
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccess to enzyme@porous organic framework biocomposites based on mechanochemical synthesis
Qing
Chen
a,
Zhi-Wei
Li
 *a,
Siming
Huang
b,
Guosheng
Chen
*a and
Gangfeng
Ouyang
a
*a,
Siming
Huang
b,
Guosheng
Chen
*a and
Gangfeng
Ouyang
a
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-sen University, Guangzhou 510006, China. E-mail: lizhw69@mail.sysu.edu.cn; chengsh39@mail.sysu.edu.cn
bGuangzhou Municipal and Guangdong Provincial Key Laboratory of Molecular Target & Clinical Pharmacology, The NMPA and State Key Laboratory of Respiratory Disease, School of Pharmaceutical Sciences, Guangzhou Medical University, Guangzhou 511436, China
First published on 24th January 2025
Abstract
Enzymes serve as highly efficient and selective biological catalysts, essential across diverse fields such as industry, medicine, and biotechnology, and are vital to green chemistry due to their efficacy at ambient temperatures. However, enzymes are sensitive to environmental conditions, which restricts their stability and reusability, limiting their broader practical applications. Porous organic frameworks encompassing metal–organic frameworks, covalent organic frameworks, and hydrogen-bonded organic frameworks have emerged as robust platforms for enzyme immobilization, offering high porosity, tailored pore structures, and strong chemical stability to safeguard encapsulated enzymes. Traditional surface immobilization methods can stabilize enzymes but often encounter low loading efficiency and enzyme leaching issues. In situ embedding methods address these challenges but typically rely on solvent-intensive, high-temperature liquid-phase syntheses that compromise enzyme functionality. This review centers on mechanochemical synthesis as a novel, green approach to creating enzyme-embedded porous organic frameworks, referred to as enzyme@porous organic frameworks. By employing mild mechanical forces, mechanochemical synthesis facilitates enzyme encapsulation under ambient conditions in the absence (or near-absence) of solvents, maintaining enzyme stability while ensuring efficient precursor-enzyme integration. We explore the mechanochemical synthesis principles, influential parameters, and advantages over liquid-phase techniques, underscoring its potential to produce multifunctional biocomposites. This review aspires to pave the way for scalable biocatalytic systems with enhanced stability and performance, advancing biocatalysis in industrial applications.
1 Introduction
Enzymes provide the necessary impetus for the speed of chemical reactions essential for sustaining life by forming a unique enzyme–substrate complex, thereby reducing the energy required for substrate conversion into products.1 Their high efficiency, specificity, and selectivity make enzymes prominent candidates in the fields of industry, medicine, and biotechnology. Additionally, their characteristic high reaction rates at ambient temperatures render them ideal catalysts for green chemistry.2 However, enzymes are sensitive to external environments, which limits their stability and reusability in practical applications. The fragility of enzymes arises from their distinct three-dimensional structure, where external stimuli such as heat, pH, organic solvents and other chemical reagents can disrupt the network of non-covalent interactions that maintain the enzyme's natural folded state, thus altering or eliminating their catalytic activity.3,4 Therefore, restricting the conformation of enzymes to a certain extent can enhance their stability.Porous organic frameworks, such as metal–organic frameworks (MOFs),5–7 covalent organic frameworks (COFs),8–10 and hydrogen-bonded organic frameworks (HOFs),11–13 have emerged as ideal platforms for enzyme immobilization due to their unique structures and properties. These porous materials not only possess high porosity and tailored pore structure that facilitate mass transfer, but also exhibit good chemical stability and mechanical strength, making them able to provide a better protective effect for loaded enzymes.3 Traditional surface immobilization techniques, such as adsorption14,15 and cross-linking methods,16 can stabilize enzymes to some extent but often suffer from low loading efficiency and the enzyme leaching issue. In contrast, in situ embedding methods,17 where the enzymes are encapsulated inside a porous organic framework as it forms, can address the limitation of the surface immobilization method. This method entails the growth of porous organic frameworks under mild conditions (e.g., without organic solvents and at ambient temperatures and pressures) to prevent structural damage to the enzymes. Unfortunately, the traditional liquid-phase synthesis methods for porous organic frameworks usually require numerous organic solvents, and in most cases the reaction conditions of high temperature and pressure are required to facilitate the formation of thermodynamically stable crystalline products, weakening the feasibility of the liquid-phase synthesis approach to encapsulate enzymes.18,19
Mechanical ball milling, as an efficient and green synthesis technique,20,21 demonstrates significant advantages in the synthesis of biocomposites. By applying mild mechanical forces, it can rapidly initiate chemical reactions at ambient temperatures and pressures without the need for large amounts of solvents,22 showing more biocompatibility to enzymes. Moreover, the mechanical milling process generates high-energy collisions and shear forces, facilitating thorough mixing and interaction between enzymes and precursors, leading to efficient enzyme encapsulation. This article aims to provide a comprehensive overview of mechanochemical synthesis for enzyme-encapsulated porous organic framework biocomposites, termed as enzyme@porous organic frameworks, with specific emphasis on the synthesis methodologies and their advantages compared to liquid-phase methods. Additionally, we also delve into the principles of mechanochemistry and the key influencing factors. It is anticipated that this review will offer useful mechanochemical methods that facilitate access to multifunctional biocomposites, advancing biocatalysis in large-scale industrial applications.
2 Introduction to mechanochemistry
Mechanochemistry refers to the process of inducing or accelerating chemical reactions in solid materials through ball milling. Compared to traditional liquid-phase methods, mechanochemical techniques do not require large amounts of solvents and can rapidly initiate chemical reactions at ambient temperatures, making them an efficient, green, and rapid synthesis technology. However, the invisibility of the reaction process and the unclear mechanisms of energy transformation make it challenging to control the ball milling process, which limits its practical applications. Therefore, in-depth understanding of the underlying mechanism and regulation of the key influencing factors are crucial for the development of mechanochemistry. This chapter outlines the reaction mechanisms of mechanochemistry and the factors influencing these reactions, aiding researchers in gaining a clearer understanding of the ball milling process to better control the chemical changes involved.2.1 Mechanism of mechanochemical transformation
In any chemical reaction, a certain amount of energy must be absorbed to overcome the energy barrier and activate the reactants. This energy is typically converted from other forms, such as light energy, electrical energy, and thermal energy, while in mechanochemistry, mechanical energy is transformed into chemical energy. During continuous mechanical processing (e.g., ball milling), three states exist-activation, equilibrium, and relaxation.23,24 Mechanochemical reactions rely on the equilibrium state (also known as mechanochemical equilibrium), which drives the reaction system into an energetically activated “steady state”. As long as the mechanical processing is uninterrupted and unaltered, this equilibrium will persist. Chemical transformations occur within this mechanochemical “steady state”. However, current mechanochemical instruments (such as planetary mills and shaker mills) operate in a completely enclosed jar that makes the “steady state” chemical changes difficult to monitor in real time.25 Thus, understanding the transformation processes of different reaction types within the instruments is crucial for revealing reaction mechanisms and the existence of intermediates.With advancements in technology, scientists have combined various detection instruments with mechanochemical equipment to achieve direct observation of these dynamic processes. Early efforts mainly relied on temperature26 and pressure27 sensors to indirectly gather information about the reactor's interior. Subsequently, the integration of time-resolved in situ (TRIS) techniques encompassing X-ray diffraction (XRD),28,29 X-ray absorption fine structure (XAFS) spectroscopy,30 Raman spectroscopy,31,32 and solid-state nuclear magnetic resonance (NMR) spectroscopy33 has provided direct insights into the structural details of mechanochemical transformations. Michalchuk et al.34 classified mechanochemical processes into three distinct periods by the TRIS method: the induction period, the reaction period, and the product period (Fig. 1a). In situ characterization of mechanochemical transformation processes using TRIS methods combined with manometry and thermometry allows for the monitoring of characteristic signal changes to represent different reactivity states (Fig. 1c). Notably, the signal intensity changes most significantly during the reaction period, which can be further divided into three stages (Fig. 1b): (1) mixing of solid materials. This stage includes bulk mixing, which involves the movement of solid particles, and microscopic mixing, which entails grinding the powder and forming flowing intermediates (such as amorphous phases, or liquid or gaseous products); (2) further mixing at the atomic or molecular level, which leads to physicochemical transformations; (3) the formation of product phases, which crystallize into solids, initially as nuclei, which then gradually grow into nanoscale or microcrystalline phases.
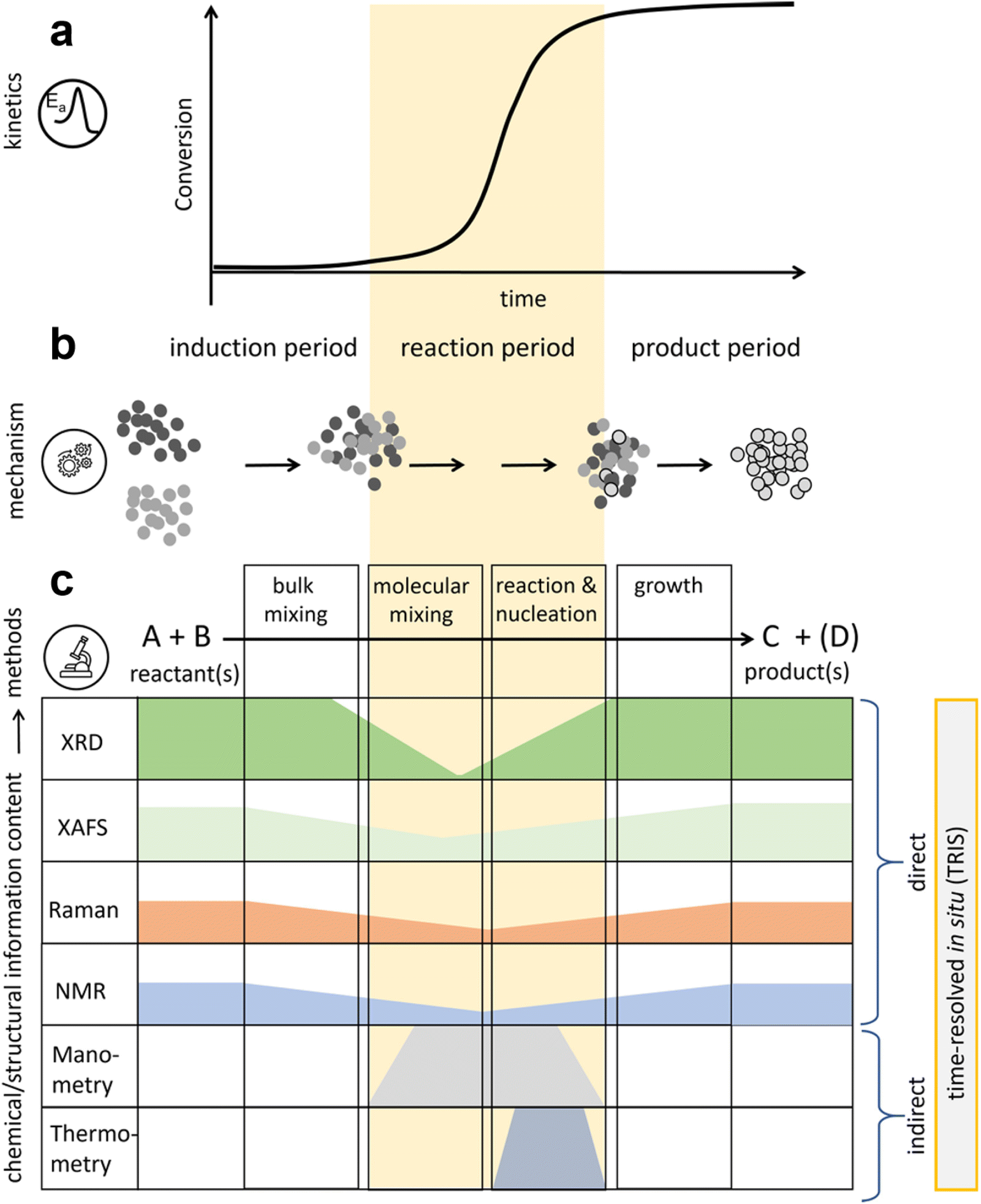 | ||
| Fig. 1 General evolution of mechanochemical transformations and the TRIS methods available to characterize the transformation processes. (a) A generic kinetic profile for a mechanochemical transformation, comprising an induction period, reaction period, and product period. (b) Schematic representation of the macroscopic mechanism describing a generic mechanochemical transformation, comprising bulk mixing, molecular mixing, reaction and nucleation, and growth phases. (c) Existing TRIS methods to characterize mechanochemical transformations, with indications of their strength to characterize different reactivity regimes.34 Copyright 2022, Wiley-VCH. | ||
During the grinding process, the reaction mainly occurs at the interfaces between metal sources and ligands. For the mechanochemical synthesis of MOFs using metal salts or metal oxides, the initial step of grinding primarily involves breaking the metal source crystals while converting mechanical energy into the potential energy of accumulated precursors. This process not only reduces particle size but also increases the number of reaction interfaces which generates numerous active sites. Subsequently, surface bond breakage, defects, and other changes occur between the high-energy precursors. Ultimately, thermodynamically stable crystals are formed. For example, Miyake's group reported the dry mechanochemical conversion of zinc oxide and imidazole ligands into ZIF-8.35 After 96 hours of grinding, a porous crystal product with a specific surface area of 1480 m2 g−1 was obtained. The authors proposed three parallel steps to explain the mechanistic synthesis of ZIF-8: (i) the fragmentation of ZnO aggregates, which generates numerous reaction interfaces exposing more active sites, (ii) the gradual conversion of ZnO at the solid interface into ZIF-8, and (iii) the aggregation of ZIF-8 nanoparticles (Fig. 2).
 | ||
| Fig. 2 Proposed mechanism for mechanochemical dry conversion of ZnO to ZIF-8,35 in which three main processes take place parallelly. Copyright 2013, Royal Society of Chemistry. | ||
2.2 Factors controlling mechanochemistry
Mechanochemical reactions depend on the change in Gibbs free energy of the reactants and the magnitude of mechanical energy, meaning that they rely on mechanical stress to initiate the reaction. By adjusting factors such as machine speed or frequency, rotation time, the addition of auxiliaries, and the size of the milling balls, the magnitude of mechanical energy can be controlled, facilitating the reaction. Generally, there is a positive correlation between speed/frequency and rotation time with the reaction outcome; that is, higher speeds and longer times increase mechanical energy, thereby enhancing the likelihood of triggering a reaction.36 However, excessive milling time may lead to poor crystal formation, deformation, or even decomposition, and also cause biomolecule inactivation.The advent of liquid-assisted grinding (LAG) has significantly accelerated reaction rates, resulting in better crystallinity while preserving the activity of biomolecules to a greater extent. For example, synthesizing ZIF-8 (ref. 37) via dry milling requires several hours for complete reaction, whereas adding a small amount of water or alcoholic solution can reduce the reaction time to mere minutes. Furthermore, incorporating a small amount of salt solution (ionic liquid-assisted grinding, ILAG) can partially function as an ionic template, inducing coordination between zinc and organic ligands, thus further accelerating the reaction.38 Recent studies have shown that adding a small amount of buffer salt solution can shorten the synthesis time of ZIF-90 to just 10 seconds.39 Here, the role of the buffer salt is to disrupt the hydration shell surrounding zinc and form chelates, facilitating the binding of the ligands of imidazole-2-carboxaldehyde (ICA) and aiding in the deprotonation of ICA, thereby lowering the activation energy of the reaction. The pre-formed amorphous Zn-ICA continues to replace the zinc coordinated water groups, with the ball milling assisting in the dehydration step (breaking Zn–O to form Zn–N), leading to the crystallization of ZIF-90. Notably, under buffer conditions, the preservation of the activity of biomolecules is more favorable than under organic solvent conditions.
The density and mass of the milling medium are also crucial for the transfer of reaction energy. In most cases, larger balls generate higher energy during collisions, which favors the formation of thermodynamically stable products. Conversely, smaller balls exhibit higher mobility and longer trajectories during stirring, allowing for better mixing and the formation of amorphous and metastable phases.40 In reactions, it is common to use a combination of ball sizes, typically with the ball-to-powder mass ratio ranging from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:3 and a filling rate of 30–35%, to ensure that reactants have sufficient space for effective milling.
:1 to 1:3 and a filling rate of 30–35%, to ensure that reactants have sufficient space for effective milling.
Condition optimization in mechanochemical synthesis usually relies on trial-and-error methods, which are both cumbersome and unpredictable. Additionally, achieving reproducible synthesis under different machine models or ball mills using the same parameters (like time and frequency) poses challenges. This limitation limits the practical application of ball milling in both research and industrial fields. Recently, Jafter et al. established an energy theory model to quantify the total energy output of the machine (Etotal) (Fig. 3).41 As long as the machine's output energy exceeds the cumulative energy standard (Etotal > Ea, where Ea is the activation energy for the reaction), the kinematic parameters (including the type and operating conditions of the ball mill) become independent of reaction kinetics, thus enhancing the reproducibility of mechanochemical experiments. At the microscopic level, the mechanical impact energy (Eimpact) generated by individual balls through systematic circular motion and collisions with the wall must exceed the threshold energy (Ethreshold) for the reaction of single reactant molecules (A + B) to overcome the energy barrier and trigger the chemical reaction. Furthermore, using the kinematic energy model, it has been demonstrated that under the same cumulative energy conditions, altering other operational parameters (such as ball mass and rotation speed) can increase reaction rates and reduce reaction times. Therefore, by calculating the mechanical output energy (Etotal), better control of mechanical parameters can be achieved to promote mechanochemical synthesis of porous organic frameworks.
 | ||
| Fig. 3 Illustration of planetary mill kinematics within the jar.41 Copyright 2024, Wiley-VCH. | ||
3 Mechanochemical synthesis of enzyme@MOF biocomposites
Metal organic frameworks (MOFs) are a class of crystalline porous materials assembled from metal nodes/clusters and organic ligands.42 Theoretically, they can be designed using various building units of metal nodes/clusters and organic ligands, providing an almost infinite variety of topologies with different pore structures.43 These characteristics have made MOFs a popular platform for enzyme immobilization.3 This section summarizes the MOF methodology for mechanochemical synthesis of enzyme@MOF biocomposites, as well as their performance advantages compared to those synthesized by traditional liquid-phase methods.3.1 Enzyme@ZIF biocomposites
Zeolitic imidazolate frameworks (ZIFs) are the dominating MOFs that enable the mechanochemical synthesis of enzyme biocomposites, owing to their facile crystallization kinetics. Although the liquid-phase ZIF method is well explored for enzyme encapsulation, it is time-consuming, and prolonged exposure of enzymes to ZIF precursors may pose the risk of conformation change.44 Meanwhile, mechanochemistry offers a more rapid and much greener alternative. Enzymes participate in the reaction in a freeze-dried state, and either no or only trace amounts of organic reagents are used as catalysts, reducing the likelihood of conformational unfolding and inactivation. Currently, two ZIFs including ZIF-8 (ref. 45 and 46) and ZIF-90 (ref. 47) have demonstrated feasibility to encapsulate enzymes under ball milling.Wei et al. first synthesized enzyme@ZIF-8 using mechanochemical methods through a simple two-step process.48 Initially, a portion of the MOF precursor was placed in a milling jar with a small amount of ethanol as a solvent, and milled at 8 hertz (Hz) for 2.5 minutes to form crystalline seeds. Subsequently, β-glucosidase (BGL) enzyme and the remaining precursor were added and milled again at the same frequency for another 2.5 minutes. Although this allowed the formation of a highly crystalline phase, the resulting BGL@ZIF-8 did not display any obvious activity, likely due to the small pore aperture of ZIF-8 (ca. 3.4 Å), which hindered substrate access to the interior enzyme. After decomposing the ZIF-8 framework under acidic conditions (pH 6.0), the released BGL retained a level of activity comparable to the natural one, indicating that the encapsulated BGL could preserve its original conformation.
Compared to ZIF-8, the interior of ZIF-90 exhibits higher hydrophilicity, contributing to keeping the conformation of the encapsulated enzyme unaltered.49 Lam and co-workers synthesized CAT@ZIF-90 with good crystallinity by mechanochemically milling imidazole-2-carboxaldehyde (ICA), nano zinc oxide (ZnO), and catalase (CAT) with trace amounts of tris buffer solution (500 mM, pH 7) or deionized water at 8 Hz for 10 seconds (Fig. 4a).39 The encapsulated CAT enzyme retained high activity, with a reaction rate constant (kobs) of 1.98 × 10−1, and it could maintain good activity even after protease treatment (kobs = 9.48 × 10−2), indicating that the ZIF-90 shell effectively shielded the enzyme against the protease. Compared to the CAT@ZIF-90 synthesized by a de novo encapsulation approach in water solution,50 regardless of whether protease K treatment is carried out, the mechanically prepared CAT@ZIF-90 exhibited higher kobs (Fig. 4b and c). This activity enhancement was attributed to the mechanical forces-induced defects in the ZIF-90 crystals, promoting mass transfer. Furthermore, this method was also adaptable to encapsulate other proteins and large-sized E. coli, highlighting the generalization.
 | ||
| Fig. 4 (a) Schematic illustration of the mechanochemical encapsulation of enzymes in ZIF-90. (b) Comparison of the catalytic activity of CAT@ZIF-90 synthesized at different milling times to that using a de novo encapsulation approach in water solution. (c) Comparison of the catalytic activity of CAT@ZIF-90 synthesized at different milling times to that using a de novo encapsulation approach in water solution, in which the samples were exposure to proteinase K after 2 hours.39 Copyright 2023, Royal Society of Chemistry. | ||
3.2 Enzyme@Zn-MOF-74 biocomposites
Although it is possible to create defects in the mechanochemically synthesized enzyme@ZIFs, the pristine narrow pore aperture dominates, posing challenges to mass transfer, especially in an enzymatic reaction with a large-sized substrate. Therefore, there is a need to develop another type of MOF with larger pore sizes for enzyme immobilization. The Zn-MOF-74 is formed by divalent metal zinc connected with 2,5-dihydroxy-1,4-benzenedicarboxylic acid, forming a three-dimensional structure with one-dimensional channels.51,52 Compared to the ZIF series (ca. 3.4 Å), it has larger pore channels (>14 Å),53 facilitating effective mass transfer and allowing substrates access to the catalytic sites of the interior enzyme while excluding larger proteases, thereby protecting the internal enzyme molecules.The feasibility to one-pot mechanochemical synthesis of enzyme@Zn-MOF-74 was demonstrated in the work reported by Wei et al.,48 in which CAT was encapsulated within Zn-MOF-74 as it formed under ball milling after 15 minutes. It has been verified that after incubation with protease K at pH 8.0, the activity of the resulting CAT@Zn-MOF-74 remains at 96.7% of that before incubation (the ratio of kobs post-incubation to kobs pre-incubation). Based on this, Ozyilmaz et al. shortened the milling time, achieving the mechanochemical synthesis of a biocatalyst in just 2.5 minutes (Fig. 5).54 The resulting CRL@Zn-MOF-74 exhibits slightly higher activity with a Michaelis constant (Km) value of 0.34 mM and a Vmax value of 130.88 U mg−1 compared to free Candida rugosa lipase (CRL) (Km = 0.44 mM, Vmax = 115 U mg−1), while CRL@ZIF-8 (Km = 0.76 mM, Vmax = 25.86 U mg−1) had a Vmax more than five times lower than that of CRL@Zn-MOF-74. These results illustrate the biocatalytic advantages conferred by the larger pore size of Zn-MOF-74. Furthermore, the authors investigated the enantioselective hydrolysis of naproxen racemic methyl ester using CRL@Zn-MOF-74, and the results indicate that CRL@Zn-MOF-74 exhibits a higher enantioselectivity (E = 245) compared to CRL@ZIF-8 (E = 136).
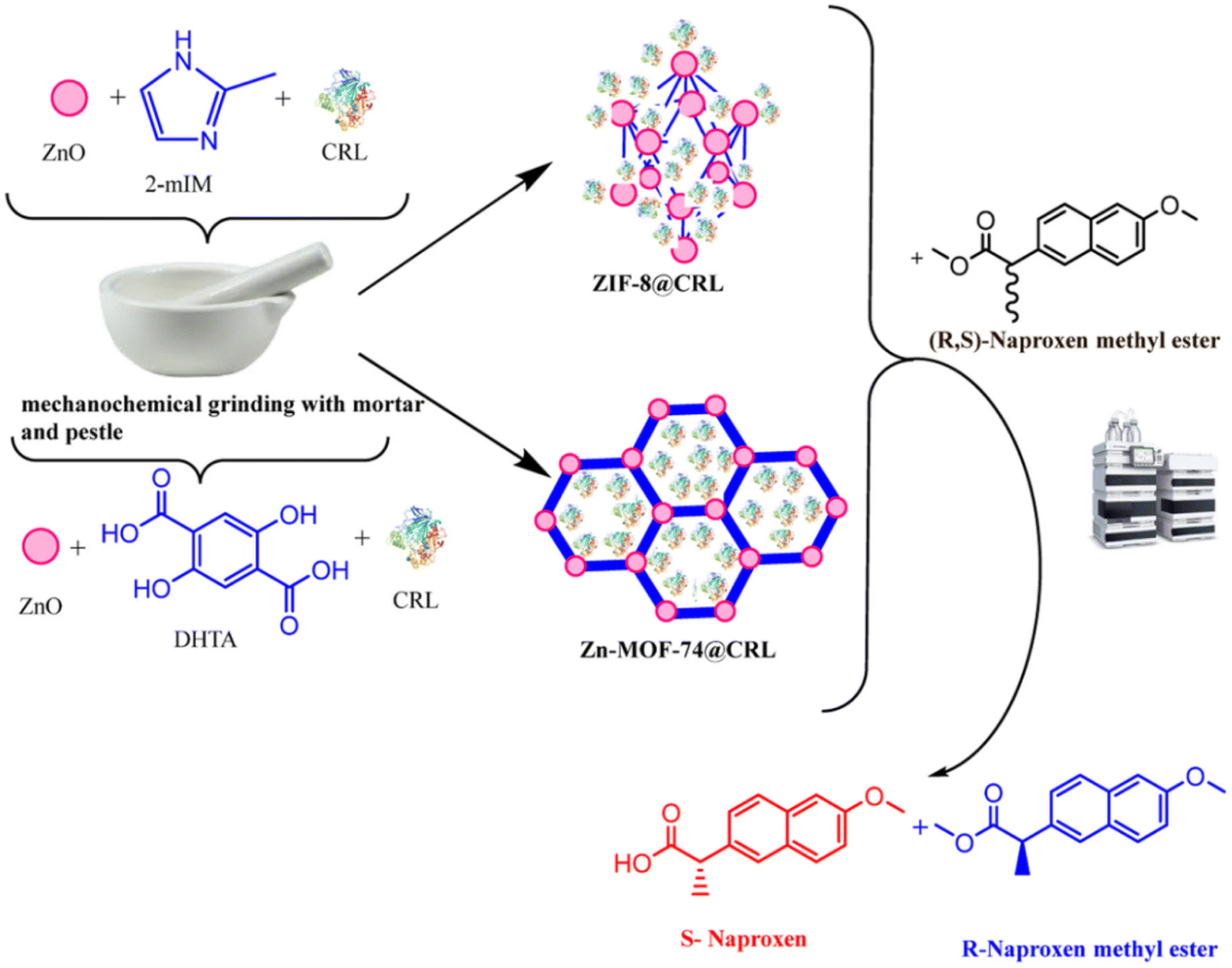 | ||
| Fig. 5 Schematic illustration of the synthesis of CRL@ZIF-8 and CRL@Zn-MOF-74 by mechanochemical methods and their utility for the enantioselective hydrolysis of (R,S)-naproxen methyl ester.54 Copyright 2023, Elsevier. | ||
3.3 Enzyme@UiO-type MOF biocomposites
UiO-66,55 a zirconium-based MOF, exhibits chemical and structural stability compared to Zn-MOFs. This enhanced stability is attributed to the strong coordination bonds formed between zirconium ions (Zr4+) as hard Lewis acids and carboxylic acid ligands as hard Lewis bases, providing good thermodynamic stability.56 The inorganic unit of UiO-66 consists of a secondary building unit (SBU) with a 12-connected Zr6O4(OH)4 core, which is bridged by terephthalic acids, forming a face-centered cubic lattice.57 This geometric arrangement of building blocks contributes to the overall stability of the framework. Furthermore, the surface chemical properties of UiO-66 can be easily modified with different components by functionalizing the terephthalic acid ligands. This not only can enhance the interactions between MOFs and enzymes,58 but also facilitates the substrate adsorption by the MOF.59 However, the liquid-phase synthesis of UiO-66 generally requires the use of strong acids, organic solvents, and elevated temperature conditions that are often detrimental to enzyme stability.60,61 As a result, current research predominantly focuses on enzyme immobilization via post-synthesis infiltration methods62 or surface binding techniques,60,63 rather than encapsulating enzymes during the in situ liquid-phase synthesis process.The mechanochemical approach presents a promising solution to these challenges by utilizing minimal solvent volumes in place of harsh acids and organic solvents, and applying mild mechanical forces instead of high temperatures. This methodology reduces the potentially harmful factors that could compromise enzyme activity. Wei et al. demonstrated the synthesis of enzyme@UiO-66-NH2 using a two-step mechanochemical method.48 In this process, ethanol was introduced during the formation of MOF seeds, initiating ball milling reactions to generate UiO-66-NH2 seeds without the enzyme. Subsequently, the enzyme and additional MOF precursors were added without introducing further solvent (Fig. 6a). By studying the effects of enzyme addition at various time points, they identified that the optimal enzyme incorporation time was after 2.5 minutes, which provided a balanced enhancement of both activity and structural integrity. Notably, the two-step method resulted in a more active composite catalyst compared to the one-step ball milling approach, with the observed rate constant (kobs) for BGL@UiO-66-NH2 increasing from 2.8 × 10−4 s−1 to 5.0 × 10−4 s−1. The catalyst exhibited excellent stability under acidic conditions (pH 6.0) and in the presence of proteases, maintaining its activity without significant loss. This resilience is attributed to the stability of UiO-66-NH2, which protects the encapsulated BGL enzyme from protease degradation. In contrast, ZIF-8 lost its protective effect under acidic conditions due to framework collapse, leading to enzyme inactivation in the presence of proteases (Fig. 6b). Furthermore, the mechanochemical method was successfully extended to encapsulate enzymes of varying molecular weights, including 270 kDa transglutaminase (Inv) and 105 kDa β-galactosidase (β-gal), demonstrating the broad applicability of this approach for enzyme immobilization.
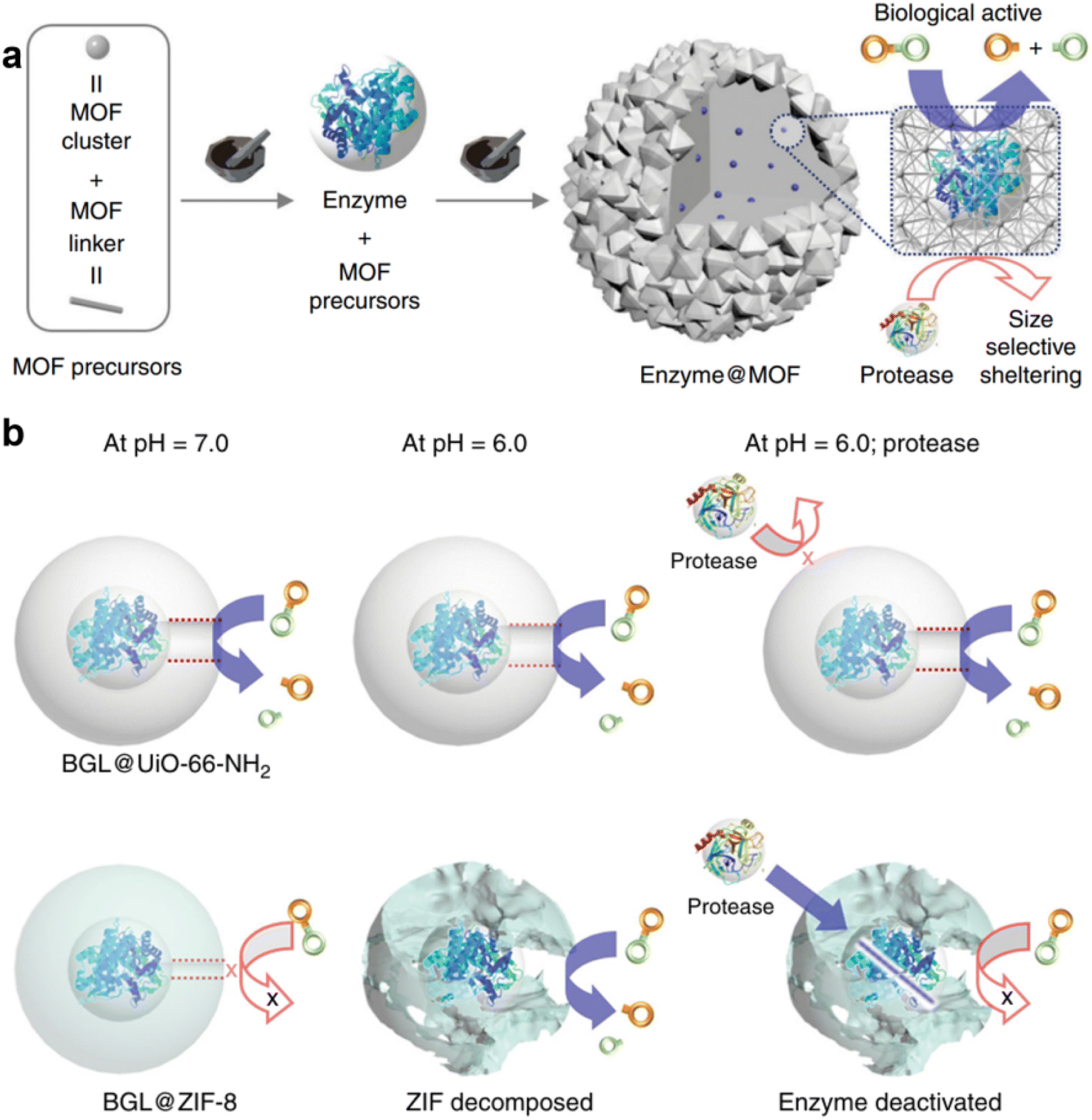 | ||
| Fig. 6 (a) Schematic illustration of the mechanochemical approach for encapsulating an enzyme within UiO-66-NH2. (b) Schematic illustration of the structural change of BCL@UiO-66-NH2 and BCL@ZIF-8.48 Copyright 2019, Springer Nature. | ||
Mechanochemistry not only facilitates the synthesis of enzyme@MOF catalysts but also enables the co-immobilization of multiple catalytic species within the same framework, thereby facilitating cascade catalytic reactions. Wang et al. were the first to develop a photobiocatalyst by encapsulating both photosensitizers and wheat germ lipase (WGL) within the UiO-67 framework.64 In this approach, a precise amount of WGL, Zr6 clusters and photosensitizer-modified units (2,2′-bipyridine-5,5′-dicarboxylic acid, BPDC) were mixed and subjected to mechanochemical milling at a frequency of 25 Hz for 90 minutes, yielding the desired photobiocatalyst (Fig. 7a). The loading amounts of both the photosensitizer and enzyme were optimized to achieve the highest catalytic performance. The optimal catalytic activity was obtained with a WGL loading of 200 mg g−1 and photosensitizer-to-BPDC ratios of 1/2 and 1/3 (Ru and Cu photosensitizers: BPDC). The optimal photobiocatalyst of WGL@UiO-67-Ru/Cu can catalyze the cross-dehydrogenation coupling of N-aryl-substituted tetrahydroisoquinoline (also known as asymmetric Mannich reaction) through a photocatalytic-enzyme cascade mechanism, as schematically presented in Fig. 7b. It exhibited excellent enantioselectivity, achieving an enantiomeric ratio of 85:15 and demonstrated remarkable recyclability, retaining up to 88% of its initial activity after ten catalytic cycles across a broad range of substrates (up to 12 different substrates) in the asymmetric Mannich reaction. Of note, this WGL@UiO-67-Ru/Cu photobiocatalyst maintained high crystallinity after ten catalytic cycles, underscoring its structural stability.
 | ||
| Fig. 7 (a) Schematical representation of integrating both photosensitizers and wheat germ lipase (WGL) into UiO-67. (b) The principle of the cross-dehydrogenation coupling reaction using the WGL@UiO-67-Ru/Cu photobiocatalyst.64 Copyright 2024, American Chemical Society. | ||
4 Mechanochemical synthesis of enzyme@COF and enzyme@HOF biocomposites
4.1 Enzyme@COFs
Covalent organic frameworks (COFs) are a class of porous materials composed of organic monomers linked by covalent bonds.65,66 COFs exhibit metal-free biocompatibility and ultra-stable covalent linkages, providing enzymes with a permeable, enclosed environment that extends their catalytic activity in biologically incompatible media.8 However, the synthesis of COFs often requires harsh conditions, such as elevated temperatures and strong acids, which can jeopardize enzyme stability during the crystallization process.67 Mechanochemical methods, by minimizing solvent usage (e.g., acids, bases, and organic solvents) and reducing reaction times, enable the activation of immobilized enzymes at room temperature. This approach effectively addresses the trade-off between COF crystallization and enzyme sensitivity.Our group has developed a mechanochemistry-guided in situ assembly strategy to construct enzyme@COF composites.68 In this method, three types of COFs-TpPa-1, TpPa-2, and TpBD-were used to encapsulate cytochrome C (Cyt C). During the mechanochemical synthesis, organic linkers and enzymes were combined with trace amounts of organic solvent and deionized water to assist the grinding process. The mixture was then ground at room temperature at 550 rpm for 1 hour to obtain the enzyme@COF biocomposites. In contrast, the solvothermal method, which requires large volumes of solvent and heating at 120 °C for 3 days, was found to be highly detrimental to enzyme viability (Fig. 8a). As a result, Cyt C@TpPa-1 synthesized using the mild mechanochemical method (Cyt c@TpPa-1 (M), where M represents mechanochemical method) retained 81.8% of the biological activity of the free enzyme (calculated as the ratio of Kcat for enzyme@COFs to that of the free enzyme), while the biocomposites produced via the solvothermal method (Cyt c@TpPa-1 (S), where S represents solvothermal method) exhibited almost complete loss of activity (Fig. 8b). To further demonstrate the versatility of this strategy, the team also immobilized horseradish peroxidase (HRP) and lipase (PS) using TpPa-1. However, the activities of HRP@TpPa-1 and PS@TpPa-1 were relatively low, with PS@TpPa-1 exhibiting only 35.5% of the activity of the free enzyme.
 | ||
| Fig. 8 (a) Synthesis of the enzyme@COF biocomposites. (b) The bioactivities of free Cyt c, TpPa-1 (M), and Cyt c@TpPa-1 biocomposites synthesized by the mechanochemistry method (Cyt c@TpPa-1 (M)) and solvothermal method (Cyt c@TpPa-1 (S)), respectively.68 Copyright 2022, Cell Press. | ||
Li et al. further optimized the synthesis protocol to further improve enzyme@COF biocomposite formation.69 To minimize the impact of organic solvents on enzyme activity, they developed a two-step synthesis method, successfully achieving enzyme@COF composites in just 5 minutes (Fig. 9a). In addition, the authors highlighted that the metal ions present in enzyme@MOF systems can interact unfavorably with the enzymes, causing conformational changes that reduce their catalytic efficiency. In contrast, the relatively weak interfacial interactions between COFs and enzymes help preserve the enzymes' native structure. Moreover, the activation effect of COF monomers on the encapsulated enzymes further enhances the catalytic activity of enzyme@COFs. For instance, lipase from Burkholderia cepacia (BCL) encapsulated in COF-TpPa-TS exhibited 1.38 times the activity of the free enzyme, and was 3.4 to 14.7 times more active than the BCL@MOF counterparts (Fig. 9b).
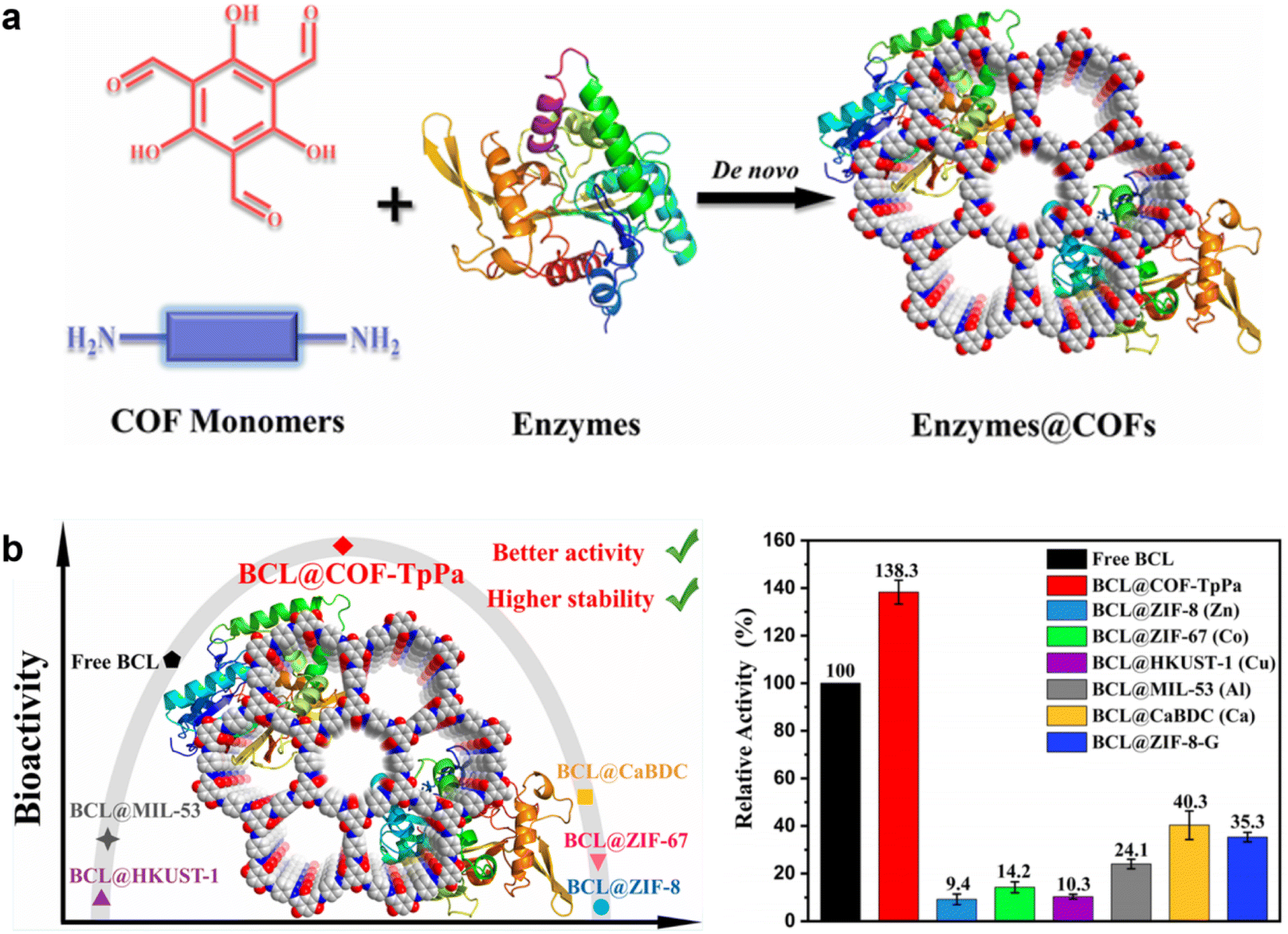 | ||
| Fig. 9 (a) Synthesis of the enzymes@COF biocomposites. (b) Catalytic activities of free BCL, BCL@COF-TpPa, and BCL@MOFs.69 Copyright 2023, American Chemical Society. | ||
4.2 Enzyme@HOFs
Hydrogen-bonded organic frameworks (HOFs) are supramolecular crystals formed by discrete molecular units that are linked in an ordered manner through hydrogen bonding.70,71 Compared to MOFs and COFs, HOFs offer the advantages of more environmentally friendly synthesis conditions and higher solvent processability. Due to these mild synthesis conditions, HOFs have garnered increasing attention for the encapsulation of enzymes using various liquid-phase assembly approaches.72–74 The concept of mechanochemical encapsulation of guest molecules such as Pd nanoparticles within a HOF has been validated by Liu's group recently.75 The authors synthesized various HOFs using mild mechanochemical methods, including previously reported PFC-1, PFC-73-Cu, PFC-76-NH2 and complex topological structures like HOF-BTB and HOF-11 as well as geometrically diverse connected frameworks such as PFC-67, PFC-68, and PFC-69 that have not been reported (Fig. 10). Computational models indicate that in solid-state organic nanoparticle milling systems, intense collisions elevate interfacial temperatures and lead to local supersaturation of monomers, facilitating the transition from amorphous to microcrystalline states through fragmentation, dissociation, and recrystallization. Subsequently, HOF monomers were milled with dispersed Pd nanoparticles (NPs) in minimal ethanol, yielding Pd@HOF composites under their respective pure HOF conditions. The resulting composites exhibited advantages such as structural stability, high catalytic activity, robust stability, and excellent adsorption properties compared to Pd NPs. These enhancements are attributed not only to the protective role of the HOF but also to the synergistic effects between the HOF and the substrates.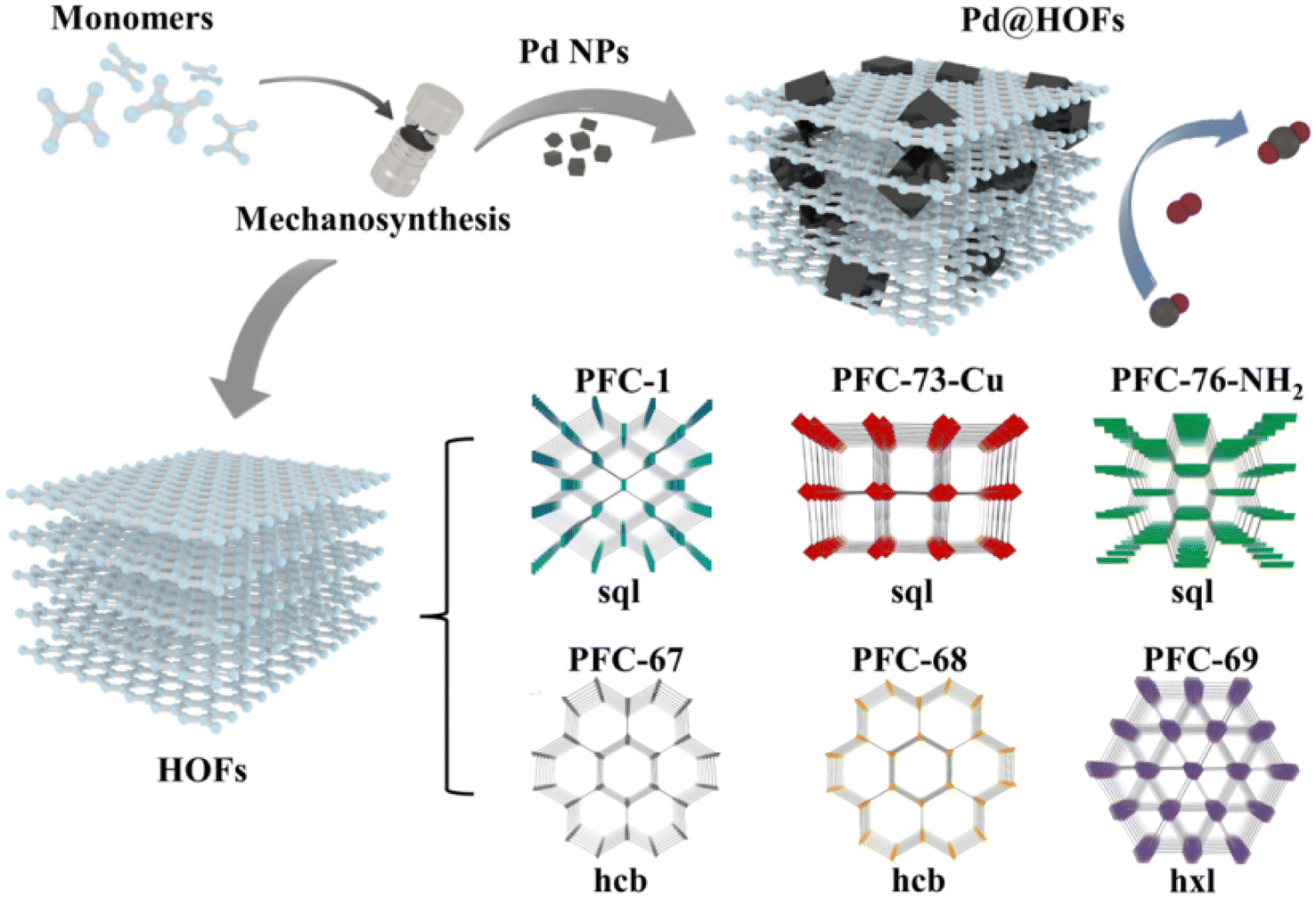 | ||
| Fig. 10 Syntheses of HOFs and Pd@HOFs by the mechanochemical method.75 Copyright 2022, Wiley-VCH. | ||
It is anticipated that this mild HOF approach featuring highly crystalline frameworks and considerable encapsulation efficiency can be adaptable to mechanochemical synthesis of enzyme@HOFs, although the feasibility has not been demonstrated yet. Compared with covalent and coordination bonds, hydrogen bonds are less stable, less directional, and more susceptible to mechanical forces,76,77 making the preparation of enzyme@HOFs more challenging. We proposed that the introduction of large aromatic groups or synthons with multiple hydrogen bonding sites into the molecular unit can effectively strengthen the intermolecular interactions, thereby making the HOFs more structurally predictable during mechanochemical synthesis and enhancing the stability of the resulting frameworks.
To provide a clearer comparison of mechanochemically synthesized MOFs, COFs, and HOFs in enzyme immobilization, Table 1 summarizes their respective advantages and disadvantages, offering valuable insights for future research and applications. Enzyme@MOF materials exhibit notable benefits, including short synthesis times, high crystallinity, and enhanced enzyme stability. However, metal ions in MOFs can impact the bioactivity of immobilized enzymes, and the most explored MOF systems, such as ZIFs, demonstrate significant acid instability, limiting their applicability in acidic environments. Mechanochemically synthesized enzyme@COF materials, such as TpPa-1, TpPa-2, and TpBD, stand out due to their metal-free nature and excellent biocompatibility. Their covalently bonded frameworks provide superior thermal and acid stability, ensuring the long-term preservation of enzyme stability and activity. Nevertheless, these systems face challenges, including extended synthesis times, the extensive use of organic solvents, and the limited range of COFs currently available for enzyme immobilization. Although there are no reports of enzyme@HOF systems synthesized via mechanochemical methods, HOFs are gaining attention for their hydrogen-bonded dynamic linkages, which enable environmentally friendly synthesis conditions, short reaction times, and high crystallinity. However, the weak intermolecular linkages of HOFs lead to structural unpredictability and inherent instability, making enzyme@HOF systems fragile and limiting their practical applications.
| Enzyme@MOF | Enzyme@COF | Enzyme@HOF | |
|---|---|---|---|
| Frameworks reported for enzyme immobilization via mechanochemistry | Zif-8, Zif-90, Zn-MOF-74, UiO-66, UiO-67 | TpPa-1, TpPa-2, and TpBD | No mechanochemically synthesized enzyme@HOFs reported yet |
| Main advantage(s) | Short synthesis time, high crystallinity; high protease stability | COF is metal-free and has good biocompatibility; covalent bonds offer high thermal and acid stability | Short synthesis time, high crystallinity, 100% atomic utilization |
| Main drawback(s) | Metal ions have a certain impact on enzyme bioactivity; poor acid stability | Long synthesis time and high organic solvent usage; limited COFs available for immobilized enzymes | HOF is inherently acid-unstable, unsuitable for enzymes requiring acidic conditions for catalysis |
5 Summary and outlook
The integration of porous organic frameworks for enzyme encapsulation opens promising avenues for developing multifunctional biocatalysts, leveraging the structural robustness of these frameworks to shield enzymes from denaturation under harsh conditions, such as acidic environments, organic solvents, and elevated temperatures. Mechanochemical encapsulation techniques, compared to traditional liquid-phase methods, offer an eco-friendly alternative characterized by ultra-short synthesis times, minimal solvent requirements, and high atomic economy. These methods also enhance biocompatibility with fragile enzymes, facilitating the formation of biocomposite materials that are often challenging to achieve via liquid-phase processes. Owing to its simple equipment and ease of operation, mechanical milling is characterized by low production costs. Moreover, it enables precise control over raw material utilization, achieving high atomic efficiency with minimal byproducts. These advantages position mechanical milling as a standout technique in the field of industrial material preparation, offering a viable and effective technological pathway for large-scale industrial production. However, critical challenges remain in optimizing these methodologies.While mechanochemistry has been widely investigated for its role in energy transformation, the precise interfacial interactions between precursors (metal sources, ligand molecules) and enzymes during self-assembly are not yet fully elucidated. Advanced time-resolved in situ spectroscopy techniques, including X-ray absorption fine structure, Raman, and solid-state NMR spectroscopy, offer valuable insights into these interactions. These insights may aid in achieving controlled enzyme immobilization, with the potential to finely regulate the spatial positioning of enzymes within the host framework to enhance catalytic efficiency.
Although encapsulation markedly enhances enzyme stability, the catalytic activity of encapsulated enzymes is often reduced relative to their free counterparts, whether mechanochemical or liquid-phase encapsulation methods are used. This limitation is largely due to diffusion barriers imposed by the material's shell, which can hinder substrate access to encapsulated enzymes. While current frameworks largely depend on their inherent porosity to mitigate this effect, the potential of surface or internal pore functionalization has been underexplored. Rational modification of surface and pore channels with specific hydrophilic and electrostatic sites may help accumulate catalytic substrates, potentially enhancing enzymatic reactions. Importantly, the flexible conformation of enzymes is sensitive to mechanical forces, necessitating careful control of parameters such as milling power and time to prevent structural damage during mechanochemical encapsulation. Additionally, the introduction of suitable buffering agents or other solutions, that can help stabilize enzymes by facilitating hydration shell formation, may reduce the risk of denaturation.
Data availability
The data supporting this article have been included in the main text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from projects of the National Natural Science Foundation of China (22174164, 22336007, 22104159), National High-Level Talents Special Support Program – Young Talents (2024WRQB006) and Guangdong Basic and Applied Basic Research Foundation (2024B1515020070).References
- T. Saleh and C. G. Kalodimos, Enzymes at work are enzymes in motion, Science, 2017, 355, 247–248 CrossRef CAS PubMed.
- R. Chapman and M. H. Stenzel, All Wrapped up: Stabilization of Enzymes within Single Enzyme Nanoparticles, J. Am. Chem. Soc., 2019, 141(7), 2754–2769 CrossRef CAS PubMed.
- S. Huang, G. Chen and G. Ouyang, Confining enzymes in porous organic frameworks: from synthetic strategy and characterization to healthcare applications, Chem. Soc. Rev., 2022, 51, 6824–6863 RSC.
- W. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C.-K. Tsung, P. Falcaro and C. J. Doonan, Metal-Organic Framework-Based Enzyme Biocomposites, Chem. Rev., 2021, 121(3), 1077–1129 CrossRef CAS PubMed.
- G. Chen, S. Huang, X. Kou, F. Zhu and G. Ouyang, Embedding Functional Biomacromolecules within Peptide-Directed Metal-Organic Framework (MOF) Nanoarchitectures Enables Activity Enhancement, Angew. Chem., Int. Ed., 2020, 59(33), 13947–13954 CrossRef CAS PubMed.
- L. Guo, R. He, G. Chen, H. Yang, X. Kou, W. Huang, R. Gao, S. Huang, S. Huang, F. Zhu and G. Ouyang, A Synergetic Pore Compartmentalization and Hydrophobization Strategy for Synchronously Boosting the Stability and Activity of Enzyme, J. Am. Chem. Soc., 2024, 146(25), 17189–17200 CrossRef CAS PubMed.
- G. Chen, X. Kou, S. Huang, L. Tong, Y. Shen, W. Zhu, F. Zhu and G. Ouyang, Modulating the Biofunctionality of Metal-Organic-Framework-Encapsulated Enzymes through Controllable Embedding Patterns, Angew. Chem., Int. Ed., 2020, 59(7), 2867–2874 CrossRef CAS PubMed.
- R. Gao, X. Kou, L. Tong, Z.-W. Li, Y. Shen, R. He, L. Guo, H. Wang, X. Ma, S. Huang, G. Chen and G. Ouyang, Ionic Liquid-Mediated Dynamic Polymerization for Facile Aqueous-Phase Synthesis of Enzyme-Covalent Organic Framework Biocatalysts, Angew. Chem., Int. Ed., 2024, 63(8), e202319876 CrossRef CAS PubMed.
- Y. Zheng, S. Zhang, J. Guo, R. Shi, J. Yu, K. Li, N. Li, Z. Zhang and Y. Chen, Green and Scalable Fabrication of High-Performance Biocatalysts Using Covalent Organic Frameworks as Enzyme Carriers, Angew. Chem., Int. Ed., 2022, 61(39), e202208744 CrossRef CAS PubMed.
- H. Liu, Y. Zhou, J. Guo, R. Feng, G. Hu, J. Pang, Y. Chen, O. Terasaki and X.-H. Bu, Reticular Synthesis of Highly Crystalline Three-Dimensional Mesoporous Covalent-Organic Frameworks for Lipase Inclusion, J. Am. Chem. Soc., 2023, 145(42), 23227–23237 CrossRef CAS PubMed.
- W. Huang, H. Yuan, H. Yang, X. Ma, S. Huang, H. Zhang, S. Huang, G. Chen and G. Ouyang, Green synthesis of stable hybrid biocatalyst using a hydrogen-bonded, Π-Π-stacking supramolecular assembly for electrochemical immunosensor, Nat. Commun., 2023, 14(1), 3644 CrossRef CAS PubMed.
- Z. Tang, X. Li, L. Tong, H. Yang, J. Wu, X. Zhang, T. Song, S. Huang, F. Zhu, G. Chen and G. Ouyang, A Biocatalytic Cascade in an Ultrastable Mesoporous Hydrogen-Bonded Organic Framework for Point-of-Care Biosensing, Angew. Chem., Int. Ed., 2021, 60(44), 23608–23613 CrossRef CAS PubMed.
- G. Chen, S. Huang, X. Ma, R. He and G. Ouyang, Encapsulating and stabilizing enzymes using hydrogen-bonded organic frameworks, Nat. Protoc., 2023, 18(7), 2032 CrossRef CAS PubMed.
- J. C. Quilles Junior, A. L. Ferrarezi and J. P. Borges, et al. , Hydrophobic adsorption in ionic medium improves the catalytic properties of lipases applied in the triacylglycerol hydrolysis by synergism, Bioprocess Biosyst. Eng., 2016, 39(12), 1933–1943 CrossRef PubMed.
- S. Pang, Y. Wu, X. Zhang, B. Li, J. Ouyang and M. Ding, Immobilization of laccase via adsorption onto bimodal mesoporous Zr-MOF, Process Biochem., 2016, 51(2), 229–239 CrossRef CAS.
- M. Mohammadi, M. Shahedi and F. Ahrari, et al. , Isocyanide-based multi-component reactions for carrier-free and carrier-bound covalent immobilization of enzymes, Nat. Protoc., 2023, 18(5), 1641–1657 CrossRef CAS PubMed.
- T. Wu, S. Huang, H. Yang, N. Ye, L. Tong, G. Chen, Q. Zhou and G. Ouyang, Bimetal Biomimetic Engineering Utilizing Metal-Organic Frameworks for Superoxide Dismutase Mimic, ACS Mater. Lett., 2022, 4(4), 751–757 CrossRef CAS.
- H. An, M. Li, J. Gao, Z. Zhang, S. Ma and Y. Chen, Incorporation of biomolecules in Metal-Organic Frameworks for advanced applications, Coord. Chem. Rev., 2019, 384, 90–106 CrossRef CAS.
- S. Huang, X. Kou, J. Shen, G. Chen and G. Ouyang, Armor-Plating Enzymes with Metal-Organic Frameworks (MOFs), Angew. Chem., Int. Ed., 2020, 59(23), 8786–8798 CrossRef CAS PubMed.
- J. Andersen and J. Mack, Mechanochemistry and organic synthesis: from mystical to practical, Green Chem., 2018, 20(7), 1435–1443 RSC.
- S. Głowniak, B. Szczęśniak, J. Choma and M. Jaroniec, Mechanochemistry: Toward green synthesis of metal-organic frameworks, Mater. Today, 2021, 46, 109–124 CrossRef.
- F. Afshariazar and A. Morsali, The unique opportunities of mechanosynthesis in green and scalable fabrication of metal-organic frameworks, J. Mater. Chem. A, 2022, 10(29), 15332–15369 RSC.
- O. Lapshin and O. Ivanova, Macrokinetics of mechanochemical synthesis in heterogeneous systems: Mathematical model and evaluation of thermokinetic constants, Mater. Today Commun., 2021, 28, 102671 CrossRef CAS.
- G. Heinicke and H. P. Hennig, Tribochemistry. Akademie-Verlag, 1984 Search PubMed.
- S. Lukin, L. S. Germann, T. Friscic and I. Halasz, Toward Mechanistic Understanding of Mechanochemical Reactions Using Real-Time In Situ Monitoring, Acc. Chem. Res., 2022, 55(9), 1262–1277 CrossRef CAS PubMed.
- K. Uzarevic, N. Ferdelji, T. Mrla, P. A. Julien, B. Halasz, T. Friscic and I. Halasz, Enthalpy vs. friction: heat flow modelling of unexpected temperature profiles in mechanochemistry of metal-organic frameworks, Chem. Sci., 2018, 9(9), 2525–2532 RSC.
- I. Brekalo, W. Yuan, C. Mottillo, Y. Lu, Z. Y. hang, J. Casaban, K. T. Holman, S. L. James, F. Duarte and P. A. Williams, et al. , Manometric real-time studies of the mechanochemical synthesis of zeolitic imidazolate frameworks, Chem. Sci., 2020, 11(8), 2141–2147 RSC.
- I. Halasz, S. A. Kimber, P. J. Beldon, A. M. Belenguer, F. Adams, V. Honkimaki, R. C. Nightingale, R. E. Dinnebier and T. Friscic, In situ and real-time monitoring of mechanochemical milling reactions using synchrotron X-ray diffraction, Nat. Protoc., 2013, 8(9), 1718 CrossRef PubMed.
- T. Friscic, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. Kimber, V. Honkimaki and R. E. Dinnebier, Real-time and in situ monitoring of mechanochemical milling reactions, Nat. Chem., 2013, 5(1), 66 CrossRef CAS PubMed.
- P. F. M. de Oliveira, A. A. L. Michalchuk, A. G. Buzanich, R. Bienert, R. M. Torresi, P. H. C. Camargo and F. Emmerling, Tandem X-ray absorption spectroscopy and scattering for in situ time-resolved monitoring of gold nanoparticle mechanosynthesis, Chem. Commun., 2020, 56(71), 10329–10332 RSC.
- M. Tireli, M. Juribasic Kulcsar, N. Cindro, D. Gracin, N. Biliskov, M. Borovina, M. Curic, I. Halasz and K. Uzarevic, Mechanochemical reactions studied by in situ Raman spectroscopy: base catalysis in liquid-assisted grinding, Chem. Commun., 2015, 51(38), 8058–8086 RSC.
- X. Ma, W. Yuan, S. E. J. Bell and S. L. James, Better understanding of mechanochemical reactions: Raman monitoring reveals surprisingly simple ‘pseudo-fluid’ model for a ball milling reaction, Chem. Commun., 2014, 50, 1585–1587 RSC.
- J. G. Schiffmann, F. Emmerling, I. C. B. Martins and L. Van Wullen, In-situ reaction monitoring of a mechanochemical ball mill reaction with solid state NMR, Solid State Nucl. Magn. Reson., 2020, 109, 101687 CrossRef CAS PubMed.
- A. A. L. Michalchuk and F. Emmerling, Time-Resolved In Situ Monitoring of Mechanochemical Reactions, Angew. Chem., Int. Ed., 2022, 61(21), e202117270 CrossRef CAS PubMed.
- S. Tanaka, K. Kida, T. Nagaoka, T. Ota and Y. Miyake, Mechanochemical dry conversion of zinc oxide to zeolitic imidazolate framework, Chem. Commun., 2013, 49(72), 7884–7886 RSC.
- W. Wang, M. Chai, M. Y. Bin Zulkifli, K. Xu, Y. Chen, L. Wang, V. Chen and J. Hou, Metal-organic framework composites from a mechanochemical process, Mol. Syst. Des. Eng., 2023, 8(5), 560 RSC.
- M. Taheri, I. D. Bernardo, A. Lowe, D. R. Nisbet and T. Tsuzuki, Green Full Conversion of ZnO Nanopowders to Well-Dispersed Zeolitic Imidazolate Framework-8 (ZIF-8) Nanopowders via a Stoichiometric Mechanochemical Reaction for Fast Dye Adsorption, Cryst. Growth Des., 2020, 20(4), 2761 CrossRef CAS.
- C.-A. Tao and J.-F. Wang, Synthesis of Metal Organic Frameworks by Ball-Milling, Crystals, 2021, 11(1), 15 CrossRef CAS.
- P. K. Lam, T. H. Vo, J.-H. Chen, S.-W. Lin, C.-L. Kuo, J.-J. Liao, K.-Y. Chen, S.-R. Huang, D. Li and Y.-H. Chang, et al., A green and ultrafast one-pot mechanochemical approach for efficient biocatalyst encapsulation in MOFs: insights from experiments and computation, J. Mater. Chem. A, 2023, 11(45), 24678 RSC.
- M. AmanNejad and K. Barani, Effects of Ball Size Distribution and Mill Speed and Their Interactions on Ball Milling Using DEM, Miner. Process. Extr. Metall. Rev., 2020, 42(6), 374 CrossRef.
- O. F. Jafter, S. Lee, J. Park, C. Cabanetos and D. Lungerich, Navigating Ball Mill Specifications for Theory-to-Practice Reproducibility in Mechanochemistry, Angew. Chem., Int. Ed., 2024, e202409731 CAS.
- H. C. Zhou and S. Kitagawa, Metal-organic frameworks (MOFs), Chem. Soc. Rev., 2014, 43(16), 5415–5418 RSC.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Development of a Cambridge Structural Database Subset: A Collection of Metal-Organic Frameworks for Past, Present, and Future, Chem. Mater., 2017, 29(7), 2618–2625 CrossRef CAS.
- G. Chen, S. Huang, X. Kou, F. Zhu and G. Ouyang, Embedding Functional Biomacromolecules within Peptide-Directed Metal-Organic Framework (MOF) Nanoarchitectures Enables Activity Enhancement, Angew. Chem., Int. Ed., 2020, 59(33), 13947–13954 CrossRef CAS PubMed.
- O. Yaghi, M. O'Keeffe and N. Ockwig, et al., Reticular synthesis and the design of new materials, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- X. C. Huang, Y. Y. Lin, J. P. Zhang and X. M. Chen, Ligand-directed strategy for zeolite-type metal-organic frameworks: zinc (II) imidazolates with unusual zeolitic topologies, Angew. Chem., Int. Ed., 2006, 45(10), 1557–1559 CrossRef CAS PubMed.
- W. Morris, C. J. Doonan and H. Furukawa, et al., Crystals as molecules: postsynthesis covalent functionalization of zeolitic imidazolate frameworks, J. Am. Chem. Soc., 2008, 130(38), 12626–12627 CrossRef CAS PubMed.
- T. H. Wei, S. H. Wu, Y. D. Huang, W. S. Lo, B. P. Williams, S. Y. Chen, H. C. Yang, Y. S. Hsu, Z. Y. Lin and X. H. Chen, et al., Rapid mechanochemical encapsulation of biocatalysts into robust metal-organic frameworks, Nat. Commun., 2019, 10(1), 5002 CrossRef PubMed.
- D. Ge, M. Li, D. Wei, N. Zhu, Y. Wang, M. Li, Z. Zhang and H. Zhao, Enhanced activity of enzyme encapsulated in hydrophilic metal-organic framework for biosensing, Chem. Eng. J., 2023, 469, 144067 CrossRef CAS.
- F. K. Shieh, S. C. Wang, C. I. Yen, C. C. Wu, S. Dutta, L. Y. Chou, J. V. Morabito, P. Hu, M. H. Hsu and K. C. Wu, et al., Imparting functionality to biocatalysts via embedding enzymes into nanoporous materials by a de novo approach: size-selective sheltering of catalase in metal-organic framework microcrystals, J. Am. Chem. Soc., 2015, 137(13), 4276–4279 CrossRef CAS PubMed.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, Rod Packings and Metal-Organic Frameworks Constructed from Rod-Shaped Secondary Building Units, J. Am. Chem. Soc., 2005, 127(5), 1504–1518 CrossRef CAS PubMed.
- P. A. Julien, K. Uzarevic, A. D. Katsenis, S. A. Kimber, T. Wang, O. K. Farha, Y. Zhang, J. Casaban, L. S. Germann and M. Etter, et al., In Situ Monitoring and Mechanism of the Mechanochemical Formation of a Microporous MOF-74 Framework, J. Am. Chem. Soc., 2016, 138(9), 2929–2932 CrossRef CAS PubMed.
- P. H. Hsu, C. C. Chang, T. H. Wang, P. K. Lam, M. Y. Wei, C. T. Chen, C. Y. Chen, L. Y. Chou and F. K. Shieh, Rapid Fabrication of Biocomposites by Encapsulating Enzymes into Zn-MOF-74 via a Mild Water-Based Approach, ACS Appl. Mater. Interfaces, 2021, 13(44), 52014–52022 CrossRef CAS PubMed.
- E. Ozyilmaz and O. Caglar, Rapid mechanochemical production of biocomposites by encapsulating enzymes to zinc based-two metal organic frameworks (ZIF-8 and Zn-MOF-74) for enantioselective hydrolysis reaction of racemic Naproxen methyl ester, Process Biochem., 2023, 134, 276–285 CrossRef CAS.
- J. H. Cavka, S. Jakobsen and U. Olsbye, et al., A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability, J. Am. Chem. Soc., 2008, 130(42), 13850–13851 CrossRef PubMed.
- X. Liu, Metal-organic framework UiO-66 membranes, Front. Chem. Sci. Eng., 2019, 14(2), 216–232 CrossRef.
- S. Vandenbrande, T. Verstraelen, J. J. Gutierrez-Sevillano, M. Waroquier and V. Van Speybroeck, Methane Adsorption in Zr-Based MOFs: Comparison and Critical Evaluation of Force Fields, J. Phys. Chem. C, 2017, 121(45), 25309–25322 CrossRef CAS PubMed.
- W. Liang, H. Xu, F. Carraro, N. K. Maddigan, Q. Li, S. G. Bell, D. M. Huang, A. Tarzia, M. B. Solomon and H. Amenitsch, et al., Enhanced Activity of Enzymes Encapsulated in Hydrophilic Metal-Organic Frameworks, J. Am. Chem. Soc., 2019, 141(6), 2348–2355 CrossRef CAS PubMed.
- F. Yang, H. H. Xie, F. Du, X. Hou and S. F. Tang, Insight into the efficient loading and enhanced activity of enzymes immobilized on functionalized UiO-66, Int. J. Biol. Macromol., 2024, 279, 135557 CrossRef PubMed.
- Y. Hu, L. Dai, D. Liu and W. Du, Rationally designing hydrophobic UiO-66 support for the enhanced enzymatic performance of immobilized lipase, Green Chem., 2018, 20(19), 4500–4506 RSC.
- P. K. Tuned to Perfection, Ironing Out the Defects in Metal-Organic Framework UiO-66, Chem. Mater., 2014, 26(14), 4068–4071 CrossRef.
- J. Cui, C. Zhang, H. Liu, L. Yang, X. Liu, J. Zhang, Y. Zhou, J. Zhang and X. Yan, Pulmonary Delivery of Recombinant Human Bleomycin Hydrolase Using Mannose-Modified Hierarchically Porous UiO-66 for Preventing Bleomycin-Induced Pulmonary Fibrosis, ACS Appl. Mater. Interfaces, 2023, 15(9), 11520–11535 CrossRef CAS PubMed.
- R. Ahmad, J. Shanahan, S. Rizaldo, D. S. Kissel and K. L. Stone, Co-immobilization of an Enzyme System on a Metal-Organic Framework to Produce a More Effective Biocatalyst, Catalysts, 2020, 10(5), 499 CrossRef CAS.
- S. Wang, W. Liu, J. Wang, J. Yu, F. Wang, C. Jin, K. Wang, P. Cheng, Z. Zhang and Y. Chen, Mechanochemical Encapsulation of Enzymes into MOFs for Photoenzymatic Enantioselective Catalysis, ACS Mater. Lett, 2024, 6(7), 2609–2616 CrossRef CAS.
- A. P. Côté, et al., Porous, Crystalline, Covalent Organic Frameworks, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- N. Huang, P. Wang and D. Jiang, Covalent organic frameworks: a materials platform for structural and functional designs, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- M. Li, S. Qiao, Y. Zheng, Y. H. Andaloussi, X. Li, Z. Zhang, A. Li, P. Cheng, S. Ma and Y. Chen, Fabricating Covalent Organic Framework Capsules with Commodious Microenvironment for Enzymes, J. Am. Chem. Soc., 2020, 142(14), 66756681 Search PubMed.
- R. Gao, N. Zhong, L. Tong, X. Kou, W. Huang, H. Yang, S. Huang, J. Wu, G. Chen and G. Ouyang, Mechanochemistry-guided reticular assembly for stabilizing enzymes with covalent organic frameworks, Cell Rep. Phys. Sci., 2022, 3(12), 101153 CrossRef CAS.
- W. J. Li, Y. M. Li, H. Ren, C. Y. Ji and L. Cheng, Improving the Bioactivity and Stability of Embedded Enzymes by Covalent Organic Frameworks, ACS Appl. Mater. Interfaces, 2023, 15(37), 43580–43590 CrossRef CAS PubMed.
- V. A. Russell, C. C. Evans and W. J. Li, et al., Nanoporous Molecular Sandwiches: Pillared Two-Dimensional Hydrogen-Bonded Networks with Adjustable Porosity, Science, 1997, 276, 575–579 CrossRef CAS PubMed.
- R. B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Multifunctional porous hydrogen-bonded organic framework materials, Chem. Soc. Rev., 2019, 48(5), 1362–1389 RSC.
- G. Chen, S. Huang, Y. Shen, X. Kou, X. Ma, S. Huang, Q. Tong, K. Ma, W. Chen and P. Wang, et al., Protein-directed, hydrogen-bonded biohybrid framework, Chem, 2021, 7(10), 2722–2742 CAS.
- W. Huang, H. Yuan, H. Yang, L. Tong, R. Gao, X. Kou, J. Wang, X. Ma, S. Huang and F. Zhu, et al., Photodynamic Hydrogen-Bonded Biohybrid Framework: A Photobiocatalytic Cascade Nanoreactor for Accelerating Diabetic Wound Therapy, JACS Au, 2022, 2(9), 2048–2058 CrossRef CAS PubMed.
- S. Huang, J. Li, Y. Lin, L. Tong, N. Zhong, A. Huang, X. Ma, S. Huang, W. Yi and Y. Shen, et al., Hydrogen-Bonded Supramolecular Nanotrap Enabling the Interfacial Activation of Hosted Enzymes, J. Am. Chem. Soc., 2024, 146(3), 1967–1976 CrossRef CAS PubMed.
- W. K. Qin, D. H. Si, Q. Yin, X. Y. Gao, Q. Q. Huang, Y. N. Feng, L. Xie, S. Zhang, X. S. Huang and T. F. Liu, et al., Reticular Synthesis of Hydrogen-Bonded Organic Frameworks and Their Derivatives via Mechanochemistry, Angew. Chem., Int. Ed., 2022, 61(27), e202202089 CrossRef CAS PubMed.
- H. Chen, D. Feng, F. Wei, F. Guo and A. K. Cheetham, Hydrogen-Bond-Regulated Mechanochemical Synthesis of Covalent Organic Frameworks: Cocrystal Precursor Strategy for Confined Assembly, Angew. Chem., Int. Ed., 2024, e202415454 Search PubMed.
- Y. Hu, W.-K. Han, Y. Liu, R.-M. Zhu, X. Yan, H. Pang and Z.-G. Gu, Mechanochemical Transition from a Hydrogen-Bonded Organic Framework to Covalent Organic Frameworks, ACS Mater. Lett., 2023, 5(9), 2534 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |