
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic enhancement of luminescence and ferroelectricity driven by (Z)-clipping of a tetraphenylethene†
Sewon
Lim‡
,
Donghwan
Kim‡
,
Hee Jung
Kim
,
Hwandong
Jang
,
Sienoh
Park
and
Eunkyoung
Kim
 *
*
Department of Chemical and Biomolecular Engineering, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, South Korea. E-mail: eunkim@yonsei.ac.kr
First published on 11th March 2025
Abstract
Synergistic enhancement of luminescence and ferroelectricity (SELF) was explored in a (Z)-isomer of tetraphenylethene derivatives containing two clipping units in (Z)-configuration (TPC2-(Z)). TPC2-(Z) was synthesized utilizing a ‘body core’ precursor, which exclusively afforded (Z)-configuration. High-resolution transmission electron microscopy measurements indicated that TPC2-(Z) formed a layered morphology in film, with well-ordered crystalline structures, which was ascribed to the (Z)-clipped self-assembled structures. The film exhibited good photoluminescence performances with 45.6% quantum yield. Simultaneously, the film exhibited high ferroelectricity as inferred from high remnant polarization (Pr = 2.54 μC cm−2) and saturated polarization (3.56 μC cm−2) along with a longitudinal piezoelectric coefficient (d33 = −23.8 pm V−1), indicating that TPC2-(Z) exhibits excellent SELF. Owing to its fluorescence and thermal stability, we fabricated light-emitting electrochemical cells (LEC) that exhibited maximum 890 cd m−2 at Von of 3.9 V. This was more than 40% enhanced performance compared to that of the (E)/(Z) mixture. A new self-powered, stimuli-sensitive electroluminescent device was demonstrated with TPC2-(Z), where the piezoelectrically tunable LECs effectively ‘switched on’ luminescence, showing 120-fold increased brightness after 254 bending at 1 Hz, compared to the ‘off’ state without bending. These results underscore that Z-clipping is an effective method for enhancing SELF and could create new self-powered, stimuli-sensitive electroluminescent devices.
New conceptsPreviously, the concept of ‘synergistic enhancement of luminescence and ferroelectricity (SELF)’ was reported by our group. However, an (E)/(Z) stereomixture of TPC2 lacks ferroelectric properties, whereas TPC1 and TPC4 exhibited excellent SELF properties. Here, we report a straightforward synthesis of the (Z)-isomer of TPC2 utilizing a ‘body core’ precursor, which afforded only the (Z) isomer of TPE (TPC2-(Z)). TPC2-(Z) was capable of forming films through a solution process upon Z-clipping of the self-assembling units of the ‘DOS’ group in the film. The TPC2-(Z) thin films exhibited exceptional properties with a high fluorescence and recorded high remnant polarization along with a high longitudinal piezoelectric coefficient. This multifunctional property of TPC2-(Z) was demonstrated in a piezoelectric device coupled with a light-emitting electrochemical cell (LEC) structure. The electroluminescence of TPC2-(Z) responded to piezoelectric stimuli generated by the TPC2-(Z) film, where the piezoelectrically tunable LECs effectively ‘switched on’ luminescence, showing a 120-fold increase in brightness after 254 bending at 1 Hz, compared to the ‘off’ state without bending. This indicates that the Z-clipping could be a promising way to realize SELF properties and create a new self-powered, stimuli-sensitive electroluminescent device. |
1. Introduction
Displays and interactive devices have been in great demand recently, and the exploration of display materials with more than two different properties has attracted increased academic and industrial interest owing to their functional tunability and good processing ability. Currently, the coupling of strong and clear luminescence emission, with other physicochemical properties, such as thermal conductivity,1 ferroelectricity,2,3 semiconductivity4 and photochromic properties,5,6 is being researched to endow luminescent organic materials with multi-functionality. Particularly, materials combining ferroelectric and luminescent properties may be applied to self-powered displays,7,8 photoelectric sensors,9 actuators for optical and memory devices,10,11 and energy harvesting materials.12 The demand for dual functionality is growing because of its multiple advantages. If a single material possesses multiple properties with diverse functionality, it can replace multiple materials required to cover this functionality. This finding can contribute to the ‘miniaturization’ of devices, while reducing manufacturing costs. In addition, application of new synergistic properties can be expected. For example, human-interactive sensing displays have developed from a combination of two or more devices into a single device, enabling scaling-down and simplifying the display. Technology with molecular-level synthesis precision is essential for future technological advancements. Accordingly, the development of electrically conductive and piezoelectric materials is underway. These materials can enable the scaling down of devices by decreasing their weight and power consumption.13In particular, materials showing synergistic enhancement of luminescence and ferroelectricity (SELF) have recently gained increasing attention, due to their ability to enable electric field-controlled luminescence, and vice versa. These materials show promising applications, including non-volatile memory with optical readout14–16 and optoelectronic devices like LEDs and modulators, where polarization tunes emission properties.17 Ferroelectric-luminescent materials also enhance electric field-tunable sensors and photovoltaic or photocatalytic energy conversion.18–20 While doped and composite materials such as Nd3+-doped PMNT ferroelectric crystals21 and Tm/Yb co-doped nanocomposites22 have been explored, they face structural incompatibilities and charge defects by dopants, disrupting polarization switching.23,24
Furthermore, ferroelectric and luminescence behaviour are challenging to realize simultaneously because of their trade-off relationship, thus, very few materials have been shown to possess such dual functionality.2,3,25 Such a relationship between these properties is explained by the semiconducting characteristics of the materials, which contribute to their luminescence, often causing depolarization, thus compromising the ferroelectric properties of the materials.26 Recently, a dual functionality ‘SELF’ was reported in metal-free pure organic single molecules and its performance in an electroluminescent (EL) device and energy harvesters using tetraphenylethene (TPE) derivatives was reported for the first time.27 TPE and its derivatives exhibit aggregation-induced emission (AIE) enhancement, in which the non-planar four-winged propeller shape of TPE restricts intramolecular rotation and motions that leads to non-radiative quenching, thus enhancing fluorescence in a highly aggregated state.28,29 In addition, a long alkoxy chain facilitates a highly ordered self-assembly of molecules.27,30 For example, decyloxy-substituted conjugated tristilbene and its derivatives are known to form a self-assembled supramolecular structure with a unique honeycomb shape motif.30–32 TPCn–TPE coupled with n clips (TPCn) has been reported to exhibit SELF. Among TPCn, TPC4 achieved an electroluminescence intensity of over 1290 cd m−2, far exceeding the indoor readability (200–300 cd m−2),33 when employed in a light-emitting electrochemical cell (LEC). Simultaneously, it exhibited ferroelectric properties, with a remnant polarization of 2.27 μC cm−2.27 However attaching four clips to the TPE core is challenging as it requires a significant amount of clips and its ferroelectric properties need improvement for practical device applications. To address this, we investigated the potential impact of isomeric structures on improving ferroelectric performance, utilizing a double-substituted TPE isomer (TPC2), in which two DOS groups are attached to the TPE core.
In the previous report TPC2 contains (E) and (Z) mixtures, resulting in poor ferroelectric behaviour.27 The intramolecular dipole of the (E) isomer is generally weaker than that of the (Z) isomer because in the former, high-priority substituents point in opposite directions, cancelling their dipole moments to some extent.34 In contrast, the direction of the dipole moment of the (Z) isomer could be focused in the direction in which substituents are facing. Similarly, azobenzene is well known for its cis–trans configuration conversion under UV irradiation. The dipole moment of trans-azobenzene (E) is 0 D, but that of cis-azobenzene (Z), formed under UV irradiation, is 3.0 D.35 Our group has previously calculated the theoretical dipole moment of TPC2-(E) and TPC2-(Z) as 0.08 D and 3.32 D, respectively.27 Therefore, the pure (Z) isomer of TPC2 is expected to exhibit enhanced ferroelectric properties. Furthermore, when the dipole moments of individual molecules are organized to form a macro-dipole through supramolecular assembly, this arrangement could further amplify ferroelectric behaviour in a film.36 Such (Z)-clippings could induce TPE body aggregation to improve luminescence through AIE, thus enabling the dual function of the materials.37 For fully-substituted TPEs (4 substitutions), intermolecular TPE-to-TPE core interaction could be hindered by the spatial overlap of free substituents.38
Moreover, the double bond-containing (Z) isomer is more advantageous than (E) for spatial intramolecular interaction or conjugation owing to the small distance between cis-position substituents.39,40 This also leads to enhanced intramolecular charge transfer (ICT), resulting in higher quantum yield.39,41 Specifically, in TPE-based molecules, each protruding phenyl group and bonded substituent is twisted, forming a through-space conjugation. This type of intramolecular conjugation is known to stabilize ICT states by minimizing non-radiative charge recombination and decay,42 which is applicable to host–guest coordination or ion-detection systems.39,43 In addition, a (Z) isomer in a solid state has been reported to exhibit a photoluminescence (PL) quantum yield five times higher than that of the (E) isomer, primarily because of different energy levels in excited states. Therefore, although the (E) isomer should form more ordered self-organized structures and exhibit higher thermal stability,44 the (Z) isomer appears promising for exploring new SELF molecules.
However, the synthesis of the (Z) isomer with a long clip is challenging because (Z) to (E) isomerization is favourable in solution or under light.45,46 This in situ isomerization of the energetically less stable (Z) into a more stable (E) isomer occurs significantly upon conversion of the central double bond of the (Z) isomer to a single-bond, and that state enables nonradiative intramolecular rotations and vibrations into (E). Thus, the McMurry coupling reaction often results in a mixture of (E) and (Z) isomers when TPE derivatives are asymmetrically substituted.46
Herein, we report the synthesis of the (Z) isomer of TPC2 utilizing a ‘body core’ precursor, which afforded the pure (Z) isomer of TPE (TPC2-(Z)). TPC2-(Z) was used to prepare films through a solution process upon Z-clipping of the self-assembling units of the 4-[3,5-bis-(3-decyloxy-styryl)-styryl]-phenyl (DOS) group. The obtained thin films exhibited exceptional SELF properties with a high fluorescence and significantly enhanced ferroelectric properties as compared to the isomer mixture. The SELF properties of TPC2-(Z) were demonstrated in a piezoelectric device coupled with an LEC structure. The results indicate that the Z-clipping of tetraphenylethenes could be a promising way to realize SELF properties and create new stimuli-sensitive EL devices.
2. Results and discussion
2-1. Synthesis and characterization of TPC2-(Z)
TPC2-(Z) was synthesized by attaching two equivalents of ‘clip’ to the TPE2PhCHO-(Z) core (Fig. 1a). To obtain the pure (Z)-specific geometry of a TPE derivative, we utilized a synthetic strategy involving the substitution of a (Z)-specific precursor with a clipping DOS group while maintaining the (Z) structure.47 First, the TPE2PhCHO-(Z) precursor was synthesized by constructing a (Z)-form stilbene derivative. We constructed a ‘body core’ that would be chemically extended with fixed Z-geometry. This was achieved via Miyaura borylation reaction between diphenyl acetylene and bis(pinacolato)diboron. Diphenyl acetylene reacted with bis(pinacol)diborane over tetrakis(triphenylphosphine)platinum(0) as a catalyst in N,N-dimethylmethanamide (DMF), yielding a white solid, (Z)-1,2-diphenyl-1,2-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)ethene, in good yield (88%), which was comparable to the previously reported values for the synthesis from bedaquiline analogs.48 | ||
| Fig. 1 (a) Synthetic scheme of TPC2-(Z) and its structure. (b) 1H NMR spectra of TPC2-(Z) around aromatic regions. Inset is the 2D-NOESY spectrum of TPE2-(Z). (c) TGA result of TPC2-(Z) under nitrogen at 10 K min−1 of heating rate. (d) UV-Vis absorption (dotted line) and the normalized PL spectra of 10−5 M solution of TPC2-(Z) (black) and TPC2-(E + Z) in CHCl3. | ||
Next, 4-formyl-4′-bromobiphenyl (referred to as ‘leg’) was synthesized via Suzuki–Miyaura coupling49,50 of 1,4-bromoiodobenzene with 4-formylphenylboronic acid over tetrakis(triphenylphosphine)palladium(0) as a catalyst in good yield (75%). The (Z) configuration of the purified TPE2PhCHO-(Z) was verified through 1H NMR following the previously reported methods51 (Fig. S1, ESI†). The synthesized (Z)-form ‘body core’ precursor and 4-formyl-4′-bromobiphenyl legs were then conjugated via Suzuki–Miyaura coupling with [1,1 bis-(diphenylphosphino)ferrocene]dichloropalladium(II) as a catalyst in 1,4-dioxane, yielding a deep-yellow solid, TPE2PhCHO-(Z) (56%) (Fig. 1a). To synthesize the clip, which is a basic motif of DOS, 3-hydroxybenzaldehyde was alkylated to decylbromide under basic conditions in DMF. Consequently, 3-decyloxybenzaldehyde was obtained in good yield (81%) using the previously reported method.31,52 After purification, it reacted with triethyl phosphite in the presence of potassium tert-butoxide (t-BuOK) in anhydrous tetrahydrofuran (THF), yielding the clip as a white solid.31 Finally, two equivalents of clip were coupled with TPE2PhCHO-(Z) via the Horner–Wadsworth–Emmons reaction, yielding a pale green solid, TPC2-(Z). The overall synthetic scheme is given in Fig. S2 (ESI†).
Spectroscopic methods and matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry were used to characterize TPC2-(Z) (Fig. S3 and S4, ESI†). In 1H NMR spectra, no noticeable differences were observed in aliphatic regions of the (E)/(Z) isomers. Despite the presence of multiple aromatic protons from the ring and substituents, which could potentially overlap, it is notable that the peaks between 7.0 and 7.7 ppm were distinguishable from other aromatic protons (ArH). These peaks displayed clear shifts between the isomers (Fig. S5a, ESI†).
The multiple shifts at δ = 7.57 ppm in Fig. 1b, as well as Fig. S5a (ESI†) for the E and Z mixture, correspond to the resonances of the proton Hf due to the electron-withdrawing property of the neighbouring stilbene group of the clip. The (E) and (Z) isomers differ in their spatial arrangements around the double bond, leading to significant changes in the spatial proximity between substituents. Protons Hc, Hd, and He are positioned in such a way that their chemical environments change substantially between the two isomers. As compared in Fig. S5b (ESI†), the peaks at 7.45, 7.3, and 7.2 differ significantly between the (Z) and (E) isomer mixture, indicating that these contain isomer-sensitive protons Hc, Hd, and He.
In the E isomer, protons from different rings are in closer proximity due to the trans arrangement of bulky clip groups, which allows for stronger nuclear Overhauser enhancement (NOE) interactions between the aromatic rings of TPE. This leads to enhanced cross-peak intensity for the NOE of the (E) and (Z) mixture in the nuclear Overhauser enhancement spectroscopy (NOESY) (Fig. S5b, ESI†). In particular, the cross-peak intensity between protons He and Ha,b,c,g and between Hd and Ha,b,c,g is increased as compared to the pure (Z) isomer (Fig. S5b, red and black circles, ESI†). The cross-peak intensity between protons He and Hf could be identified (yellow dashed circle) in the mixture. Conversely, in the (Z) isomer, bulky clip groups are on the same side, resulting in steric hindrance that forces parts of the molecule further apart, reducing NOE interactions and consequently decreasing the cross-peak intensity. In particular, the cross-peak intensity between protons He and Ha,b,c,g disappeared in the Z isomer, supporting the proposed assignment of this compound. Thus, combined analysis of the 1H NMR and 2D NOESY spectra enabled the assignment of protons within the Z isomer (Fig. 1b). Such (E)/(Z) isomer-sensitive protons and NOE interactions were matched to the previous studies on TPE.51,53–55
Thermogravimetric analysis (TGA) was performed to test thermal stability of the synthesized material (Fig. 1c, Fig. S6, ESI†). The temperature onset for 5% weight loss (Td,5%) was 243 °C, which was considerably lower than that of the (E)- and (Z)- mixture (389 °C, as previously reported).27TPC2-(Z) is thermally less stable than TPC2-(E + Z), because the bulky clip groups are on the same side, resulting in steric hindrance in the (Z) form. To further identify physical properties with temperature change, differential scanning calorimetry (DSC) was performed. In the scanning range of −10–200 °C, an endothermal phase transition is observed at ∼120 °C in the heating cycle for TPC2-(Z), which matched to the structural phase transition temperature (Tsp) for the TPC2-(E + Z) reported in our previous paper.27 The Tsp for the TPC2-(E + Z) mixture was irreversible and no peak was identified in the cooling cycle. Interestingly, in the pure TPC2-(Z) (Fig. S7, ESI†), a peak at ∼103 °C was identified during the cooling accompanied by broadening in the cooling cycle. This suggests the thermal transition in TPC2-(Z) is reversible with a 17 K thermal hysteresis. This aligns with similar observations reported for organic ferroelectric materials, where transition temperatures are shifted in the cooling cycles.56–59 Interestingly, the Tsp of TPC2-(E + Z) was 125 °C,27 which was similar to that of TPC2-(Z), as opposed to large differences in TGA analysis. This result indicates that structural differences between (E) and (Z) contribute primarily to stability but not to phase transition behaviour.
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were determined via cyclic voltammetry in a chloroform solution containing 0.1 M of tetrabutylammonium hexafluorophosphate as the electrolyte, with a platinum disk, a platinum wire, and Ag/AgCl as the working, counter, and reference electrodes, respectively (Fig. S8, ESI†). The onset potential for the oxidation (Eonset) of TPC2-(Z) was 1.19 V versus Ag/AgCl. HOMO and LUMO energy levels were determined using the relationships: EHOMO (eV) = −[(4.8 − eVox,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) onset,Fc/Fc+) + eVox,onset]60 and ELUMO (eV) = EHOMO + Eg, where Eg = 1240/λonset, with λonset derived from the UV-vis absorption spectrum. The EHOMO and ELUMO were −5.53 eV and −2.49 eV, respectively.
onset,Fc/Fc+) + eVox,onset]60 and ELUMO (eV) = EHOMO + Eg, where Eg = 1240/λonset, with λonset derived from the UV-vis absorption spectrum. The EHOMO and ELUMO were −5.53 eV and −2.49 eV, respectively.
2-2. Optical and AIE properties of TPC2-(Z)
As previously reported by our group, TPCn type molecules are highly soluble in many organic solvents,27 such as toluene, chloroform, and THF. Thus, optical and spectroscopic properties of TPC2-(Z) were measured in various organic solvents. The TPC2-(E + Z) mixture in chloroform exhibited maximum absorption at 325 nm in the UV spectrum (Fig. 1d), which matched to a previous report.27TPC2-(Z) exhibited a similar UV spectrum as the TPC2-(E + Z) mixture (Fig. 1d). However, the fluorescence spectra of the TPC-(Z) and TPC2-(E + Z) mixture differed significantly. Unlike the mixture, the pure TPC2-(Z) exhibited an emission peak in the 500 nm region corresponding to AIE. Emission spectra in chloroform exhibited a sharp peak at 424 nm (Fig. 1d), originating from the DOS units.27 Although weak, an obvious emission band at approximately 500 nm was observed in CHCl3 solution, indicating that aggregated TPE cores exist in the solution even in the absence of a poor solvent such as water.The absorption bands of TPC2-(Z) in the THF–water mixture (Fig. 2a) appeared in the range of 350–400 nm and broadened significantly as the water fraction (ƒw) increased from ƒw = 50% to 90%. The intensity at the absorbance tail beyond 400 nm increased with ƒw, indicating the formation of nanoparticles that cause Mie scattering.61 This aggregated emission is further supported by emission measurement in water (Fig. 2b), where water and THF are used as poor and good solvents, respectively, with ƒw in THF ranging from 0% to 90% and the TPC2-(Z) concentration maintained at 10−5 M. Particles remained well-dispersed without any signs of precipitation in these solutions. As shown in the inset of Fig. 2b, TPC2-(Z) starts to aggregate at ƒw = 50%, which can be observed as increasing opaqueness even without UV exposure. This trend persists as the water fraction increases. Upon UV exposure, two emission colours were observed depending on ƒw: from 0% to 30% the emission appeared pale blue, and above 50%, it shifted to bright green. This observation was further confirmed by the PL spectra (Fig. 2c). As ƒw increases from 30% to 50%, the emission peak in the normalized PL spectra shifts by approximately 100 nm towards longer wavelengths (Fig. S9, ESI†). The emission intensity also abruptly increases with this shift. This finding was attributed to the aggregation of TPE units in the core of TPC2-(Z), eventually inducing strong AIE.
 | ||
| Fig. 2 (a) UV-Vis absorption spectra of TPC2-(Z) with different water fractions (0% (black dotted), 10% (red), 30% (blue), 50% (magenta), 70% (green), 80% (navy), and 90% (purple) at a concentration of 10−5 M). (b) Images of the TPC2-(Z) solutions with increasing water fraction (ƒw): from left (0%) to right (90%) in a THF–water mixture. Before UV irradiation (top) and under 254 nm UV irradiation (bottom). (c) PL spectra of TPC2-(Z) with different water fractions (0% (black dotted), 10% (red), 30% (blue), 50% (magenta), 70% (green), 80% (navy), and 90% (purple) at an excitation wavelength of 330 nm; at a concentration of 10−5 M). (d) Plot of EAIE and Eclip against the water fraction (ƒw) in a THF–water mixture (black: TPC1, red: TPC2-(E + Z), blue: TPC4, and green: TPC2-(Z)). (e) The average diameter of particles of TPC2-(Z) in the THF–water mixture with varying fw measured by dynamic light scattering (DLS). (f) 50000× magnification FE-SEM images of TPC2-(Z) samples obtained from the solutions with various water fractions (ƒw) 50%, 70%, 80%, and 90%. The white scale bar represents 500 nm. | ||
From a structural point of view, the TPE and DOS unit in a molecule contribute differently to emission as the solution composition changes. When supramolecular aggregation occurs via DOS units, the clipping of these units induces aggregation caused quenching (ACQ) at a short wavelength (λclip(max) = 405 nm). In contrast, the aggregation of TPE units enhances emission intensity at a longer wavelength (λAIE(max) = 496 nm) through AIE. Because the emission region between λclip and λAIE was quite well separated, the AIE effect of the TPEs did not cancel out by the ACQ of the DOS units. The competing two effects exerted by different molecular units are confirmed by calculating Eclip and EAIE (Fig. 2d, bottom). Eclip was based on the PL spectra of tris(4-(dodecyloxy)phenyl)stilbene (TSB-C12), which was known to be structurally similar to the clipping DOS unit and was thus used as a reference to examine the self-assembly effect of DOS units.27 TSB-C12 in THF (ƒw = 0) exhibited an emission band at around 405 nm, so Eclip is defined as the ratio of emission intensity at a given ƒw to the intensity at 0% water (I0) at 405 nm. The Eclip for the ƒw ranging from 0% to 30% was not changed for TPC2 derivatives, unlike for TPC1 and TPC4. A significant decrease of Eclip was observed at ƒw ≥ 30%, while TPC1 and TPC4 decreased at ƒw ≥ 50%, indicating that clipping in TPC2 derivatives occurs effectively even with a smaller water content than other TPCn derivatives.
The efficiency of AIE (EAIE) is defined as the ratio of Iw to I0 at 500 nm. The bright green emission was attributed to a 20-fold increase in the strength of the AIE effect. Furthermore, the EAIE at fw = 90%, was approximately 50-fold. In contrast to Eclip, EAIE for TPC2-(Z) increases in a staircase pattern with increasing ƒw (Fig. 2d, top). Although the DOS clipping effect (Eclip) could not distinguish TPC2-(Z) from that of the mixture containing (E) isomer, EAIE for TPC2-(Z) was significantly increased at ƒw ≥ 30%, distinguishing TPC2-(Z) from the TPC2-(E + Z) mixture, and other TPCn derivatives. Such a high AIE effect of TPC2-(Z), surpassing those of other TPCn molecules as water fraction increases, indicates that the TPE–TPE intermolecular interaction in TPC2-(Z) was stronger than in other TPCs owing to the specific orientation of DOS units in the (Z) configuration. Due to the AIE effect, the PLQY of TPC2-(Z) 5.2% in solution was increased to 45.6% in a drop casted film. The PLQY in the TPC2-(Z) film enhanced by 8.8 times compared to the value in the solution, and this increased ratio (rQY) was higher than those of the other TPCns (rQY = 5.8 and 6.3 for TPC1 and TPC4, respectively).
The size of the aggregated particles formed in different THF–water solutions was determined using dynamic light scattering (DLS) at room temperature. As shown in Fig. 2e, the average particle size at ƒw = 50% was 806.5 nm, which was decreased to ∼300 nm with increasing water content in the solution. The size distribution results were also visualized using field emission scanning electron microscopy (FE-SEM; Fig. 2f). Under 50000× magnification, granular particles are observed to become finer with increasing ƒw. At ƒw = 50%, the granular particles in the range of 50–80 nm size were primarily observed. As ƒw increases, small and closely spaced aggregates become more irregular in shape.
TPC2-(Z) powder samples were obtained from a solution with ƒw = 90% and analyzed using confocal microscopy. The as-prepared TPC2-(Z) powders exhibited strong emission in the confocal microscopic image (Fig. 3a-i). Similarly, in the TPC2-(Z) thin film, prepared from a water/THF mixture (ƒw = 90%), bright emissive aggregates were observed in the confocal microscopic image (Fig. 3a-ii). This suggests that the aggregated particles seen in the SEM image (Fig. 2f) primarily consist of TPC2-(Z) particles. The aggregation behaviour of TPC2-(Z) was further visualized using transmission electron microscopy (TEM) (Fig. S10, ESI†). The observed granular particles coalesced into giant aggregates with a wide size distribution. The molecular packing structure was elucidated using high-resolution TEM (HR-TEM) and the corresponding selected area diffraction (SAD) (Fig. 3b–d). TPC2-(Z) was dissolved in a THF–water mixture (ƒw = 90%), where it exhibited a highly assembled morphology. The confocal image of this sample (Fig. 3a-iii) exhibited higher emission intensity than the film (Fig. 3a-ii), possibly because of the crystalline nature of TPC2-(Z). Indeed, several dots forming a circular pattern in the SAD patterns indicate the presence of crystalline TPC2-(Z) with a distinct interplanar distance (Fig. 3c and Fig. S11, ESI†). The HR-TEM image of TPC2-(Z) shows lattice fringes with an average distance of 1.54 nm (Fig. 3c and d), indicating that the molecules are highly oriented in frameworks. The magnified image shows evident dark domains with an average size of approximately 0.82 nm (Fig. 3d) separated by bright streaks of 2.5 nm. As the TPE area was determined in the DOS-rich region surrounding the assembled TPE blocks, the size of the TPE blocks from the top view is 1.37 nm, matching the powder X-ray diffraction (XRD) and density functional theory (DFT) calculations for dimers with the TPE–TPE interaction discussed below (Table S1, ESI†).
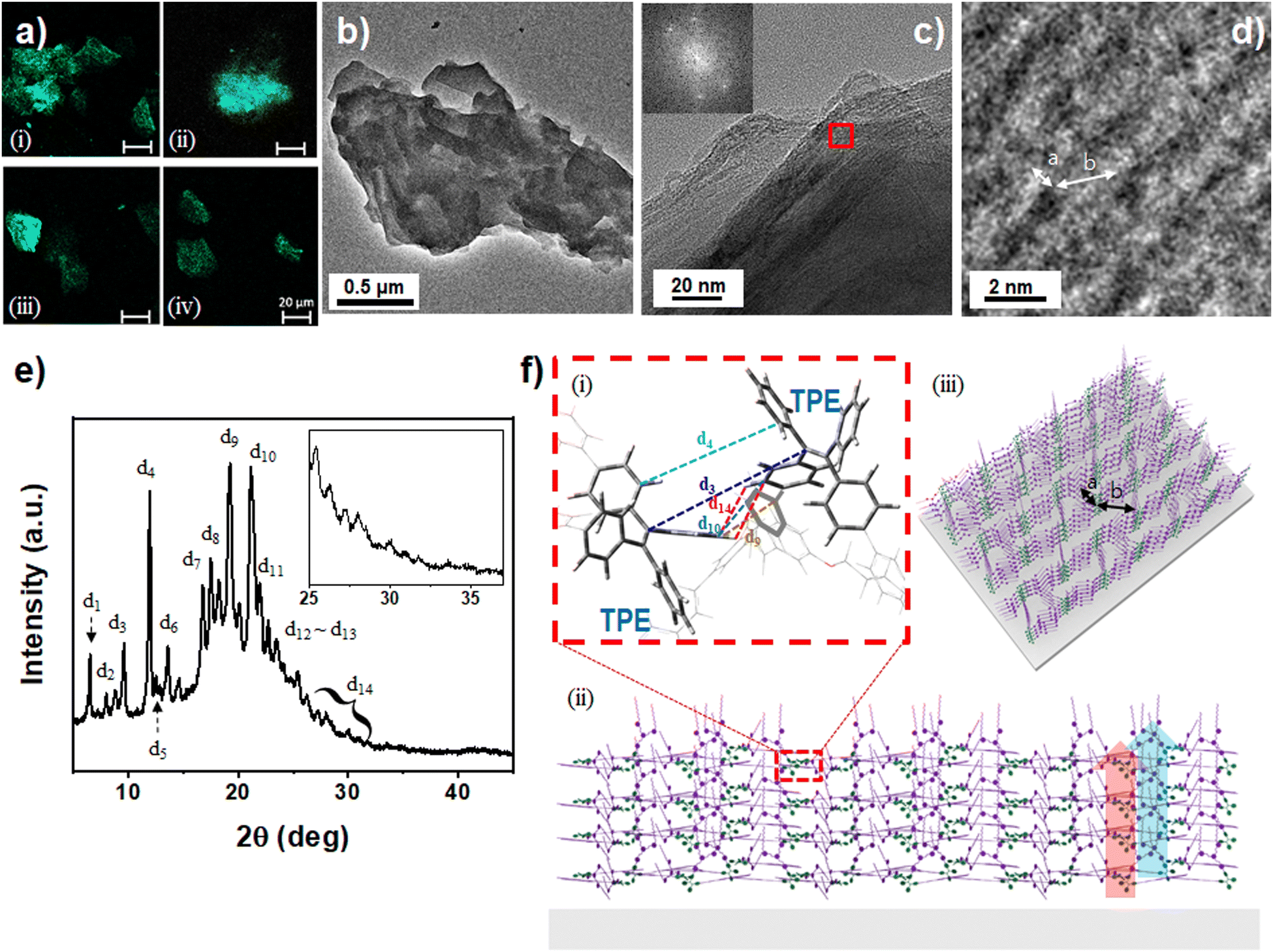 | ||
| Fig. 3 (a) Confocal images of the (i) TPC2-(Z) powders obtained from a solution with ƒw = 90%, (ii) dried TPC2-(Z) film on a slide glass obtained from the solution with ƒw = 90%, (iii) dried TPC2-(Z) sample on a TEM grid obtained from the solution with ƒw = 90%, and (iv) an LEC film containing TPC2-(Z), a host (p-CBP), and an electrolyte (THABF4) with a host:guest-ratio of 7.9:1. The white scale bar represents 20 μm. (b)–(d) TEM images of TPC2-(Z) in DW/THF (ƒw = 90%) solution. The dark area in the TEM image represents the area where molecules are densely populated. (e) Powder XRD pattern of pristine TPC2-(Z) particles. (f) (i) A simulated molecular structure from DFT calculations through Gaussian 16 showing the interaction between two molecules via TPE centers, with distances d1 to d14 marked in Fig. 2e. (ii) and (iii) Graphic representation of the molecular arrangement on a substrate, expanded from the DFT-calculated structure of two molecules (Fig. S13, ESI†) to multiple molecules through TPE aggregation (olive) and DOS unit clipping (purple), generated using Rhinoceros software. The diagram shows the molecular distribution and interactions, with enhanced luminescence indicated by TPE aggregation (red arrow) and ferroelectricity indicated by DOS unit clipping (blue arrow). | ||
2-3. Molecular packing structure of TPC2-(Z)
The XRD pattern shows sharp peaks between 2θ of 5–35° (Fig. 3e and Fig. S12, ESI†), indicating the high crystallinity of the TPC2-(Z) sample. The characteristic peaks were observed at 2θ ≈ 6.5° (d1 = 13.6 Å), 11.9° (d4 = 7.43 Å), 13.5° (d6 = 6.53 Å), 16.8° (d7 = 5.29 Å), 19.2 (d9 = 4.61 Å), and 21.1° (d10 = 4.21 Å), and small peaks at 2θ = 27°–32° (d14). These characteristic interplanar spacings were analysed and compared to the optimized molecular structures of TPC2-(Z) and the TPC2-(Z)–TPC2-(Z) dimer. These structures were derived using DFT calculations through the Gaussian 1662 software using the B3LYP method with a 6-31G** basis set (Fig. S13, ESI†). The structure of TPC2-(Z) consists of a hard TPE centre and relatively soft DOS groups attached to TPE. Therefore, the calculations for the interaction between two molecules were divided into two cases: one where the two molecules interact through the TPE centres (TPE–TPE, Fig. S13a and d, ESI†) and another where the interaction occurs between the DOS groups (clip–clip, and Fig. S13b and d, ESI†).The optimized structure of TPC2-(Z) consisted of peripheral phenyl groups at TPE units and two orthogonally oriented DOS groups, indicating that the entire molecule was asymmetric. The molecular dipole was calculated as 3.86 Debye, with the centre located between the neighbouring stilbene units. The EHOMO and ELUMO were calculated as −5.07 eV and −1.71 eV, respectively. These values were 0.47 eV and 0.78 eV, higher than those obtained from cyclic voltammetry, respectively. The length of the DOS unit, from the terminal C of decyl to the C of the connecting benzene of the TPE, C(πB)–C(decyl), was calculated as 13.8 Å, and that to the first decyl carbon was 11.6 Å, respectively, which matched to d1 and d2 obtained through XRD. The size of TPE was approximately 8 Å.
For TPE–TPE interactions, one phenyl face (C(π)) of the TPE interacts with a phenyl hydrogen of the other TPE, resulting in a packed structure via the C–H⋯π interaction. The C(π)–H distances were in the range of 2.85 Å (d12)–3.06 Å, matching the interaction distance for TPEs stacked via C–H⋯π interactions63 and aligned well with the d-spacing (d14) obtained through XRD (Fig. 3e). The closest C⋯π interaction at the TPE phenyls of TPC2-(Z) was 3.89 Å, which corresponded to the d-spacing (d11) in the XRD results. The sharp peaks at 19.2 (d9 = 4.61 Å) and 21.1° (d10 = 4.21 Å) correspond to the intermolecular C(π)–C(π) distances between the interacting TPEs (Fig. S12 and Table S1, ESI†). The distance for C(π)–C(π) interactions between two ethene centres and two phenyl centres of the two TPC2-(Z) molecules were calculated to be 10.28 Å and 7.82 Å, respectively, matching d-spacings d3 and d4, respectively, obtained through XRD.
The TPE interactions left DOS units in the outer face, where they interact through clipping with the DOS unit of the next molecules. The dipole moment increases to 7.77 D when three of the TPC2-(Z) molecules interact (Fig. S13c, ESI†). The length of the decyl unit and intramolecular distance from the terminal C of decyl to C of the connecting benzene of TPE, C(πB)–C(decyl), was relatively constant: 11.52 Å–11.70 Å and 13.84 Å–14.03 Å. Thus, d1 and d2 in the XRD pattern could be assigned to the decyloxy unit of DOS (13.6 Å and 11.1 Å, respectively). The presence of these sharp peaks indicates the ordered clipping of DOS units in the TPC2-(Z) solid powder. These d-spacing values match well with those of TPC2-(E + Z) and TPC4 (13.5 and 13.7 Å, respectively).27,30 Although small, the shorter d1 of TPC2-(Z) indicates that the molecules are more tightly assembled. Furthermore, the shortest C(σ)–C(σ) spacing for TPC2-(Z) was approximately 0.7 Å longer than that in TPC4, possibly because of the weaker steric hindrance in the TPC2-(Z) assemblies, than those in the TPC4 assemblies. This result could be matched to the enhanced EAIE of TPC2-(Z). Overall, the d-spacing values computed through DFT match well with those obtained through XRD (Table S1, ESI†).
The results of HR-TEM, SAD, and XRD indicated the high crystallinity of TPC2-(Z). Furthermore, DFT calculation showed that clipping DOS units were near-orthogonally branched with an angle of 65.57°. Thus, the interactions between TPC2-(Z)s in an assembled structure might not cancel out the dipole of TPC2-(Z). Indeed, the results of DFT calculations support the fact that the dipole moment of TPC2-(Z) increased to 6.07 D and 7.77 D (Fig. S13, ESI†) through TPE–TPE interactions and clipping, respectively. These values are considerably higher than the dipole moment of a single TPC2-(Z) molecule, 3.32 D.27 Therefore TPC2-(Z) formed a macro-dipole through the ordered aggregation in 3D aggregation, indicating potential ferroelectricity in the film. Due to the near-orthogonal DOS units, molecular packing can result in the formation of various crystalline assemblies in the film, including a packed row structure and a vesicle-like structure. A molecular arrangement on a substrate was graphically represented, expanding from the DFT-calculated structure of two molecules to a larger system of multiple molecules through TPE aggregation (olive) and DOS unit clipping (purple). The side view of the 3D aggregation illustrates the enhanced luminescence attributed to TPE aggregation and the generation of macro-dipoles resulting from DOS unit clipping (Fig. 3f-ii), which feature SELF properties. The 3D arrangement (Fig. 3f-iii) was comparable to the HR-TEM image analysis.
2-4. Ferroelectric properties of the TPC2-(Z) thin film
Ferroelectric materials change their inherent polarization direction under an external electric field of sufficient strength. Thus, the ferroelectric properties of TPC2-(Z) were examined by measuring polarization–electric field (P–E) characteristics of a TPC2-(Z) film capacitor (Al/TPC2-(Z)/ITO) in the Sawyer–Tower circuit.64 When a voltage was applied, the polarization of the thin TPC2-(Z) film was observed from 2 V. The polarization increases with voltage, saturating at 5 V (Fig. S16, ESI†). The film shows characteristic P–E loops and hysteresis (Fig. 4a, black) upon the reversal of applied voltage. Remnant polarization (Pr) was determined as 2.01 μC cm−2, and the coercive field was 21.6 MV m−2 for a pristine film at driving voltages in the range of −5 V to +5 V. | ||
| Fig. 4 (a) Polarization–electric field loops of TPC2-(Z) before (black) and after (red) annealing. (b) PFM phase and (c) PFM amplitude loops of TPC2-(Z) at 5 arbitrary points. (d) AFM height (top), PFM phase (middle) and PFM amplitude (bottom) images of TPC2-(Z) under different tip bias (scale: 5 × 5 μm2). (e) The UV-vis absorption (dotted line) and normalized PL spectra (solid line) of TPC2-(Z) (blue), TPC2-(Z) with CBP and THABF4 (red) and (p-CBP + THABF4) (black) thin film on the glass surface. (f) Structural representation of the LEC device and FE-SEM images of the device surface (50000×, scale bar: 500 nm) along with a cross-cut image (100000×, scale bar: 100 nm). (g) Photographs of pure TPC2-(Z) film (left), the emitting layer of an LEC (middle) under 365 nm UV irradiation, and light emission from the LEC device using TPC2-(Z) in the emitting layer under the VCL mode (right) (scale bar: 2.5 cm). (h) Electroluminescence spectra of the LEC containing TPC2-(Z) (1.82 μmol) at different voltages near Vmax under a VCL driving measurement. (10.8 V (black), 10.9 V (red), 11 V (blue), 11.1 V (magenta), 11.2 V (green), and 11.3 V (navy)). | ||
Because the polarization properties of the film might be caused by aggregated TPC2-(Z), we investigated the P–E properties of a thermally annealed film. According to the DSC results (Fig. S7, ESI†), phase transition occurs at 120 °C (Tsp). Thus, the pristine TPC2-(Z) film was annealed at 150 °C for 1 h, which is slightly higher than the Tsp of TPC2-(Z), and then cooled to ambient temperature. The annealed film afforded an increased Pr value of 2.54 μC cm−2 under the driving voltage in the range of −5 V to +5 V (Fig. 4a, red). This finding indicates that the aggregated molecular structure on a glass film is further organized through annealing, which was predicted by the DSC phase transition curve. Thus, the polarization effect originated through a specific intermolecular three-dimensional structure and could be altered by phase transition. The saturated polarization (Ps) value was 3.56 μC cm−2. Because the film from the (Z)/(E) mixture did not show P–E hysteresis, the high polarization results were quite promising. Furthermore, Pr after annealing is the largest among the TPC analogs (Table 1).
| Sample | λ PL,film [nm] | Φ f,film [%] | λ EL [nm] | Lvmaxd [Cd m−2] | P r [μC cm−2] | E c [MV m−2] | d 33 [pm V−1] | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Maximum PL wavelength on film. b PLQY on film. c Maximum EL wavelength. d Maximum EL intensity. e Remnant polarization at RT, = −5 V to +5 V unless otherwise specified. f Coercive field. g Piezoelectric coefficient. h After annealing = −5 V to +5 V. i Saturated polarization. j 4-(1,2,2-Triphenylvinyl)benzaldehyde. | ||||||||
| TPC2-(Z) | 496 | 45.6 | 496 | 891 (LEC) | 2.01 (2.54h, 3.56i) | 21.6 | −23.8 | This work |
| TPC1 | 481 | 100 | 422 | 528 (LEC) | 1.93 (0.36h, 2.73i) | 22.1 | −9.3 | 27 |
| TPC2-(E/Z) | 495 | 85.8 | 478 | 686 (LEC) | — | — | — | 27 |
| TPC4 | 508 | 78.3 | 504 | 1294 (LEC) | 2.27 (1.73h, 3.58i) | 20.2 | −17.4 | 27 |
| Cz-2TPE (2,7) | 483 | 47.5 | — | 1129 (OLED) | — | — | — | 65 |
| Multi-TPE (TPE5) | 494 | 18 | — | 2808 (OLED) | — | — | — | 65 |
| TPE–CHOj | — | — | — | 10.8 (LEC) | — | — | — | 66 |
| [(ATHTP) (18-crown-6)]BF4 | — | — | — | — | 3.18 | 1.01 | — | 67 |
| PVDF/PVDF-TrFE 60/40 wt% | — | — | — | — | 7.7 | 32 | — | 68 |
| BTA-C6 | — | — | — | — | 3.5 | 180 | — | 68 |
| PVDF | — | — | — | — | 7.0 | 50 | — | 69 |
| 6TP | — | — | — | — | 40 | 40 | — | 25 |
In addition, the surface morphology and piezoelectric phase and amplitude images were obtained using piezoresponse force microscopy (PFM) (Fig. 4b–d). Hysteresis loops were measured using a conductive cantilever with a tip bias from −10 V to 10 V. Upon the application of electrical bias, the amplitude of electrochemical coupling increases for both signs (Fig. 4c). The PFM phase loop in Fig. 4b demonstrates significant changes in polarization orientation, as evidenced by the abrupt phase switching at −10 V and 10 V. This sharp 180° phase flip indicates polarization reversal, characteristic of ferroelectric-like switching behaviour. Similarly, the PFM amplitude loop in Fig. 4c exhibits a butterfly-shaped response, where the amplitude decreases near 0 V and increases symmetrically at higher positive and negative biases. This PFM result, combined with sharp diffraction peaks (d1, d2, …, d14) in powder XRD (Fig. 3e and f), non-centrosymmetric stacking in DFT (Fig. S13c, ESI†), thermal transition in DSC (Fig. S7, ESI†), and spontaneous polarization in the P–E loop (Fig. 4a and Fig. S13, ESI†), confirms that TPC2-(Z) exhibits ferroelectric behaviour with switchable dipoles and domain realignment under an external electric field. The longitudinal piezoelectric coefficient (d33) was −23.8 pm V−1 for TPC2-(Z) from the PFM amplitude (S-4, ESI†); it was substantially higher than those of other TPCn molecules.27 This magnitude of d33 was comparable to those of known organic piezoelectric molecules, such as trialkylbenzene-1,3,5-tricarboxamide with d33 up to 20 pm V−1.36 This value also far exceeds those of other organic single molecules such as 4,5-dibromo-2-methyl-1H-imidazole,65 croconic acid, 3-hydroxy-1H-phenalen-1-one, and phenylmalonaldehyde with moderate d33 (Table S2, ESI†).66
The strong agreement between XRD analysis and DFT calculations provides a foundation for identifying the polarization inversion unit and symmetry-breaking mechanism in TPC2-(Z). Specifically, the polarization inversion unit in TPC2-(Z) could be governed by the interplay between TPE–TPE interactions and the spatial confinement of DOS units, which together enforce critically the non-centrosymmetric packing arrangement. XRD analysis shows that the d1 = 13.6 Å peak corresponds to the ordered DOS arrangement, while d4 = 7.43 Å represents intermolecular distances between DOS unit confinement. However, the near-orthogonal alignment of DOS units (65.57°) only emerges when TPE–TPE interactions provide a rigid structural framework, ensuring a stable polarization switching mechanism. Therefore, a key symmetry-breaking mechanism arises from TPE–TPE interactions constraining DOS clipping orientation, enforcing a non-centrosymmetric packing arrangement. Without these interactions, the clipped DOS units would adopt a centrosymmetric structure, preventing polarization inversion. This is confirmed by the increase in dipole moment when both interactions coexist (7.77 D), compared to TPE–TPE alone (3.86 D) or DOS clipping alone (6.07 D) (Fig. S13c, ESI†). Thus, the TPE–TPE framework acts as the polarization unit, enabling symmetry breaking and polarization alignment. This molecular-level alignment provides the necessary interactions for ferroelectric switching, further validated by the P–E hysteresis loop, DSC phase transition behaviour, and high crystallinity observed in HR-TEM and XRD.
2-5. Electroluminescence properties of TPC2-(Z)
TPC2-(Z) exhibits good thermal stability and strong emission in the film state (Fig. 4e), allowing the fabrication of an EL device without the decomposition of the active material. We produced LECs because of their simple structure and fabrication.67,68 On a patterned indium tin oxide (ITO) glass, poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate (PEDOT:PSS) was coated as a hole transporting layer. The emitting layer consisted of para-4,4′-bis(9H-carbazol-9-yl)biphenyl (CBP) as an electron-donating host, TPC2-(Z) as a light-emitting guest and tetrahexylammonium tetrafluoroborate (THABF4) as an electrolyte (Fig. 4f). The emitting layer is uniformly coated on the PEDOT:PSS layer, as confirmed via FE-SEM (Fig. 4f). Lastly, an aluminum layer with a thickness of approximately 100 nm was deposited over the emitting layer as a cathode. During the application of current, ions in the emitting layer were redistributed owing to external voltage, and guest molecules were electrochemically doped by the host. Consequently, an in situ p–n junction is formed in the emitting layer, and injected holes and electrons combine to become excitons, thus emitting light.69,70The composition of the light emitting layer was varied with the CBP-to-TPC2-(Z) molar ratio. The sum of the weights of CBP and TPC2-(Z) was fixed at 10.14 mg (25.35 mg mL−1) and the molar ratio between those was varied from 5.4:1 to 21.7:1. The amount of THABF4 was fixed at 2.36 mg (5.90 mg mL−1, 17.9 mM) for all samples. The host–guest ratio and LEC performance are summarized in Tables S3–S5 (ESI†). Considering the HOMO (= −5.53 eV) and LUMO (= −2.49 eV) energy levels and bandgap (3.50 eV) of CBP,27 CBP can cover fully that of TPC2-(Z). Thus, energy transfer from host to guest could occur favourably. The thin-film emitting layer exhibited primarily PL of TPC2-(Z), confirming the distinct host–guest interaction. Furthermore, the confocal image (Fig. 3a-iv) shows that the emission colours of TPC2-(Z) and LEC are similar, indicating that the host and electrolyte did not quench the PL of TPC2-(Z). Compared to the PL spectra of TPC2-(Z) pure film centred at 496 nm, the mixture of TPC2-(Z), CBP, and THABF4 at a ratio of 7.8:61.6:30.6, resulted in a 10 nm blue shift in thin film state (Fig. 4e). The photographic images also reveal a noticeable colour difference between the films (Fig. 4f). In the EL spectrum of the LEC device, the highest emission peak is centred at 496 nm, which is inherent to the AIE of TPC2-(Z) (Fig. 4h). EL spectra of LECs with different host–guest compositions of LEC are listed (Fig. S14, ESI†). Generally, the λmax of the spectra shifts to shorter wavelengths to emit blueish luminescence as the host-to-guest ratio decreases, indicating that there must be a sufficient amount of guest material to fully convert the transferred charge into excitons. Simultaneously, the left shoulder peak at approximately 400 nm reflects the competing effect of DOS units and the host. The peaks in Fig. 4h were deconvoluted, showing the area percentages of 7% and 93% for 407 nm and 496 nm, respectively (Fig. S15, ESI†), indicating that the energy transfer from the host to aggregated TPC2-(Z) is quite efficient for the emitting layer with a CBP:TPC2-(Z) molar ratio of = 7.9:1.
We measured the LEC performance in two different modes: voltage–current driving LEC (VCL) and pulse–current driving LEC (PCL, 50% cycle, 235 Hz) as shown in Fig. 5a and b, respectively. In the VCL mode, the luminescence gradually increased with driving voltage, but upon reaching turn on voltage (Von), the rise becomes sharper. In addition, the current efficiency was relatively lower compared to those of PCL. LEC with 1.82 μmol TPC2-(Z) shows an overall superior performance with luminescence of 627.1 Cd m−2 and 890.1 Cd m−2 at Von of 9.9 V and 3.9 V, under VCL and PCL, respectively. This was more than approximately 40% enhanced performance compared to that of (E)/(Z) mixtures (Table 1). Luminescence and current efficiency change substantially at approximately 10 mol% TPC2-(Z) (Fig. 5c). Therefore, at least 10 mol% of guest is required to enhance the overall LEC performances. Based on results reported by our group for other TPCn compounds, TPC2-(Z) exhibited higher luminescence than the TPC2-(E + Z) stereomixture. This result was consistent with the PL results, signifying that (Z) isomer, which was aggregated to a greater degree than the (E) isomer, demonstrated excellent SELF properties. Among other TPCn, TPC1 and TPC4 possess both electroluminescence and piezoelectric activity (Fig. 5d), the emission wavelength of TPC2-(Z) was between those of TPC1 and TPC4, as an increasing number of DOS units conjugated to the TPE core led to the narrow bandgap (Eg). Although TPC4 shows high EL intensity through PCL mode, its Pr value (2.27 μC cm−2) was lower than that of TPC2-(Z). Notably, this figure shows TPCn could be geometrically tuned to enhance Pr while simultaneously revealing EL properties, thereby achieving SELF properties. In contrast, many other materials are either electroluminescent or ferroelectric, but not both, and thus do not exhibit SELF.
 | ||
| Fig. 5 (a) I–V–L curve of TPC2-(Z) LEC under VCL (black) and PCL (red) driving mode. (b) Current efficiency curve of TPC2-(Z) LEC under VCL (black) and PCL (red) driving modes, with long-term stability under PCL driving (inset). (c) Variation in luminescence and current efficiency of LEC with different mole percentages of TPC2-(Z) guest versus CBP host material. (d) Plot of remnant polarization (Pr) plot of TPCn versus maximum EL intensity (Lv) of devices: (1) multi-TPE (TPE5),71 (2) Cz-2TPE (2,7),71 (3) TPE–CHO,72 (4) [(ATHTP)crown-6]BF4,73 (5) BTA-C6,74 and (6) PVDF.75 (e) Change in luminescence in response to piezoelectric switching of the TPC2-(Z) LEC device. (inset) Output voltage of TPC2-(Z) PENG during physical bending. | ||
2-6. Self-powered and piezoelectrically operated EL devices
Materials exhibiting high P–E hysteresis can be utilized not only in ferroelectric devices, such as switches and memory, but also in energy harvesting applications. These include piezoelectric harvesters76 and triboelectric generators,77 which enable self-powering or low energy consumption of the resulting electronics and sensors.67 As SELF materials feature luminescent and ferroelectric properties, they can be applied as sensors that are visually perceivable, without additional display devices, or as displays that can respond to mechanical movements or frequency changes in the surrounding environment without the need to use additional sensor devices. Ultimately SELF materials may be used to fabricate interactive devices with a simple structure, taking advantage of sensitivity to stimuli and luminescence properties. To explore this possibility, an emissive piezoelectric nanogenerator (PENG) was fabricated using TPC2-(Z).A 2 wt% solution of TPC2-(Z) in chlorobenzene was spin-coated onto ITO/polyester substrate with an area of 2.5 × 3.5 cm2. An Al top electrode with a thickness of 100 nm is then thermally deposited on TPC2-(Z) (Fig. S17a, ESI†). The PENG based on TPC2-(Z) film was subjected to repetitive convex bending at a frequency of 1 Hz to explore its self-powering capacity. The piezoelectric output of the PENG was reproducibly generated, as monitored in real time. The neat thin films of TPC2-(Z) were mechanically stable over repetitive bending, without crack formation. The output voltage generated by the PENG is up to 0.17 V (Fig. 5e, inset), which is higher than that of TPC4 (0.13 V). The PENGs exhibited a current density of up to 4.07 nA cm−2. The maximum attained power density, calculated based on the highest output voltage and highest current observed, was 6.9 mW m−2.78 This value was lower than those of inorganic-based devices,79–81 but was at a similar level or slightly lower than that of PVDF-based devices.82–85
Finally, we fabricated an EL device that utilizes the piezoelectric properties of TPC2-(Z), to realize a piezoelectrically operated EL device. To operate the LEC utilizing piezoelectric energy generated by the TPC2-(Z) PENG film, the alternating current from PENG was converted into direct current (DC), using 4 bridge diodes. The DC voltage generated by bending at a frequency of 1 Hz was stored in the capacitor and amplified to 3.2 V (Fig. S17c, ESI†). The LEC was fabricated with a solution of 7 mg of p-CBP, 3.14 mg of TPC2-(Z), and 2.36 mg of THABF4 in 0.4 mL of cyclohexanone (61.6:7.8:30.6 molar ratio). This device exhibited a luminance of 1 cd m−2 at 10 V, when the PENG film was stationary and in an almost “switched off” state. Upon repeated bending of the PENG at a frequency of 1 Hz, the luminance of the LEC increased. The LEC effectively “switched on” luminescence, showing a 120-fold increase in brightness after 254 bending at 1 Hz, compared to the “off” state (Fig. 5e). These findings evidently demonstrated that TPC2-(Z) holds promising potential for applications in self-powered sensor displays and interactive devices. Overall, the Z-clipping could be a promising way to realize synergistic enhancement of luminescence and ferroelectricity, leading to a new self-powered, stimuli-sensitive electroluminescent device.
3. Conclusion
A new (Z)-specific TPC2 was synthesized through a unique ‘attaching legs to body core’ straightforward synthetic strategy. With good solubility to various organic solvents, TPC2-(Z) was capable of forming films through a solution process upon Z-clipping of the self-assembling units. This TPC2-(Z) thin film exhibited exceptional properties with a high fluorescence (PLQY = 45.6%). Because of its high thermal stability and optical functionality, TPC2-(Z) was applied in an LEC, and the device exhibited an overall superior EL performance among the TPCns, with a brightness of 890 Cd m−2, at a Von of 3.9 V, which was a 40% enhancement in performance compared to that of (E)/(Z) mixtures. The P–E hysteresis loop of the neat TPC2-(Z) film exhibited a Pr of 2.54 μC cm−2 at a driving voltage in the range of −5 V to +5 V. The PFM amplitude and phase images showed a piezoelectric response with a d33 of −23.8 pm V−1 for TPC2-(Z), which is the highest value among TPCns and is comparable to those of highly organic piezoelectric molecules such as trialkylbenzene. The ferroelectricity of the TPC2-(Z) film was effectively applied to fabricate a PENG, which generated a piezoelectric output voltage of up to 0.17 V. Finally, the fabricated TPC2-(Z) LEC exhibited a substantial increase in EL intensity upon the repeated bending of the TPC2-(Z) PENG film up to 254 times. It entailed a 120-fold increase in brightness compared to that of the ‘off’ state before bending. These results clearly indicate that the Z-clipping of AIE materials could be a promising way to realize synergistic enhancement of luminescence and ferroelectricity, opening a new path to multi-functionality on a device level.4. Experimental section
4-1. Materials
1,3,5-Bromomethyl benzene, triethylphosphite, 3-hydroxybenzaldehyde, 1-bromodecane, t-BuOK, sodium carbonate, cesium carbonate, 4-formylphenylboronic acid, 1,1,2,2-tetraphenylethene, tetrakis(triphenylphosphine)palladium(0), tetrakis(triphenylphosphine)platinum(0), [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), DMF, THF, toluene, ethanol, and 1,4-dioxane, were purchased from Sigma-Aldrich Chemicals and used without further purification. PEDOT:PSS and patterned ITO glass substrates were purchased from Ossila and Wooyang GMS, respectively. THABF4 was purchased from Sigma-Aldrich. CBP was obtained from TCI. Sigma-Aldrich provided cyclohexanone, which was utilized as a solvent.4-2. Instruments and characterization
1H-NMR spectra were recorded using a Bruker Avance II 400 spectrometer at a frequency of 400 MHz. Chemical shifts were given in parts per million and were reported relative to tetramethylsilane using the residual solvent chloroform-d as an internal standard. MALDI-TOF/TOF mass spectra were recorded on a Bruker Daltonics Autoflex III mass spectrometer in the reflection mode using a laser (λ = 355 nm); the sample for this test was prepared with tert-butylphenyl-2-methyl-2-propenylidene malononitrile as a matrix. The UV-vis spectra were recorded using a PerkinElmer LAMBDA 750 UV/VIS/NIR spectrophotometer. A PerkinElmer LS-55 fluorescence spectrometer was used to record the photoluminescence emission spectra. The pulsed current system was operated at 235 Hz and 50% duty cycle using a Keithley 2400 source meter controlled by a MATLAB code. The voltage of TPCn LEC was measured using a current ramp, and the EQE was recorded using Lambertian emission. HR-TEM images were obtained using a JEM-F200, JEOL, with 200 mesh copper grid. The accelerating voltage was 200 kV. Confocal microscopic images were obtained using a LSM 900, Carl Zeiss. Resulting images were processed via Zen Blue 3.3 imaging software.4-3. Synthesis of TPC2-(Z) (Fig. S1, ESI†)
Under a nitrogen atmosphere, the solution of the clip (868 mg, 1.16 mmol, 2.1 equiv.) in anhydrous THF (20 mL) was cooled to 0 °C. t-BuOK (218 mg, 3.5 equiv.) was slowly added to the solution, after which the mixture was stirred for 10 min, followed by the addition of TPE2PhCHO (300 mg, 0.555 mmol, 1 equiv.). The reaction was allowed to proceed for an additional 30 min at 0 °C and overnight at room temperature. The resulting mixture was quenched with brine, extracted with dichloromethane (DCM), washed with brine, and then dried over MgSO4. After filtration, the solvent was evaporated under reduced pressure. The residue was purified via silica gel column chromatography (DCM:hexane = 1:1) and recrystallized, yielding 140 mg of a pale green solid (14.7%).
1H NMR (400 MHz, CDCl3, δ): 7.60 (m, 10H), 7.44 (m, 4H), 7.31 (m, 12H), 7.21 (d, 4H), 7.18 (m, 26H), 6.86 (m, 4H, J = 8.28 Hz), 4.04 (t, 8H), 1.84 (m, 8H), 1.29 (m, 56H), and 0.89 (m, 12H).
13C NMR (100 MHz, CDCl3, δ): 159.40, 143.78, 142.91, 140.71, 139.85, 138.58, 138.00, 136.15, 131.92, 131.48, 129.67, 129.30, 128.83, 128.50, 128.25, 127.90, 127.71, 127.10, 126.98, 126.10, 125.96, 123.99, 119.20, 114.08, 112.33, 68.01, 31.97, 29.72, 29.60, 29.42, 29.35, 26.09, 22.71, and 14.14.
Mass analysis (MALDI-TOF/TOF): calculated m/z for C126H144O4 [M]+ 1721.11, measured 1722.45.
4-4. Fabrication of LEC
Patterned ITO glass substrates were cleaned with 0.5 wt% aqueous Hellmanex, deionized water, and acetone for 20 min respectively in an ultrasonic bath and dried for 1 h at 80 °C in an oven after washing. The patterned ITO glasses were subjected to UV-ozone treatment for 20 min and spin coated with PEDOT:PSS at 4000 rpm for 1 min. The ITO glasses were then annealed for 1 h at 130 °C on a hot plate and coated with an active solution in an N2-filled glove box, followed by additional annealing for 1 h at 50 °C. The active layers were then spin-coated for 60 s at 2000 rpm. Solutions for the active layer were prepared by dissolving 7.00 mg of p-CBP, 2.3 mg or 3.14 mg of TPC2-(Z), and 2.36 mg of THABF4 in 0.4 mL of cyclohexanone. To improve the properties of LEC mentioned in the main text, the amount of TPC2-(Z) was increased to 3.14 mg. Thermal evaporation under a high vacuum (2 × 10−5 torr) through a shadow mask was used to deposit aluminum nanoparticles on the active layer.4-5. Preparation of ITO–ferroelectric–Al capacitors and ferroelectric characterization
The patterned ITO glass substrates were ultrasonicated with 1 wt% aqueous Hellmanex III, deionized water and acetone, for 20 min with each. The substrates were then dried in an oven at 80 °C for 1 h to completely evaporate the residual solvent. The solutions for coating the ferroelectric layers were prepared by dissolving TPC2-(Z) derivatives in cyclohexanone to obtain a concentration of 2 wt%. The ferroelectric layers were then spin-coated at 2000 rpm for 60 s. Finally, Al electrodes (100 nm) were deposited on the ferroelectric layers via thermal evaporation through a shadow mask under a high vacuum (2 × 10−5 torr). The area used for ferroelectric measurement was 0.3 × 0.3 cm2. The polarization–electric field (P–E) loops were evaluated using the Sawyer–Tower circuit. To avoid displacement and leakage input in the P–E curve, the double wave method was used to subtract the non-switching current from the initial signal. The input voltage was applied through triangular-wave bias, using a function generator (Agilent 33220A 20 MHz Function/Arbitrary Waveform Generator) and a power amplifier (Pendulum Instruments, Dual Channel High Voltage Linear Amplifier A400D). The device response was recorded using a digital oscilloscope (Tektronics Digital Phosphor Oscilloscope, DPO4104B).4-6. PFM measurement
The surface morphology, and piezoelectric phase and amplitude images, were measured using atomic force microscopy (ParkSystems, NX10) with a Pt-coated tip (radius of <30 nm, resonance frequency of 75 kHz, force constant of 2.8 N m−1, Nanosensors). The applied force was approximately 80 nN during 256 line scans with a scan rate of 0.7 Hz. The PFM hysteresis loops were measured using a triangle pulse (tip bias) from −10 V to 10 V.Author contributions
E. Kim directed the research project, supervised the experimental design, and wrote the manuscript. S. Lim performed the synthesis and optical characterization of materials. D. Kim performed the fabrication of films, calculated the molecular structures, and measured the ferroelectric properties of materials. H. Kim performed the structural analysis of materials, theoretical calculations, and polarization mechanism of molecules. H. Jang performed the electrical characterization and image analysis of films. S. Park contributed to the simulated image analysis and discussion.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation (NRF) grant funded by the Korean government (Ministry of Science, ICT & Future Planning, MSIP) through the Creative Materials Discovery Program (2018M3D1A1058536) and the NRF grant (No. RS-2023-00302697).References
- N. Luo, J. Xu, X. Cheng, Z. Li, Y. Huang, M.-H. Whangbo, S. Deng and M. Hong, J. Mater. Chem. C, 2021, 9, 14439–14443 RSC.
- C. Sun, Y. Li, J. Yin, D. Li, C. Wu, C. Zhang and H. Fei, Angew. Chem., Int. Ed., 2024, 63, e202407102 CrossRef CAS PubMed.
- W. Zhang, S. Jiao, M. Zhao, C. Xu, Z. Yang, D. Li, Z. Tang, Y. Lu, H.-L. Cai and X. S. Wu, J. Phys. Chem. C, 2023, 127, 17261–17268 CAS.
- A. K. Srivastava, W. Zhang, J. Schneider, J. E. Halpert and A. L. Rogach, Adv. Sci., 2019, 6, 1901345 CrossRef CAS PubMed.
- Z. Cui, Q. Yang, X. Qu, X. Zhang, Y. Liu and G. Yang, J. Mater. Chem. C, 2022, 10, 672–679 CAS.
- Y. Huang, H. K. Bisoyi, S. Huang, M. Wang, X. M. Chen, Z. Liu, H. Yang and Q. Li, Angew. Chem., Int. Ed., 2021, 60, 11247–11251 CAS.
- J. Y. Kim, S. Lee, S. Lee, K. Lee, Y. Huh, Y. E. Kim, J. W. Lee, C. E. Lee, D. Kim and B. J. Yim, Cell Rep. Phys. Sci., 2022, 3, 101006 CAS.
- Y. Zhou, J. Fu, Z. Chen, F. Zhuge, Y. Wang, J. Yan, S. Ma, L. Xu, H. Yuan, M. Chan, X. Miao, Y. He and Y. Chai, Nat. Electron., 2023, 6, 870–878 Search PubMed.
- D. Peng, H. Zou, C. Xu, X. Wang, X. Yao, J. Lin and T. Sun, AIP Adv., 2012, 2, 042187 Search PubMed.
- J.-X. Gao, Z.-X. Wang, Y.-Y. Tang, Y.-L. Liu, L. Zhou, X.-G. Chen, W.-Y. Zhang and R.-G. Xiong, J. Mater. Chem. C, 2019, 7, 11022–11028 CAS.
- R. Yang, Nat. Electron., 2020, 3, 237–238 Search PubMed.
- Y. Zhang, X.-J. Song, Z.-X. Zhang, D.-W. Fu and R.-G. Xiong, Matter, 2020, 2, 697–710 Search PubMed.
- S. Yu, T. H. Park, W. Jiang, S. W. Lee, E. H. Kim, S. Lee, J. E. Park and C. Park, Adv. Mater., 2023, 35, 2204964 CAS.
- R. C. G. Naber, C. Tanase, P. W. M. Blom, G. H. Gelinck, A. W. Marsman, F. J. Touwslager, S. Setayesh and D. M. de Leeuw, Nat. Mater., 2005, 4, 243–248 CAS.
- Y. Hu, H. Zhang, J. Zhou, J. Shen, B. Chen, A. Li and W. Chen, RSC Adv., 2023, 13, 11432–11440 CAS.
- X.-X. Cao, S.-R. Ding, G.-W. Du, Z.-Y. Jing, Y.-A. Xiong, Z.-J. Feng, H.-R. Ji, Q. Pan and Y.-M. You, Adv. Mater., 2025, 37, 2416837 CAS.
- H. Tan, G. Castro, J. Lyu, P. Loza-Alvarez, F. Sánchez, J. Fontcuberta and I. Fina, Mater. Horiz., 2022, 9, 2345–2352 CAS.
- G. Canu, G. Bottaro, M. T. Buscaglia, C. Costa, O. Condurache, L. Curecheriu, L. Mitoseriu, V. Buscaglia and L. Armelao, Sci. Rep., 2019, 9, 6441 CrossRef PubMed.
- B. Wen, Y. Zhu, D. Yudistira, A. Boes, L. Zhang, T. Yidirim, B. Liu, H. Yan, X. Sun, Y. Zhou, Y. Xue, Y. Zhang, L. Fu, A. Mitchell, H. Zhang and Y. Lu, ACS Nano, 2019, 13, 5335–5343 CrossRef CAS PubMed.
- S. Li, Y. Wang, M. Yang, J. Miao, K. Lin, Q. Li, X. Chen, J. Deng and X. Xing, Mater. Adv., 2022, 3, 5735–5752 RSC.
- L. Xu, Z. Wang, B. Su, J. Xiong, X. Yang, R. Su, C. He and X. Long, Chem. Commun., 2021, 57, 488–491 RSC.
- E. Pan, G. Bai, J. Zhou, L. Lei and S. Xu, Nanoscale, 2019, 11, 11642–11648 RSC.
- A. Chouprik, E. Savelyeva, E. Korostylev, E. Kondratyuk, S. Zarubin, N. Sizykh, M. Zhuk, A. Zenkevich, A. M. Markeev, O. Kondratev and S. Yakunin, Nanomaterials, 2023, 13, 3063 CrossRef CAS PubMed.
- H. Tan, S. Estandía, F. Sánchez and I. Fina, ACS Appl. Electron. Mater., 2023, 5, 6630–6639 CrossRef CAS.
- T. Takeda, J. Wu, N. Ikenaka, N. Hoshino, I. Hisaki and T. Akutagawa, ChemistrySelect, 2018, 3, 10608–10614 CrossRef CAS.
- C. Park, K. Lee, M. Koo and C. Park, Adv. Mater., 2021, 33, 2004999 CAS.
- D. Kim, H. T. T. Thuy, B. Kim, Y. Auh, M. Rémond, K. K. Leong, T. Liang, J. Kim and E. Kim, Adv. Funct. Mater., 2023, 33, 2208157 CrossRef CAS.
- Z. He, C. Ke and B. Z. Tang, ACS Omega, 2018, 3, 3267–3277 CAS.
- N. L. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 20, 15349–15353 CAS.
- D. Bléger, D. Kreher, F. Mathevet, A. J. Attias, G. Schull, A. Huard, L. Douillard, C. Fiorini-Debuischert and F. Charra, Angew. Chem., Int. Ed., 2007, 46, 7404–7407 Search PubMed.
- D. Bléger, D. Kreher, F. Mathevet, A. J. Attias, I. Arfaoui, G. Metgé, L. Douillard, C. Fiorini-Debuisschert and F. Charra, Angew. Chem., Int. Ed., 2008, 47, 8412–8415 Search PubMed.
- V. Cheltsov, Proceedings of LFNM 2005. 7th International Conference on Laser and Fiber-Optical Networks Modeling, IEEE, 2005, pp. 131–135 DOI:10.1109/LFNM.2005.1553210.
- M. Anandan, J. Soc. Inf. Disp., 2008, 16, 287–310 CAS.
- R. F. Standaert and S. B. Park, J. Org. Chem., 2006, 71, 7952–7966 CAS.
- E. Merino and M. Ribagorda, Beilstein J. Org. Chem., 2012, 8, 1071–1090 CAS.
- I. Urbanaviciute, X. Meng, M. Biler, Y. Wei, T. D. Cornelissen, S. Bhattacharjee, M. Linares and M. Kemerink, Mater. Horiz., 2019, 6, 1688–1698 CAS.
- L. Xu, Z. Zhu, X. Zhou, J. Qin and C. Yang, Chem. Commun., 2014, 50, 6494–6497 CAS.
- Y. Huang, G. Zhang, R. Zhao and D. Zhang, Chem. – Eur. J., 2023, 29, e202300539 CAS.
- Y. Xie and Z. Li, Chem. – Asian J., 2019, 14, 2524–2541 CAS.
- M. J. Hurlock, Y. Kan, T. Lécrivain, J. Lapka, K. L. Nash and Q. Zhang, Cryst. Growth Des., 2018, 18, 6197–6203 CAS.
- X. Jiawei, Z. Chong, W. Xunchang, J. Jiaxing and W. Feng, Acta Chim. Sin., 2017, 75, 473–478 CrossRef.
- P. Shen, Z. Zhuang, X.-F. Jiang, J. Li, S. Yao, Z. Zhao and B. Z. Tang, J. Phys. Chem. Lett., 2019, 10, 2648–2656 CrossRef CAS PubMed.
- J. Zhang, L. Hu, K. Zhang, J. Liu, X. Li, H. Wang, Z. Wang, H. H. Sung, I. D. Williams and Z. Zeng, J. Am. Chem. Soc., 2021, 143, 9565–9574 CrossRef CAS PubMed.
- J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS PubMed.
- Y. Wang, Z. Qian, X. Li, A. Qin, Y. Guo and B. Tang, J. Mater. Chem. C, 2021, 9, 12681–12693 RSC.
- Z. Wang, X. Cheng, A. Qin, H. Zhang, J. Z. Sun and B. Z. Tang, J. Phys. Chem. B, 2018, 122, 2165–2176 CrossRef CAS PubMed.
- W. Tian, T. Lin, H. Chen and W. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 6302–6314 CrossRef CAS PubMed.
- D. L. Priebbenow, L. Barbaro and J. B. Baell, Org. Biomol. Chem., 2016, 14, 9622–9628 RSC.
- A. Kamal, J. R. Tamboli, V. L. Nayak, S. Adil, M. Vishnuvardhan and S. Ramakrishna, Bioorg. Med. Chem., 2014, 22, 2714–2723 CrossRef CAS PubMed.
- A. Kamal, M. K. Reddy, T. B. Shaik, Y. Srikanth, V. S. Reddy, G. B. Kumar and S. V. Kalivendi, Eur. J. Med. Chem., 2012, 50, 9–17 CrossRef CAS PubMed.
- L. Li, L. He, X. Liu, H. Liu, L. Hu, P. Guo and W. Bu, RSC Adv., 2017, 7, 38581–38585 CAS.
- J. De, W.-Y. Yang, I. Bala, S. P. Gupta, R. A. K. Yadav, D. K. Dubey, A. Chowdhury, J.-H. Jou and S. K. Pal, ACS Appl. Mater. Interfaces, 2019, 11, 8291–8300 CAS.
- T. Huang, T. Chen and L.-B. Han, J. Org. Chem., 2018, 83, 2959–2965 CAS.
- J.-C. Poupon, D. Marcoux, J.-M. Cloarec and A. B. Charette, Org. Lett., 2007, 9, 3591–3594 CAS.
- M. Wakioka, M. Ikegami and F. Ozawa, Macromolecules, 2010, 43, 6980–6985 CAS.
- Y.-Z. Tang, Y.-M. Yu, J.-B. Xiong, Y.-H. Tan and H.-R. Wen, J. Am. Chem. Soc., 2015, 137, 13345–13351 CAS.
- R. Kumari, A. De, A. J. Bhattacharyya and T. N. G. Row, Mater. Adv., 2024, 5, 3411–3423 CAS.
- Z. Fu, W. Xia, W. Chen, J. Weng, J. Zhang, J. Zhang, Y. Jiang and G. Zhu, Macromolecules, 2016, 49, 3818–3825 CAS.
- Y. Chen, Y. Niu, P. Gong, Z. Xiao and G. Li, Macromol. Rapid Commun., 2017, 38, 1700401 CrossRef PubMed.
- R. G. da Costa, F. R. Farias, L. Maqueira, C. Castanho, L. S. Carneiro, J. Almeida, C. D. Buarque, R. Q. Aucélio and J. Limberger, J. Braz. Chem. Soc., 2019, 30, 81–89 CAS.
- B. Z. Tang, Y. Geng, J. W. Y. Lam, B. Li, X. Jing, X. Wang, F. Wang, A. B. Pakhomov and X. Zhang, Chem. Mater., 1999, 11, 1581–1589 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- Q. Huang, T. Yu, Z. Xie, W. Li, L. Wang, S. Liu, Y. Zhang, Z. Chi, J. Xu and M. P. Aldred, J. Mater. Chem. C, 2017, 5, 11867–11872 RSC.
- H. Zhu, T. Miyashita and M. Mitsuishi, Polym. J., 2019, 51, 795–801 CrossRef CAS.
- M. Owczarek, K. A. Hujsak, D. P. Ferris, A. Prokofjevs, I. Majerz, P. Szklarz, H. Zhang, A. A. Sarjeant, C. L. Stern and R. Jakubas, Nat. Commun., 2016, 7, 13108 CrossRef CAS PubMed.
- S. Horiuchi, J. y Tsutsumi, K. Kobayashi, R. Kumai and S. Ishibashi, J. Mater. Chem. C, 2018, 6, 4714–4719 RSC.
- K. Schlingman, Y. Chen, R. S. Carmichael and T. B. Carmichael, Adv. Mater., 2021, 33, 2006863 CrossRef CAS PubMed.
- S. Tang and L. Edman, Photoluminescent Materials and Electroluminescent Devices, 2017, pp. 375–395 Search PubMed.
- E. Fresta and R. D. Costa, J. Mater. Chem. C, 2017, 5, 5643–5675 CAS.
- S. B. Meier, D. Tordera, A. Pertegas, C. Roldan-Carmona, E. Orti and H. J. Bolink, Mater. Today, 2014, 17, 217–223 CAS.
- S. Biswas, D. Jana, G. S. Kumar, S. Maji, P. Kundu, U. K. Ghorai, R. P. Giri, B. Das, N. Chattopadhyay and B. K. Ghorai, ACS Appl. Mater. Interfaces, 2018, 10, 17409–17418 CAS.
- W.-L. Gong, B. Wang, M. P. Aldred, C. Li, G.-F. Zhang, T. Chen, L. Wang and M.-Q. Zhu, J. Mater. Chem. C, 2014, 2, 7001–7012 CAS.
- L. He, K. Xu, P. P. Shi, Q. Ye and W. Zhang, Adv. Electron. Mater., 2022, 8, 2100635 CAS.
- I. Urbanaviciute, S. Bhattacharjee, M. Biler, J. A. Lugger, T. D. Cornelissen, P. Norman, M. Linares, R. P. Sijbesma and M. Kemerink, Phys. Chem. Chem. Phys., 2019, 21, 2069–2079 RSC.
- L. Ruan, D. Zhang, J. Tong, J. Kang, Y. Chang, L. Zhou, G. Qin and X. Zhang, Preparation and device applications of ferroelectric β-PVDF films, IntechOpen, 2018 Search PubMed.
- L. Lu, W. Ding, J. Liu and B. Yang, Nano Energy, 2020, 78, 105251 CrossRef CAS.
- J. W. Lee, H. J. Cho, J. Chun, K. N. Kim, S. Kim, C. W. Ahn, I. W. Kim, J.-Y. Kim, S.-W. Kim and C. Yang, Sci. Adv., 2017, 3, e1602902 CrossRef PubMed.
- S. K. Karan, R. Bera, S. Paria, A. K. Das, S. Maiti, A. Maitra and B. B. Khatua, Adv. Energy Mater., 2016, 6, 1601016 CrossRef.
- X. Yang and W. A. Daoud, Adv. Funct. Mater., 2016, 26, 8194–8201 CrossRef CAS.
- A. Waseem, I. V. Bagal, A. Abdullah, M. A. Kulkarni, H. Thaalbi, J. S. Ha, J. K. Lee and S. W. Ryu, Small, 2022, 18, 2200952 CrossRef CAS PubMed.
- G. Cheng, Z.-H. Lin, L. Lin, Z.-L. Du and Z. L. Wang, ACS Nano, 2013, 7, 7383–7391 CrossRef CAS PubMed.
- D. W. Jin, Y. J. Ko, C. W. Ahn, S. Hur, T. K. Lee, D. G. Jeong, M. Lee, C. Y. Kang and J. H. Jung, Small, 2021, 17, 2007289 CrossRef CAS PubMed.
- X. Li, W. Yu, X. Gao, H. Liu, N. Han and X. Zhang, Mater. Adv., 2021, 2, 6011–6019 RSC.
- J. Zhu, Y. Zhu and X. Wang, Adv. Mater. Interfaces, 2018, 5, 1700750 CrossRef.
- P. Sengupta, P. Sadhukhan, S. Saha, S. Das and R. Ray, Nano Energy, 2023, 109, 108277 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh01620c |
| ‡ S. Lim and D. Kim contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |