
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison between mechanochemical and solution synthesis of Zn and Cu complexes containing pyridine and p-halogen substituted benzoates†
Giorgio
Cagossi
 ab,
Paolo P.
Mazzeo
ac,
Alessia
Bacchi
ac and
Paolo
Pelagatti
*ab
ab,
Paolo P.
Mazzeo
ac,
Alessia
Bacchi
ac and
Paolo
Pelagatti
*ab
aDepartment of Chemical Science, Live Science and Environmental Sustainability, University of Parma, Parco Area delle Scienze 17/A, 43124 Parma, Italy. E-mail: paolo.pelagatti@unipr.it
bInteruniversity Consortium of Chemical Reactivity and Catalysis (CIRCC), Via Celso Ulpiani 27, 70126 Bari, Italy
cBiopharmanet-tec, Parco Area delle Scienze 27/A, 43124 Parma, Italy
First published on 6th March 2025
Abstract
Mechanochemistry can be an essential tool for coordination chemistry, demonstrating significant advantages over solution protocols, enabling highly selective, efficient, and rapid syntheses with conversion of the reagents achieved within minutes of grinding. The mechanochemical synthesis of heteroleptic Zn(II) and Cu(II) complexes containing non-chelating ligands like pyridine and p-halogen-substituted benzoates showcased the potential of this technique, with a detailed comparison to solution-based synthetic methods. Structural analyses via X-ray diffraction confirm that the crystalline phases produced mechanochemically are identical to those obtained in solution. The synthesis of anhydrous complexes under dry mechanochemical conditions was also achieved without specialized equipment, highlighting the versatility of this approach even for moisture-sensitive compounds.
Introduction
The mechanochemical synthesis of molecular metal complexes is nowadays well documented.1–3 Its development has been mainly triggered by the advantages offered by mechanochemistry compared to the more conventional solution based protocols, related to the reduced amount of solvent used during the grinding process and the more favoured kinetics. These aspects positively impact the sustainability profile of the mechanochemical approach.4,5 However, the perception that a “conventional” synthetic chemist has towards a mechanochemical reaction is often that of a highly heterogeneous system, where the correct combination of the reagents to give the desired product occurs more by chance than based on a well-defined reaction pathway. For this reason, it is important that synthetic mechanochemists insist on demonstrating that mechanochemistry can lead to clean, fast, and selective reactions. Examples dealing with the synthesis of metal complexes containing only monodentate ligands are particularly relevant in this regard. In fact, the lack of chelation can in principle lead to different stoichiometries and coordination geometries, especially in the presence of coordinatively labile metal ions. This is particularly true in the case of Zn2+ ions, whose d10 electronic configuration leads to a significant structural variability owing to the absence of a dominant ligand field stabilization energy.6 With the idea of testing the selectivity of mechanochemistry in the synthesis of coordination metal complexes, we directed our attention to heteroleptic Zn2+ complexes containing simple ligands, such as benzoic acids and pyridine. Although the selected ligands must be considered “very simple”, the absence of chelators may in principle lead to different coordination polyhedra, like a tetrahedron, a trigonal bipyramid or an octahedron. On the other hand, the construction of a heteroleptic complex is in itself a sophisticated process, since two ligands must self-assemble around the metal with a well-defined stoichiometry, likely through a multistep process. Moreover, the innate coordination isomerism of the benzoate anion, such as κ1-monodentate, κ2-chelating or μ-bridging, makes the final coordination geometry even more unpredictable, thus making the mechanochemical synthesis of Zn-complexes even more challenging.In this regard, it is worth noting that most of the mechanochemically synthesized molecular Zn-complexes reported in the literature are homoleptic complexes containing chelating ligands,7,8 corresponding to bis-chelate complexes9 or mono-chelate complexes where the coordination environment of the metal is completed by coordinated anions.10,11
As part of our ongoing research program aimed at developing sustainable synthetic approaches for the synthesis of coordination compounds,12 hybrid materials,13 and co-crystals,14,15 here we report the mechanochemical synthesis of a series of Zn2+ complexes obtained by grinding Zn(OAc)2·2H2O with three different 4-halogenated benzoic acids (4-Cl, 4-Br or 4-I-benzoic acid) and pyridine. The basic salt and the 4-halogenated benzoic acids were selected to assure a facile deprotonation of the acidic ligands with consequent elimination of the acetate anion as acetic acid, thus avoiding the use of additional bases. Pyridine was preferred over substituted pyridines to avoid the presence of additional coordination donors.
A search on the CCDC database restricted to the structural fragment Zn(py)(4-X-C6H4COO)2 (X = Cl, Br and I) returned only one structure corresponding to the tetrahedral complex Zn(py)2(4-Cl-C6H4COO)2, where the two carboxylates are κ1-monodentate.16 No structures were found with X = Br or I. For this reason, the reactions involving 4-Br and 4-I-benzoic acid were also conducted in solution with the aim to isolate X-ray quality single crystals for structural characterization. This parallel approach allows for a direct comparison of the results obtained for the same reactions conducted in solution and by grinding, an aspect which is always of great interest among the mechanochemistry community. Finally, to extend the scope of the approach, the same reactions were conducted using Cu(OAc)2·H2O in place of the Zn salt. For the copper complexes, several structures can be found in the CCDC database, derived from reactions carried out in solution. For all three carboxylate ligands, hydrate pentacoordinate complexes of formula Cu(py)2(4-X-C6H4COO)2(H2O) (X = Cl, Br and I) are known.17,18 With X = Br, an anhydrous complex of formula Cu(py)2(4-X-C6H4COO)2 is also known.18 The hydrate complexes form polar crystals, useful for optical second-order harmonic generation.17 This structural knowledge allows for a quick structural check of the ground materials by powder X-ray diffraction analysis, making an easy determination of the selectivity control exerted by mechanochemistry possible.
Experimental
Materials and methods
All the reagents were of high purity and used as received if not otherwise indicated. Solvents were dried over activated 3 Å molecular sieves under nitrogen.Dehydration of Zn(OAc)2·2H2O was carried out by heating at 120 °C for 24 h. The solvents used as LAG (Liquid Assisted Grinding) agents were used as received. All the syntheses were carried out with a Retsch MM400 ball mill, using a stainless-steel jar equipped with balls of the same material.
Crystallography
Crystal structures were obtained by collecting the diffracted intensities on suitable single crystals with a Photon III 2D detector on a Bruker D8 Venture diffractometer equipped with a kappa goniometer and an Oxford Cryostream. Data collections were performed by using microfocused Mo Kα radiation (λ = 0.71073 Å). Lorentz polarization and absorption correction were applied, while data reduction was performed using APEX v5 software. All structures were solved by direct methods using SHELXT19 and refined by using full-matrix least-squares on all F2 using SHELXL20 as implemented in Olex2,21 using anisotropic thermal displacement parameters for all non-hydrogen atoms. Hydrogen atoms were added in calculated positions. Crystal data and structure refinement parameters are reported in the ESI.† The cif files of complexes 1s, 2s, 3s and 8s have been deposited at the Cambridge Crystallographic Data Centre, with ref. codes 2411964–2411967. These data can be obtained free of charge by using the joint CCDC and FIZ Karlsruhe Access Structure service.PXRD data were collected in the Bragg–Brentano geometry with Cu Kα radiation on a Rigaku SmartLab XE diffractometer equipped with a solid-state Hypix3000 2D detector. The samples were placed on a zero-background sample holder with a scan rate of 10° min−1 (5° ≤ 2θ ≤ 50°). A length-limiting slit of 15 mm was used as a compromise to improve X-ray flux over the sample; 5° Soller slits allowed improvement of the peak profile and limited the overlapping of reflections. Quantitative phase analysis was conducted by Rietveld refinement, using GSAS-II software22 and yields were calculated based on the weight percentage of complex 2s relative to the total solid content in the reaction mixture (see the ESI†). VT-PXRD measurements were carried out on a Rigaku SmartLab XE diffractometer equipped with an Anton-Paar TTK600 nonambient chamber with a flat copper sample holder. Data were collected in the Bragg–Brentano geometry under isothermal conditions, letting the sample thermalize before starting the data acquisition. A firing profile similar to that used for the calorimetric analyses was applied for consistency.
Spectroscopy
FT-IR spectra were recorded by means of a PerkinElmer Spectrum Two FT-IR spectrophotometer coupled with a PerkinElmer UATR accessory and diamond crystal plate in the range of 400–4000 cm−1.Thermal analyses
TGA analyses were performed with a PerkinElmer TGA 8000 instrument (mass of the sample: 1–3 mg) by means of a Pt crucible under air flux (30 mL min−1) in the temperature range of 30–500 °C at 10 °C min−1. Higher temperatures were not applied to avoid possible crucible damage due to the high metal content of the samples.Elemental analyses were conducted by means of a Thermo Fisher Flash Smart instrument, with gas-chromatographic separation.
Synthesis
Solution syntheses of the hydrate Zn complexes
Yield: 63%. Elemental analysis calcd. for C24H20Cl2N2O5Zn (found): C 52.12 (52.13), H 3.65 (3.69), N 5.07 (5.11)%. FTIR (cm−1): 3182, 1587, 1540, 1486, 1400, 1280, 1219, 1163, 1140, 1088, 1046, 1013, 854, 771, 689, 616, 560, 522, 471, 423.
Yield: 82%. Elemental analysis calcd. for C24H20Br2N2O5Zn (found): C 44.93 (45.25), H 3.14 (3.26), N 4.37 (4.72)%. FTIR (cm−1): 3357, 3069, 1585, 1535, 1486, 1442, 1411, 1394, 1275, 1216, 1169, 1138, 1091, 1067, 1040, 1009, 853, 819, 768, 757, 713, 697, 627, 560, 485, 462, 422.
Yield: 78%. Elemental analysis calcd. for C24H20I2N2O5Zn (found): C 39.19 (39.68), H 2.74 (2.85), N 3.81 (3.81)%. FTIR (cm−1): 3071, 1579, 1551, 1486, 1442, 1393, 1217, 1172, 1135, 1069, 1039, 1004, 952, 855, 824, 761, 696, 625, 455, 421.
Synthesis of the anhydrous complexes [Zn(py)2(4-X-C6H4COO)2] (X = Cl, Br), 7s–8s
Anhydrous zinc acetate (91.5 mg, 0.5 mmol) and p-X-benzoic acid (1 mmol, 156 mg for X = Cl, 201 mg for X = Br) were mixed in 10 mL of dry THF under a constant flow of N2, until complete dissolution. Then excess anhydrous pyridine (500 μL, 6.2 mmol) was added. The mixture was refluxed for 3 hours under an inert N2 atmosphere and solvent was completely removed under vacuum. The product was washed with three portions of cold n-hexane (1-2mL), weighed and characterized.Yield: 45%. Elemental analysis calcd. for C24H18Br2N2O4Zn (found): C 46.23 (46.13), H 2.91 (2.70), N 4.49 (4.13)%. FTIR (cm−1): 3061, 2925, 1716, 1608, 1585, 1564, 1479, 1487, 1447, 1393, 1354, 1275, 1242, 1215, 1163, 1129, 1087, 1064, 1045, 1008, 964, 852, 841, 765, 692, 651, 640, 562, 484, 465, 421.
Conversion of anhydrous 5s into hydrate 2s
A small aliquot of the anhydrous complex 5s was placed in a 10 mL glass vial and dissolved in dichloromethane (5 mL). The vial was left open and after 2 days single crystals suitable for diffraction were collected, washed with n-hexane and characterized by means of FT-IR ATR and SCXRD, confirming the conversion to the hydrate complex 2s.VT-PXRD of hydrate 2m
Variable temperature powder X-ray diffraction (VT-PXRD) analysis was performed on complex 2m using a Rigaku SmartLab XE diffractometer equipped with an Anton-Paar TTK600 non-ambient chamber. The sample was placed on a flat copper sample holder and heated to each temperature step under isothermal conditions, allowing the sample to thermalize for 1 minute before acquiring the diffraction pattern. Measurements were collected at the following temperature steps: 25 °C, 50 °C, 70 °C, 80 °C, 90 °C, 100 °C, 110 °C, 120 °C, and 130 °C, and finally cooled back to 25 °C to monitor structural reversibility.Results and discussion
A structural search conducted on the Cambridge Crystallographic Data Centre (CCDC) based on the fragment [Zn(py)(4-X-C6H4COO)] with X = Cl, Br and I, gave back only one structure, corresponding to the complex [Zn(py)2(4-Cl-C6H4COO)2].16 Here Zn is tetrahedrally surrounded by two pyridines and two κ1-monodentate carboxylate ligands. Unfortunately, the synthetic details are not clearly reported in the original paper, although it appears that an initial grinding of ZnCl2 and 4-Cl-benzoic acid was followed by the addition of pyridine. To initiate our study, we then tried to establish robust protocols for the isolation of well-defined complexes. First, we reacted Zn(OAc)2·2H2O with the three benzoic acids and pyridine under identical experimental conditions, with the aim to isolate crystalline materials suitable for single crystal X-ray analysis (Scheme 1, path a). The knowledge of the crystalline structure of possible target products is of fundamental importance when performing new mechanochemical reactions intended to lead to a precise crystalline phase. | ||
| Scheme 1 – The different synthetic paths adopted in this work to obtain zinc complexes: (a) and (c) solution syntheses for complexes 1s–3s and 7s–8s; (b) and (d) mechanochemical syntheses for complexes 1m–3m and 7m–8m (see Experimental for details). | ||
Nicely, crystals suitable for X-ray analysis were obtained for all three complexes (see Experimental for details).
In all cases, the structural resolution revealed the formation of octahedral complexes of formula [Zn(py)2(4-X-C6H4COO)2(H2O)] (1s–3s in Scheme 1, path a), where two pyridine ligands occupy the axial positions while the equatorial plane features a κ2-chelating and a κ1-coordinated benzoate ligand, and a water molecule (Fig. 1). The inclusion of water was not surprising, owing to the hydrate nature of the metal salt and the use of wet THF.
 | ||
| Fig. 1 Ortep drawing of complex 3s. All non-hydrogen atoms are shown as ellipsoids at the 50% probability level. H atoms (isotropically refined) are reported in the ball-and-stick style for the sake of clarity. Colour code: red = O, blue = N, grey = C, white = H, and purple = I. Complexes 1s and 2s are reported in the ESI.† | ||
All three complexes exhibit isostructural characteristics and crystallize in the monoclinic P21/n space group. The crystal packing features four complexes per unit cell. Within the structure, water molecules participate in hydrogen bonding, bridging two complex molecules. These interactions involve the uncoordinated oxygen of the κ1-coordinated COO group and one oxygen atom of the bridging COO group of a neighbouring molecule. This arrangement results in a hydrogen-bonded chain of complex molecules running along the b axis. Parallel chains are held together by dispersive contacts involving the aromatic rings (see the ESI†).
We then tried to reproduce the same products under mechanochemical conditions. It should be noted that the synthesis of the complexes under grinding must necessarily occur through several distinct steps, since a reactive event involving four different reagents in a single impact is highly unlikely, if not impossible. The feasibility of the mechanochemical approach was initially evaluated in the synthesis of 2m, grinding the solids for 30 min at 20 Hz using a single stainless steel ball. Under these conditions, the PXRD analysis evidenced the formation of the complex but also the incomplete conversion of 4-Br-benzoic acid and zinc acetate (Fig. 2). An optimization of the grinding conditions was then undertaken, including frequency, milling time, the number of grinding balls and the use of a LAG. The PXRD traces obtained for the different experiments are collected as shown in Fig. 2, together with the diffractograms of 4-Br-benzoic acid and Zn(OAc)2·2H2O. To conduct an estimation of the yields, the PXRD traces of the reaction products were analysed by Rietveld phase quantification (see the ESI†). To avoid possible decomposition of the complex during milling, two different times were chosen, 5 and 30 minutes, while three different frequencies were tested, corresponding to 20, 25 and 30 Hz. The effect of the use of one or two stainless steel balls was also evaluated, as well as the use of THF, used in the solution syntheses, in the LAG procedure.23,24
 | ||
| Fig. 2 Comparison between the PXRD traces of the mechanochemical syntheses of 2m under different grinding conditions; the calculated patterns of 2s (top) and of the reagents (bottom) are also reported. | ||
Irrespective of frequency, using a single ball, complex formation already occurred in the first 5 minutes of grinding, with yields not higher than 65%. No other side products were detected, indicating high selectivity towards the formation of the target complex. With a frequency of 20 or 25 Hz, the extension of the reaction time to 30 minutes did not have a substantial effect, whereas at 30 Hz a yield of about 82% was reached. Encouraged by this trend, retaining a frequency of 30 Hz, the milling time was extended to 120 minutes reaching a conversion of about 87%. The addition of small amounts of THF (η parameter = 0.3 μL mg−1) at different milling times (5, 30, and 120 minutes) had significant effects after 30 and 120 minutes, leading to yields of 92 and 97%, respectively. The results indicate an eight-fold reduction of the reaction time necessary to reach yields equivalent or higher than those obtained by solution synthesis.
Although THF has a beneficial effect on the final yield, the results indicate that the reaction also proceeds under solvent-free mechanochemical conditions, likely due to the presence of the liquid reagent pyridine and the water of crystallization of the metal salt.25 The introduction of a second ball into the reaction jar did not result in any notable increase in yield, suggesting that a single ball provides adequate energy transfer and mixing. Even testing an excess of reagents, particularly pyridine, did not further enhance the yield. In all reactions, the final product could be purified by recrystallization from THF, with a recrystallization yield of 85% (see the ESI†).
A similar behaviour was observed in the synthesis of the other two complexes containing 4-Cl and 4-I benzoic acids, with which the almost complete conversion of the reagents could be obtained by grinding at 30 Hz for 30 minutes (see the ESI†). Again, pure crystalline solids could be isolated after recrystallization in THF, yielding 1m and 3m with 81 and 87% yield, respectively (see the ESI†).
We then turned our attention to copper complexes. A search on the CCDC database returned four structures of interest for the fragment [Cu(py)(4-X-C6H4COO)2], with X = Cl, Br or I. Three structures correspond to pentacoordinate complexes of formula [Cu(py)2(4-X-C6H4COO)2(H2O)],17,18 indicated here as 4s–6s (Scheme 2). Although their stoichiometry is equivalent to those of complexes 1s–3s, in these cases the metal has a square pyramidal coordination, defined by two trans-κ1-monodentate carboxylate anions, two pyridines and a molecule of water. An alternative structure is represented by the octahedral complex [Cu(py)2(4-Br-C6H4COO)2], where two κ2-chelating carboxylates and two trans-arranged pyridines surround the metal centre (Scheme 2).18 The mechanochemical reactions were conducted under the same conditions applied for the standardized synthesis of 1m–3m (see Scheme 2).
 | ||
| Scheme 2 – The mechanochemical process used to synthesize the hydrated copper complexes 4m–6m (see Experimental for details) along with the potential alternative bis-chelate complex that does not form under mechanochemical conditions. | ||
In all cases, the mechanochemical products were exactly corresponding to the mononuclear pentacoordinate complexes 4s–6s obtained in solution, as inferred by the PXRD traces, with complete conversion of the reagents within 30 minutes of grinding (Fig. 3). No traces of the bis–chelate complex were found.
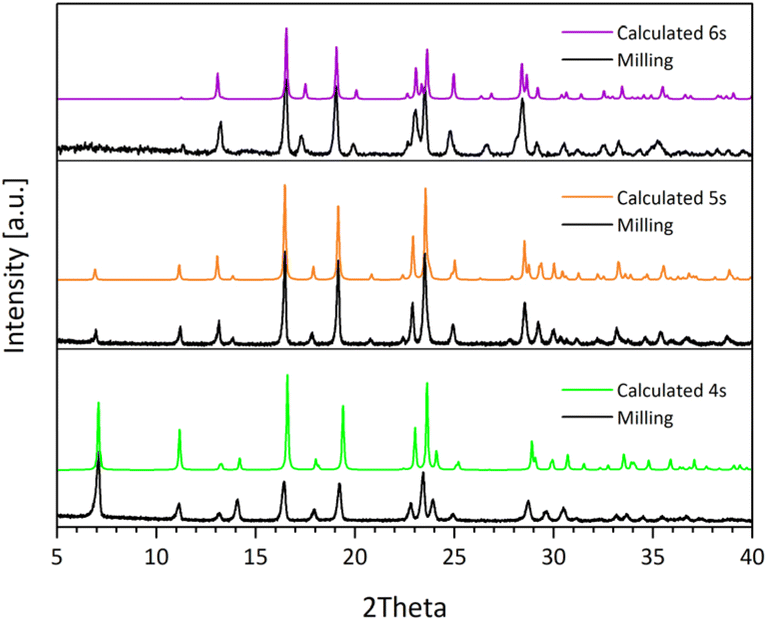 | ||
| Fig. 3 Comparison between the PXRD traces of the mechanochemical products obtained (4m–6m) without further purification, and those calculated from X-ray single structures of 4s–6s. | ||
To fully appreciate the efficiency of the mechanochemical approach, a comparison of yields and processing times is essential. Mechanochemistry significantly reduces the reaction time and enormously simplifies the workup procedure. The set-up of the reaction includes only a few minutes to weigh the reagents and load the jar, followed by 30 minutes of milling to have a high conversion of the reagents.
In contrast, the traditional reflux method requires hours of reaction and the need to remove the solvent to isolate the products, with yields no higher than 82%. These advantages underscore the exceptional efficiency of the mechanochemical route, which in all cases demonstrated high selectivity.
Because of the possibility of obtaining anhydrous phases, such as complex 7s [Zn(py)2(4-Cl-C6H4COO)2],16 we became interested in verifying the possibility of synthesizing anhydrous complexes by conducting the mechanochemical reaction under anhydrous conditions. We first tried to synthesize the bromine-containing complex [Zn(py)2(4-Br-C6H4COO)2] (8s) in solution, with the aim of isolating X-ray quality single crystals. For this purpose, anhydrous Zn(OAc)2 was refluxed in anhydrous THF with a two-fold excess of 4-Br-C6H4COOH and a large excess of pyridine, under a nitrogen atmosphere (Scheme 1, path c). Nicely, well-shaped single crystals were isolated from a dichloromethane/n-hexane mixture. The structural resolution revealed the formation of the target complex, where two κ1-coordinated carboxylates and two pyridines are tetrahedrally surrounding the metal centre, as can be inferred from Fig. 4.
 | ||
| Fig. 4 Ortep drawing of complex 8s. All non-hydrogen atoms are shown as ellipsoids at the 50% probability level. H atoms (isotropically refined) are reported in the ball-and-stick style for the sake of clarity. Colour code: red = O, blue = N, grey = C, white = H, and orange = Br. | ||
The complex adopts a tetrahedral coordination geometry and crystallizes in the monoclinic C2/c space group. The crystal packing is dictated by weak interactions involving aromatic hydrogens and carboxylate oxygens (see the ESI†).
The same reaction was then repeated under mechanochemical conditions. To assure a dry environment, Zn(OAc)2·2H2O was thermally treated for 24 hours at 120 °C to obtain anhydrous Zn(OAc)2. The anhydrous character was confirmed by FTIR spectroscopy and TGA analysis. The jar and the ball were left at 95 °C for 16 hours and then cooled in a desiccator containing anhydrous CaCl2. Once removed from the desiccator, the jar was quickly charged with 4-Br-C6H4COOH and anhydrous pyridine, using the same molar ratio applied for the wet synthesis, without the addition of any LAG agent (Scheme 1, path d). The closure of the jar was done without any precaution to exclude moisture. After 30 minutes of milling at 20Hz the jar was opened and the solid 8m was quickly subjected to PXRD analysis. The experimental diffractogram was perfectly in agreement with the one calculated from the solid state structure of 8s (Fig. 5). This transformation occurs with reduction of the hapticity of one of the carboxylate groups, form κ2-chelating to κ1-monodentate, a behaviour not unexpected based on the known plasticity of the coordinated COO group.26,27 The same procedure was extended to the mechanochemical synthesis of 7m, again observing the quantitative conversion of the reagents into the target product (Fig. 5).
 | ||
| Fig. 5 Comparison between the PXRD traces of the mechanochemical products obtained using the “dry milling” protocol and those calculated from the single crystal structures: 8m, top; 7m, bottom. | ||
The anhydrous zinc complexes exhibited a quantitative yield under dry milling conditions, in contrast to its hydrated counterpart. The elimination of water from the metal salt appears to facilitate a more efficient reaction pathway, possibly preventing competitive coordination of water molecules and favouring the direct formation of the anhydrous product.
It is worth mentioning that dissolution of complex 8m in dichloromethane followed by slow evaporation of the solvent in air, gave signs of the formation of complex 2s, as inferred by PXRD analysis (see the ESI†). This finding makes the success of the mechanochemical synthesis of 8m even more straightforward, considering the simple experimental setup adopted for its preparation. We finally investigated the possibility of thermally converting the hydrate complex 2m into the anhydrous 8m. The dehydration was followed by VT-PXRD, from 25 °C to 130 °C (see the ESI†). The trace of the starting complex 2m remains intact up to 80 °C, the temperature at which the signals belonging to the anhydrous phase 8m start to appear. These remain visible until 110 °C, while higher temperatures lead to the appearance of new peaks not corresponding to known phases. These new signals could be derived from the temperature induced loss of pyridine, in agreement with the TGA trace recorded for the hydrate complex (see the ESI†).
Conclusions
This work demonstrates that the mechanochemical synthesis of hydrate heteroleptic mononuclear complexes of Zn2+ and Cu2+ with pyridine and benzoic acids proceeds with high yields within 30 minutes of grinding at room temperature. In the case of Cu complexes, the complete conversion of the reagents has been observed, whereas a recrystallization is required for the hydrate zinc complexes to remove the traces of unreacted reagents. In contrast, dry milling enables the direct and quantitative formation of an anhydrous zinc complex. All reactions exhibited high selectivity, yielding the same crystalline phases obtained via solution synthesis but with shorter reaction times and significantly simplified experimental procedures. The ability to obtain an anhydrous complex under mechanochemical conditions without the need for specialized equipment, such as a glove box or a Schlenk line, further underscores the potential of mechanochemistry as a powerful and practical tool for the selective synthesis of metal complexes.28Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1s, 2s, 3s and 8s have been deposited at the CCDC under 2411964–2411967.Author contributions
GC: investigation and writing-original draft; PPM: validation, review and editing; AB: supervision; PP: supervision, conceptualization and writing, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Centro di Strutturistica Chimica M. Nardelli of the University of Parma for X-ray data collections. D. Balestri (University of Parma) is thanked for technical assistance. This work has benefited from the equipment and framework of the COMP-R Initiative, funded by the ‘Departments of Excellence’ program of the Italian Ministry for University and Research (MUR, 2023–2027). This work also benefitted of the 2020 and 2022 call of the PRIN: Research Projects of Relevant Interest program of the Italian Ministry for University and Research (PRIN2020Y2CZJ2 and 202224KAX8).Notes and references
- A. Beillard, X. Bantreil, T. X. Métro, J. Martinez and F. Lamaty, Chem. Rev., 2019, 119, 7529–7609 CrossRef CAS PubMed.
- O. Bento, F. Luttringer, T. Mohy El Dine, N. Pétry, X. Bantreil and F. Lamaty, Eur. J. Org. Chem., 2022, 2202101516 Search PubMed.
- J. F. Reynes, F. Leon and F. García, ACS Org. Inorg. Au, 2024, 4, 432–470 CrossRef CAS PubMed.
- P. Sharma, C. Vetter, E. Ponnusamy and E. Colacino, ACS Sustain. Chem. Eng., 2022, 10, 5110–5116 CrossRef CAS.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 Search PubMed.
- R. Brahma and J. B. Baruah, ACS Omega, 2020, 5, 3774–3785 Search PubMed.
- L. Leoni, A. Carletta, L. Fusaro, J. Dubois, N. A. Tumanov, C. Aprile, J. Wouters and A. D. Cort, Molecules, 2019, 24, 1–10 Search PubMed.
- T. E. Shaw, L. Mathivathanan and T. Jurca, Organometallics, 2019, 38, 4066–4070 Search PubMed.
- R. C. Pooja Sethi and R. Khare, Asian J. Chem., 2020, 32, 2594–2600 Search PubMed.
- D. Feng, X. Hao, Z. Fei, P. Huang and F. Guo, Organometallics, 2024, 43, 1640–1646 Search PubMed.
- R. J. Allenbaugh, J. R. Zachary, J. R. Williams, A. L. Shaw, J. D. Bryson, A. N. Underwood and E. E. O'Donnell, Inorg. Chim. Acta, 2018, 477, 102–108 CrossRef CAS.
- C. Gazzurelli, M. Solzi, F. Cugini, P. P. Mazzeo, A. Bacchi and P. Pelagatti, Inorg. Chim. Acta, 2022, 539, 121010 CrossRef CAS.
- C. Gazzurelli, A. Migliori, P. P. Mazzeo, M. Carcelli, S. Pietarinen, G. Leonardi, A. Pandolfi, D. Rogolino and P. Pelagatti, ACS Sustain. Chem. Eng., 2020, 8, 14886–14895 CrossRef CAS.
- P. P. Mazzeo, M. Prencipe, T. Feiler, F. Emmerling and A. Bacchi, Cryst. Growth Des., 2022, 22, 4260–4267 CrossRef CAS PubMed.
- A. Bacchi and P. P. M. Mazzeo, Crystallogr. Rev., 2021, 27, 102–123 CrossRef CAS.
- A. Karmakar, R. J. Sarma and J. B. Baruah, Inorg. Chem. Commun., 2006, 9, 1169–1172 CrossRef CAS.
- K. Takahashi, N. Hoshino, T. Takeda, K. Satomi, Y. Suzuki, S. I. Noro, T. Nakamura, J. Kawamata and T. Akutagawa, Dalton Trans., 2016, 45, 3398–3406 RSC.
- R. E. Del Sesto, A. M. Arif and J. S. Miller, Inorg. Chem., 2000, 39, 4894–4902 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr. Sect. A Found. Crystallogr., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr. Sect. C Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 Search PubMed.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed.
- L. E. Wenger and T. P. Hanusa, Chem. Commun., 2023, 59, 14210–14222 RSC.
- G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755–1775 RSC.
- J. Seo, C. Bonneau, R. Matsuda, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 9005–9013 CrossRef CAS PubMed.
- K. Kubota, R. Takahashi and H. Ito, Chem. Sci., 2019, 10, 5837–5842 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR spectra of all complexes, PXRD traces, Rietveld refinements, VT-PXRD, TGA trace of 5m, and ORTEP views and packing contacts of 1–3 and 8. CCDC 2411964–2411967. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4mr00150h |
| This journal is © The Royal Society of Chemistry 2025 |