
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphoproteomics guides low dose drug combination of cisplatin and silmitasertib against concurrent chemoradiation resistant cervical cancer†
Irene A.
George‡
ab,
Janani
Sambath‡
ab,
R. E.
Dhawale
c,
Manisha
Singh
d,
Vinita
Trivedi
d,
R.
Venkataramanan
ef,
Richa
Chauhan
*d and
Prashant
Kumar
 *abefgh
*abefgh
aManipal Academy of Higher Education (MAHE), Manipal, 576104, Karnataka, India
bInstitute of Bioinformatics, Bangalore, 560066, Karnataka, India. E-mail: prashant@ibioinformatics.org
cVedantaa Hospital & Research Centre, Palghar, 401606, Maharashtra, India
dMahavir Cancer Sansthan and Research Centre (MCSRC), Patna, 801505, Bihar, India. E-mail: chauhan_richa@outlook.com
eKarkinos Foundation, Mumbai, 400086, Maharashtra, India
fKarkinos Healthcare Pvt Ltd, Navi Mumbai, 400705, Maharashtra, India
gDatar Cancer Genetics, Nashik, 422010, Maharashtra, India
hCentre of Excellence for Cancer – Gangwal School of Medical Sciences and Technology, Indian Institute of Technology Kanpur, Kanpur, 208016, Uttar Pradesh, India
First published on 27th November 2024
Abstract
Cisplatin-based concurrent chemoradiotherapy (CCRT) is the standard treatment for cervical patients with locally advanced disease. Despite the improved survival rates and prognosis observed in patients undergoing CCRT, over 30–40% do not achieve complete response and are at risk of locoregional recurrence. Targeting crucial molecules that confer resistance may improve the clinical outcomes of the treatment resistant patient cohort. Herein, we employed a liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based phosphoproteomic approach to identify the altered phosphophorylation events, activated kinases and dysregulated pathways involved in treatment resistance. We quantified 2531 unique phosphopeptides mapping to 1099 proteins of which 74 proteins were differentially phosphorylated between the cohorts. Pathway analysis revealed dysregulation of the DNA repair pathway and the proteins involved in DNA repair in the non-responder cohort. Additionally, we identified kinase signature associated with CCRT resistance. Kinases such as CSNK2A1, PRKDC, PLK-1, NEK2, ATM and CDK1 are predicted to be activated in non-responders. In particular, we showed that CSNK2A1 is involved in oncogenesis of cervical cancer and pharmacological inhibition led to reduced cell proliferation, migration and colony formation. Moreover, the combination of the CSNK2A1 inhibitor, silmitasertib with cisplatin demonstrated synergism (combination index < 1) and yielded a beneficial reduction in dosage. The dose reduced combination potentially reduced the proliferative, migratory and colony formation ability in vitro. Our findings highlight the potential of phosphoproteomics to identify clinically significant targets and pathways implicated in CCRT resistance. Our study also indicates that combination therapy could serve as an effective treatment strategy to improve the efficacy of patients undergoing CCRT.
1. Introduction
Cervical cancer ranks as the fourth most prevalent cancer in females, contributing to 341![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 831 deaths worldwide (GLOBOCAN, 2022). The primary risk factor for cervical cancer is persistent infection with high-risk subtypes of the human papillomavirus (HPV). Although HPV vaccination and screening for premalignant lesions has reduced the cervical cancer incidence in developed countries, it is still a major healthcare problem in low- and middle-income countries (LMICs). In India, cervical cancer stands as thesecond most common cancer in women, making up around 10% of cases of female cancers.1,2 According to the 2020 report from the National Cancer Registry Programme, 60% of cervical cancer cases were diagnosed at an advanced local stage.2 Locally advanced cervical cancer (stages IIB–IVA) (LACC) has poor prognosis compared to stages IA and IB.3 Currently, cisplatin-based concurrent chemoradiation therapy is regarded as the standard therapy for LACC patients.
831 deaths worldwide (GLOBOCAN, 2022). The primary risk factor for cervical cancer is persistent infection with high-risk subtypes of the human papillomavirus (HPV). Although HPV vaccination and screening for premalignant lesions has reduced the cervical cancer incidence in developed countries, it is still a major healthcare problem in low- and middle-income countries (LMICs). In India, cervical cancer stands as thesecond most common cancer in women, making up around 10% of cases of female cancers.1,2 According to the 2020 report from the National Cancer Registry Programme, 60% of cervical cancer cases were diagnosed at an advanced local stage.2 Locally advanced cervical cancer (stages IIB–IVA) (LACC) has poor prognosis compared to stages IA and IB.3 Currently, cisplatin-based concurrent chemoradiation therapy is regarded as the standard therapy for LACC patients.
Concurrent chemoradiotherapy (CCRT) has been shown to offer greater advantages over radiation therapy alone in LACC patients.4,5 CCRT leads to a 5-year overall survival of 66% and disease-free survival of 58% by reducing the local, distant recurrence and progression of the disease.6 CCRT treatment involves weekly administration of cisplatin with concurrent radiation therapy. Cisplatin is the most potent chemotherapeutic agent in treating locally advanced as well as metastatic cancer.5,7–9 Nevertheless, resistance to the therapy leads to relapses in numerous instances. About 30–50% of patients fail to respond to CCRT.10 Additionally, various studies have shown the toxicity associated with cisplatin not only affect the treatment but also impact the prognosis of cervical cancer patients.4,11 In the case of LACC, the effectiveness of radiotherapy also depends on the size of the tumor.12 The inclusion of additional chemotherapeutic agents along with cisplatin in CCRT clinical settings led to acute toxicity in patients with LACC.13 Furthermore, no added advantages in terms of tumor response or progression-free survival were observed.13 For example, concurrent chemoradiotherapy with cisplatin and fluorouracil (FU) doublet therapy didn’t show any survival benefits or increase in complete response rate.14 This necessitates the identification of potential drug targets that can be utilized either individually or in combination with cisplatin, particularly for the treatment-resistant patient population.
With the development of high throughput technologies, molecular mechanisms underlying tumorigenesis, therapy resistance and prognosis can be reconnoitered.15–18 Increasing studies suggest that phosphoproteomics offers high-throughput and systematic exploration of activated kinases and altered signaling networks involved in treatment resistance.19,20 We employed mass spectrometry-based phosphoproteomics to decipher the molecular alterations to uncover novel biomarkers and therapeutic targets. We carried out phosphoproteomic analysis of tumor biopsy samples from treatment naive cervical cancer patients. We employed a TMT-based quantitative approach along with IMAC-based phosphopeptide enrichment to identify the dysregulated phosphorylation events and kinases involved in treatment resistance. Our comprehensive phosphoproteomic analysis identified the involvement of the DNA repair pathway in CCRT resistance. Furthermore, kinase substrate analysis identified the kinases involved in treatment resistance. Our findings indicate the potential for combination treatment strategies to enhance sensitivity to CCRT, potentially reducing the dosage of cisplatin and associated toxicity.
2. Materials and methods
2.1. Patient tumor sample collection
For the phosphoproteomic analysis, we used the tumor biopsy samples from 5 responders and 5 non-responders from patients with stage IIIB cervical cancer. Representative images from the histopathological analysis confirming squamous cell carcinoma have been provided in Fig. S1 (ESI†). All the samples were collected before the start of the therapy. The samples were collected from patients enrolled in Mahavir Cancer Sansthan, Patna, Bihar, India. All the patients have given written consent for participating in the study. The study was approved by the institutional ethics committee, Mahavir Cancer Sansthan (MCS/IEC No. 01/05/2018/413). The study was carried out as per the national ethical guidelines for biomedical and health research involving human participants, by the Indian Council of Medical Research (ICMR), India. Informed consent was obtained from the patients. The samples were stored at −80 °C until use. The clinicopathological details of the samples used are included in Table S1 (ESI†). All the patients underwent concurrent chemoradiation therapy and the treatment response was recorded according to RECIST (response evaluation criteria in solid tumors) criteria. For the immunohistochemistry experiments, formalin fixed paraffin embedded samples from 16 responders and 16 non-responders were used.2.2. Sample preparation, protein digestion and TMT labeling
Protein extraction was carried out and was estimated using bicinchoninic acid (BCA) protein assay (Pierce, ThermoFisher Scientific). Equal amount of protein was reduced, and alkylated using 100 mM dithiothreitol (DTT) and 20 mM iodoacetamide (IAA), respectively. Ice cold acetone was added and incubated overnight at −20 °C. Precipitated protein was then reconstituted in TEABC and urea buffer, and digestion with trypsin (1:20, Promega) was carried out for 12–16 h at 37 °C. The digested peptides were then cleaned using a Sep-Pak C18 Plus Light cartridge (Waters, Catalog # WAT023501). Peptide estimation was carried out using a bicinchoninic acid assay (BCA) peptide estimation kit and equal amount of the peptides was used for TMT labeling. 10-Plex TMT (tandem mass tags) labels from ThermoFisher Scientific were used according to the manufacturer's protocol. The reaction was quenched by adding 5% hydroxylamine. After checking the labeling efficacy, equal amount of protein from each sample was pooled.
2.3. Fractionation and IMAC-based phosphopeptide enrichment
Labelled peptides were reconstituted in basic reverse phase liquid chromatography (bRPLC) solvent (10 mM triethylammonium bicarbonate buffer (TEABC) pH 8.4), and were fractionated using high-pH, reverse-phase XBridge C18 columns (5 μm, 250 × 4.6 mm; Waters Corporation, Milford, MA, USA), with an increasing gradient of bRPLC solvent B (10 mM TEABC in 90% acetonitrile (ACN), pH 8.4). Fractionation was carried out using an Agilent 1100 high-pH reverse-phase liquid chromatography (RPLC) system. 96 fractions were collected, which were then pooled into 11 fractions. The pooled fractions were vacuum dried and subjected to phosphopeptide enrichment. Immobilized metal affinity chromatography (IMAC)-based enriched peptides were eluted with 40 μL of 50% ACN and 0.1% trifluoroacetic acid (TFA). The peptides were resuspended in 30 μL of 0.1% TFA and desalted using C18 Stage Tips (Thermo Fisher Scientific).2.4. Mass spectrometry analysis, data analysis and statistical analysis
The eluted fractions were then subjected to LC-MS/MS analysis using a QExactive HF-X Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Scientific, Bremen, Germany) interfaced with a Dionex Ultimate 3000 nanoflow liquid chromatography system. Each of the fractions was then reconstituted in 0.1% formic acid (Solvent A) and loaded in a trap column (Thermo Fisher Scientific Acclaim PepMap100 75 μm × 2 cm nanoViper 2Pk). The peptides were then resolved on an analytical column (Thermo Scientific PepMap RSLC C18 2 μm, 100 Å, 75 μm × 50 cm), at a flow rate of 300 nL min−1. A linear gradient range of 8–32% solvent B (0.1% formic acid in 80% acetonitrile) for 100 min was used and the total run time was 120 min for the chromatographic run. The precursor scan MS scan (350–1700 m/z) was acquired in the Orbitrap at a resolution of 120000 at 200 m/z. The most intense ions with charge state ≥2 were isolated and fragmented using HCD fragmentation with 32% normalized collision energy and detected at a mass resolution of 45000 at 200 m/z. The automated gain control target value for MS/MS was set as 1 × 105 and the ion filling time was set at 100 ms, while dynamic exclusion was set for 20 s. The data-dependent acquisition was implemented where the most intense precursor ions were detected. Following the data acquisition, a data search was carried out against the Human RefSeq protein database (version 81, containing 43225 experimentally validated protein entries with common contaminants) using the SEQUEST search algorithm through the Proteome Discoverer platform (version 2.1, Thermo Scientific). The search parameters employed for Proteome Discoverer analysis were as follows: two missed cleavages allowed, trypsin as the cleavage enzyme, a tolerance of 10 ppm on the precursors, and 0.02 Da on the fragment ions. Carbamidomethylation at cysteine, TMT 10-plex (+229.163) modification at the N-terminus of peptide and lysine were kept as fixed modifications and the phosphorylations of serine, threonine, and tyrosine; the deamidation of asparagine and glutamine and the oxidation of methionine were selected as dynamic modifications. Data were also searched against a decoy database and filtered with a 1% false discovery rate (FDR). Data are available via ProteomeXchange with identifier PXD055164. PTM-Pro 2.0 was used to fetch high confident post-translational modification (PTM) sites at the peptide and protein level based on >75% site localization probability for further analysis.21 Two samples “t-test” (p < 0.05) and fold change calculation were calculated as the ratio of phosphorylation levels of resistant vs. sensitive cohort.
To compare the mRNA expression of the kinases identified in our data with The Cancer Genome Atlas (TCGA), we utilized the GEPIA tool (https://gepia.cancer-pku.cn/detail.php).
2.5. Pathway analysis, kinome map and kinase substrate analysis
Pathway analysis was carried out using the web-based tool WEB-based GEne SeT AnaLysis (https://www.webgestalt.org/). The identification of enriched kinases in the data was conducted using KinMap, and the kinome map was subsequently generated through using the same tool (https://www.kinhub.org/kinmap/index.html). Kinase substrate analysis was carried out using kinase-substrate enrichment analysis (KSEA) (casecpb.shinyapps.io/ksea/).2.6. Immunohistochemistry
For the immunohistochemistry (IHC) analysis, FFPE tissue sections were obtained from Mahavir Cancer Sansthan, Patna Bihar, India. IHC was carried out as previously described.22 The collected tissue sections were deparaffinized and for antigen retrieval, the sections were incubated in antigen retrieval buffer (0.01 M trisodium citrate buffer, pH 6) for 20 minutes. To quench the action of endogenous peroxidases, a solution containing methanol and chloroform (1:1) was used. Washes were carried out using phosphate-buffered saline (PBS) plus 0.05% Tween-20. Subsequently, to avoid non-specific binding, blocking was carried out using 5% goat serum to avoid non-specific binding of primary antibody for 30 minutes. Incubation with primary phosho-SMC1A (S957) (Abcam, Cambridge, United Kingdom) was done overnight at 4 °C in a humidified chamber. The sections were washed with wash buffer and incubated with appropriate horseradish peroxidase conjugated secondary antibodies for 30 minutes at room temperature. The sections were then incubated with 3,3′-diaminobenzidine (DAB) substrate. The signal was then developed by employing DAB chromogen from DAKO, based in Glostrup, Denmark, and counterstained with hematoxylin. The slides were scored by an experienced pathologist. Images were taken at 10× on an Olympus DP-21 microscope.
2.7. Cell culture, chemicals and cell culture assays
Cervical cancer cell line CaSki was grown in Dulbecco's modified Eagle's medium (DMEM; with high glucose, HIMEDIA), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were maintained under humidified conditions at 37 °C and 5% CO2. Cisplatin and silmitasertib were used in this study.Cell viability and proliferation assays were carried out using a (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyltetrazolium bromide) (MTT) assay. For the cell viability assay, cells were seeded in a 96 well plate (3 × 103 cells per well) and incubated for 48 hours. For determining the combination effect of cisplatin and inhibitor, the cells were treated with varying concentrations of cisplatin and a fixed concentration of the inhibitor (IC50). The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Catalogue number: M6494) assay was carried out after 48 h of incubation at 37 °C and 5% CO2. For determining the combination index and dose reduction index, the cells were treated with a constant-ratio of cisplatin and the inhibitor. The cells were then incubated at 37 °C and 5% CO2 for 48 hours. MTT reagent was added according to the manufacturer's instructions. The combination index and dose reduction index were calculated using the CompuSyn software (https://compusyn.updatestar.com/). For the proliferation assay, cells were treated with inhibitor alone, drug alone or a combination of both. The readings were taken at 0 hour, 24 hours, 48 and 72 hours. For migration, 15 × 103 cells were seeded in a 24 well plate in triplicate. A wound scratch was made once the cells reached 90–95% confluency. The cells were gently washed with PBS and then incubated with media containing the respective agents. The cells were imaged at 0 h and 18 h. For the colony formation assay, the cells were seeded in 12 or 6 well plates, and the cells were treated with cisplatin, inhibitor or a combination. Cells were plated for 10 days.
3. Results
3.1. Discovery of the patient sample cohort used for phosphoproteomic analysis
For the phosphoproteomic analysis, we collected tissue biopsy samples from treatment naïve cervical cancer patients. The patients were clinically confirmed for stage IIIB disease and later underwent cisplatin-based concurrent chemoradiation therapy. All patients underwent a treatment protocol comprising external beam radiotherapy (EBRT) concurrent with weekly cisplatin (40 mg m−2), succeeded by intracavitary radiotherapy (ICRT). A dose of 50 Gy in 25 fractions@2 Gy per fraction was delivered daily from Monday to Friday for 5 weeks using the 3D conformal radiotherapy technique (3DCRT). This was followed by intracavitary irradiation after a gap of 3–5 days. Three applications of high dose rate (HDR) brachytherapy were done with a dose of 7 Gy per fraction weekly. The patients were followed up monthly for the first three months, trimonthly for the next 2 years, biannually for the next 3 years and then annually if required. Clinical responses were evaluated using the response evaluation criteria in solid tumors (RECIST 1.1).23 The responses were classified as complete responders (CR), when there was disappearance of all signs of cancer in response to treatment, and as non-responders (NR), if they had a partial response, progressive, or stable disease after treatment. We included 5 responders and 5 non-responders for the phosphoproteomic analysis. The treatment timeline and sample collection points are depicted in Fig. 1A. In this pilot study, we utilized baseline tissue samples to understand the correlation with CCRT resistance. | ||
| Fig. 1 Treatment timeline and workflow carried out in phosphoproteomic analysis of CCRT resistant cervical cancer patients. (A) Sample collection and treatment time line. Tissue samples were collected before the start of the therapy. All the patients underwent concurrent chemoradiotherapy (CCRT), and their response was evaluated at the end of the treatment cycle through subsequent follow-ups. For the study, 5 samples from responders and 5 samples from the non-responders were used. (B) Workflow for the phosphoproteomic analysis of CCRT resistant and sensitive cervical cancer patients. Proteins were extracted and digested using trypsin, and digested peptides were then labelled with a 10-plex tandem mass tags (TMT) labelling kit. Labelled samples were then enriched for phosphopeptides using IMAC and analysed in a Q Exactive HF-X Hybrid Quadrupole–Orbitrap mass spectrometer. (C) Phosphoproteomic data analysis led to the identification of serine, threonine and tyrosine phosphorylation sites. Pie chart representing the number of serine, threonine and tyrosine phosphorylation sites identified in our study. | ||
To explore the phosphoproteome of CCRT resistant cervical cancer, we carried out multiplexed TMT and LC-MS/MS analysis. The tissue samples were digested, and labelled with 10 plex TMT tags. The labelled samples were then pooled, fractionated and enriched for the phosphopeptides. The detailed methodology followed in the experiment is explained in the methodology section. Fig. 1B depicts the phosphoproteomic workflow and validation experiments conducted in the study. The phosphoproteomic analysis led to the identification of 5524 (Table S2, ESI†) and quantification of 2531 unique phosphopeptides mapping to 1099 proteins among the samples (Table S3 and Fig. S2, ESI†). Among the quantified phosphosites, 5005 correspond to serine, 367 to threonine, and 7 are tyrosine phosphorylation (Fig. 1C).
3.2. Altered phosphorylation involved in CCRT resistance
Analyzing dysregulated phosphoproteins between responders and non-responders provided key insights into altered phosphorylation events predisposed in treatment evasion. We further compared the phosphoprofiles between the CCRT-resistant and the sensitive cohort. Our statistical analysis of the quantified phosphopeptides revealed significantly phosphorylated peptides in responders and non-responders (Table S4, ESI†). Fig. 2A represents the volcano plot of the quantified phosphopeptides in responders and non-responders. We observed significant phosphorylation of 144 phosphopeptides corresponding to 123 proteins. The hierarchical clustering with significantly phosphorylated phosphopeptides resulted in distinct phosphoprofiles between the responders and non-responder cohorts (p-value < 0.05) (Fig. 2B). Furthermore the fold change cut off resulted in identification of dysregulation of 88 phosphopeptides corresponding to 74 proteins, of which 40 phosphopeptides (mapping to 40 proteins) are hyperphosphorylated (>1.5-fold, p-value < 0.05) and 48 phosphopeptides (mapping to 36 proteins) are hypophosphorylated (<1.5-fold, p-value < 0.05). We observed hyperphosphorylation (>1.5-fold, p-value < 0.05) of proteins such as structural maintenance of chromosomes 1A (SMC1A), integrin subunit alpha 3 (ITGA3), LEM domain containing 3 (LEMD3), O-6-methylguanine-DNA methyltransferase (MGMT), and high mobility group nucleosome binding domain 1 (HMGN1) in the non-responder cohort. The phosphopeptides corresponding to proteins such as keratin, secreted phosphoprotein 1 (SPP1), Rab11 family-interacting protein 1 (Rab11-FIP1), tight junction protein 2 (TJP2), and phosphoglycerate mutase 1 (PGAM1) were downregulated (<1.5-fold, p-value < 0.05) in non-responders compared to responders.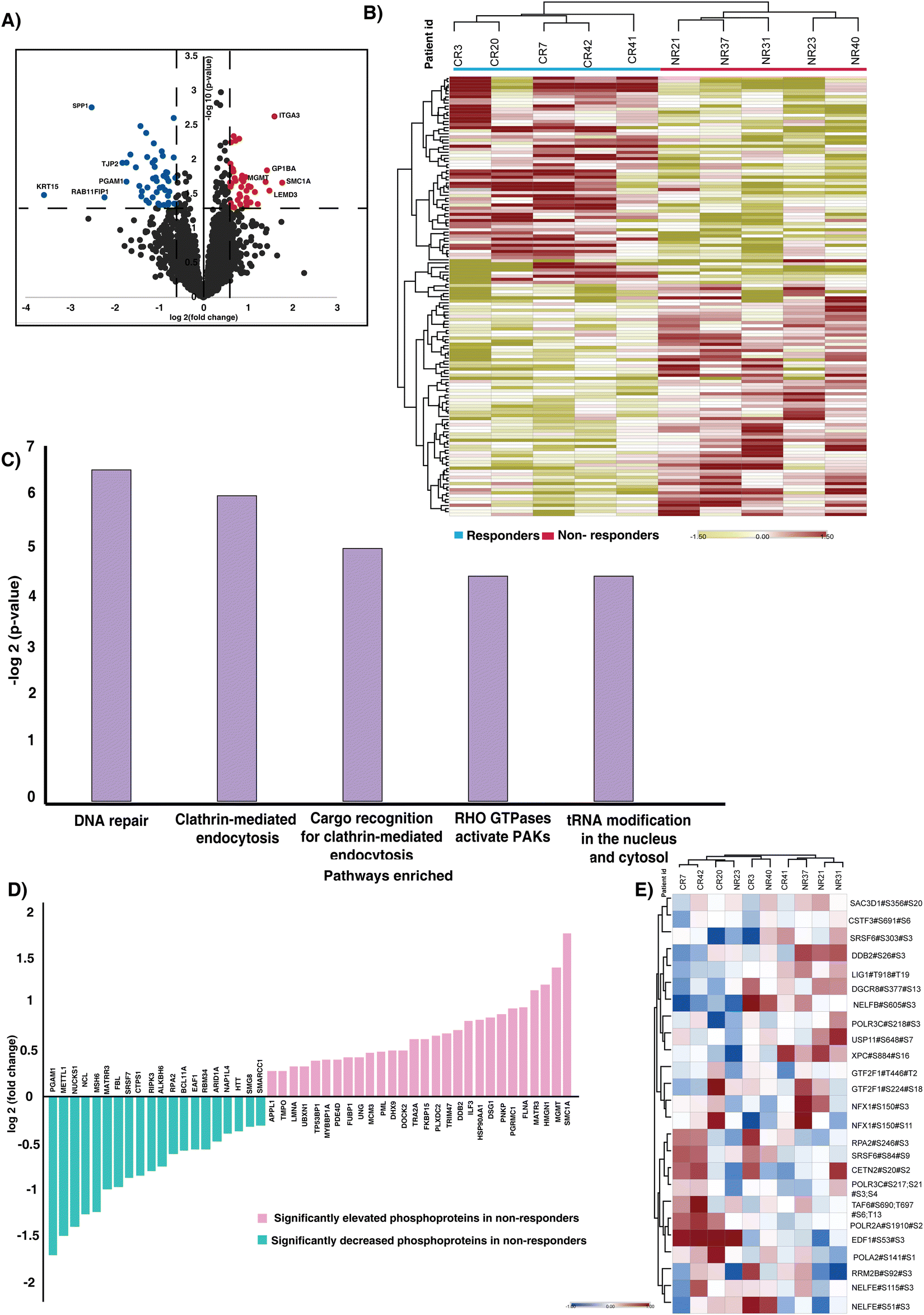 | ||
| Fig. 2 Phosphoproteome of CCRT resistant and sensitive cervical cancer patients. (A) Volcano plot of the phosphoproteome of CCRT resistant and sensitive cohorts. Log fold change of phosphorylation of the proteins plotted against the t-test p-values (−log10). Significant levels are represented with dashed lines. Fold change was calculated as the ratio of resistant versus sensitive samples. Data points in blue color are significantly hypophosphorylated phosphopeptides and data points in red are hyperphosphorylated phosphopeptides in the CCRT resistant cohort (fold change cut off > 1.5; p-value < 0.05). (B) Unsupervised clustering of significantly phosphorylated phosphopeptides in responders and non-responders. Abundance values of significantly phosphorylated phosphopeptides are represented in the heatmap. (C) Top five pathways enriched in non-responders. WEB-based GEne SeT AnaLysis Toolkit (webgestalt) based pathway analysis revealed pathways enriched in the cohort. (D) Proteins involved in the DNA repair pathway which are enriched in our data. The X-axis represents the proteins and Y-axis represents the log fold change of the phosphorylation. (E) Heatmap representing the phosphorylation of (MSigDB) hallmark genes identified in our data. Rows represent the hallmark genes enriched and the columns correspond to the individual samples. | ||
3.3. Molecular pathways in CCRT resistance
The pathway analysis of significantly regulated phosphoproteins was carried out to understand the mechanism involved driving treatment resistance in cervical cancer patients. The analysis revealed enrichment of pathways such as DNA repair (p-value = 0.011), clathrin-mediated endocytosis (p-value = 0.015), Cargo recognition for clathrin-mediated endocytosis (p-value = 0.032), Rho GTPase activated PAKs (p-value = 0.047) and tRNA modification in the nucleus and cytosol (p-value = 0.047) as significantly enriched (Fig. 2C) (Table S5, ESI†). We proceeded to assess the enrichment of phosphoproteins that were previously identified as associated with DNA repair. Fig. 2D represents the significantly phosphorylated proteins associated with the DNA repair. SMC1A, MGMT, HMGN1, Matrin 3 (MATR3) and Filamin A (FLNA) are the top 5 hyperphosphorylated proteins involved in the pathway. The immunohistochemistry staining showed high expression of phosphorylated SMC1A in non-responders compared to responders (Fig. S3, ESI†). We further checked the phosphorylation of DNA hallmark genes from the molecular signatures database (MSigDB) and identified differential expression of 26 phosphopeptides corresponding to 22 proteins (Fig. 2E).The GO-term analysis further revealed enrichment in the nucleus, membrane and membrane enclosed lumen as the top three. Additionally, the protein binding, nucleic acid binding and ion binding were active in non-responders (Fig. S4, ESI†).
3.4. Kinases driving the phosphoprofiles of CCRT resistant patients
Kinases play a pivotal role in driving signaling mechanisms that contribute to the initiation of malignant transformation, relapse and resistance.24 We sought to identify the kinases that were enriched in our data set. We identified enrichment of 50 kinases in our data set (Fig. S5A, ESI†). Out of the 50 kinases, two kinases, receptor-interacting protein kinase 3 (RIPK3) and adaptor-associated protein kinase 1 (AAK1), were hyperphosphorylated in responders. Downregulation of RIPK3 was previously reported to be involved in cisplatin resistance via the DNA repair pathway.25 Additionally, we examined the expression of RIPK3 and AAK1 in normal and tumor samples using TCGA data. We observed high expression of both the kinases in tumor samples compared to normal (Fig. S5B and C, ESI†).Kinase substrate enrichment analysis (KSEA) was used to map the phosphorylated proteins to the corresponding kinases. The enrichment of kinases such as casein kinase II subunit alpha (CSNK2A1), DNA-dependent protein kinase, catalytic subunit (PRKDC), polo-like kinase 1 (PLK-1), NIMA related kinase 2 (NEK2), ataxia-telangiectasia mutated (ATM) and cyclin-dependent kinase 1 (CDK1) in the non-responder cohort (Fig. 3A). Most of these kinases actively involved in DNA damage repair and their role in therapy resistance are previously reported in different cancers.26–31 CSNK2A1, the top most enriched kinase is reported to be involved in tumorigenesis in several cancers.32 The mRNA expression of CSNK2A1 in cervical cancer was significantly high in tumor samples compared to the normal tissues of TCGA and GTEx data (Fig. 3B). CSNK2A1 interacts with several proteins and phosphorylates several substrates. We further sought to identify the interacting hub proteins, and substrates enriched in our data using Harmonizome 3.0 and kinase enrichment analysis 3 (KEA3), respectively (Fig. 3C) (Table S6, ESI†). In the non-responder cohort we observed hypophosphorylation of key hub proteins, including replication protein A2 (RPA2), serine/arginine repetitive matrix protein 2 (SRRM2), tight junction protein 2 (TJP2), among others, and hyperphosphorylation of hub proteins such as transformer-2 alpha (TRA2A), interleukin enhancer binding factor 3 (ILF3), and heat shock protein 90 alpha family class A member 1 (HSP90AA1), among others. Additionally, the proteins such as SMC1A, HMGN1, MGMT, and several other substrates of CSNK2A1 were hyperphosphorylated in the non-responders.
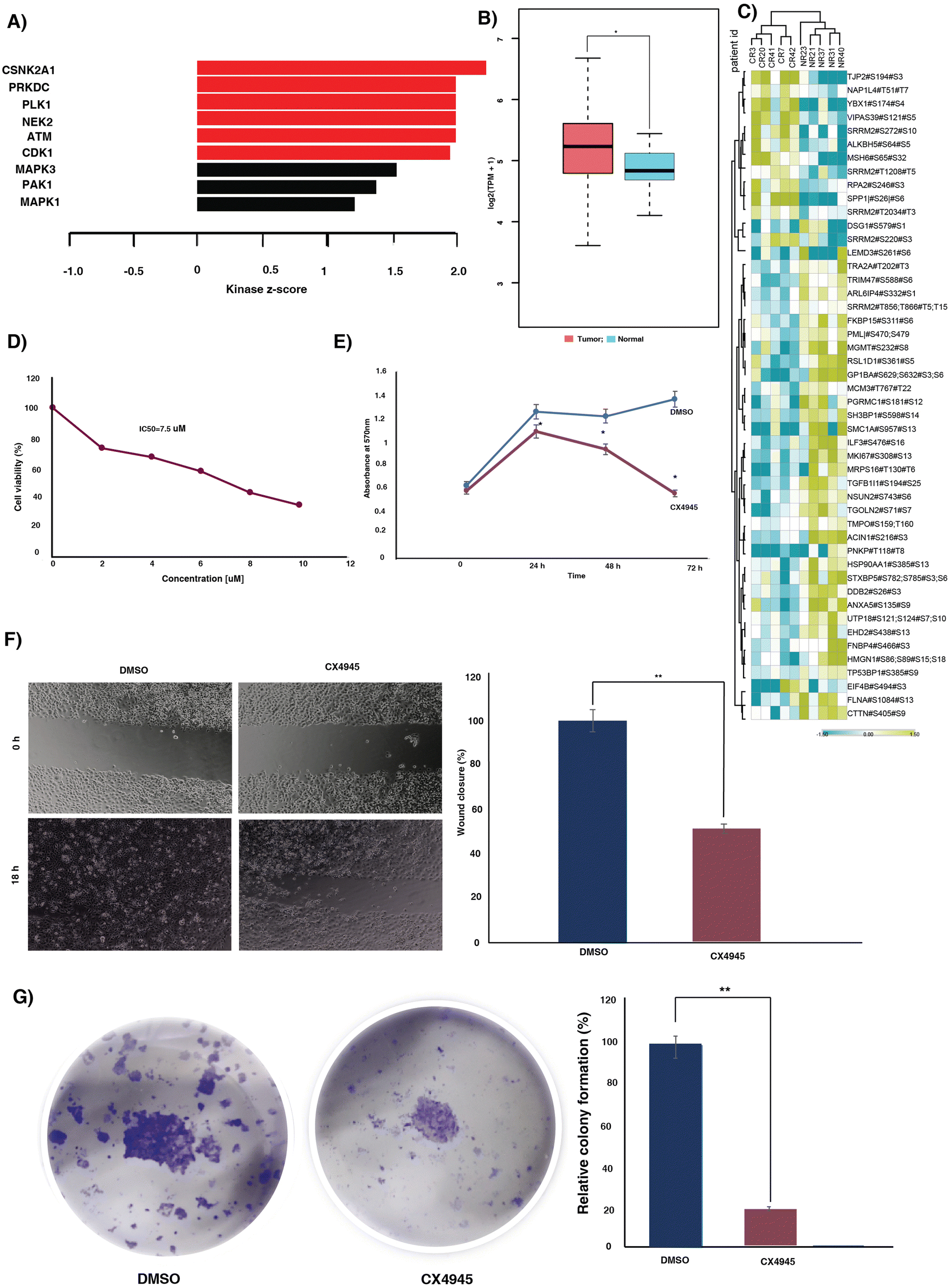 | ||
| Fig. 3 Kinases enriched in the CCRT resistant patient cohort. (A) The kinase substrate analysis predicted the enriched kinases in the CCRT resistant patients. Bar chart representing the kinases predicted to be activated in the CCRT resistant cohort. (B) Expression level of CSNK2A1 in cervical cancer and normal tissue samples in The Cancer Genome Atlas database. The expression of CSNK2A1 is higher than in the normal tissues. (C) Heat map showing the phosphorylation intensities of the substrates and hub proteins of CSNK2A1. This figure shows hyperphosphorylated peptides and hypophosphorylated peptides in yellow and blue, respectively. (D) Graph representing the cell viability and IC50 of the CaSki cell line following treatment with CX4945. An MTT assay was carried out after 48 h of treatment with various concentrations of CX4945. The X-axis represents the concentration of CX4945 and y-axis shows the percentage of cell viability. The data highlights the sensitivity of the cell line to the treatment and the IC50 was calculated from the dose response curve. (E) Line graph showing the proliferation of CaSki with and without CX4945. CX4945 was added after 24 h of cell seeding. OD values at different time points are plotted for the growth curve. (F) Cell migration (wound healing assay) of CaSki in the presence and absence of CX4945. Representative images of cell migration of CaSki at 0 h and 18 h. Bar diagram representing the wound closure of untreated and treated cell lines. Wound closure percentage was calculated as the ratio of final cell free area to initial cell free area. (G) Colony formation assay of cells treated with or without CX4945. Cells were seeded in a 24 well plate. The cells were treated with DMSO or CX4945. Colonies were visualized upon staining with crystal violet. Bar graph representing the relative colony formation upon treatment with CX4945. | ||
To evaluate the oncogenic potential of the top most enriched kinase CSNK2A1, we further treated the cervical cancer cell line, CaSki, with varying doses of CSNK2A1 inhibitor, silmitasertib (CX4945). We observed a dose-dependent impact of the inhibitor on the viability of the cell line (Fig. 3D). Furthermore, we evaluated the effect of inhibition of CSNK2A1 on the migration, proliferation and colony formation ability. Inhibition of CSNK2A1 further resulted in significant reduction in proliferation, migration and colony formation of the cell line (Fig. 3E–G).
3.5. CSNK2A1 inhibition sensitizes cells to cisplatin
We sought to understand the role of CSNK2A1 in mediating cisplatin (CDDP) sensitivity and the feasibility of the CSNK2A1 inhibitor silmitasertib (CX4945) in combination treatment approaches. We treated the CaSki cell line with varying concentrations of cisplatin alone or in combination with a constant dosage of CX4945. A significant decrease in viability of the cells was observed upon the addition of CX4945 along with cisplatin compared to cisplatin alone (Fig. S7, ESI†). Furthermore, to determine the combination index (CI) and the dose reduction (DRI) of cisplatin and CX4945, the cells were further treated with a constant ratio of both the drugs at their IC50 (Fig. 4A). The DRI and CI for the drug combination were calculated using the software CompuSyn. The combination showed a synergistic effect for the fractional inhibition range of 0.45 to 0.97 with CI ranging from 0.89 to 0.49 (Table S6, ESI†). Additionally, we computed the dose reduction index (DRI) required to achieve a fraction of affected (Fa) = 0.5. In the subsequent experiments we utilized lower concentrations of cisplatin and the inhibitor as determined using the DRI. | ||
| Fig. 4 Combination treatment and assays in CaSki cells. (A) Graphical representation of constant ratio treatment of CX4945 in combination with cisplatin. CaSki cells were treated with a constant ratio of CX4945 and cisplatin at their IC50 value for 48 h. Absorbance was taken and percentage cell viability was calculated. Combination index (CI) and dose reduction index (DRI) of the combination were calculated using CompuSyn software. The CI and DRI for Fa = 0.5 are represented in the table. (B) Migration assay of CaSki cells followed by treatment with DMSO, CX4945 (dose reduced), cisplatin (dose reduced) and the combination. Graphical representation comparing the single agent treatments, combination and the untreated samples. (C) Proliferation of CaSki cells at different time points upon treatment with DMSO, CX4945 (dose reduced), cisplatin (dose reduced) and the combination. (D) Graphical representation of the colony formation assay of CaSki cells at different time points upon treatment with DMSO, CX4945 (dose reduced), cisplatin (dose reduced) and the combination. | ||
3.6. Combination treatment with reduced dose of the agents gave decreased proliferation and migration
We further extended our experiments to assess the impact of the dose-reduced combination of cisplatin and the inhibitor on migration, proliferation and colony formation. Cell migration was assessed using a scratch wound assay. The CaSki cells were treated with single agents or in combination for 18 hours. Monotherapy with reduced dose of cisplatin and the inhibitor significantly reduced the migration of CaSki compared to the DMSO control. However, the dose-reduced combination of cisplatin and the inhibitor exhibited the most significant effect on CaSki migration (Fig. 4B).Treatment with reduced dose of cisplatin and CX4945 caused a significant decrease in cell proliferation compared to single agent treatment. CaSki was treated with reduced concentration of CX4945, cisplatin or in combination. The combination treatment yielded a significant reduction in cell proliferation at 24 h, compared to both the single agent treatments and the control, with no significant difference between the single agent treatments and the control. Furthermore, at 48 h also the dose-reduced combination treatment exhibited the most significant effect on proliferation. However, at 72 h there was no significant difference between the CX4945 alone or dose-reduced combination. Our results suggest combination treatment results in an early and significant effect on proliferation compared to the single agent treatment. Moreover, prolonged exposure to a low dose of CX4945 resulted in a significant reduction in cell proliferation (Fig. 4C).
Furthermore, we evaluated the impact of both single-agent monotherapy and combination therapy on the colony formation of CaSki cells. We observed significant reduction in clone formation in single agent treatment with cisplatin and the inhibitor compared to the DMSO control. Interestingly, we observed that cisplatin had a greater impact on clone formation than the inhibitor. Furthermore, the dose-reduced combination treatment led to the absence of colony formation (Fig. 4D).
4. Discussion
Navigating the patient heterogeneity is crucial in clinical oncology, as making treatment decisions based on the molecular characteristics yields optimal clinical outcomes. Predictive biomarker-based patient selection and identification of druggable targets prior to the therapy can combat resistance, toxicities and side effects that are commonly associated with conventional therapeutics. In locally advanced cervical cancer, CCRT is the standard treatment option as it improves local disease control and survival of the patients.33 Cisplatin based chemoradiation therapy showed superior disease-free survival in cervical cancer patients compared to neoadjuvant chemotherapy followed by radical surgery.34 However, treatment failure and acute toxicities in the patients treated with CCRT remain a major challenge in managing the locally advanced diseases.35 Therefore, alternative approaches to improve the clinical outcome of these patients have to be explored. Integration of targeted agents could potentially improve the efficacy of chemoradiotherapy without excessive toxicity. In this study we performed mass spectrometry based phosphoproteomics to assess the altered phosphorylation events, activated kinases and pathways associated with CCRT resistance in cervical cancer patients. Our phosphoproteomics analysis of treatment naïve tissue samples provided several insights about the innate mechanisms involved in CCRT response.An aberrant phosphorylation process has been observed to play a significant role in conferring resistance to therapeutic interventions in cancer.36,37 Comparison of the phosphoproteome of treatment sensitive and resistant cohorts revealed distinct phosphorylation signature between the cohorts. Our study identified 2531 phosphopeptides corresponding to 1099 proteins in cervical cancer patients. Our statistical analysis led to the identification of 77 differentially phosphorylated proteins between the cohorts providing potential predictive candidate biomarkers. SMC1A is the top most phosphorylated protein in the non-responder cohort and is correlated with worse prognostic outcome in hepatocellular carcinoma.38 Phosphorylation of SMC1A (pSMC1A) on serine 957 by ataxia-telangiectasia mutated (ATM) kinase after ionization radiation is reported as a crucial event for maintaining chromosomal stability and cell survival after DNA damage.39,40 The kinase substrate enrichment analysis also predicted the activation of ATM kinase among the identified kinases. Previously, it has been reported that high levels of ATM prior to irradiance correlated with radioresistance in cervical cancer.41 Immunohistochemistry-based validation experiments further supported the high expression of pSMC1A in non-responder cohorts. The stratification of patients prior to treatment can be achieved through assessment of expression of the pSMC1A.
To understand the mechanisms involved in treatment resistance, we further carried out pathway analysis with significantly phosphorylated proteins. The pathway analysis revealed involvement of the DNA repair pathway in driving treatment resistance of cervical cancer. DNA repair proteins were reported to be strongly involved in resistance to various treatment modalities. In cisplatin-based chemoradiotherapy, the cisplatin–DNA adducts enhance the radiation induced double stranded breaks (DSBs) and/or prevent repair.42 Our data suggests that activation of the DNA repair pathway in non-responder cohorts might be conferring chemoradiation resistance in non-responder cohorts. We observed dysregulated phosphorylation of several proteins involved in the DNA repair pathway. For example, hyperphosphorylation of DDB2 is observed in non-responder cohorts compared to the responder cohorts. Phosphorylation of DDB2 is reported to be essential to facilitate the initiation of nuclear excision repair.43 O6-methylguanine-DNA methyltransferase (MGMT) promotes resistance to radiation therapy as well as cisplatin-based chemotherapy.44 Elevated PNKP phosphorylation results in stabilization of the protein and mediates DNA repair.45 Increased phosphorylation of PNKP was observed in non-responders. In esophageal cancers, downregulation of RIPK3 is associated with cisplatin resistance primarily through the activation of the DNA repair pathway.25 In the non-responder cohort, we identified decreased phosphorylation of RIPK3, and it was reported that the activity of RIPK3 is correlated with its phosphorylation.46 Our data suggest that targeting the DNA repair pathway could potentially improve the efficacy of CCRT. Fig. 5 represents a data driven pathway involved in CCRT resistance in cervical cancer.
 | ||
| Fig. 5 Data driven pathway of CCRT resistance in cervical cancer patients. Pathway analysis revealed enrichment of the DNA repair pathway in CCRT resistant patients. Activation of kinases, and dysregulation of phosphorylation in substrates involved in the DNA repair pathway were observed in our data. | ||
Incorporating small molecule inhibitors that target kinases may enhance the treatment's effectiveness while reducing toxicity, compared to adding chemotherapy in combination with CCRT. Our kinase substrate analysis predicted the activation of several kinases that could be investigated as potential drug targets. Kinases such as CSNK2A1, PRKDC, PLK-1, NEK2, ATM and CDK1 were predicted to be enriched in non-responders. Involvement of these kinases was previously reported in treatment resistance in several cancers. These kinases are also involved in the DNA repair pathway at different levels. CSNK2A1 mediates tumorigenesis and is regarded as a prognostic as well as therapeutic target in several cancers.47 HMGA1 a downstream substrate of CSNK2A1 was reported to be hyperphosphorylated and was reported to be involved in cisplatin resistance.48 Our data suggests that the enrichment of CSNK2A1 in the CCRT resistant cohort mediates resistance. We further inhibited CSNK2A1 in CaSki with small molecule inhibitor CX4945. CX4945 is an ATP-competitive inhibitor currently under clinical trial for various cancers.49 Inhibition of CSNK2A1 significantly reduced the proliferation, migration and colony formation ability. Our preclinical data on a cervical cancer cell line indicate the oncogenic role of CSNK2A1 in cervical cancer.
Several pieces of evidence suggest enhanced efficacy of combination treatment compared to monotherapy. The synergistic or addictive action of the therapeutic agents increases the efficacy by targeting multiple pathways and potentially reducing the drug resistance. Recently, the FDA approved the usage of pembrolizumab with chemoradiotherapy in patients with stage III–IVA cervical cancer.50 Patients treated with pembrolizumab plus chemoradiotherapy showed 41% lower chance of disease progression or mortality.50 Integration of targeted therapeutic agents could also reduce the dosage of the chemotherapeutic agent and thereby the toxicity associated with a high dosage. Here, we further investigated the potential of combining the CSNK2A1 inhibitor along with cisplatin in a preclinical model. Combining the inhibitor along with cisplatin reduced the viability of cells compared to cisplatin treatment alone. Additionally, the combination index (CI) of cisplatin and CX4945 was calculated using CompuSyn software indicating synergism with CI values ranging from 0.77 to 0.49 for Fa = 0.5 ∼ 0.9. Moreover, our results also indicated favorable dose reduction for the combination treatment. We sought to investigate the effect of dose-reduced combination of cisplatin and the inhibitor on the proliferation, migration and colony formation ability of the cell line. Cisplatin along with the inhibitor resulted in better effect on cell proliferation, migration and colony formation, which further confirmed the effectiveness of this combination therapy in treating cervical cancer.
Our results reveal the differential phosphoprofiles in CCRT resistant and sensitive cervical cancer patients. Activation of the DNA repair pathway and differential phosphorylation of the proteins involved in the pathway might be conferring resistance in the non-responder cohort. Our IHC-based validation of pSMC1A between the responder and non-responders suggests the potential of pSMC1A in predicting the treatment response. Furthermore, we confirmed the role of activated kinase CSNK2A1 in oncogenesis of cervical cancer and the synergistic effects of the CSNK2A1 inhibitor in combination with cisplatin. Our results suggest that combination of CX4945 along with cisplatin could potentially be implemented to treat CCRT resistant cervical cancer patients. However, further multicentric validation studies have to be carried out.
5. Conclusions
In this study, we employed phosphoproteomics to explore the treatment resistance signature in cervical cancer patients. Our results suggest the preconditioned differences in phosphorylation events and activated kinases in treatment naïve patients contribute to therapy evasion in the resistant cohort. Our results reveal the potential of pSMC1A as a predictive biomarker for CCRT resistance. Further validation experiments on samples collected from multiple centers should be carried out to consolidate the results. Our study also indicates the activation of the DNA repair pathway and dysregulated phosphorylation of several proteins involved in the pathway in the non-responder cohort. Our results suggest that targeting CSNK2A1 along with cisplatin could be an effective strategy to improve the efficacy of CCRT in cervical cancer.Author contributions
PK and RC conceptualized the study. RC, MS, and VT carried out sample collection and recorded the clinicopathological details of the patients. IAG carried out the experiments and data analysis. IAG and PK were involved in data interpretation. IAG, JS and PK were involved in manuscript writing and figure preparation. PK, RED and RC critically reviewed and edited the manuscript. All the authors have read and approved the final manuscript.Data availability
The data supporting this article have been included as part of the ESI.† Data are available via ProteomeXchange with identifier PXD055164.Conflicts of interest
There are no conflicts to declare.Acknowledgements
IAG is a recipient of Senior Research Fellowship from Council of Scientific and Industrial Research (CSIR), Government of India. We would like to acknowledge Vivek Ghose for his assistance in drafting the mass spectrometry methodology section and providing essential technical support.References
- V. Kulothungan, K. Sathishkumar, S. Leburu, T. Ramamoorthy, S. Stephen, D. Basavarajappa, N. Tomy, R. Mohan, G. R. Menon and P. Mathur, Burden of Cancers in India - Estimates of Cancer Crude Incidence, YLLs, YLDs and DALYs for 2021 and 2025 Based on National Cancer Registry Program, BMC Cancer, 2022, 22(1), 527, DOI:10.1186/s12885-022-09578-1.
- P. Mathur, K. Sathishkumar, M. Chaturvedi, P. Das, K. L. Sudarshan, S. Santhappan, V. Nallasamy, A. John, S. Narasimhan and F. S. Roselind, ICMR-NCDIR-NCRP Investigator Group. Cancer Statistics, 2020: Report From National Cancer Registry Programme, India, JCO Global Oncol., 2020, 6, 1063–1075, DOI:10.1200/GO.20.00122.
- Prognosis and Survival for Cervical Cancer. https://cancer.ca/en/cancer-information/cancer-types/cervical/prognosis-and-survival#:~:text=Early%20stage%20cervical%20cancer%20has,is%20only%20in%20the%20cervix.
- J. A. Green, J. M. Kirwan, J. F. Tierney, P. Symonds, L. Fresco, M. Collingwood and C. J. Williams, Survival and Recurrence after Concomitant Chemotherapy and Radiotherapy for Cancer of the Uterine Cervix: A Systematic Review and Meta-Analysis, Lancet, 2001, 358(9284), 781–786, DOI:10.1016/S0140-6736(01)05965-7.
- Chemoradiotherapy for Cervical Cancer Meta-Analysis Collaboration, Reducing Uncertainties about the Effects of Chemoradiotherapy for Cervical Cancer: A Systematic Review and Meta-Analysis of Individual Patient Data from 18 Randomized Trials, J. Clin. Oncol., 2008, 26(35), 5802–5812, DOI:10.1200/JCO.2008.16.4368.
- L. Kumar and S. Gupta, Integrating Chemotherapy in the Management of Cervical Cancer: A Critical Appraisal, Oncology, 2016, 91(suppl. 1), 8–17, DOI:10.1159/000447576.
- P. G. Rose, B. N. Bundy, E. B. Watkins, J. T. Thigpen, G. Deppe, M. A. Maiman, D. L. Clarke-Pearson and S. Insalaco, Concurrent Cisplatin-Based Radiotherapy and Chemotherapy for Locally Advanced Cervical Cancer, N. Engl. J. Med., 1999, 340(15), 1144–1153, DOI:10.1056/NEJM199904153401502.
- K. Scatchard, J. L. Forrest, M. Flubacher, P. Cornes and C. Williams, Chemotherapy for Metastatic and Recurrent Cervical Cancer, Cochrane Database Syst. Rev., 2012, 10(10), CD006469, DOI:10.1002/14651858.CD006469.pub2.
- P. M. Fracasso, J. A. Blessing, J. Wolf, T. F. Rocereto, J. S. Berek and S. Waggoner, Phase II Evaluation of Oxaliplatin in Previously Treated Squamous Cell Carcinoma of the Cervix: A Gynecologic Oncology Group Study, Gynecol. Oncol., 2003, 90(1), 177–180, DOI:10.1016/s0090-8258(03)00253-1.
- S. Markovina, K. A. Rendle, A. C. Cohen, L. M. Kuroki, S. Grover and J. K. Schwarz, Improving Cervical Cancer Survival–A Multifaceted Strategy to Sustain Progress for This Global Problem, Cancer, 2022, 128(23), 4074–4084, DOI:10.1002/cncr.34485.
- A. Dueñas-González, J. J. Zarbá, F. Patel, J. C. Alcedo, S. Beslija, L. Casanova, P. Pattaranutaporn, S. Hameed, J. M. Blair, H. Barraclough and M. Orlando, Phase III, Open-Label, Randomized Study Comparing Concurrent Gemcitabine plus Cisplatin and Radiation Followed by Adjuvant Gemcitabine and Cisplatin versus Concurrent Cisplatin and Radiation in Patients with Stage IIB to IVA Carcinoma of the Cervix, J. Clin. Oncol., 2011, 29(13), 1678–1685, DOI:10.1200/JCO.2009.25.9663.
- J. J. Kovalic, C. A. Perez, P. W. Grigsby and M. A. Lockett, The Effect of Volume of Disease in Patients with Carcinoma of the Uterine Cervix, Int. J. Radiat. Oncol., Biol., Phys., 1991, 21(4), 905–910, DOI:10.1016/0360-3016(91)90728-m.
- S. Varghese, T. Ram, S. Pavamani, E. Thomas, V. Jeyaseelan and P. Viswanathan, Concurrent Chemo-Irradiation with Weekly Cisplatin and Paclitaxel in the Treatment of Locally Advanced Squamous Cell Carcinoma of Cervix: A Phase II Study, J. Cancer Res. Ther., 2014, 10(2), 330, DOI:10.4103/0973-1482.136621.
- Y. S. Kim, S. S. Shin, J.-H. Nam, Y.-T. Kim, Y.-M. Kim, J. H. Kim and E. K. Choi, Prospective Randomized Comparison of Monthly Fluorouracil and Cisplatin versus Weekly Cisplatin Concurrent with Pelvic Radiotherapy and High-Dose Rate Brachytherapy for Locally Advanced Cervical Cancer, Gynecol. Oncol., 2008, 108(1), 195–200, DOI:10.1016/j.ygyno.2007.09.022.
- B. Deb, V. N. Puttamallesh, K. Gondkar, J. P. Thiery, H. Gowda and P. Kumar, Phosphoproteomic Profiling Identifies Aberrant Activation of Integrin Signaling in Aggressive Non-Type Bladder Carcinoma, Jpn. Clin. Med., 2019, 8(5), 703, DOI:10.3390/jcm8050703.
- G. Sathe, I. A. George, B. Deb, A. P. Jain, K. Patel, B. Nayak, S. Karmakar, A. Seth, A. Pandey and P. Kumar, Urinary Glycoproteomic Profiling of Non-Muscle Invasive and Muscle Invasive Bladder Carcinoma Patients Reveals Distinct N-Glycosylation Pattern of CD44, MGAM, and GINM1, Oncotarget, 2020, 11(34), 3244–3255, DOI:10.18632/oncotarget.27696.
- K. Gondkar, G. Sathe, N. Joshi, B. Nair, A. Pandey and P. Kumar, Integrated Proteomic and Phosphoproteomics Analysis of DKK3 Signaling Reveals Activated Kinase in the Most Aggressive Gallbladder Cancer, Cells, 2021, 10(3), 511, DOI:10.3390/cells10030511.
- A. Kumar, D. S. Nayakanti, K. K. Mangalaparthi, V. Gopinath, N. V. N. Reddy, K. Govindan, G. Voolapalli, P. Kumar and L. D. Kumar, Quantitative Proteome Profiling Stratifies Fibroepithelial Lesions of the Breast, Oncotarget, 2021, 12(5), 507–518, DOI:10.18632/oncotarget.27889.
- D. O. Debets, K. E. Stecker, A. Piskopou, M. C. Liefaard, J. Wesseling, G. S. Sonke, E. H. Lips and M. Altelaar, Deep (Phospho)Proteomics Profiling of Pre- Treatment Needle Biopsies Identifies Signatures of Treatment Resistance in HER2+ Breast Cancer, Cell Rep. Med., 2023, 4(10), 101203, DOI:10.1016/j.xcrm.2023.101203.
- I. A. George, G. Sathe, V. Ghose, A. Chougule, P. Chandrani, V. Patil, V. Noronha, R. Venkataramanan, S. Limaye, A. Pandey, K. Prabhash and P. Kumar, Integrated Proteomics and Phosphoproteomics Revealed Druggable Kinases in Neoadjuvant Chemotherapy Resistant Tongue Cancer, Front. Cell Dev. Biol., 2022, 10, 957983, DOI:10.3389/fcell.2022.957983.
- A. H. Patil, K. K. Datta, S. K. Behera, S. Kasaragod, S. M. Pinto, S. G. Koyangana, P. P. Mathur, H. Gowda, A. Pandey and T. S. K. Prasad, Dissecting Candida Pathobiology: Post-Translational Modifications on the Candida Tropicalis Proteome, OMICS: J. Integr. Biol., 2018, 22(8), 544–552, DOI:10.1089/omi.2018.0093.
- S.-W. Kim, J. Roh and C.-S. Park, Immunohistochemistry for Pathologists: Protocols, Pitfalls, and Tips, J. Pathol. Transl. Med., 2016, 50(6), 411–418, DOI:10.4132/jptm.2016.08.08.
- E. A. Eisenhauer, P. Therasse, J. Bogaerts, L. H. Schwartz, D. Sargent, R. Ford, J. Dancey, S. Arbuck, S. Gwyther, M. Mooney, L. Rubinstein, L. Shankar, L. Dodd, R. Kaplan, D. Lacombe and J. Verweij, New Response Evaluation Criteria in Solid Tumours: Revised RECIST Guideline (Version 1.1), Eur. J. Cancer, 2009, 45(2), 228–247, DOI:10.1016/j.ejca.2008.10.026.
- J. Cicenas, E. Zalyte, A. Bairoch and P. Gaudet, Kinases and Cancer, Cancers, 2018, 10(3), 63, DOI:10.3390/cancers10030063.
- Y. Sun, L. Zhai, S. Ma, C. Zhang, L. Zhao, N. Li, Y. Xu, T. Zhang, Z. Guo, H. Zhang, P. Xu and X. Zhao, Down-Regulation of RIP3 Potentiates Cisplatin Chemoresistance by Triggering HSP90-ERK Pathway Mediated DNA Repair in Esophageal Squamous Cell Carcinoma, Cancer Lett., 2018, 418, 97–108, DOI:10.1016/j.canlet.2018.01.022.
- U. K. Hussein, A. G. Ahmed, Y. Song, K. M. Kim, Y. J. Moon, A.-R. Ahn, H. S. Park, S. J. Ahn, S.-H. Park, J. R. Kim and K. Y. Jang, CK2α/CSNK2A1 Induces Resistance to Doxorubicin through SIRT6-Mediated Activation of the DNA Damage Repair Pathway, Cells, 2021, 10(7), 1770, DOI:10.3390/cells10071770.
- Y. Chen, Y. Li, J. Xiong, B. Lan, X. Wang, J. Liu, J. Lin, Z. Fei, X. Zheng and C. Chen, Role of PRKDC in Cancer Initiation, Progression, and Treatment, Cancer Cell Int., 2021, 21(1), 563, DOI:10.1186/s12935-021-02229-8.
- M. Wu, Y. Wang, D. Yang, Y. Gong, F. Rao, R. Liu, Y. Danna, J. Li, J. Fan, J. Chen, W. Zhang and Q. Zhan, A PLK1 Kinase Inhibitor Enhances the Chemosensitivity of Cisplatin by Inducing Pyroptosis in Oesophageal Squamous Cell Carcinoma, EBioMedicine, 2019, 41, 244–255, DOI:10.1016/j.ebiom.2019.02.012.
- H. Xu, L. Zeng, Y. Guan, X. Feng, Y. Zhu, Y. Lu, C. Shi, S. Chen, J. Xia, J. Guo, C. Kuang, W. Li, F. Jin and W. Zhou, High NEK2 Confers to Poor Prognosis and Contributes to Cisplatin-Based Chemotherapy Resistance in Nasopharyngeal Carcinoma, J. Cell. Biochem., 2019, 120(3), 3547–3558, DOI:10.1002/jcb.27632.
- I. A. George, R. Chauhan, R. E. Dhawale, R. Iyer, S. Limaye, R. Sankaranarayanan, R. Venkataramanan and P. Kumar, Insights into Therapy Resistance in Cervical Cancer, Adv. Cancer Biol.: Metastasis, 2022, 6, 100074, DOI:10.1016/j.adcanc.2022.100074.
- N. Bansal, D. C. Marchion, E. Bicaku, Y. Xiong, N. Chen, X. B. Stickles, E. A. Sawah, R. M. Wenham, S. M. Apte, J. Gonzalez-Bosquet, P. L. Judson, A. Hakam and J. M. Lancaster, BCL2 Antagonist of Cell Death Kinases, Phosphatases, and Ovarian Cancer Sensitivity to Cisplatin, J. Gynecol. Oncol., 2012, 23(1), 35, DOI:10.3802/jgo.2012.23.1.35.
- S. W. Strum, L. Gyenis and D. W. Litchfield, CSNK2 in Cancer: Pathophysiology and Translational Applications, Br. J. Cancer, 2022, 126(7), 994–1003, DOI:10.1038/s41416-021-01616-2.
- O. Cho and M. Chun, Management for Locally Advanced Cervical Cancer: New Trends and Controversial Issues, Radiat. Oncol. J., 2018, 36(4), 254–264, DOI:10.3857/roj.2018.00500.
- S. Gupta, A. Maheshwari, P. Parab, U. Mahantshetty, R. Hawaldar, S. Sastri Chopra, R. Kerkar, R. Engineer, H. Tongaonkar, J. Ghosh, S. Gulia, N. Kumar, T. S. Shylasree, R. Gawade, Y. Kembhavi, M. Gaikar, S. Menon, M. Thakur, S. Shrivastava and R. Badwe, Neoadjuvant Chemotherapy Followed by Radical Surgery Versus Concomitant Chemotherapy and Radiotherapy in Patients With Stage IB2, IIA, or IIB Squamous Cervical Cancer: A Randomized Controlled Trial, J. Clin. Oncol., 2018, 36(16), 1548–1555, DOI:10.1200/JCO.2017.75.9985.
- F. Coutinho, M. Gokhale, C. Doran, M. Monberg, K. S. Yamada and L. Chen, Treatment Patterns and Outcomes among Locally Advanced Cervical Cancer Patients Receiving Concurrent Chemoradiotherapy, J. Clin. Oncol., 2023, 41(suppl. 16), e17511, DOI:10.1200/JCO.2023.41.16_suppl.e17511.
- H. Johnson, S. Narayan and A. K. Sharma, Altering Phosphorylation in Cancer through PP2A Modifiers, Cancer Cell Int., 2024, 24(1), 11, DOI:10.1186/s12935-023-03193-1.
- S. Mouron, M. J. Bueno, A. Lluch, L. Manso, I. Calvo, J. Cortes, J. A. Garcia-Saenz, M. Gil-Gil, N. Martinez-Janez, J. V. Apala, E. Caleiras, P. Ximénez-Embún, J. Muñoz, L. Gonzalez-Cortijo, R. Murillo, R. Sánchez-Bayona, J. M. Cejalvo, G. Gómez-López, C. Fustero-Torre, S. Sabroso-Lasa, N. Malats, M. Martinez, A. Moreno, D. Megias, M. Malumbres, R. Colomer and M. Quintela-Fandino, Phosphoproteomic Analysis of Neoadjuvant Breast Cancer Suggests That Increased Sensitivity to Paclitaxel Is Driven by CDK4 and Filamin A, Nat. Commun., 2022, 13(1), 7529, DOI:10.1038/s41467-022-35065-z.
- Y. Zhang, F. Yi, L. Wang, Z. Wang, N. Zhang, Z. Wang, Z. Li, X. Song, S. Wei and L. Cao, Phosphorylation of SMC1A Promotes Hepatocellular Carcinoma Cell Proliferation and Migration, Int. J. Biol. Sci., 2018, 14(9), 1081–1089, DOI:10.7150/ijbs.24692.
- S.-T. Kim, B. Xu and M. B. Kastan, Involvement of the Cohesin Protein, Smc1, in Atm-Dependent and Independent Responses to DNA Damage, Genes Dev., 2002, 16(5), 560–570, DOI:10.1101/gad.970602.
- R. Kitagawa, C. J. Bakkenist, P. J. McKinnon and M. B. Kastan, Phosphorylation of SMC1 Is a Critical Downstream Event in the ATM-NBS1-BRCA1 Pathway, Genes Dev., 2004, 18(12), 1423–1438, DOI:10.1101/gad.1200304.
- F. Roossink, H. W. Wieringa, M. G. Noordhuis, K. A. ten Hoor, M. Kok, L. Slagter-Menkema, H. Hollema, G. H. de Bock, E. Pras, E. G. E. de Vries, S. de Jong, A. G. J. van der Zee, E. Schuuring, G. B. A. Wisman and M. A. T. M. van Vugt, The Role of ATM and 53BP1 as Predictive Markers in Cervical Cancer, Int. J. Cancer, 2012, 131(9), 2056–2066, DOI:10.1002/ijc.27488.
- P. B. Tchounwou, S. Dasari, F. K. Noubissi, P. Ray and S. Kumar, Advances in Our Understanding of the Molecular Mechanisms of Action of Cisplatin in Cancer Therapy, J. Exp. Pharmacol., 2021, 13, 303–328, DOI:10.2147/JEP.S267383.
- N. Roy, S. Bagchi and P. Raychaudhuri, Damaged DNA Binding Protein 2 in Reactive Oxygen Species (ROS) Regulation and Premature Senescence, Int. J. Mol. Sci., 2012, 13(9), 11012–11026, DOI:10.3390/ijms130911012.
- S.-H. Chen, W.-T. Huang, W.-C. Kao, S.-Y. Hsiao, H.-Y. Pan, C.-W. Fang, Y.-L. Shiue, C.-L. Chou and C.-F. Li, O6-Methylguanine-DNA Methyltransferase Modulates Cisplatin-Induced DNA Double-Strand Breaks by Targeting the Homologous Recombination Pathway in Nasopharyngeal Carcinoma, J. Biomed. Sci., 2021, 28(1), 2, DOI:10.1186/s12929-020-00699-y.
- J. L. Parsons, S. V. Khoronenkova, I. I. Dianova, N. Ternette, B. M. Kessler, P. K. Datta and G. L. Dianov, Phosphorylation of PNKP by ATM Prevents Its Proteasomal Degradation and Enhances Resistance to Oxidative Stress, Nucleic Acids Res., 2012, 40(22), 11404–11415, DOI:10.1093/nar/gks909.
- K. Moriwaki and F. K.-M. Chan, RIP3: A Molecular Switch for Necrosis and Inflammation, Genes Dev., 2013, 27(15), 1640–1649, DOI:10.1101/gad.223321.113.
- R. Wu, W. Tang, K. Qiu, P. Li, Y. Li, D. Li and Z. He, An Integrative Pan-Cancer Analysis of the Prognostic and Immunological Role of Casein Kinase 2 Alpha Protein 1 (CSNK2A1) in Human Cancers: A Study Based on Bioinformatics and Immunohistochemical Analysis, Int. J. Gen. Med., 2021, 14, 6215–6232, DOI:10.2147/IJGM.S330500.
- Z. Shi, D. Wu, H. Xu, J. Yang and X. Sun, CSNK2A1-Mediated Phosphorylation of HMGA2 Modulates Cisplatin Resistance in Cervical Cancer, FEBS Open Bio, 2021, 11(8), 2245–2255, DOI:10.1002/2211-5463.13228.
- C. D’Amore, C. Borgo, S. Sarno and M. Salvi, Role of CK2 Inhibitor CX-4945 in Anti-Cancer Combination Therapy - Potential Clinical Relevance, Cell. Oncol., 2020, 43(6), 1003–1016, DOI:10.1007/s13402-020-00566-w.
- D. Lorusso, Y. Xiang, K. Hasegawa, G. Scambia, M. Leiva, P. Ramos-Elias, A. Acevedo, V. Sukhin, N. Cloven, A. J. Pereira De Santana Gomes, F. Contreras Mejía, A. Reiss, A. Ayhan, J.-Y. Lee, V. Saevets, F. Zagouri, L. Gilbert, J. Sehouli, E. Tharavichitkul, K. Lindemann, R. Lazzari, C.-L. Chang, R. Lampé, H. Zhu, A. Oaknin, M. Christiaens, S. Polterauer, T. Usami, K. Li, K. Yamada, S. Toker, S. M. Keefe, S. Pignata and L. R. Duska, Pembrolizumab or Placebo with Chemoradiotherapy Followed by Pembrolizumab or Placebo for Newly Diagnosed, High-Risk, Locally Advanced Cervical Cancer (ENGOT-Cx11/GOG-3047/KEYNOTE-A18): A Randomised, Double-Blind, Phase 3 Clinical Trial, Lancet, 2024, 403(10434), 1341–1350, DOI:10.1016/S0140-6736(24)00317-9.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mo00147h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |