
MobiChIP: a compatible library construction method of single-cell ChIP-seq based droplets†
Xianhong
Yu‡
*ab,
Guantao
Zheng‡
b,
Liting
Xu‡
c,
Weiyi
Guo‡
ac,
Guodong
Chen
b,
Yiling
Zhu
c,
Tingting
Li
b,
Mingming
Rao
b,
Linyan
Wang
d,
Rong
Cong
c and
Hao
Pei
*c
aThe Key Laboratory for Biomedical Photonics of MOE at Wuhan National Laboratory for Optoelectronics – Hubei Bioinformatics and Molecular Imaging Key Laboratory, Department of Biomedical Engineering, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan 430074, China
bShanghai MobiDrop Co., Ltd., Room 351, Building 1, Guoshoujing Road, Shanghai Free Trade Pilot Zone, Shanghai, 200000, China. E-mail: xianhongyu@mobidrop.com
cMobiDrop (Zhejiang) Co., Ltd., No. 1888 Longxiang Avenue, Tongxiang, Zhejiang Province 314500, China. E-mail: haopei@mobidrop.com
dEye Center, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Zhejiang Provincial Key Laboratory of Ophthalmology, Zhejiang Provincial Clinical Research Center for Eye Diseases, Zhejiang Provincial Engineering Institute on Eye Diseases, Hangzhou, Zhejiang, China
First published on 18th October 2024
Abstract
To illustrate epigenetic heterogeneity, versatile tools of single-cell ChIP-seq (scChIP-seq) are essential for both convenience and accuracy. We developed MobiChIP, a compatible ChIP-seq library construction method based on current sequencing platforms for single-cell applications. MobiChIP efficiently captures fragments from tagmented nuclei across various species and allows sample mixing from different tissues or species. This strategy offers robust nucleosome amplification and flexible sequencing without customized primers. MobiChIP reveals regulatory landscapes of chromatin with active (H3K27ac) and repressive (H3K27me3) histone modification in peripheral blood mononuclear cells (PBMCs) and accurately identifies epigenetic repression of the Hox gene cluster, outperforming ATAC-seq. Meanwhile, we also integrated scChIP-seq with scRNA-seq to further illustrate cellular genetic and epigenetic heterogeneity.
Single-cell sequencing can reveal the complex heterogeneity of individual cells and detect rare populations.1–4 It has enabled transcriptome sequencing, DNA methylation sequencing, DNase sequencing, and ATAC-seq for open chromatin.5–14 While ATAC and DNase provide genome-wide views of chromatin accessibility, studying chromatin modifications and TF binding sites offers deeper insights into epigenomic heterogeneity and cell states. ChIP-seq, a powerful tool for studying protein-DNA interactions both in vivo and in vitro, is used to identify transcription factor binding sites or histone modification sites across the genome.15–18 Methods like CoBATCH, CUT&Tag, and ACT-seq, using protein A-Tn5 (PATn5), have been applied to study protein–DNA interactions, cell fate trajectories, and population heterogeneities in developmental and pathological processes, but are labor-intensive.19–24 To achieve high-throughput scChIP-seq, microfluidic chips have been developed, but these methods require customized sequencing strategies.25,26
We developed a scChIP-seq method called MobiChIP, which uses a modified adaptor to capture tagmented chromatin with protein A-Tn5. MobiChIP eliminates the need for customized sequencing primers and is compatible with current sequencing platforms. Using MobiChIP, we investigated the epigenome of healthy PBMCs. Histone modifications H3K27ac and H3K27me3 revealed different landscapes of active and repressive genomic regions. We compared MobiChIP to ATAC-seq and integrated the epigenetic data with transcriptomic data to identify distinct heterogeneous populations. This study provides a method and bioinformatics pipeline for exploring epigenetic regulation and gene expression in complex tissues, offering a multi-omics approach to better understand epigenetic heterogeneity in PBMCs.
High-throughput MobiChIP profiling of single cells
To perform MobiChIP on a large scale of cells to reveal the epigenetic heterogeneity, we combine the CoBATCH and microfluidic to generate the high-throughput single cell ChIP-seq based droplets (Fig. 1A). To enable the sequencing of scChIP-seq library agilely on current platform, we modified the capture adaptor of MobiChIP to sequence with pooled sample from others. We mixed equal number of human K562 cells and mouse L929 cells, and sequenced the resultant library. The data showed collision rate of ∼5.1%, suggesting a successful labeling of single cells (Fig. 1B). Total 350 mouse L929 cells and 469 human K562 cells were identified, clustered into two populations (Fig. 1C). The results showed that the mouse-derived cells and the human-derived cells were well separated, proving that captured signals of H3K27ac were useful for clustering analysis. Meanwhile, we further analyzed the number of valid fragments captured in different cells and found that the separation of cell clusters was independent to the number of captured valid fragments (Fig. 1D). In order to meet the sample mixing in some distinctive usage scenario, we also applied MobiChIP to map the histone modification with different samples in the same tube. Sample indexes of adaptor assembled into PATn5 were used to label different samples. Human K562, mouse 3T3 and hamster CHO were mixed to perform MobiChIP, and 97%, 96% and 95% labeling ratio was achieved respectively (Fig. 1E).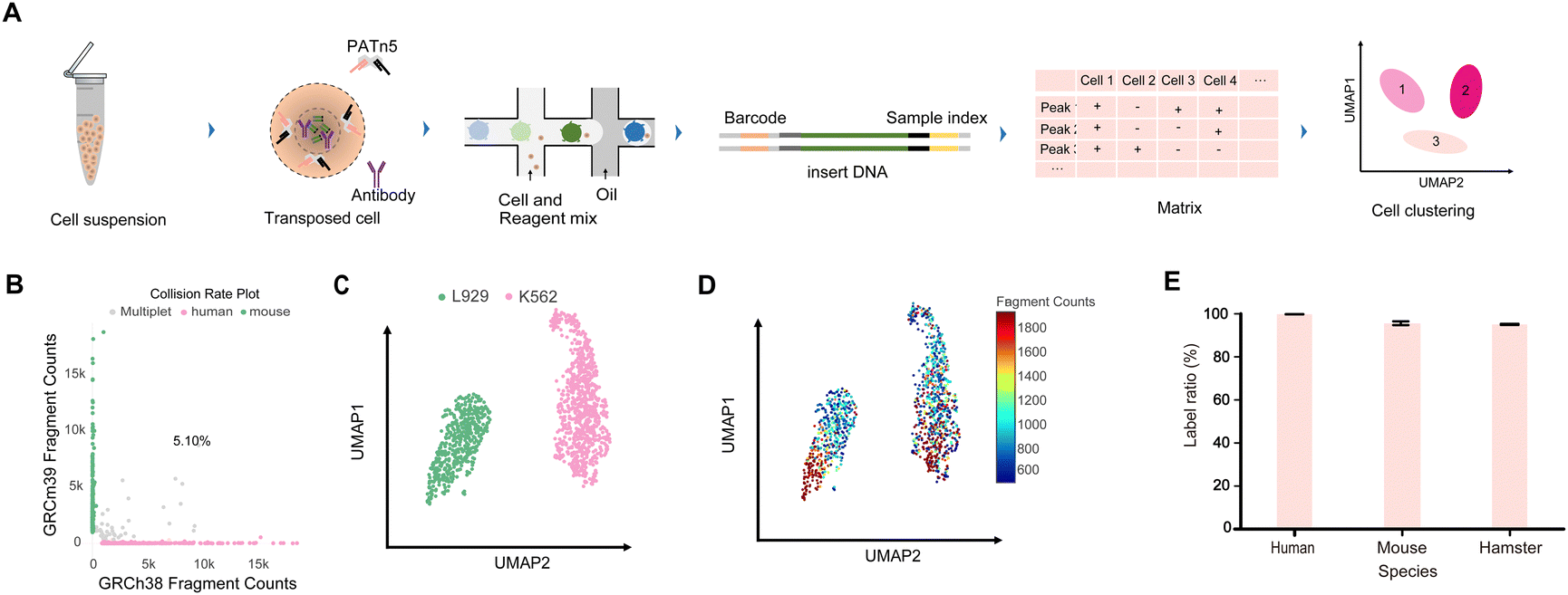 | ||
Fig. 1 High-throughout MobiChIP profling of single cells in different species. (A) Schematic of the MobiChIP workflow. This MobiChIP experiment contains the following procedure: sample preparation, ChIP and tagmentation, encapsulation, library preparation, sequencing and data analysis. (B) Scatterplots showing collision rate 5.10% for each unique barcode combination. Mouse L929 and human K562 cells were mixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. (C) UMAP projection of cells from (B) colored by cell clustering. (D) UMAP projection of cells from (B) colored by valid fragments. (E) The label ratio of human, mouse and hamster cells with barcoded PATn5 transposome. :1. (C) UMAP projection of cells from (B) colored by cell clustering. (D) UMAP projection of cells from (B) colored by valid fragments. (E) The label ratio of human, mouse and hamster cells with barcoded PATn5 transposome. | ||
Single-cell profiles of several histone modifications in several samples
To validate the performance of MobiChIP, we performed experiments and quality control in several samples (extended data Fig. 1A). we did a reproducibility validation in PBMCs with H3K27ac, and the results of two biological replicates showed that clustering and cell ratios were consistent (extended data Fig. 1B and C). To further illustrate the accuracy of MobiChIP, we compared the results of MobiChIP and scCUT&Tag, and the data showed that the results of the two methods were comparable (extended data Fig. 1D). Altogether, we performed MobiChIP with various histone modifications in K562 cell (H3K4me3) and health PBMCs (H3K4me1, H3K27ac, H3K27me3) respectively. Single cells were identified based on the number of fragments and fraction of fragments falling into peak regions, called from merged bulk data. Altogether, we obtained MobiChIP profiles of various histone modifications for 15104 single cells, with the median unique fragments per cell: 1024 (H3K27ac), 1553 (H3K4me3), 1086 (H3K27me3) and 1457 (H3K4me1) (Fig. 2A). Total 25% to 58% of fragments fell within narrow peak regions and was consistent with those reported results, indicating a low level of background (Fig. 2B).23,25 The fragment length distribution was consistent with the capture of nucleosome fragments, as well as mono-, di- and tri-nucleosomes for all modifications (Fig. 2C).
 | ||
| Fig. 2 Single-cell profiles of several histone modifications in human samples. (A) Comparison of number of unique fragments per anti-body per cell in MobiChIP. H3K4me1: n = 3522 cells; H3K4me3: n = 1918 cells; H3K27me3: n = 4534 cells; H3K27ac: n = 5130 cells. (B) Comparison of percent-age of fragments falling into peak regions per antibody per cell in MobiChIP. Peaks were obtained by peak calling in merged bulk datasets. Cell number is same as in (A). (C) Distribution of fragment lengths in MobiChIP per anti-body. | ||
Profiling of H3K27ac and H3K27me3 in single cells
To verify the fidelity of the MobiChIP, we mixed all single cells incubated with H3K27me3 antibody and compared the result with public data. By using the IGV snapshots, we found that the merged single-cell data showed a high consistency with the public data at the location of Hox gene cluster (Fig. 3A). We aggregated 500 single cells randomly sorted from PBMCs, the distribution of H3K27me3 signal was consistent with the aggregate one at Hox gene cluster region (Fig. 3A). Meanwhile, the consistence suggested the quality of captured fragments from single cell was qualified to perform bioinformatic analysis. Since the histone modification H3K27me3 is a repressive epigenetic marker, the data suggests that gene expression in the Hox gene cluster is suppressed in PBMC cells. As a key gene for somite development, the expression of Hox family is significantly altered during blood cells mature. To demonstrate the epigenetic signal around the Hox gene cluster, we also performed MobiChIP experiments with H3K27ac antibody in PBMCs (Fig. 3B). We also accessed the H3K27ac ChIP-seq data as well as the ATAC-seq data from the public databases. Via the comparison we found that the ATAC-seq data indicated more chromatin open regions around the Hox gene cluster, while the MobiChIP and public ChIP-seq data indicated that the signal of H3K27ac around the Hox gene cluster was weak at a low level. In addition to that, site-specific analysis showed that the signal of H3K27ac was mainly present at promoter and enhancer regions (Fig. 3C), and the signals in TSS region were consistent with reported one (Fig. 3D).27 By combining with single-cell RNA-seq data, we found that clusters were specific and the native marker CD79A was expressed normally in B-cells, but HOXA1, HOXA3 and HOXA9 genes were not detected significantly in all cell types (Fig. 3E). Based on the above data, it is easy to conclude that two epigenetic modifications, H3K27me3 and H3K27ac, play an important role in regulating the expression of related genes around the Hox gene cluster of PBMCs in blood, and also proves that MobiChIP is a more accurate tool to reveal the regulation of genes expression and cell fate decision than ATAC-seq.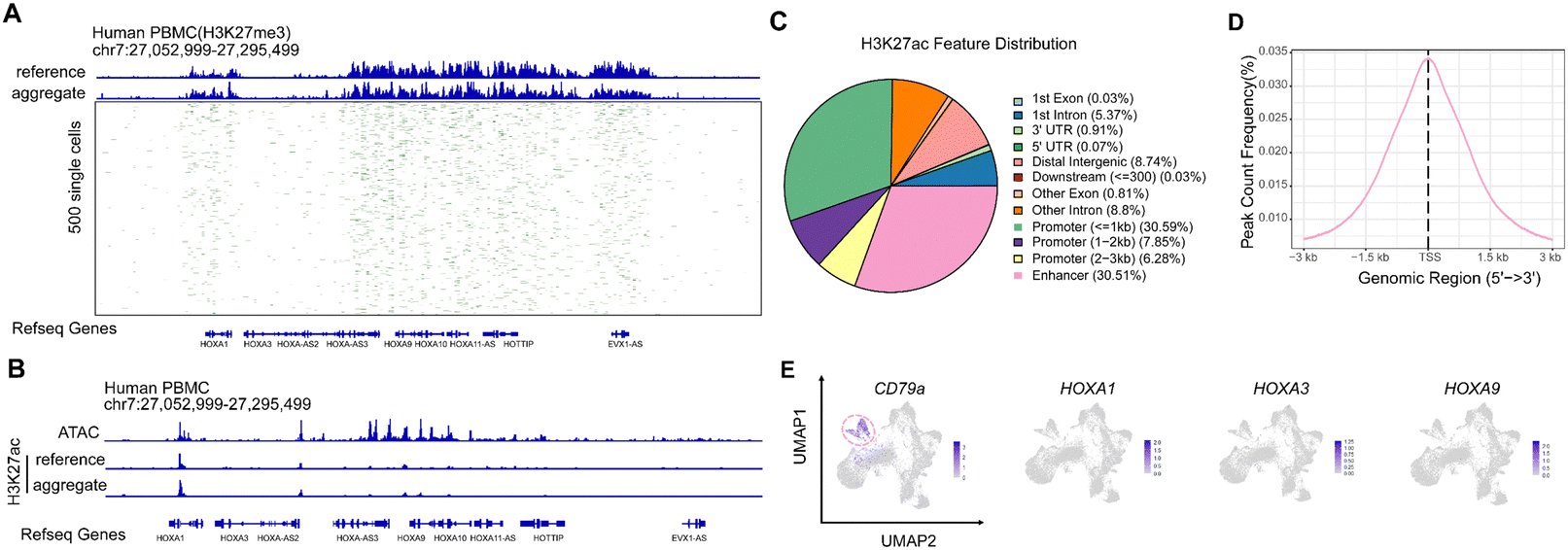 | ||
| Fig. 3 Profiling of H3K27ac and H3K27me3 in single cells. (A) Track plot representation of the MobiChP signal for H3K27me3 in PBMCs. The reference was downloaded from published per 500 bp windows and binarized. (B) Comparison of the MobiChP signal for H3K27ac with ATAC-seq signal. The reference (H3K27ac) and ATAC-seq was downloaded from published data. Peaks were obtained by peak calling in merged bulk datasets. (C) Distribution of H3K27ac feature at different elements of genome. (D) Peak count frequency of H3K27ac feature at TSS ± 3 kb. (E) Feature plot of CD79a, HOXA1, HOXA3, and HOXa9 genes in map projection of PBMCs with scRNA-seq. B cells were colored by red ring dashed line. | ||
MobiChIP reveals the RNA expression and gene regulation of PBMCs
We performed histone modification of H3K4me1 in healthy human PBMCs and obtained total 10031 cells and 1467 unique DNA fragments per cell. By ArchR clustering and dimension reduction, we obtained five distinct cell populations, including B cells, CD4-positive T cells, CD8-positive T cells, NK cells and monocytes (Fig. 4A) with cell type specific genes (extended data Fig. 1E). In order to validate that the data quality of MobiChIP is reliable, we projected MobiChIP data with bulk ChIP-seq data (Fig. 4A). We accessed bulk ChIP-seq data for each cell type in PBMC cells from the database. Via mapping analysis, we found that the cell type annotated from the MobiChIP data corresponded well to the public data without bias. Furthermore, we found the annotation results of subpopulations were consistent with the projection of published single-cell RNA-seq data (Fig. 4B). Since single-cell ChIP-seq is used to study cellular heterogeneity at the level of gene regulation, we performed single-cell RNA-seq sequencing on this sample in order to visualize the combination of RNA expression information and DNA regulation information, and to make multi-omics comparisons of the cells at different dimensions. Single-cell RNA-seq sequencing yielded 14453 cells with a median 1889 genes per cell. By integrating and analyzing the data from single-cell ChIP-seq and single-cell RNA-seq through Signac, we found that the multi-omics data containing two different dimensions of information can be perfectly matched together (Fig. 4C). Through further subpopulations annotation, those data corroborated one another (Fig. 4D). Ultimately, our results showed that data analysis of MobiChIP can be unified with data from bulk ChIP-seq, and integrated with single-cell RNA-seq. This strategy provides a powerful tool to illustrate cellular epigenetic and gene expression heterogeneity from different dimensions.
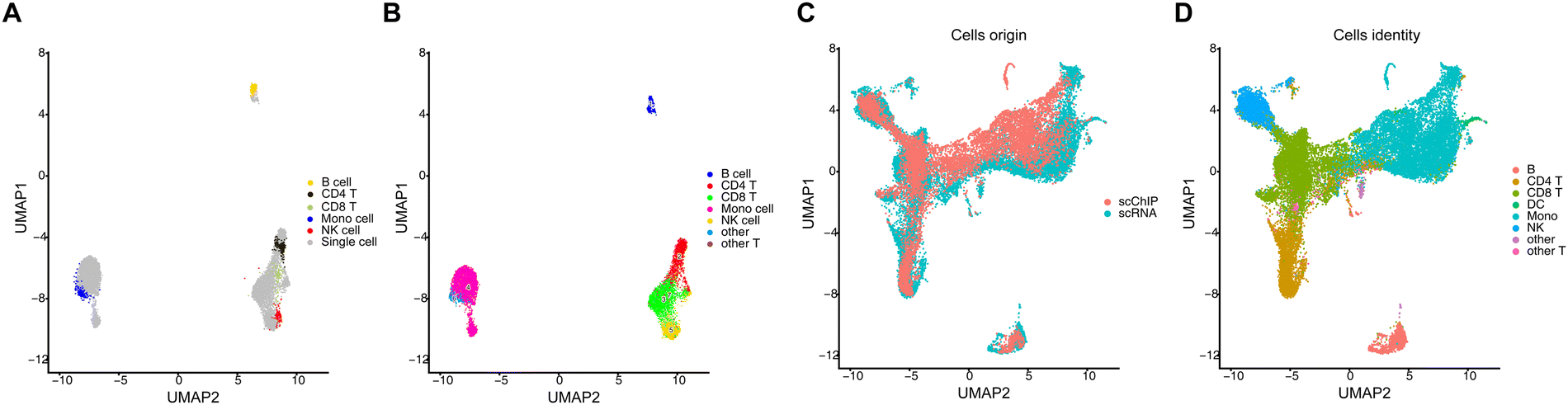 | ||
| Fig. 4 MobiChIP reveals epigenetic heterogeneity of health PBMCs. (A) Single cell ChIP-seq data of MobiChIP was projected with bulk ChIP data in terms of H3M4me1 in PBMCs. (B) Single cell ChIP-seq data of MobiChIP was projected with single cell RNA-seq data of PBMCs. (C) Co-embedding of scChIP-seq data and scRNA-seq data. (D) The integration and annotation of scRNA-seq data and scChIP-seq data. | ||
Discussion
We developed a new sequencing technology that is seamlessly compatible with existing sequencing platforms and without customized sequencing primers. It also enables the mixing of samples from different species, which greatly reduces experimental costs and increases throughput of cells.Cell heterogeneity of different tissues and organs plays an important role in disease development and progression. However, the relationship of gene expression and gene regulation relies on the specific ChIP-seq technology to accurately explain. Via performing MobiChIP and RNA-seq on PBMCs, we identified a strong signal for H3K27me3 (a repressive marker) around the Hox gene cluster in PBMCs, while the signal of H3K27ac (an active marker) was weak at a low level, suggesting that the alteration of epigenetic modification plays an important role in the maturation process of PBMCs. We also fully investigated the data of ATAC-seq, and the chromatin accessibility still remains around the Hox gene cluster, suggesting the ATAC-seq data not corresponded with the repression of Hox gene. Therefore, MobiChIP has a higher precision and can distinguish the differences in cellular epigenetic heterogeneity. Meanwhile, combining the scRNA-seq data demonstrated that the genes in the Hox gene cluster, whose expression is gradually closed with the differentiation of blood cells.
Since cell fate decisions are induced by the regulation of multiple dimensions, recently multi-omics technologies have also been developed. In order to enable the MobiChIP technology more applicable, we also build a set of process for multi-omics analysis. By performing MobiChIP and scRNA-seq sequencing with the same sample of PBMCs, we found that the data from MobiChIP can match well with the data from single-cell RNA-seq. The data from two dimensions can corroborate each other in terms of clustering and annotation. It also proves that the multi-omics analysis process we developed is fully capable of data union in multiple dimensions.
The limitation of MobiChIP platform is insufficient depth of bioinformatics analysis for the time being. Using MobiChIP to unveil the development trajectory, cell specific regulatory elements and some new regulatory targets need be further studied. However, as a versatile tool, we foresee that MobiChIP would help to get more biological information from DNA regulation level, to reveal epigenetic and genetic heterogeneity coupled with 3′ transcriptome, 5′ transcriptome, VDJ and MobiCITE of MobiDrop. We also hope that researchers using this technology together to promote this platform to further improve the experiment and bioinformatics process.
Author contributions
X. Y., H. P. and R. C. conceived and designed the study. X. Y. designed and performed all experiments with the help from L. W. G. Z. and L. X. performed the computational analyses. W. G. and G. C. provided technical support supervised by X. Y. X. Y. wrote the paper with input from all other authors. All authors participated in data discussion and interpretation.Data availability
Raw data are deposited in GEO under accession number GSE273350. The following publicly available datasets were used in this study: GSE157910 (H3K27me3 and H3K27ac scCUT&Tag of PBMC), GSE226267 (ATAC-seq from PBMC of healthy control), ENCODE ChIP-seq (ENCSR000ASJ, ENCSR000AUP, ENCSR007HLH, ENCSR138DOM, ENCSR391EQV).Conflicts of interest
The authors declare no competing interests.Acknowledgements
X. Y. and H. P. were supported by the Microfluidic Bioassay Technology Venture Team Program of China (2022R02005), the Automatic Digital PCR All-in-One Machine Program of China (2021C03199). We thank all members of MobiDrop for critical comments on this manuscript. We thank the Mingma Technologies (Shanghai, China) for help of next sequencing.References
- J. Kalucka, L. Rooij, J. Goveia, K. Rohlenova, S. J. Dumas, E. Meta, N. V. Conchinha, F. Taverna, L. Teuwen, K. Veys, M. García-Caballero, S. Khan, V. Geldhof, L. Sokol, R. Y. Chen, L. Treps, M. Borri, P. de Zeeuw, C. Dubois, T. K. Karakach, K. D. Falkenberg, M. Parys, X. K. Yin, S. Vinckier, Y. X. Du, R. A. Fenton, L. Schoonjans, M. Dewerchin, G. Eelen, B. Thienpont, L. Lin, L. Bolund, X. R. Li, Y. L. Luo and P. Carmeliet, Single-Cell Transcriptome Atlas of Murine Endothelial Cells, Cell, 2020, 180, 764–779 CrossRef CAS.
- X. P. Han, Z. M. Zhou, L. J. Fei, H. Y. Sun, R. Y. Wang, Y. Chen, H. D. Chen, J. J. Wang, H. N. Tang, W. H. Ge, Y. C. Zhou, F. Ye, M. M. Jiang, J. Q. Wu, Y. Y. Xiao, X. N. Jia, T. Y. Zhang, X. J. Ma, Q. Zhang, X. L. Bai, S. J. Lai, C. X. Yu, L. J. Zhu, R. Lin, Y. C. Gao, M. Wang, Y. Q. Wu, J. M. Zhang, R. Y. Zhan, S. Y. Zhu, H. L. Hu, C. C. Wang, M. Chen, H. Huang, T. B. Liang, J. H. Chen, W. L. Wang, D. Zhang and G. J. Guo, Construction of a human cell landscape at single-cell level, Nature, 2020, 581, 303–309 CrossRef CAS.
- A. N. Tikhonova, I. Dolgalev, H. Hu, K. K. Sivaraj, E. Hoxha, Á. Cuesta-Domínguez, S. Pinho, I. Akhmetzyanova, J. Gao, M. Witkowski, M. Guillamot, M. C. Gutkin, Y. T. Zhang, C. Marier, C. Diefenbach, S. Kousteni, A. Heguy, H. Zhong, D. R. Fooksman, J. M. Butler, A. Economides, P. S. Frenette, R. H. Adams, R. Satija, A. Tsirigos and I. Aifantis, The bone marrow microenvironment at single-cell resolution, Nature, 2019, 569, 222–228 CrossRef CAS.
- M. Litviňuková, C. Talavera-López, H. Maatz, D. Reichart, C. L. Worth, E. L. Lindberg, M. Kanda, K. Polanski, M. Heinig, M. Lee, E. R. Nadelmann, K. Roberts, L. Tuck, E. S. Fasouli, D. M. DeLaughter, B. McDonough, H. Wakimoto, J. M. Gorham, S. Samari, K. T. Mahbubani, K. Saeb-Parsy, G. Patone, J. J. Boyle, H. Zhang, H. Zhang, A. Viveiros, G. Y. Oudit, O. A. Bayraktar, J. G. Seidman, C. E. Seidman, M. Noseda, N. Hubner and S. A. Teichmann, Cells of the adult human heart, Nature, 2020, 588, 466–472 CrossRef.
- C. Kuppe, M. M. Ibrahim, J. Kranz, X. T. Zhang, S. Ziegler, J. Perales-Patón, J. Jansen, K. C. Reimer, J. R. Smith, R. Dobie, J. R. Wilson-Kanamori, M. Halder, Y. X. Xu, N. Kabgani, N. Kaesler, M. Klaus, L. Gernhold, V. G. Puelles, T. B. Huber, P. Boor, S. Menzel, R. M. Hoogenboezem, E. M. J. Bindels, J. Steffens, J. Floege, R. K. Schneider, J. Saez-Rodriguez, N. C. Henderson and R. Kramann, Decoding myofibroblast origins in human kidney fibrosis, Nature, 2021, 589, 281–286 CrossRef CAS PubMed.
- C. Cochain, E. Vafadarnejad, P. Arampatzi, J. Pelisek, H. Winkels, K. Ley, D. Wolf, A. E. Saliba and A. Zernecke, Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis, Circ. Res., 2018, 122, 1661–1674 CrossRef CAS.
- P. Zhu, H. S. Guo, Y. X. Ren, Y. Hou, J. Dong, R. Li, Y. Lian, X. Y. Fan, B. Q. Hu, Y. Gao, X. Y. Wang, Y. Wei, P. Liu, J. Yan, X. L. Ren, P. Yuan, Y. F. Yuan, Z. Q. Yan, L. Wen, L. Y. Yan, J. Qiao and F. C. Tang, Single-cell DNA methylome sequencing of human preimplantation embryos, Nat. Genet., 2018, 50, 12–19 CrossRef CAS PubMed.
- H. Q. Liu, J. T. Zhou, W. Tian, C. Y. Luo, A. Bartlett, A. Aldridge, J. Lucero, J. K. Osteen, J. R. Nery, H. M. Chen, A. Rivkin, R. G. Castanon, B. Clock, Y. E. Li, X. M. Hou, O. B. Poirion, S. Preissl, A. Pinto-Duarte, C. O'Connor, L. Boggeman, C. Fitzpatrick, M. Nunn, E. A. Mukamel, Z. Z. Zhang, E. M. Callaway, B. Ren, J. R. Dixon, M. M. Behrens and J. R. Ecker, DNA methylation atlas of the mouse brain at single-cell resolution, Nature, 2021, 598, 120–128 CrossRef CAS PubMed.
- W. F. Jin, Q. S. Tang, M. M. Wan, K. R. Cui, Y. Zhang, G. Ren, B. Ni, J. Sklar, T. M. Przytycka, R. Childs, D. Levens and K. J. Zhao, Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples, Nature, 2015, 528, 142–146 CrossRef CAS PubMed.
- J. Cooper, Y. Ding, J. Z. Song and K. J. Zhao, Genome-wide mapping of DNase I hypersensitive sites in rare cell populations using single-cell DNase sequencing, Nat. Protoc., 2017, 12, 2342–2354 CrossRef CAS PubMed.
- C. A. Lareau, F. M. Duarte, J. G. Chew, V. K. Kartha, Z. D. Burkett, A. S. Kohlway, D. Pokholok, M. J. Aryee, F. J. Steemers, R. Lebofsky and J. D. Buenrostro, Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility, Nat. Biotechnol., 2019, 37, 916–924 CrossRef CAS PubMed.
- A. M. Ranzoni, A. Tangherloni, I. Berest, S. G. Riva, B. Myers, P. M. Strzelecka, J. R. Xu, E. Panada, I. Mohorianu, J. B. Zaugg and A. Cvejic, Integrative Single-Cell RNA-Seq and ATAC-Seq Analysis of Human Developmental Hematopoiesis, Cell Stem Cell, 2021, 28, 472–487 CrossRef CAS PubMed.
- K. Zhang, J. D. Hocker, M. Miller, X. M. Hou, J. Chiou, O. B. Poirion, Y. J. Qiu, Y. E. Li, K. J. Gaulton, A. Wang, S. Preissl and B. Ren, A single-cell atlas of chromatin accessibility in the human genome, Cell, 2021, 184, 5985–6001 CAS.
- W. Xu, Y. Wen, Y. Y. Liang, Q. S. Xu, X. F. Wang, W. F. Jin and X. Chen, A plate-based single-cell ATAC-seq workflow for fast and robust profiling of chromatin accessibility, Nat. Protoc., 2021, 16, 4084–4107 CAS.
- P. J. Park, ChIP-seq: advantages and challenges of a maturing technology, Nat. Rev. Genet., 2009, 10, 669–680 CrossRef CAS.
- D. S. Johnson, A. Mortazavi, R. M. Myers and B. Wold, Genome-wide mapping of in vivo protein-DNA interactions, Science, 2007, 316, 1497–1502 CrossRef CAS.
- C. D. Schmid and P. Bucher, ChIP-Seq data reveal nucleosome architecture of human promoters, Cell, 2007, 131, 831–833 CrossRef CAS PubMed.
- A. Rotem, O. Ram, N. Shoresh, R. A. Sperling, A. Goren, D. A. Weitz and B. E. Bernstein, Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state, Nat. Biotechnol., 2015, 33, 1165–1172 CrossRef CAS.
- Q. H. Wang, H. Q. Xiong, S. S. Ai, X. H. Yu, Y. X. Liu, J. J. Zhang and A. B. He, CoBATCH for High-Throughput Single-Cell Epigenomic Profiling, Mol. Cell, 2019, 76, 206–216 CrossRef CAS.
- H. Q. Xiong, Y. J. Luo, Q. H. Wang, X. H. Yu and A. B. He, Single-cell joint detection of chromatin occupancy and transcriptome enables higher-dimensional epigenomic reconstructions, Nat. Methods, 2021, 18, 652–660 CrossRef CAS PubMed.
- H. S. Kaya-Okur, S. J. Wu, C. A. Codomo, E. S. Pledger, T. D. Bryson, J. G. Henikoff, K. Ahmad and S. Henikoff, CUT&Tag for efficient epigenomic profiling of small samples and single cells, Nat. Commun., 2019, 10, 1930 CrossRef PubMed.
- B. J. Carter, W. L. Ku, J. Y. Kang, G. Q. Hu, J. Perrie, Q. S. Tang and K. J. Zhao, Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq), Nat. Commun., 2019, 10, 3747 CrossRef.
- C. X. Zhu, Y. X. Zhang, Y. E. Li, J. Lucero, M. M. Behrens and B. Ren, Joint profiling of histone modifications and transcriptome in single cells from mouse brain, Nat. Methods, 2021, 18, 283–292 CrossRef CAS PubMed.
- S. Gopalan, Y. Q. Wang, N. W. Harper, M. Garber and T. G. Fazzio, Simultaneous profiling of multiple chromatin proteins in the same cells, Mol. Cell., 2021, 81, 4736–4746 CrossRef CAS.
- M. Bartosovic, M. Kabbe and G. Castelo-Branco, Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues, Nat. Biotechnol., 2021, 39, 825–835 CrossRef CAS PubMed.
- S. J. Wu, S. N. Furlan, A. B. Mihalas, H. S. Kaya-Okur, A. H. Feroze, S. N. Emerson, Y. Zheng, K. Carson, P. J. Cimino, C. D. Keene, J. F. Sarthy, R. Gottardo, K. Ahmad, S. Henikoff and A. P. Patel, Single-cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression, Nat. Biotechnol., 2021, 39, 819–824 CrossRef CAS PubMed.
- D. Hu, L. Abbasova, B. M. Schilder, A. Nott, N. G. Skene and S. J. Marzi, CUT&Tag recovers up to half of ENCODE ChIP-seq peaks in modifications of H3K27, bioRxiv, preprint, 2023 DOI:10.1101/2022.03.30.486382.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mo00111g |
| ‡ Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |