
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring tribochemical transduction pathways for dehydrogenation of molecular hydrides†
Esmeralda
Muñoz-Cortés
 a,
Fabrice
Leardini
a,
Marcello
Conte
b,
Adolfo del
Campo
c,
Eduardo
Flores
d,
Jose Ramon
Ares
a and
Roman
Nevshupa
*e
a,
Fabrice
Leardini
a,
Marcello
Conte
b,
Adolfo del
Campo
c,
Eduardo
Flores
d,
Jose Ramon
Ares
a and
Roman
Nevshupa
*e
aMIRE Group, Department of Physics of Materials, Autonomous University of Madrid, Madrid, Spain
bRTec Instruments SA, Orbe, Switzerland
cInstitute of Ceramics and Glass, CSIC, Madrid, Spain
dApplied Physics Department, Centre for Research and Advanced Studies (CINVESTAV), Mérida 97310, Mexico
eEduardo Torroja Institute of Construction Sciences, CSIC, Madrid, Spain. E-mail: r.nevshupa@csic.es; Tel: +34-911035746
First published on 4th January 2025
Abstract
Recent research has shown that mechanical energy can trigger dehydrogenation (hydrogen release) from metal and complex hydrides at room temperature, offering an alternative to traditional heat-based methods. This study investigates whether the tribochemical approach can also be effective to release hydrogen from molecular hydrides such as ethane 1,2-diamineborane (EDAB). Surprisingly, despite dehydrogenating at a lower temperature than metal and complex hydrides, EDAB exhibited faint hydrogen release under mechanical stress. To understand this behavior, the tribochemical decomposition pathways of EDAB were investigated using operando Mechanically Stimulated Gas Emission Mass Spectrometry in combination with other surface and material characterization techniques. The lack of hydrogen emission from EDAB is attributed to a combination of strong intramolecular bonds (covalent and dative bonds) within the molecule, and weak intermolecular interactions (hydrogen bonds and van der Waals forces) between EDAB molecules.
1. Introduction
The growing focus on renewable and low-carbon technologies in the current energy transition has placed significant hope on hydrogen. Hydrogen's versatility and near-zero emissions during use make it a promising candidate across various applications. Governmental strategies also highlight its potential for bridging the gap between renewable energy generation and consumption.1 It can act as a storage vector alongside batteries, enabling energy transport and mitigating seasonal variations in energy production and demand.However, widespread adoption of hydrogen technology faces several challenges. These include improving the efficiency and effectiveness of hydrogen storage and transportation. Storing hydrogen through chemical bonds in condensed-phase materials offers a viable approach for on-demand power generation, especially for mobile applications. This method can meet established targets for range, passenger space, and cargo capacity.2 However, faster hydrogenation/dehydrogenation kinetics are needed to meet refueling time expectations. Additionally, optimizing thermodynamics is crucial to minimize dehydrogenation temperatures and enhance overall performance.3
In this context, ammonia borane (AB) emerges as a promising candidate due to its 12 wt% hydrogen content and relatively low dehydrogenation temperature range (107–140 °C).4,5 However, its room-temperature volatilization and rapid sublimation under vacuum limit its practicality (see also ESI†).6 A notable improvement of AB thermal stability was achieved through adduction of alkane, which substitutes one of the protic hydrogens.7 This modification not only reduces dehydrogenation exothermicity and stabilizes the B–N bond, but also minimizes volatile nitrogen impurities like ammonia, a significant drawback for practical applications.2 Ethane 1,2-diamineborane (EDAB), the dimer form of AB methyl adduct, with the chemical formula BH3NH2(CH2)2NH2BH3 appears to strike the good balance between thermal stability and low room-temperature volatility offering a 9.4 wt% H2 storage capacity.
Balancing optimal thermodynamics between storage temperature stability and low dehydrogenation temperatures presents a key limitation in solid-state hydrogen storage. The current approach necessitates heating the storage material to release hydrogen, which is energy-intensive. An alternative approach could involve non-thermal dehydrogenation pathways.8 Several studies explored the effectiveness of tribochemical dehydrogenation for light metal (MgH2)9 and complex (NaAlH4)10 hydrides. These studies demonstrated successful dehydrogenation of these stable hydrides at room temperature and under vacuum using mild tribological activation (rubbing and shearing). This finding holds significant practical promise. It opens the possibility of utilizing readily available mechanical energy sources like vibrations, wind, and waves to extract hydrogen from hydrides without the need for energy transducers or thermal steps, potentially leading to a reduction in the cost of delivered hydrogen.
The precise molecular mechanisms of these tribochemical reactions remain unclear, but are likely associated with factors such as structural and phase transformations,11 generation of stress and concentration gradients,12 and formation of voids and defects within the crystal structure. Since these mechanisms depend heavily on the hydride's specific crystalline structure and bonding, a key question arises: are other hydride types, such as molecular hydrides like EDAB, susceptible to tribochemical dehydrogenation? This work aims to explore the feasibility of triggering EDAB dehydrogenation through a tribochemical mechanism and investigate the potential steps involved in such reactions.
2. Materials and methods
Cylindrical pellets 13 mm in diameter and ca. 2 mm thick were obtained by compacting, under uniaxial pressure of 65 MPa, of commercial powder of EDAB (Boron Specialties, 97% purity). The pellets were prepared and conserved under a static Ar atmosphere before and after the tests. X-ray diffraction (Cu Kα radiation, θ/2θ configuration, 10°–60° 2θ range) was used to analyze the pellet structure. Thermal gravimetric analysis coupled with mass spectrometry (TGA-MS) under argon flow (heating rate: 10 °C min−1) was used to evaluate thermal stability and the composition of emitted gases. During the isothermal test the required temperature was attained at a heating rate 10 °C min−1 and then maintained constant for 90–380 min depending on the temperature. The ion currents were measured in the range of mass-to-charge ratios from 1 to 90.Micro-Raman confocal spectrometry (incident laser: 532 nm, power: 5.6 mW) and FTIR microscopy was used to analyze the chemical structure of mechanically affected zones compared to pristine areas. It was conducted under normal atmosphere. In-plane thermal conductivity of EDAB pellets was determined using van der Pauw method.13
Nanoindentation with a Berkovich diamond tip (load: 5–20 mN, peak load hold time: 120 s, loading/unloading rates: 4–40 mN min−1, 10 measurements for each load) was used to characterize mechanical properties.
Tribochemical reactions under vacuum were studied in real time using Mechanically Stimulated Gas Emission Mass Spectrometry (MSGE-MS) which was described in detail elsewhere.14–16Operando mass-spectrometry of gases emitted due to tribological solicitation has been used as a powerful tool to investigate into the pathways of complex tribochemical processes in lubricants,16–25 thin films,8,26–28 engineering materials16,29–36 and minerals.37–41 This technique combines a mass-spectrometry, a nearly zero-emission ultrahigh vacuum friction cell and a dynamic gas expansion system. The latter allows for accurate quantification of emitted gas rates with the detection limit around 1 pmol s−1.15,16,31,42 The tribostimulation of the pellets was done using an alumina ball, 3 mm in diameter, under normal load 0.44–1.1 N and the frequency of reciprocating motion 1 full cycle (forward and backward strokes) per second. The additional arrangements which were made to avoid undesirable gas emission sources other than triboemission from mechanically affected zones of the sample are described in ESI.† The tribostimulated gas emission was determined from the mass-spectrometry signal and benchmarked against the steady background gas pressure with the indenter standing still. The Differential Mass-Spectra (DMS) were derived by subtracting the reference spectrum from the mass-spectra measured during tribostimulation. The statistically insignificant components of DMS were filtered out following a previously developed procedure.16,23,43,44 The behavioural analysis of mass-spectroscopic components was employed to figure out the plausible gaseous precursors for the mass-spectrometer signals.21,25 For a detailed description of the MSGE-MS system and the principles of DMS analysis, refer to ref. 14, 16, 21, 25, 28, 30 and 31.
A separate experiment to investigate possible chemical changes in the EDAB under applied mechanical deformation was conducted using Nuclear Magnetic Resonance (NMR). For this experiment about 0.5 g of EDAB powder was grounded in a porcelain mortar just prior to the NMR analysis. A portion of the ground EDAB was analyzed using solid state Magic Angle Spinning (MAS) 1H and 11B NMR, while the other one was analyzed using 1H and 11B solution RMN using deuterated dimethyl sulfoxide (DMSO-d6) as a solvent.
Magic angle spinning nuclear magnetic resonance analysis (MAS-NMR) was conducted with a Bruker AVANCE 400 spectrometer (9.4 T magnetic field) at 128.38 and 400.13 MHz for 11B and 1H, respectively. Spectra were obtained after single pulse irradiation of 2 μs with a recycling time of 10 s for 11B and a pulse of 3 μs and 20 s of recycling for 1H signals. Samples were spun in 4 mm ZrO2 rotors at 10 kHz around an axis inclined at 54°44′ with respect to the magnetic field. The number of scans was chosen to get signal/noise ratios higher than 10 (40 and 16 for 11B and 1H, respectively). Values of 11B and 1H chemical shifts were reported in ppm relative to 0.3 M H3BO3 aqueous solutions and tetramethylsilane (TMS) respectively.
3. Results and discussion
3.1. Initial structural characterization
XRD showed that the compacted pellets retained the same orthorhombic Pbca structure as the original EDAB powder (see Fig. S1 in ESI†).2 This outcome was expected as the applied compressive stress during pellets' compaction (554 MPa) was below a pressure threshold (1 GPa) required to induced an orthorhombic – to – monoclinic phase transition.453.2. Analysis of mechanically stimulated gas emission
The DMS spectrum showing the statistically significant components, hereinafter referred to as filtered DMS, is presented in Fig. 1. Notably, the intensity of the H2+ signal was surprisingly low, with only a slight increase above the noise level observed during rubbing. This is in stark contrast to previous studies involving significantly more stable metal hydrides like MgH2 and NaAlH4,9,10,16,46 where hydrogen was the predominant emission component.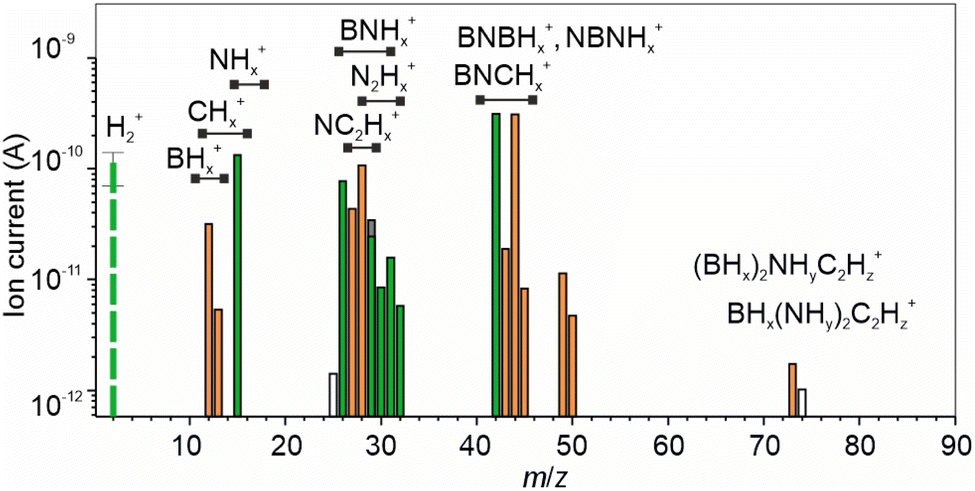 | ||
| Fig. 1 Mean differential mass-spectrum (DMS) of volatile products emitted from EDAB pellets subjected to rubbing under vacuum at room temperature. The species following coupled and uncoupled behaviours are highlighted in orange and green, correspondingly. | ||
Our study did not detect any molecular ions of EDAB (m/z 88) in the DMS. This is interesting because Massimi et al.47 reported faint signals at m/z 87 and 88 during the initial stages of EDAB pyrolysis at 70 °C, which disappeared at higher temperatures. These signals were potentially linked to EDAB molecules. The absence of a signal at m/z 88 in our study most likely indicates that tribostimulation was not a cause of EDAB sublimation. It's worth noting that no peaks suggestive of EDAB sublimation were observed in the analysis of residual gases at room temperature, i.e. when the EDAB pellet was placed in vacuum without rubbing (see Fig. S7 in ESI†).
The principal components in the DMS were m/z 42 and 44. The peak at m/z 42 has been previously assigned to ionized fragments of molecules with a B–N–B (aminodiborane)48 and/or N–B–N backbone. The m/z 44 peak, which appeared as the main component of EDAB thermolysis,4,5 is attributed to splitting of a C–C bond. The components at m/z 12, 13, and 15 primarily arose from BHx+ ions encompassing both 10B and 11B isotopes with a minor contribution from CHx+ and NHx+. Within the range of signals at m/z 25 to 32 the associations can be made with 11BNHx+, CNHx+ and C2Hx+ ions.49 The lack of signals at m/z 24 and 23, which correspond to 11B2H2+ and 11B2H+, effectively rules out the existence of diborane. It's noteworthy that in the reference mass spectrum of diborane, the signal at m/z 24 is about 90% of the main component 11B2H4+ (m/z 26). The component at m/z 32 is more likely associated with hydrazine (N2H4+). Lastly, the signals observed at m/z 73 and 74 can be attributed to EDAB ions originated from scission of one B–N bond such as 11BH3NH2(CH2)2NH2+, or analogues species. The presence of both BHx+ (x = 1,2) and 11BH3NH2(CH2)2NH2+ supports this hypothesis. The association of faint signals at m/z 49 and 50 remains unclear at this point and requires further investigation. The assignments of the DMS constituents are detailed in Table S1 in ESI.†
Behavioural analysis (BA) of the mass spectrometry time-series provided further insight into the origin and composition of the emitted gases. The BA has been proven a powerful tool for analyzing gas emission processes of different dynamics.21,22,25,27,28 In this study, the time series displayed two distinct patterns depending on their response to tribostimulation. Tribostimulation can trigger either coupled or uncoupled gas emission behavior. Coupled behavior, as illustrated in Fig. 2a, shows gas emission starting with the onset of tribostimulation and stopping when it ceases. Conversely, uncoupled behavior (Fig. 2b) is characterized by an initial burst of gas emission followed by a decline even as tribostimulation continues. This uncoupled behavior might be explained by the material in the affected zone becoming activated during the very first rubbing cycle, with subsequent rubbing having little additional effect, or by the presence of surface layers or thin films.16,30 In contrast, coupled behavior likely relates to the ongoing process of shear deformation, which persists throughout tribostimulation.
 | ||
| Fig. 2 Representative time series of mass spectrometric signals: (a) coupled behaviour showing continued emission throughout the tribostimulation; (b) uncoupled behaviour, which is triggered by the very first rubbing cycles. After the end of rubbing the signal decays gradually. | ||
The DMS in Fig. 1 highlights signals exhibiting coupled and uncoupled behavior with orange and green colours, respectively. Components showing coupled behaviour likely correspond to aminoborane, ammonia borane, ethylamine, and the EDAB decomposition products resulting from B–N bond (BHx and BN2C2Hy) and C–C bond cleavage. In contrast, uncoupled behaviour is likely linked to hydrazine, molecules with an N–B–N backbone, and hydrogen. These compounds are potentially formed through bond scission and recombination.
3.3. Surface and chemical characterization of the mechanically affected zone
A high-resolution secondary electron image of the Mechanically Affected Zone (MAZ) (Fig. 3) reveals a markedly irregular surface morphology. Additionally, periodic micrometer-scale wrinkles are observed, oriented perpendicular to the sliding direction. These wrinkles bear a strong resemblance to Schallamach waves, which are known to form in front of a smooth, hard solid sliding against a soft, adhesive elastomer.50,51 The formation mechanism of Schallamach waves involves a buckling instability of the deformed elastomer surface, followed by detachment of a pressed elastomer surface layer. | ||
| Fig. 3 Secondary electron image of the sample after the friction test obtained under low vacuum. Dashed lines show the limits of the Mechanically Affected Zone (MAZ). The structures indicated by the blue arrows are associated with plastic flow. The inset shows a cross-section of the MAZ along x measured using a white-light confocal microscope. aw and dw are the half width and the depth of the MAZ, respectively. | ||
The observed surface morphology suggests that EDAB experienced intensive plastic deformation throughout the MAZ. This deformation likely involved a combination of shearing processes within the surface layer and compressive flow within microscopic wrinkles. This finding is particularly noteworthy considering that EDAB pellets, obtained through powder pressing, typically exhibit weak inter-granular bonding. Consequently, plastic deformation in such materials is usually localized within individual grains, with fracture occurring along grain boundaries. Notably, the pile-up features observed on the sides of the MAZ were significantly less pronounced compared to those observed for metals,30 light metal hydrides,9,16 and complex hydrides.10,46 This further strengthens the hypothesis that the wear behaviour of EDAB exhibits greater similarity to elastomers than to crystalline solids.
The volume of the groove generated on the pellet surface due to rubbing was used as a low-bound approximation of the total deformed volume. The average cross-sectional area of the groove, Aw, was determined by calculating the mean value of numerical integral on cross-sectional profiles (as depicted in the inset of Fig. 3) obtained at various locations along the MAZ. The average volume of displaced or worn material, Vw, was then calculated as the product of Aw and the groove length, l. For the groove produced after 2000 rubbing cycles, Vw was found to be 1.9 × 10−5 cm3. Subsequently, the mass of the removed material, mw, was estimated by multiplying Vw by the density, ρ, of EDAB (ρ = 0.82 g cm−3), resulting in a value of approximately 15.6 μg.
Fig. 4(b) shows representative Raman spectra acquired from the surface layer (blue) and the interior (red) of the pristine region of the pellet. Fig. 4a depicts the corresponding spectral mapping across the pristine region's cross-section. Both spectra exhibit similar vibrational bands, which have been previously assigned to the primary vibrational modes of the EDAB molecule.4,5,52,53 However, slight variations in the relative intensities of specific bands are observed. Notably, the surface layer exhibits a decrease in the intensity of bands located at 2292 cm−1, 1606 cm−1, and 1372 cm−1, which are associated with υB–H, δNH2, and δNH3/νB–N vibration modes, respectively.54
 | ||
| Fig. 4 (a) Mapping of Raman features along the cross section of pristine surface of the EDAB pellet obtained using micro-Raman confocal spectrometry. The red and blue colours represent zones with dominant Raman spectra (i) and (ii), respectively.; (b) the representative Raman spectra corresponding to the surface layer (blue), volume (red) and MAZ (black). | ||
Interestingly, these observed changes in the surface layer mirror the transformations documented during ammonia borane heating above 95 °C, a stage preceding the initial hydrogen release at 109 °C.52,55 Similar transformations have also been reported under high pressures exceeding 1 GPa in a diamond anvil cell.45 However, it's highly improbable that the residual stress in a soft material like EDAB could remain after cold pressing as high as 1 GPa. Furthermore, several bands in the surface layer spectra exhibit significant broadening and shoulders on the blue side. Collectively, these findings suggest that the surface layer likely formed during the cold pressing of EDAB powder and might possess a distorted and strained crystalline structure. It can potentially be described as a “frozen” metastable state resembling the state observed just before the first thermal decomposition step.
In contrast, no significant alterations in the Raman spectrum were observed within the MAZ compared to the bulk EDAB. This finding is unexpected considering the substantial plastic deformation revealed by microscopy analysis. The only minor changes detected were increased intensities of bands within the 1000–1650 cm−1 range, which are attributed to BH, BN and NH vibrations. A more detailed examination of the MAZ surface revealed the presence of two distinct zone types: one characterized by sharp bands and another with broad bands (further details provided in Fig. S8 at the ESI†). These zones might potentially correspond to regions with varying degrees of strain.
The FTIR spectra revealed bands consistent with those of unprocessed EDAB (see Fig. S9 at the ESI for details†).56–58 Notably, no significant variations were observed that could be directly attributed to the mechanical treatment.
1H (400 MHz, DMSO-d6) and 11B (128 MHz, DMSO-d6) NMR spectroscopy of unprocessed (as received) EDAB and the mechanically treated EDAB aligned well with previously reported structures (see ESI for details†).59–62 Notably, the NMR spectra of the ground EDAB did not reveal any substantial changes indicative of alterations in the EDAB structure. For example, the 11B NMR spectrum of the ground material displayed only a broad resonance band at a chemical shift of δ = −20.6 ppm, which corresponds to the BH3 unit of EDAB.63,64 The resonance bands associated with BH4−, BH2 and N–B–N backbone molecules (diammoniates) were absent.2,63,64
3.4. Mechanical properties
The elastic modulus, Eit, and hardness, Hit, of the EDAB pellets were measured as a function of nanoindentation depth and are presented in Fig. 5(a). The elastic modulus exhibited a decrease from 3.7 GPa to 2.9 GPa as the penetration depth increased from 1.3 μm to 2.9 μm (corresponding to a load increase from 2 mN to 20 mN). Conversely, the hardness displayed a linear increase from 57 MPa to 143 MPa. Notably, the obtained values for both Hit and Eit fall within the corresponding ranges reported for the compressive yield strength and modulus of elasticity of thermosetting polymers such as polyamide-6,6, epoxy, and polyester.65,66Fig. 5(b) presents the plastic work, elastic work, and plasticity index of EDAB determined as the ratio of plastic work to the total work expended during nanoindentation. The plasticity index exhibits a gradual decrease from 0.9 to 0.8 with increasing penetration depth. This trend correlates with the observed substantial increase in hardness. In simpler terms, as the indenter penetrates deeper, the material becomes stiffer, requiring less plastic deformation to accommodate the indentation. However, it's important to note that plastic deformation remained the dominant deformation mode throughout the entire investigated range of penetration depths. To the best of our knowledge, this study represents the first report on the mechanical properties of EDAB. | ||
| Fig. 5 (a) Hardness and instrumental elastic modulus obtained from nanoindentation tests as function of the penetration depth. (b) Elastic, We, and plastic, Wpl, work expended during nanoindenation of EDAB pellets and corresponding plasticity index. | ||
3.5. The role of frictional heating
The transitional rise in temperature at the contact surfaces was estimated using Tian and Kennedy's heuristic formula derived from Blok's hypothesis for moving heat sources.67–69Table 1 summarizes the experimental parameters along with the mechanical and thermal properties of EDAB and alumina (the indenter material) employed in the calculations. The thermal conductivity of EDAB (λ2) measured in this study fell below the instrument's lowest measurable limit of 2 W (m−1 K−1). However, based on the observed similarities between EDAB and thermosetting polymers as well as ammonia borane, we estimated λ2 to be within the range of 0.1–0.45 W (m−1 K−1).65,66,70,71 Taking the load into account, the resulting flash temperature was estimated to range from 7.7 K to 15.5 K above ambient temperature, translating to values well below 315 K. This estimated temperature increase remains significantly lower than the threshold temperature required for the initial decomposition step4 of EDAB (394 K). Additionally, the short duration of the “hot spot” on the pellet surface (approximately 10 ms) renders it unlikely that frictional heating could significantly contribute to EDAB sublimation.44,723.6. Thermal gravimetric analysis (TGA) and mass spectrometry (MS) of emitted gases
TGA-MS was conducted to contrast the results of triboinduced EDAB dehydrogenation and to determine the thermodynamic parameters controlling EDAB thermolysis using an isothermal method. TGA-MS data for a constant heating rate are presented in the ESI.† The results generally align with previously reported data. They also provide some additional details regarding the influence of EDAB foaming on both the TGA data and the composition of the evolved gas species. Thermal decomposition agrees with the reactions proposed by Neiner et al.2 as shown in Scheme 1. | ||
| Scheme 1 Reactions of EDAB thermal decomposition.2 | ||
Fig. 6 shows the results of isothermal TGA-MS tests performed at 90, 100, and 110 °C. H2, ArH+ and water vapours were the main components detected in the mass-spectrometry data, in accordance with literature.4 These tests reveal that hydrogen emission starts well before the temperature reaches the point of decomposition observed during temperature ramping experiments (Fig. S4 in ESI†). For target temperature 90 °C, hydrogen emission begins to rise sharply between 75–85 °C and plateaus once the target temperature is reached. At 100 °C the H2 emission continued to grow gradually during the isothermal stage. The inflection point occurred at approximately 90 minutes and then H2 pressure linearly decreased. At 110 °C, hydrogen emission doesn't plateau at all. It continues to increase and reached the inflection point at about 25 minutes before rapidly decreasing to the background level. The time to inflection and the maximum pressure decreased with increasing temperature.
 | ||
| Fig. 6 The results of isothermal TGA-MS at 90 °C, 100 °C and 110 °C: (a) relative mass decrease, (b) variation of H2 partial pressure. The dashed lines in (a) depict the expected behaviours without foaming and bubbling. | ||
The pattern of H2 emission observed at 100 and 110 °C suggests that precursors involved in the initial solid-state EDAB decomposition reaction (shown in Scheme 1) are depleted along the tests. This idea is supported by calculations of the relative amount of H2 emitted per milligram of EDAB. To simplify comparison of H2 yield at different temperatures, the emitted H2 was quantified by integrating the mass spectrometry signal (H2+ ion current) over time. The relative amount of H2 per mass of EDAB is presented in the table of Fig. 6b, in units of nA min mg−1. Interestingly, the calculated values for 100 °C and 110 °C are very similar. However, the value at 90 °C is significantly lower, suggesting that the first-step dehydrogenation reaction wasn't complete at this temperature during the 330 minutes test. It's worth noting that after 40 min. TGA did not plateau, but the mass continued to decrease from 4.3% to 4.9%, indicating a slow, ongoing release of hydrogen as observed by mass spectrometry.
Another striking result of the isothermal tests is that EDAB decomposition occurred without induction period. In case of ammonia borane, below 120 °C the induction period was notable. It increased with decreasing temperature and reached 120 min at 90 °C.75
The total mass loss follows a similar trend for the initial 20 minutes, before foaming and bubbling significantly affect the measurement. The step-like changes and sharp peaks likely correspond to the formation and collapse of small bubbles accompanied by the release of molten EDAB droplets or solid particles. Similarly, the slow increase and decrease in mass loss observed at 100 and 110 °C can be attributed to the inflation and deflation of larger bubbles. Fig. 6a depicts the expected mass change behaviour without bubbling, represented by the dashed lines.
An isoconversional model-free method75,76 was employed to determine the activation energy, Ea, of the decomposition reaction. This method avoids the need to assume a specific reaction model. The reaction extent, α, i.e. the fraction of emitted H2 at a given time to the total amount of H2 emitted during the test, was calculated from mass-spectrometry data for each isothermal test (see Fig. 7a). These plots allowed us to determine the time, t, required to reach a given α values (between 0.05 and 0.5) at the three different temperatures. Next, Arrhenius plots were constructed for each α value (see Fig. S6 in ESI†). These plots depict ln(t) versus the reciprocal of temperature. The Ea for each α value was then obtained from the slope, b, of the corresponding linear fit in the Arrhenius plot:
| Ea = bR | (1) |
 | ||
| Fig. 7 (a) α-Time plots of the first step of EDAB decomposition, (b) activation energy for the first-step hydrogen emission obtained for various values of α using the isoconversional method. The red line is interpolation of the measured data using a fifth-degree polynomial function. The dashed line depicts the possible extrapolated trend of the data towards α = 0. | ||
As the decomposition reaction proceeds as indicated by increasing α values, the Ea gradually approaches 140 kJ mol−1 that is not so far from the reported values obtained using the Kissinger method (see Table 2). However, at the initial stages of decomposition (low α values), the Ea exhibits a significant decrease. At an α = 5%, the Ea is as low as 59 ± 5 kJ mol−1. Extrapolating this trend to the very beginning of the reaction (α = 0%), the Ea would theoretically reach around 21 kJ mol−1. The low decomposition temperatures for EDAB (75–95 °C) justify such low values of activation energy at low α. These results are consistent with the calorimetry data, which showed a shoulder in the range 75–112 °C.2
| Hydride | Enthalpy change: 1st/2nd step (kJ mol−1) | Activation energy (kJ mol−1) |
|---|---|---|
| a Kissinger method. b This study. c Integrating area under isothermal DSC curve. d Arrhenius plot. e Kissinger plot for nonisothermal DSC. f Isoconversional (model-free fitting) treatment of isothermal data. | ||
| EDAB | −(26 ± 4)/−(9 ± 3)4 | 185 ± 7/180 ± 3 (ref. 4)a |
| −(20 ± 1.5)/−(7.5 ± 1.2)2 | 172/—2 | |
| −20/−14 (ref. 5) | 59–132b | |
| AB | −21 (ref. 77) | 184 (ref. 78)c |
| 183 (ref. 79)d | ||
| 160 (ref. 80)e | ||
| 147.5/144.5 (ref. 75)a | ||
| 102/22.9 (ref. 75)f | ||
However, this behaviour contrasts with the findings reported for parent AB,75 where the highest activation barrier was observed at low α. This discrepancy can be attributed to the presence of an induction period in the case of AB. In contrast, EDAB does not exhibit such an induction period, leading to a lower initial energy barrier for decomposition. Gangal et al.75 attributed the progressive decrease of activation energy to the autocatalytic effect of exothermal reaction of AB decomposition. Since both AB and EDAB possess similar enthalpy changes for the first decomposition step (as shown in Table 2), this explanation seems unlikely for EDAB.
The shapes of the plots in Fig. 7a indicate that the solid-state reaction is of nearly constant rate, which tended to accelerate slightly with increasing temperature.76 Picking an appropriate reaction model is not easy because of the large variation of the Ea with α, which may indicate the existence of various parallel reactions. The distribution of activation energy values might be more likely related to the presence of defects within EDAB crystalline structure, which could potentially act as nucleation sites for decomposition.
3.7. Possible tribochemical processes
The analysis of minor emission gas components, which account for less than 6% (by ion current) of H2 emission, can be useful to elucidate the mechanisms of underlying tribochemical processes. The mass spectra of gases released during tribochemical decomposition differed significantly from those observed during thermal dehydrogenation.Additionally, vibrational spectroscopy and NMR analysis revealed no chemical changes in the material.
The 11B spectra showed a slight downfield shift of the broad resonance from −23.4 ppm (as-received EDAB) to −25.3 ppm (ground EDAB). In the 1H spectra, mechanical activation resulted in a decrease in intensity and broadening of the main EDAB resonances,81 along with the appearance of two additional resonances at 5 ppm and −0.5 ppm, assigned to pore water and adsorbed water, respectively.82 This is expected, as EDAB is highly hygroscopic. Grinding under ambient air for approximately two minutes resulted in water adsorption. The observed line broadening is consistent with residual stress induced by grinding.831H spin–lattice (T1) and spin–spin (T2) relaxation times were also measured for solid EDAB (Table S2 in ESI†). The values for untreated EDAB are consistent with literature values.84 Grinding decreased both T1 and T2, which can be attributed to increased disorder.85
These findings suggest that tribochemical degradation of EDAB doesn't follow the same pathway as thermal dehydrogenation (outlined in Scheme 1). The results obtained from MSGE-MS indicate the two different tribochemical processes (see Scheme 2). The first one is associated with the coupled behaviour of gas emission during rubbing. The release of gaseous products from EDAB can be attributed to bond scission, most likely involving C–C and N → B bonds. The homolytic N → B cleavage is barrierless as only the dative bond is elongated during the bond breaking. Its endothermic cleavage leads to neutral molecules (1) and (2).86,87 Conversely, homolytic cleavage of the covalent C–C bond can result in either a pair of radicals (3) or a pair of oppositely charged ions. Cleavage of C–C bond is less likely due to its much higher bond dissociation enthalpy (89.8 ± 0.5 kcal mol−1) compared to the N → B bond (31.1 kcal mol−1). For C–N bond dissociation energy is around 3 kcal mol−1 higher than for C–C.88,89 While radical (3) can be relatively stable due to hyperconjugation and potentially detectable by mass spectrometry after entering the gas phase, radicals formed through C–N bond cleavage might undergo 1,2 elimination or proton–shift reactions. These secondary reactions could lead to the formation of: (i) ammonia borane (molar mass 31) and unsaturated C2NBH8 or (ii) aminoborane (molar mass 29) and saturated C2NBH10. The latter scenario appears more likely because, similar to ethenamine, C2NBH8 is expected to be highly reactive.90 This hypothesis is further supported by the observation of dominant coupled components at m/z 27 and 28 in the DMS, while the component at m/z 29 was weaker and followed the uncoupled behaviour.
 | ||
| Scheme 2 Possible coupled reactions of EDAB tribochemical decomposition. | ||
The uncoupled emission likely comprises gaseous species containing the B–N–B backbone and ammonia borane (the DMS components at m/z 29–31 and 42). The appearance of B–N–B backbone-containing molecules can be tentatively attributed to secondary reactions involving EDAB molecules and radicals (·C2NBH9 and/or ·NBH5) resulting from C–N bond cleavage. Despite the insights provided by mass spectrometry, obtaining more precise information on the composition of emitted gases or radicals remains challenging at this stage because of the given level of uncertainty of mass spectrometry measurements and the lack of reference data on the specific cracking patterns of potential EDAB decomposition products. Nonetheless, one clear observation emerges: most products of tribochemical reactions must be volatile leaving no residue behind since vibrational spectroscopy and NMR analysis did not detect any significant changes in the chemical structure of EDAB on the mechanically affected zones compared to bulk EDAB.
While the coupled gas emission can be rationally attributed to chemical processes occurring within the bulk material, the uncoupled gas emission likely originates from the surface layer. This decoupling from the indenter movement can be explained by mechanical removing of the material of the surface layer from the MAZ. Vibrational spectrometry suggested that this layer has the same chemical structure as the bulk EDAB, but exhibits a considerably higher degree of disorder, possibly formed during cold hydrostatic pressing. Interestingly, applying shear stress to the pristine EDAB (bulk) through reciprocating rubbing did not induce equivalent structural disorder. Similarly, grinding, which combines compressive strain and shearing forces, had no effect on boron environments according to NMR measurements. This is surprising because many researchers have attributed shearing to be a critical factor in driving tribochemical reactions.
When the activation factor comes into play the uncoupled gas emission starts almost immediately. Conversely, the slow decrease in emission likely reflects that some kind of transport phenomenon, e.g. diffusion from deeper layers beneath the mechanically affected surfaces, or slow thermally driven relaxation, e.g. of residual stress, are involved. This behaviour differs from the coupled gas emission, which decays much faster in an exponential manner. The rate of decay for coupled gas is roughly 5 times higher than that of the uncoupled mode.
The pre-existing crystallographic disorder in the surface layer might be essential for triggering the uncoupled tribochemical reactions. Considering the van der Waals91 and dihydrogen92 forces holding EDAB molecules together, we can infer that the disordered surface layer likely possesses quite low residual stress, which should not exceed the EDAB hardness (150 MPa). Such stress is negligibly low compared to the stress levels required to direct cleavage of strong covalent and dative bonds within the EDAB molecule, but it can contribute to lowering the activation free energy according to Eyring expression:
| Ea = Ea,0 − σΔV | (2) |
Activation volume is usually interpreted as the molecular volume change between the initial state and the transition state so that the reaction rate constant is enhanced by the external force exerted on the reactants. If the activation volume is high enough (depending on the activation barrier), tribochemical reactions can be activated even at low stress levels. Recently, Li and Szlufarska93 revisited the interpretation of the activation volume using density functional theory calculations. They proposed that mechanochemical coupling does not solely arise from local deformation of chemical bonds at the reaction site but also includes contributions from the deformation of the surrounding bulk material. This contribution is particularly significant for materials with low stiffness, such as EDAB and other hydrides.
The value of activation volume required to cause the same H2 emission rate from EDAB under mechanical activation as under thermal desorption at given temperature can be found from the following expression (refer to ESI for more details†):
 | (3) |
When applying eqn (3) to EDAB under the conditions of thermal decomposition initiation (Tt = 355 K, Ea,0 = 59 kJ mol−1) and the tribochemical process (σ = 100 MPa, Ttch = 310 K) the calculated minimal required ΔV is 124 Å3. We assumed that the maximum stress level under plastic flow cannot exceed the material hardness. This value of ΔV is significantly larger than those typically reported for tribochemical processes involving bond dissociation, which usually fall within the range 2.2–11 Å3. Considering that an EDAB molecule has a linear structure and is only about 10 Å long,91 the required change of the activation volume markedly surpasses its bond deformation capability.
It can be suggested that the combination of relatively strong intramolecular bonds (covalent and dative) and weak intermolecular bonds (van der Waals and hydrogen) could explain the low susceptibility of EDAB to tribochemical activation. The low stiffness and hardness of EDAB, which are controlled by intermolecular bonds, allow for plastic flow at a low stress level, which is not sufficient to strain the intramolecular bonds to a level at which their cleavage can be activated. In contrast, ionic and complex hydrides, which do not have a similar mechanism for stress relaxation, are prone to strong tribochemical activation.
4. Conclusions
The tribochemical processes occurring in EDAB under mechanical solicitation were analysed using operando Mechanically Stimulated Gas Emission Mass Spectrometry. In contrast to the previous studies, which reported intensive hydrogen release from metal and complex hydrides subjected to similar mechanical solicitation under vacuum and room temperature, faint hydrogen emission was found in this study. This finding is striking since, in comparison with metal and complex hydrides, EDAB has much lower initial activation energy ≤59 ± 5 kJ mol−1 and its dehydrogenation is an exothermic reaction.The analysis of the emitted gases points at the possible bond dissociation within EDAB molecules, most likely involving C–C and N → B bonds. Spectroscopic techniques and NMR did not show any notable chemical changes on the mechanically affected surfaces. This suggests that the products of tribochemical reactions were highly volatile.
Nanoindentation tests showed that EDAB's mechanical properties are close to those of thermosetting polymers: hardness 57–143 MPa and elastic modulus 2.9–3.7 GPa.
Microscopy revealed significant changes of surface morphology on the mechanically affected zones. Periodic microscopic wrinkles perpendicular to the motion direction resembling Schallamach waves were observed. These features are usually observed on worn surfaces of elastomers and it can be associated with the intensive plastic flow, which was the possible driving force for the observed tribochemical reactions.
The lack of hydrogen emission was attributed to a combination of strong covalent and dative intramolecular forces and weak hydrogen and van der Waals intermolecular forces holding molecules together. It was suggested that weak intermolecular interaction provided a channel of mechanical energy dissipation through plastic flow without generating significant stress levels which could trigger intramolecular bond dissociation.
Data availability
The data that support the findings of the following study: are openly available in digital.csic.es, https://doi.org/10.20350/digitalCSIC/16448.Author contributions
Conceptualization and methodology, E. M., J. A., and R. N.; investigation, all the authors; writing—original draft preparation, R. N.; writing—review and editing, E. M., J. A., and R. N.; all authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank to J. F. Fernández and M. Manso Silvan from UAM for valuable discussion. The technical assistance of S. Guerra is also acknowledged. This work was supported by the Ministry of Science and Innovation of Spain (grants PID2019-111063RB-I00, PID2020-112770RB-C22, PID2020-117573GB-I00, RTI2018-099794-B-I00, and TED2021-129950B-I00).References
- Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the regions, A Hydrogen Strategy for a Climate-Neutral Europe, Report 52020DC0301, European Commission, Brussels, 2020 Search PubMed.
- D. Neiner, A. Karkamkar, M. Bowden, Y. Joon Choi, A. Luedtke, J. Holladay, A. Fisher, N. Szymczak and T. Autrey, Energy Environ. Sci., 2011, 4, 4187–4193 RSC.
- S. E. Hosseini and B. Butler, Int. J. Green Energy, 2020, 17, 13–37 CrossRef CAS.
- F. Leardini, M. J. Valero-Pedraza, E. Perez-Mayoral, R. Cantelli and M. A. Bañares, J. Phys. Chem. C, 2014, 118, 17221–17230 CrossRef CAS.
- F. Leardini, D. Mirabile Gattia, A. Montone, F. Cuevas, E. Perez-Mayoral, M. J. Valero-Pedraza, M. A. Bañares and R. Cantelli, Int. J. Hydrogen Energy, 2015, 40, 2763–2767 CrossRef CAS.
- H. C. Kelly and J. O. Edwards, J. Am. Chem. Soc., 1960, 82, 4842–4846 CrossRef CAS.
- W. H. Schechter, R. M. Adams and G. F. Huff, Production of dimethylamine-borane, US3122585, 1964.
- J.-R. Ares, R. Nevshupa, E. Muñoz-Cortés, C. Sánchez, F. Leardini, I.-J. Ferrer, V. Minh Huy Tran, F. Aguey-Zinsou and J.-F. Fernández, ChemPhysChem, 2019, 20, 1248–1262 CrossRef CAS PubMed.
- R. Nevshupa, J. R. Ares, J. F. Fernández, A. del Campo and E. Roman, J. Phys. Chem. Lett., 2015, 6, 2780–2785 CrossRef CAS PubMed.
- E. Muñoz-Cortés, O. L. Ibryaeva, M. Manso Silvan, B. Zabala, E. Flores, A. Gutierrez, J. R. Ares and R. Nevshupa, Phys. Chem. Chem. Phys., 2023, 25, 494–508 RSC.
- J. G. Hernández and T. Friščić, Tetrahedron Lett., 2015, 56, 4253–4265 CrossRef.
- A. E. M. Beedle, M. Mora, C. T. Davis, A. P. Snijders, G. Stirnemann and S. Garcia-Manyes, Nat. Commun., 2018, 9, 3155 CrossRef PubMed.
- M. V. Morales-Gallardo, A. M. Ayala, M. Pal, M. A. Cortes Jacome, J. A. Toledo Antonio and N. R. Mathews, Chem. Phys. Lett., 2016, 660, 93–98 CrossRef CAS.
- R. A. Nevshupa, M. Conte, A. Igartua, E. Roman and J. L. de Segovia, Tribol. Int., 2015, 86, 28–35 CrossRef.
- R. A. Nevchoupa, J. de Segovia and E. A. Deulin, Vacuum, 1999, 52, 73–81 CrossRef CAS.
- E. Jimenez-Relinque, M. Sapiña, R. Nevshupa, E. Roman and M. Castellote, Chem. Eng. J., 2016, 286, 560–570 CrossRef CAS.
- C. Kajdas and K. Hiratsuka, Proc. Inst. Mech. Eng., Part J, 2009, 223, 827–848 CrossRef.
- S. Mori, Appl. Surf. Sci., 1987, 27, 401–410 CrossRef CAS.
- X. Wu, P. Cong, H. Nanao, K. Kobayashi and S. Mori, Langmuir, 2002, 18, 10122–10127 CrossRef CAS.
- R. Lu, S. Mori, K. Kobayashi and H. Nanao, Appl. Surf. Sci., 2009, 255, 8965–8971 CrossRef CAS.
- R. Nevshupa, M. Conte, S. Guerra and E. Roman, Lubricants, 2017, 5, 27 CrossRef.
- M. Mahrova, M. Conte, E. Roman and R. Nevshupa, J. Phys. Chem. C, 2014, 118, 22544–22552 CrossRef CAS.
- R. Nevshupa, M. Conte, A. del Campo and E. Roman, Tribol. Int., 2016, 102, 19–27 CrossRef CAS.
- V. Totolin, M. Conte, E. Berriozábal, F. Pagano, I. Minami, N. Dörr, J. Brenner and A. Igartua, Lubr. Sci., 2014, 26, 514–524 CrossRef CAS.
- A. Igartua, E. Berriozabal, R. Nevshupa, E. Roman, F. Pagano, L. Pleth Nielsen, S. Louring and L. Muntada, Tribol. Int., 2017, 114, 192–200 CrossRef CAS.
- R. Nevshupa, J. Caro, A. Arratibel, R. Bonet, A. Rusanov, J. R. Ares and E. Roman, Tribol. Int., 2019, 129, 177–190 CrossRef CAS.
- A. Rusanov, R. Nevshupa, J.-M. Martin, M. Á. Garrido and E. Roman, Diamond Relat. Mater., 2015, 55, 32–40 CrossRef CAS.
- A. Rusanov, R. Nevshupa, J. Fontaine, J.-M. Martin, T. Le Mogne, V. Elinson, A. Lyamin and E. Roman, Carbon, 2015, 81, 788–799 CrossRef CAS.
- P. Řepa, Vacuum, 1992, 43, 367–371 CrossRef.
- R. Nevshupa, K. Cruz, I. Martinez, S. Ramos, I. Llorente and E. Roman, Tribol. Int., 2016, 97, 360–370 CrossRef CAS.
- R. Nevshupa, K. E. Grinkevich, I. Martinez and E. Roman, Mater. Constr., 2016, 66, e099 Search PubMed.
- M. Gouider, Y. Berthier, P. Jacquemard, B. Rousseau, S. Bonnamy and H. Estrade-Szwarckopf, Wear, 2004, 256, 1082–1087 CrossRef CAS.
- J. T. Dickinson, L. C. Jensen, S. C. Langford and R. P. Dion, J. Polym. Sci., Part B:Polym. Phys., 1993, 31, 1441–1449 CrossRef CAS.
- J. T. Dickinson, L. C. Jensen and R. P. Dion, J. Appl. Phys., 1993, 73, 3047–3054 CrossRef CAS.
- J. T. Dickinson, L. C. Jensen, S. C. Langford and R. P. Dion, J. Polym. Sci., Part B:Polym. Phys., 1994, 32, 993–999 CrossRef CAS.
- M. R. Louthan Jr, G. R. Caskey Jr, J. A. Donovan and D. E. Rawl Jr, Mater. Sci. Eng., 1972, 10, 357–368 CrossRef.
- E. Muñoz-Cortés, J. Sánchez-Prieto, B. Zabala, C. Sanchez, E. Flores, A. Flores, E. Roman, J. R. Ares and R. Nevshupa, RSC Mechanochemistry, 2024, 1, 196–210 Search PubMed.
- A. G. Peressadko, R. A. Nevshupa and E. A. Deulin, Vacuum, 2002, 64, 451–456 CrossRef CAS.
- J. T. Dickinson, L. C. Jensen, S. C. Langford and J. P. Hirth, J. Mater. Res., 1991, 6, 112–125 CrossRef CAS.
- J. T. Dickinson, L. C. Jensen and S. C. Langford, Phys. Rev. Lett., 1991, 66, 2120–2123 CrossRef CAS PubMed.
- J. T. Dickinson, L. C. Jensen, S. C. Langford, P. E. Rosenberg and D. L. Blanchard, Phys. Chem. Miner., 1991, 18, 320–325 CrossRef CAS.
- R. A. Nevchoupa, J. L. De Segovia and E. A. Deulin, Vacuum, 1999, 52, 73–81 CrossRef CAS.
- R. A. Nevshupa, E. Roman and J. L. de Segovia, J. Vac. Sci. Technol., A, 2008, 26, 1218–1223 CrossRef CAS.
- R. Nevshupa, J. Frict. Wear, 2009, 30, 118–126 CrossRef.
- R. Guan, P. Wang, Y. Song and V. N. Staroverov, J. Phys. Chem. C, 2021, 125, 18614–18622 CrossRef CAS.
- E. Muñoz-Cortés, O. L. Ibryaeva, M. Manso Silvan, B. Zabala, E. Flores, A. Gutierrez, J. R. Ares and R. Nevshupa, Phys. Chem. Chem. Phys., 2023, 25, 494–508 RSC.
- L. Massimi, M. G. Betti, S. Caramazza, P. Postorino, C. Mariani, A. Latini and F. Leardini, Nanotechnology, 2016, 27, 435601 CrossRef PubMed.
- F. Baitalow, J. Baumann, G. Wolf, K. Jaenicke-Rößler and G. Leitner, Thermochim. Acta, 2002, 391, 159–168 CrossRef CAS.
- H. A. McGee Jr and C. T. Kwon, Inorg. Chem., 1970, 9, 2458–2461 CrossRef.
- M. Barquins, Mater. Sci. Eng., 1985, 73, 45–63 CrossRef CAS.
- L. Martínez, R. Nevshupa, D. Felhös, J. L. De Segovia and E. Román, Tribol. Int., 2011, 44, 996–1003 CrossRef.
- M. J. Valero-Pedraza, V. Gascón, M. A. Carreón, F. Leardini, J. R. Ares, Á. Martín, M. Sánchez-Sánchez and M. A. Bañares, Microporous Mesoporous Mater., 2016, 226, 454–465 CrossRef CAS.
- K. M. Dreux, L. E. McNamara, J. T. Kelly, A. M. Wright, N. I. Hammer and G. S. Tschumper, J. Phys. Chem. A, 2017, 121, 5884–5893 CrossRef CAS PubMed.
- S. F. Parker, RSC Adv., 2018, 8, 23875–23880 RSC.
- R. Hinojosa-Nava, E. V. Mejía-Uriarte, A. R. Vázquez-Olmos and R. Y. Sato-Berrú, Spectrochim. Acta, Part A, 2023, 284, 121776 CrossRef CAS PubMed.
- M. Rueda, L. M. Sanz-Moral, J. J. Segovia and Á. Martín, Chem. Eng. J., 2016, 306, 164–173 CrossRef CAS.
- J. Goubeau and H. Schneider, Chem. Ber., 1961, 94, 816–821 CrossRef CAS.
- J. Baumann, F. Baitalow and G. Wolf, Thermochim. Acta, 2005, 430, 9–14 CrossRef CAS.
- J. S. Wang and R. A. Geanangel, Inorg. Chim. Acta, 1988, 148, 185–190 CrossRef CAS.
- W. J. Shaw, J. C. Linehan, N. K. Szymczak, D. J. Heldebrant, C. Yonker, D. M. Camaioni, R. T. Baker and T. Autrey, Angew. Chem., Int. Ed., 2008, 47, 7493–7496 CrossRef CAS PubMed.
- M. Sugie, H. Takeo and C. Matsumura, J. Mol. Spectrosc., 1987, 123, 286–292 CrossRef CAS.
- P. V. Ramachandran and A. S. Kulkarni, Dalton Trans., 2016, 45, 16433–16440 RSC.
- A. C. Stowe, W. J. Shaw, J. C. Linehan, B. Schmid and T. Autrey, Phys. Chem. Chem. Phys., 2007, 9, 1831–1836 RSC.
- O. Gunaydin-Sen, R. Achey, N. S. Dalal, A. Stowe and T. Autrey, J. Phys. Chem. B, 2007, 111, 677–681 CrossRef CAS PubMed.
- C. Li, E. Coons and A. Strachan, Acta Mech., 2014, 225, 1187–1196 CrossRef.
- F. Amato, et al. , J. Hazard. Mater., 2014, 275, 31–36 CrossRef CAS PubMed.
- X. Tian and F. E. Kennedy Jr, J. Tribol., 1994, 116, 167–174 CrossRef.
- F. Kennedy, Modern Tribology Handbook, 2001, vol. 1, pp. 235–272 Search PubMed.
- D. G. Bansal and J. L. Streator, Wear, 2012, 278–279, 18–27 CrossRef CAS.
- P. Niazi, M. Javanbakht, M. Karevan and M. R. Tavakoli, Polym. Bull., 2023, 80, 4387–4406 CrossRef CAS.
- Y. Yang, T. G. Voskuilen, T. L. Pourpoint, D. R. Guildenbecher and J. P. Gore, Int. J. Hydrogen Energy, 2012, 37, 5128–5136 CrossRef CAS.
- R. Nevshupa, E. Roman and J. L. de Segovia, Int. J. Mater. Prod. Technol., 2010, 38, 57–65 CrossRef CAS.
- R. E. Bolz, Handbook of Tables for Applied Engineering Science, 1970 Search PubMed.
- M. M. Robins, R. W. Rennell and R. D. Arnell, J. Phys. D: Appl. Phys., 1984, 17, 1349 CrossRef.
- A. C. Gangal and P. Sharma, Int. J. Chem. Kinet., 2013, 45, 452–461 CrossRef CAS.
- M. E. Brown and A. K. Galwey, Thermochim. Acta, 1979, 29, 129–146 CrossRef CAS.
- G. Wolf, J. Baumann, F. Baitalow and F. P. Hoffmann, Thermochim. Acta, 2000, 343, 19–25 CrossRef CAS.
- A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578–3582 CrossRef CAS PubMed.
- X. Kang, Z. Fang, L. Kong, H. Cheng, X. Yao, G. Lu and P. Wang, Adv. Mater., 2008, 20, 2756–2759 CrossRef CAS PubMed.
- S. Sepehri, B. B. Garcia and G. Cao, J. Mater. Chem., 2008, 18, 4034–4037 RSC.
- K. Turani-I-Belloto, M.-J. Valero-Pedraza, R. Chiriac, F. Toche, D. Granier, D. Cot, E. Petit, P. G. Yot, J. G. Alauzun and U. B. Demirci, ChemistrySelect, 2021, 6, 9853–9860 CrossRef CAS.
- E. G. Sorte, J. M. Rimsza and T. M. Alam, Molecules, 2020, 25, 933 CrossRef CAS PubMed.
- M. Jochum, U. Werner-Zwanziger and J. W. Zwanziger, J. Chem. Phys., 2008, 128, 052304 CrossRef CAS PubMed.
- E. C. Reynhardt and C. F. Hoon, J. Phys. C: Solid State Phys., 1983, 16, 6137 CrossRef CAS.
- K. L. Murty, K. Detemple, O. Kanert and J. T. M. Dehosson, Metall. Mater. Trans. A, 1998, 29, 153–159 CrossRef.
- O. Arias-Cuevas, Z. Li, R. Lewis and E. A. Gallardo-Hernandez, Wear, 2010, 268(3–4), 543–551 CrossRef CAS.
- A. Haaland, Angew Chem., Int. Ed., 1989, 28, 992–1007 CrossRef.
- S. Mori, H. Nanao and K. Takiwatari, Langmuir, 2024, 40, 7257–7267 CrossRef CAS PubMed.
- X. Wu, P. Cong and S. Mori, Appl. Surf. Sci., 2002, 201, 115–122 CrossRef CAS.
- K. K. Singh, Shivani, P. Tandan and A. Misra, Astrophys. Space Sci., 2018, 363, 213 CrossRef.
- H.-Y. Ting, W. H. Watson and H. C. Kelly, Inorg. Chem., 1972, 11, 374–377 CrossRef CAS.
- T. Richardson, S. de Gala, R. H. Crabtree and P. E. M. Siegbahn, J. Am. Chem. Soc., 1995, 117, 12875–12876 CrossRef CAS.
- T. A. Aiken, J. Kwasny and W. Sha, Clean. Eng. Technol., 2021, 2, 100035 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mr00072b |
| This journal is © The Royal Society of Chemistry 2025 |