García-Jiménez and R. A. García-Muñoz, J. Nanobiotechnology, 2024, 22, 249 CrossRef PubMed.
- P. R. Salekdeh, L. Ma’mani, J. Tavakkoly-Bazzaz, H. Mousavi, M. H. Modarressi and G. H. Salekdeh, J. Nanobiotechnology, 2021, 19, 1–16 CrossRef PubMed.
- A. H. Khalbas, T. M. Albayati, N. S. Ali and I. K. Salih, S. Afr. J. Chem. Eng., 2024, 50, 261–280 Search PubMed.
- C. Trayford and S. van Rijt, Biomater. Sci., 2024, 12, 5450–5467 RSC.
- Y. Zhang, X. Lin, X. Chen, W. Fang, K. Yu, W. Gu, Y. Wei, H. Zheng, J. Piao and F. Li, Int. J. Nanomed., 2024, 19, 5859 CrossRef PubMed.
- A. M. Kaczmarek, M. Suta, H. Rijckaert, A. Abalymov, I. Van Driessche, A. G. Skirtach, A. Meijerink and P. Van Der Voort, Adv. Funct. Mater., 2020, 30, 2003101 CrossRef CAS.
- N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC.
- Q. Ouyang, G. Li, X. Zhang, X. Zhao, S. Fu and L. Li, Small, 2024, 20, 2305793 CrossRef CAS PubMed.
- S. S. Park, M. Santha Moorthy and C.-S. Ha, N. P. G. Asia .Mater., 2014, 6, e96 CrossRef CAS.
- Y. Hu, K. Qian, P. Yuan, Y. Wang and C. Yu, Mater. Lett., 2011, 65, 21–23 CrossRef CAS.
- N. Hao, H. Wang, P. A. Webley and D. Zhao, Microporous Mesoporous Mater., 2010, 132, 543–551 CrossRef CAS.
- X. Zhou, S. Qiao, N. Hao, X. Wang, C. Yu, L. Wang, D. Zhao and G. Q. Lu, Chem. Mater., 2007, 19, 1870–1876 CrossRef CAS.
- M. Mandal and M. Kruk, J. Mater. Chem., 2010, 20, 7506–7516 RSC.
- N. Muramoto, T. Sugiyama, T. Matsuno, H. Wada, K. Kuroda and A. Shimojima, Nanoscale, 2020, 12, 21155–21164 RSC.
- R. Cheng, R. Zou, L. He, L. Liu, C. Cao, X. Li, X. Guo and J. Xu, Energy and Fuels, 2021, 35, 10562–10574 CrossRef CAS.
-
S. C. Nunes, P. Almeida and V. de Zea Bermudez, The Sol-Gel Handbook, 2015, pp. 575–604 Search PubMed.
- J. Tripathi, M. Gupta, A. Yadav, K. Waghmode and P. More, Res. Chem. Intermed., 2024, 50, 1645–1660 CrossRef CAS.
- I. Yellapurkar, S. Bhabal, M. M. V. Ramana, K. Jangam, V. Salve, S. Patange and P. More, Res. Chem. Intermed., 2021, 47, 2669–2687 CrossRef CAS.
- P. Patil, S. Kadam, D. Patil and P. More, J. Mol. Liq., 2022, 345, 117867 CrossRef CAS.
- M. Sudharsan, S. Subramanian, A. J. Amali and D. Suresh, ChemistrySelect, 2020, 5, 6131–6140 CrossRef CAS.
- M. Valipour, S. Habibzadeh and M. Taherimehr, J. Indian Chem. Soc., 2024, 101, 101382 CrossRef CAS.
- E. A. Popova, R. E. Trifonov and V. A. Ostrovskii, Russ. Chem. Rev., 2019, 88, 644 CrossRef CAS.
- Z. Fülöp, P. Bana, M. Temesvári, J. Barabás, Z. Kazsu, Z. Béni, I. Greiner and J. Éles, Org. Process Res. Dev., 2024, 28(9), 3685–3690 CrossRef.
- S. Swami, S. N. Sahu and R. Shrivastava, RSC Adv., 2021, 11, 39058–39086 RSC.
- A. Maurya and A. Agrawal, Mini-Rev. Med. Chem., 2024, 24, 176–195 CrossRef CAS PubMed.
- M. Arshad, A. R. Bhat, S. Pokharel, J.-E. Kim, E. J. Lee, F. Athar and I. Choi, Eur. J. Med. Chem., 2014, 71, 229–236 CrossRef CAS PubMed.
- N. Dhiman, K. Kaur and V. Jaitak, Bioorg. Med. Chem., 2020, 28, 115599 CrossRef CAS PubMed.
- S. Leyva-Ramos and J. Cardoso-Ortiz, Curr. Org. Chem., 2021, 25, 388–403 CrossRef CAS.
- R. Mittal, A. Kumar and S. K. Awasthi, RSC Adv., 2021, 11, 11166–11176 RSC.
- Q. Qiu, J. Song, H. Zheng, M. Shahriari, A. F. El-kott, A. G. Alkhathami and K. Morsy, J. Organomet. Chem., 2025, 1023, 123423 CrossRef CAS.
- M. Norouzi, N. Noormoradi and M. Mohammadi, Nanoscale Adv., 2023, 5, 6594–6605 RSC.
- Z. Heidarnezhad, A. Ghorbani-Choghamarani and Z. Taherinia, Nanoscale Adv., 2024, 6, 4360–4368 RSC.
- M. Karegar and M. M. Khodaei, ChemistrySelect, 2022, 7, e202201138 CrossRef CAS.
- J. Safaei-Ghomi, S. Paymard-Samani, Z. Zahraie and H. Shahbazi-Alavi, Nanomed. Res. J., 2019, 4, 91–100 CAS.
- E. Valiey and M. G. Dekamin, Sci. Rep., 2022, 12, 18139 CrossRef CAS PubMed.
- S. Safapoor, M. G. Dekamin, A. Akbari and M. R. Naimi-Jamal, Sci. Rep., 2022, 12, 10723 CrossRef CAS PubMed.
- J. Safaei-Ghomi, S. Paymard-Samani, S. Zahedi and H. Shahbazi-Alavi, Z. Naturforsch, 2015, 70, 819–828 CrossRef CAS.
- Z. Hajizadeh, F. Hassanzadeh-Afruzi, D. F. Jelodar, M. R. Ahghari and A. Maleki, RSC Adv., 2020, 10, 26467–26478 RSC.
- S. Y. Priyanka, P. Rana, R. Bandichhor, A. Srivastava and R. K. Sharma, J. Chem. Eng., 2024, 496, 153995 CrossRef CAS.
- R. Jahanshahi and B. Akhlaghinia, RSC Adv., 2015, 5, 104087–104094 RSC.
- M. M. Maseer, T. Kikhavani and B. Tahmasbi, Nanoscale Adv., 2024, 6, 3948–3960 RSC.
- C.-Y. Hsu, A. R. AlBajalan, S. A. Awad, M. Suliman, N. Juraev, C. Rodriguez-Benites, H. AlMohamadi and A. J. Kadhim, Nanoscale Adv., 2024, 6, 6408–6419 RSC.
- E. Valiey and M. G. Dekamin, RSC Adv., 2022, 12, 437–450 RSC.
- E. Valiey and M. G. Dekamin, Nanoscale Adv., 2022, 4, 294–308 RSC.
- A. Yaghoubi, M. G. Dekamin and B. Karimi, Catal. Lett., 2017, 147, 2656–2663 CrossRef CAS.
- Z. Zhang, H. Duan, S. Li and Y. Lin, Langmuir, 2010, 26, 6676–6680 CrossRef CAS PubMed.
- A. Hassankhani, B. Gholipour, S. Rostamnia, E. Zarenezhad, N. Nouruzi, T. Kavetskyy, R. Khalilov and M. Shokouhimehr, Appl. Organomet. Chem., 2021, 35, e6346 CrossRef CAS.
- A. Akbari, M. G. Dekamin, A. Yaghoubi and M. R. Naimi-Jamal, Sci. Rep., 2020, 10, 10646 CrossRef CAS PubMed.
- M. Sam, M. G. Dekamin and Z. Alirezvani, Sci.Rep., 2021, 11, 2399 CrossRef CAS PubMed.
- A. Yaghoubi, M. G. Dekamin, E. Arefi and B. Karimi, J. Colloid Interface Sci., 2017, 505, 956–963 CrossRef CAS PubMed.
- K. Zhang, L.-L. Xu, J.-G. Jiang, N. Calin, K.-F. Lam, S.-J. Zhang, H.-H. Wu, G.-D. Wu, B. Albela and L. Bonneviot, J. Am. Chem. Soc., 2013, 135, 2427–2430 CrossRef CAS PubMed.
- A. T. Kal-Koshvandi, M. R. Ahghari and A. Maleki, NJC, 2020, 44, 12619–12632 RSC.
- M. G. Dekamin, J. Mokhtari and M. R. Naimi-Jamal, Catal. Commun., 2009, 10, 582–585 CrossRef CAS.
- C. Behloul, M. Benlahrech, F. Foubelo, C. Nájera and M. Yus, Synthesis, 2018, 50, 3430–3435 CrossRef CAS.
- P. Akbarzadeh, N. Koukabi and E. Kolvari, Res. Chem. Intermed., 2019, 45, 1009–1024 CrossRef CAS.
- Z. Hajizadeh, F. Hassanzadeh-Afruzi, D. F. Jelodar, M. R. Ahghari and A. Maleki, RSC Adv., 2020, 10, 26467–26478 RSC.
- Z. N. Tisseh, M. Dabiri, M. Nobahar, H. R. Khavasi and A. Bazgir, Tetrahedron, 2012, 68, 1769–1773 CrossRef CAS.
- S. Bondarian, M. G. Dekamin, E. Valiey and M. R. Naimi-Jamal, RSC Adv., 2023, 13, 27088–27105 RSC.
- N. Ahmed and Z. N. Siddiqui, RSC Adv., 2015, 5, 16707–16717 RSC.
- M. Khodamorady and K. Bahrami, ChemistrySelect, 2019, 4, 8183–8194 CrossRef CAS.
- M. Bakherad, R. Doosti, A. Keivanloo, M. Gholizadeh and K. Jadidi, J. Iran. Chem. Soc., 2017, 14, 2591–2597 CrossRef CAS.
- A. El-Sewedy, E. A. El-Bordany, N. F. H. Mahmoud, K. A. Ali and S. K. Ramadan, Sci. Rep., 2023, 13, 17869 CrossRef CAS PubMed.
- H. Tourani, M. R. Naimi-Jamal and M. G. Dekamin, ChemistrySelect, 2018, 3, 8332–8337 CrossRef CAS.
- A. Nanda, N. Kaur, M. Kaur, F. M. Husain, H. Han, P. K. Bhowmik and H. S. Sohal, Molecules, 2024, 29, 4339 CrossRef CAS PubMed.
- A. Nouri Parouch, N. Koukabi and E. Abdous, Res. Chem. Intermed., 2020, 46, 3295–3310 CrossRef CAS.
- M. Ara, H. Ghafuri and N. Ghanbari, Colloid Interface Sci. Commun., 2023, 53, 100704 CrossRef CAS.
- Z. Du, C. Si, Y. Li, Y. Wang and J. Lu, Int. J. Mol. Sci., 2012, 13, 4696–4703 CrossRef CAS PubMed.
- J. He, B. Li, F. Chen, Z. Xu and G. Yin, J. Mol. Catal. A:Chem., 2009, 304, 135–138 CrossRef CAS.
- X. Yuan, Z. Wang, Q. Zhang and J. Luo, RSC Adv., 2019, 9, 23614–23621 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Mohammad G.
Dekamin
,
Mohammad G.
Dekamin