
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoordination bonds as a tool for tuning photoconductance in nanostructured hybrid materials made of molecular antennas and metal nanoparticles†
Nataliia
Marchenko
a,
Deborah
Martin
a,
Adeline
Pham
a,
Seifallah
Abid
b,
Eva
Cretal
a,
Alfonso
Ibarra
 c,
Delphine
Lagarde
a,
Marine
Tassé
d,
Jacques
Bonvoisin
b,
Gwénaël
Rapenne
be,
Jérémie
Grisolia
a,
Claire
Kammerer
*b and
Simon
Tricard
*a
c,
Delphine
Lagarde
a,
Marine
Tassé
d,
Jacques
Bonvoisin
b,
Gwénaël
Rapenne
be,
Jérémie
Grisolia
a,
Claire
Kammerer
*b and
Simon
Tricard
*a
aLaboratoire de Physique et Chimie des Nano-Objets, INSA, CNRS, Université de Toulouse, Toulouse, France. E-mail: tricard@insa-toulouse.fr
bCEMES, CNRS, Université de Toulouse, Toulouse, France. E-mail: kammerer@cemes.fr
cInstituto de Nanociencia de Aragón, Universidad de Zaragoza, Zaragoza, Spain
dLaboratoire de Chimie de Coordination, CNRS, Université de Toulouse, Toulouse, France
eDivision of Materials Science, Nara Institute of Science and Technology, Ikoma, Nara, Japan
First published on 10th February 2025
Abstract
The synthesis of robust, versatile materials in which electrical conduction is enhanced by light irradiation is of prime importance for fields as varied as photodetectors, photodiodes, solar cells and light sensors. Hybrid materials offer the advantage of combining the robustness of an inorganic building block with the adaptability of a molecular subunit. Herein, we demonstrate the importance of properly investigating the nature of the chemical interactions between the constituent elements in order to optimize photoconductance within hybrid materials. To this end, platinum nanoparticle self-assemblies are synthesized in solution, including a series of zinc-porphyrins differentially functionalized with pyridine moieties in the meso position. The presence of coordinating groups on the molecular entities drastically reinforced both the structural cohesion of the system and its photoconductive properties.
New conceptsEven though hybrid materials have been developed for decades, the attention that has been paid to the chemical interactions between the constituent elements was mostly focused either on weak forces or on covalent bonds, for which fine-tuning is limited. The new concept here is that coordination bonds between molecular entities and the surface of inorganic nanoparticles can be an efficient tool to tune both the structural morphology and the functional properties of the materials. In this study, we showed that the molecular response to light irradiation was synergistically reflected on the Coulomb blockade of the inorganic nanoparticles. However, the concept of structuring hybrid materials through coordination bonds goes far beyond the example of photoconductance, and can be effective in other fields of research linked to electronics, such as memory elaboration, nanoelectronics, or neuromorphic electronics. |
Photoconductance refers to the increase in electrical conductivity of a material when exposed to light. It is commonly utilized in various electronic devices and applications, such as photodetectors, photodiodes, solar cells, and light sensors. Among the large variety of photoconductive materials, hybrid materials show the advantage of combining the conductivity and robustness of their inorganic building blocks with the versatility and tunability of their molecular building blocks.1–3 For example, synergetic interactions between conjugated organic moieties and ZnO nanosheets led to very sensitive functional photoconductor devices,4 molecular junctions formed in gold nanoparticle arrays led to resonant photoconductance,5 or photoresponsive copper phthalocyanine derivatives coupled with PbS quantum dots led to light effect transistors.6 When their constituent components are suitably chosen, hybrid materials then offer phototransport properties that far exceed those of the independent building groups.7 More recently, photoresponsive materials have gained interest for application in neuromorphic analogy, i.e. for functional behaviors inspired by the operation of the nervous system. Some photoinduced switching properties indeed mimic artificial synaptic features. For instance, controlled polarity switching has been achieved via harnessing defects in doped PdSe2,8 or via inducing ferroelectric polarization in van der Waals heterostructures.9 Furthermore, optoelectronic synaptic devices elaborated from nanostructures have been used for designing functional setups.10 As recent examples, a silicon carbide nanowire photoelectric device was used for artificial vision systems,11 oxide heterojunctions for computing applications,12 or metal halide perovskites for optical memory.13 In this context, combining inorganic nanoparticles and photoresponsive molecules can thus lead to nanostructured hybrid materials where the molecules act as antennas to harvest light and enhance the electrical properties of the nanoparticle assembly. However, although ligand–nanoparticle interaction is attracting a great deal of interest in the colloidal phase,14 most of the time, little attention is paid to such a structural interaction at the solid state in nanoparticle self-assemblies for functional materials,15–17 and particularly how it influences photoconductive properties. So far, methodologies for tuning photoconductance in hybrid materials made of metal nanoparticles have focused on modifying the length, the charge or the conjugation of the ligands.5,18 Within the frame of this study, we decided to focus on the strength of coordination between ultra-small (<2 nm) nanoparticles and porphyrins functionalized by different numbers of pyridine moieties. We thus expected some exciton delocalization from the photosensitive molecules to the metal nanoparticles, as already observed between quantum dots and carbene molecules.19
Our group has developed hybrid materials including platinum ultra-small nanoparticles some years ago, paying close attention to the ligand properties and their influence on the electrical response of the self-assemblies. Using ultra-small nanoparticles has two advantages: (1) structural, as they are in the size range of molecules, thus giving molecules as much importance as nanoparticles in their relative structuring, and (2) functional in charge transport, as they show Coulomb blockade at room temperature. The latter can be tuned by the size of the nanoparticles, by the distance between them and, more importantly within the frame of interactions with molecules, by the polarizability of these molecules.20 For example, we showed that the electrical properties of the hybrid materials could be tuned by exchanging polyoxometalates of different electrical charges,21 or within a single material using spin crossover coordination polymer where the polarizability switched with temperature.22 Finally, we observed lamellar structuration using helical peptidic polymers, in hybrid materials where the dielectric relaxation could be tuned by the temperature or by a bias voltage.23,24 In the present study, we aim to include a dye molecule, where charge separation under light irradiation will enhance the material's conductive properties.
Porphyrins have been chosen as they are well-known photoactive compounds. On the one hand, these dyes have a planar core structure with a conjugated π-system that facilitates high absorption and extinction coefficients and show high-rate charge-carrier transport in covalent organic frameworks.25 On the other hand, they contain various peripheral functional groups that can interact with the nanoparticles. In particular, chemical modification of porphyrin meso-substituents can effectively tune the properties of hybrid systems, when the porphyrins are associated with nanoparticles. For instance, coordination of meso-tetrakis(1-methylpyridinium-4-yl)porphyrin chloride to ZnO nanoparticles greatly increased the photoluminescence (PL) of the porphyrin, whereas the replacement of the methylpyridinium substituents by trimethylanilinium led to a PL intensity drop due to energy transfer to the ZnO nanoparticles.26 In other studies, smart design of nanoparticle–porphyrin colloidal systems allowed enhancing the porphyrin optical properties, e.g. with gold nanoparticles for targeted photodynamic therapy,27 with quantum dots for reduction of CO2,28 or with platinum nanoparticles for hydrogen evolution.29 Porphyrins under light irradiation then transferred energy to the surface of the nanoparticles. Here, we want to develop a chemical tool to tune such an energy transfer and thus control photoconductance in hybrid materials.
Platinum nanoparticles were synthesized by Pt2(dba)3 decomposition under 1 bar of carbon monoxide (CO) in tetrahydrofuran (THF) at room temperature with further washing with pentane to remove residual ligands.20,30 After washing, the nanoparticles were dispersed in THF. This protocol allows obtaining “naked” ultra-small Pt nanoparticles (Pt NP), without any organics, stabilized by CO and THF, with a mean size of 1.4 nm according to the TEM results (Fig. 1a). As antennas, we selected three porphyrins functionalized at the meso position with an increasing number of pyridine moieties (Fig. 1b), which present a lone electron pair that can form a coordination bond with the Pt surface. Namely, we used zinc(II) 5,10,15,20-tetraphenylporphyrin (ZTP), which does not have specific coordination sites and served as reference, zinc(II) 5-pyridyl-10,15,20-tris (3,5-di-tert-butylphenyl)porphyrin (ZMPy), which displays one pyridine meso-substituent and zinc(II) 5,15-dipyridyl-10,20-di(3,5-di-tert-butylphenyl)porphyrine (ZBPy), which carries two pyridine fragments located on opposite meso positions. Metalation of the porphyrinic macrocycles with Zn and the presence of tert-butyl groups in ZMPy and ZBPy was necessary to facilitate solubility of the molecules at the concentrations required for self-assembly in THF. ZTP was ordered from a commercial supplier. The synthesis of the mono- and dipyridyl porphyrins was adapted from well-known procedures for the ring formation and metalation steps.31–33 A statistical approach was employed for the synthesis of the A3B-porphyrin ZMPy, starting from 3,5-di-tert-butylbenzaldehyde, 4-pyridinecarboxaldehyde and pyrrole. In contrast, the trans-A2B2-porphyrin ZBPy was prepared according to a selective route relying on 5-(4-pyridyl)dipyrromethane as a key precursor. Detailed synthesis protocols and characterization of ZMPy and ZBPy are given in the ESI.† The hybrid materials were prepared by self-assembly, by mixing THF solutions of Pt NP and of ZTP, ZMPy or ZBPy molecules, respectively, and stirring the resulting mixture for two hours. The relative amount of each solution was adjusted to achieve a ligand/Pt ratio of 0.05 equivalents (corresponding to ∼5 ligands per nanoparticles and to 20 equivalents of Pt atom per porphyrinic ligand), which was the best value for this system: with less ligands, free particles were observed in microscopy; with more ligands, free ligands led to insulating samples in charge transport. The obtained self-assemblies were called SA-ZTP, SA-ZMPy and SA-ZBPy, respectively.
 | ||
| Fig. 1 (a) TEM picture and size distribution of the pristine ultra-small (1.4 ± 0.3 nm) Pt NP; (b) chemical structure of the ZTP, ZMPy and ZBPy porphyrins. | ||
Depending on the porphyrin structure, the shape of the self-assembly differed significantly (Fig. 2). For ZTP without specific coordination sites, malformed agglomerates were observed, with typical aggregation sizes between 50 and 100 nm. Some weak van der Waals interactions and π-stacking were present between Pt NP and ZTP to form such aggregates. In contrast, in the presence of pyridine-substituted porphyrins, thus adding the possibility to create coordination bonds with the Pt surface, bigger and better-defined objects were obtained: a few μm-long rod-shaped assemblies with monopyridyl ZMPy, and 100–500 nm-wide spheres with dipyridyl ZBPy. Tomography reconstructions showed that the objects were homogenous, without any specific long-range ordering between the nanoparticles. However, small angle X-ray scattering (SAXS) proved the presence of local order within the assemblies, as broad peaks were observed in the q-range between 0.1 and 0.4 Å−1 (Fig. S1 in the ESI†). These measurements gave correlation distances of 2.2 nm for SA-ZTP, 2.9 nm for SA-ZMPy and 2.7 nm for SA-ZBPy, which correspond to an edge-to-edge distance of 0.8 nm for SA-ZTP, 1.5 nm for SA-ZMPy and 1.3 nm for SA-ZBPy. As the size of the functionalized porphyrins is around 1.6 nm from one edge of a phenyl/pyridine to another, we expect that the molecules separated the nanoparticles in an anisotropic way, with “face-on” bonding mode through metal–π interactions, in addition to the coordination between the pyridyl moieties and the Pt surface of other nanoparticles. In addition, Zn(II) can coordinate pyridines in axial positions,29 thus resulting in complex, structurally interlinked systems. However, the interparticle distance within the assemblies was significantly bigger in the presence of pyridine moieties than in the reference with ZTP. From a structural point of view, the presence of coordinating pyridines on the molecular porphyrins therefore affected the self-assembly of hybrid materials, both their shape at the mesoscale and their structuration at the nanoscale.
 | ||
| Fig. 2 TEM pictures of the self-assemblies of Pt NP with porphyrins. (a) and (b) SA-ZTP, (c) and (d) SA-ZMPy and (e) and (f) SA-ZBPy – (a), (c) and (e) low magnification images, and (b), (d) and (f) tomography 3D reconstruction. | ||
Fourier-transform infrared spectroscopy was performed to prove the coordination of the ligand to the surface of the nanoparticles (Fig. 3). Indeed, taking as a reference the band value for the “naked” CO-stabilized Pt NP (2041 cm−1), a shift in the vibration band of CO means that the electronic density on the surface changed because of ligand coordination.34 Whereas almost no shift was observed for SA-ZTP (2042 cm−1), a significant shift to higher wavenumbers was found for SA-ZMPy (2046 cm−1) and SA-ZBPy (2046 cm−1), corresponding to a depletion of the electronic density of the surface. Indeed, lower electronic density at the surface implies weaker back-donation from the NPs to the antibonding orbitals of the CO molecule, and thus the appearance of a vibration band at a higher wavenumber.21,22 The presence of the pyridine meso-substituent, by creating a coordination bond, thus allowed stronger electronic communication between the porphyrins and the nanoparticles in the materials.
 | ||
| Fig. 3 Infrared spectra of the pristine Pt NP and of the SA-ZTP, SA-ZMPy and SA-ZBPy self-assemblies. (a) Full spectra and (b) zoomed-in view of the terminal CO region (the baselines are shifted for clarity – the dashed lines are a guide to the eye); peak maxima: Pt NP: 2041 cm−1, SA-ZTP: 2042 cm−1, SA-ZMPy: 2046 cm−1, and SA-ZBPy: 2046 cm−1. | ||
To study further this electronic communication, we followed the porphyrin PL quenching by an increasing amount of nanoparticles, going from 5 to 160 equivalents (eq.), where eq. corresponds to the equivalent number of platinum atoms per porphyrin molecule. We kept the concentration of the porphyrin constant at 0.0245 mM and varied the Pt NP concentration from 0.125 to 3.95 mM. We observed neither a change in intensity nor a band shift in the porphyrin absorption spectra upon addition of Pt NP (Fig. S2 in the ESI†). Irradiation of the systems was performed at 425 nm, in the Soret band of the porphyrins. Two intense emission bands were found around 600 nm and 650 nm (Fig. 4a–c). We observed that increasing the nanoparticle quantity leads to a decay of the porphyrin fluorescence. There are three main mechanisms of fluorescence decay: static quenching, dynamic quenching, and the inner filter effect.35–37 In our study we ignored the latter since we worked at moderate concentrations and used a cuvette with a short path length. Static quenching occurs when in the ground state a quencher forms a non-fluorescent complex with a fluorophore. Dynamic quenching appears when a fluorophore in the excited state transfers part of the energy to an acceptor. The acceptor can further release this energy either non-radiatively or as a photon of lower energy. There, excited state energy transfer processes will shorten the lifetime of fluorescence. Time-resolved PL (TRPL) measurements showed that the decay time upon addition of increasing amounts of Pt NP remained constantly equal to 2.0 ns for the non-modified porphyrin ZTP, like in the free molecule in solution (Fig. 4d). There was thus no dynamic quenching in SA-ZTP. In contrast, for SA-ZMPy and SA-ZBPy, a new shorter decay time became significant starting from 80 eq. of Pt atoms, at 1.0 ns for ZMPy and 0.7 ns for ZBPy, in addition to the decay time of the free molecules at 2.2 ns (Fig. 4e and f). This observation therefore suggested that the presence of coordination from antennas to nanoparticles was accompanied by dynamic energy transfer at high Pt content, in addition to the static quenching inherent in all assemblies. A possible explanation can be as follows: when there was a small amount of Pt NP, or when there was no coordinating meso-substituent on the porphyrin, only two porphyrin systems existed in a solution: the free porphyrins (luminescent) and porphyrins interacting with the surface of nanoparticles (forming non-luminescent assemblies in a static quenching regime). However, when the amount of Pt NP was in large excess and when the porphyrin had the possibility to form coordination bonds, a third system appeared – porphyrins that were in close proximity to Pt NP but did not form bonds with them. Short distances between the luminophore and the quencher allow energy transfer to proceed (1–10 nm for Förster resonance energy transfer).38,39 Therefore, at high Pt concentration, dynamic quenching became more pronounced, in addition to static quenching, always present in all assemblies. The Stern–Volmer formalism is a helpful tool to quantify PL quenching.37 If only one type of quenching occurs in a system, the relation of the PL intensities of the luminophore without and with quencher (I0/I) depends linearly on the concentration of the quencher. The Stern–Volmer plots of the self-assemblies clearly showed that it was not the case, in good agreement with the coexistence of dynamic and static quenching as described above (Fig. S3a in ESI†). However, at a low Pt concentration, (<0.5 mM–20 eq.), as a rough simplification, we assumed the dynamic quenching to be negligible and thus the quenching mechanism to be static, with only two possible states for the porphyrin fluorophore F: either free in solution, or interacting with the nanoparticle quencher Q to form the non-luminescent assembly FQ (eqn (1)). There, we were able to estimate the static association constants Ka by linearly fitting the evolution of I0/I as a function of Q following eqn (2) (Fig. S3b in ESI†), and found the following results: SA-ZTP: Ka = 0.9 ± 0.2 mM−1; SA-ZMPy: Ka = 1.8 ± 0.1 mM−1; and SA-ZBPy: Ka = 1.9 ± 0.1 mM−1. Here too, we confirmed that the presence of coordinating pyridine moieties was significant, as they led to stronger association constants. In brief, the PL results indicated that the formation of a coordination bond between Pt NP and ZMPy or ZBPy resulted in stronger association, enhanced PL quenching, and more efficient energy transfer.
 | (1) |
 | (2) |
 | ||
| Fig. 4 (a)–(c) Photoluminescence (PL) and (d)–(f) time-resolved PL spectra of (a) and (d) ZTP, (b) and (e) ZMPy and (c) and (f) ZBPy in the presence of different amounts of Pt nanoparticles; eq. corresponds to the equivalent number of platinum atoms per porphyrin molecule. τ: decay time. λexc = 425 nm. | ||
To study the electrical behavior of the hybrid materials, a drop of the assembly suspension was evaporated on a gold surface and conductive AFM was performed on the deposited assemblies. A large number of I–V curves (∼50) were measured at different positions of individual objects to ensure statistical significance. They were normalized at 2 V and averaged to compare the characteristics of one sample to another (Fig. 5a). The nonlinear behavior in the I–V characteristics was the signature of the presence of Coulomb blockade. Such a blockade occurs at room temperature in our systems thanks to the use of ultra-small nanoparticles.20–22 The presence of porphyrins induced a higher non-linearity of the I–V curves than in Pt NP, with a more pronounced effect in SA-ZMPy and SA-ZBPy than in SA-ZTP. Coulomb blockade comes from the charging energy Ec of a nanoparticle, which behaves as a capacitor at the nanoscale when it is crossed by a conductive electron. The charging energy is defined as Ec = e2/(2πεrε0d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(s/(s − d)), where e is the charge of the electron, ε0 is the permittivity of vacuum, εr is the dielectric constant of the medium surrounding the particles, d is the particle diameter, and s is the center-to-center distance between two particles. The differences in the I–V characteristics of the self-assembly mostly come from the interparticle distance s, as d and εr are comparable (Table S1 in ESI†). To compare the characteristics, we referred to the phenomenological equation I ∝ Vξ, where ξ is a scaling parameter related to the current paths in the system. SA-ZMPy and SA-ZBPy showed ξ values of up to 4, comparable to the highest values already reported in the literature.40,41 When compared to previous hybrid materials from our group, always with platinum nanoparticle but associated either with alkylthiols, arylthiols or polyoxometalates,20,21 we obtained a record Coulomb blockade associated with the highest Ec and ξ with the pyridine functionalized porphyrins (Fig. S4 in ESI†). Thus, choosing large, rigid molecules with specific coordination groups can be an effective strategy for improving Coulomb blockade, as it drives large interparticle distances. Finally, photoconductance measurements were performed by focalizing a laser on the assemblies, at the interface between the AFM tip and the substrate. On–off photoconduction measurements with a wavelength close to that of the Q-band absorption of the porphyrins (at 532 nm, 10 mW) showed a current increase under irradiation by a factor of two for Pt NP and SA-ZTP, whereas this increase raised up to nine for SA-ZMPy and to eleven for SA-ZBPy (Fig. 5b). Even if the local heating effect or a polarization of the NPs cannot be excluded in all the systems, there was a clear improvement of photoconduction using the functionalized antennas. We expect this improvement to be due to the presence of the dynamic quenching described above, which triggered energy transfer from molecules to nanoparticles under light irradiation. In addition, a pure molecular effect could also occur in the framework of charge transport limited by Coulomb blockade, with an increase in molecular polarizability under irradiation, since in the excited state, the electronic density redistribution of the porphyrins populates more antibonding orbitals.42 Large current fluctuations over time are characteristic of electrical percolation mechanisms in granular materials made up of aggregated nanoparticles. These fluctuations were described as resulting either from electromigration of metal atom filaments,43 or from conformational changes in the ligands.44 Here, we cannot exclude that their origin came from the dynamic population of the molecular excited states under irradiation, illustrated by more pronounced variations in SA-ZMPy and SA-ZBPy where dynamic quenching was observed.
ln(s/(s − d)), where e is the charge of the electron, ε0 is the permittivity of vacuum, εr is the dielectric constant of the medium surrounding the particles, d is the particle diameter, and s is the center-to-center distance between two particles. The differences in the I–V characteristics of the self-assembly mostly come from the interparticle distance s, as d and εr are comparable (Table S1 in ESI†). To compare the characteristics, we referred to the phenomenological equation I ∝ Vξ, where ξ is a scaling parameter related to the current paths in the system. SA-ZMPy and SA-ZBPy showed ξ values of up to 4, comparable to the highest values already reported in the literature.40,41 When compared to previous hybrid materials from our group, always with platinum nanoparticle but associated either with alkylthiols, arylthiols or polyoxometalates,20,21 we obtained a record Coulomb blockade associated with the highest Ec and ξ with the pyridine functionalized porphyrins (Fig. S4 in ESI†). Thus, choosing large, rigid molecules with specific coordination groups can be an effective strategy for improving Coulomb blockade, as it drives large interparticle distances. Finally, photoconductance measurements were performed by focalizing a laser on the assemblies, at the interface between the AFM tip and the substrate. On–off photoconduction measurements with a wavelength close to that of the Q-band absorption of the porphyrins (at 532 nm, 10 mW) showed a current increase under irradiation by a factor of two for Pt NP and SA-ZTP, whereas this increase raised up to nine for SA-ZMPy and to eleven for SA-ZBPy (Fig. 5b). Even if the local heating effect or a polarization of the NPs cannot be excluded in all the systems, there was a clear improvement of photoconduction using the functionalized antennas. We expect this improvement to be due to the presence of the dynamic quenching described above, which triggered energy transfer from molecules to nanoparticles under light irradiation. In addition, a pure molecular effect could also occur in the framework of charge transport limited by Coulomb blockade, with an increase in molecular polarizability under irradiation, since in the excited state, the electronic density redistribution of the porphyrins populates more antibonding orbitals.42 Large current fluctuations over time are characteristic of electrical percolation mechanisms in granular materials made up of aggregated nanoparticles. These fluctuations were described as resulting either from electromigration of metal atom filaments,43 or from conformational changes in the ligands.44 Here, we cannot exclude that their origin came from the dynamic population of the molecular excited states under irradiation, illustrated by more pronounced variations in SA-ZMPy and SA-ZBPy where dynamic quenching was observed.
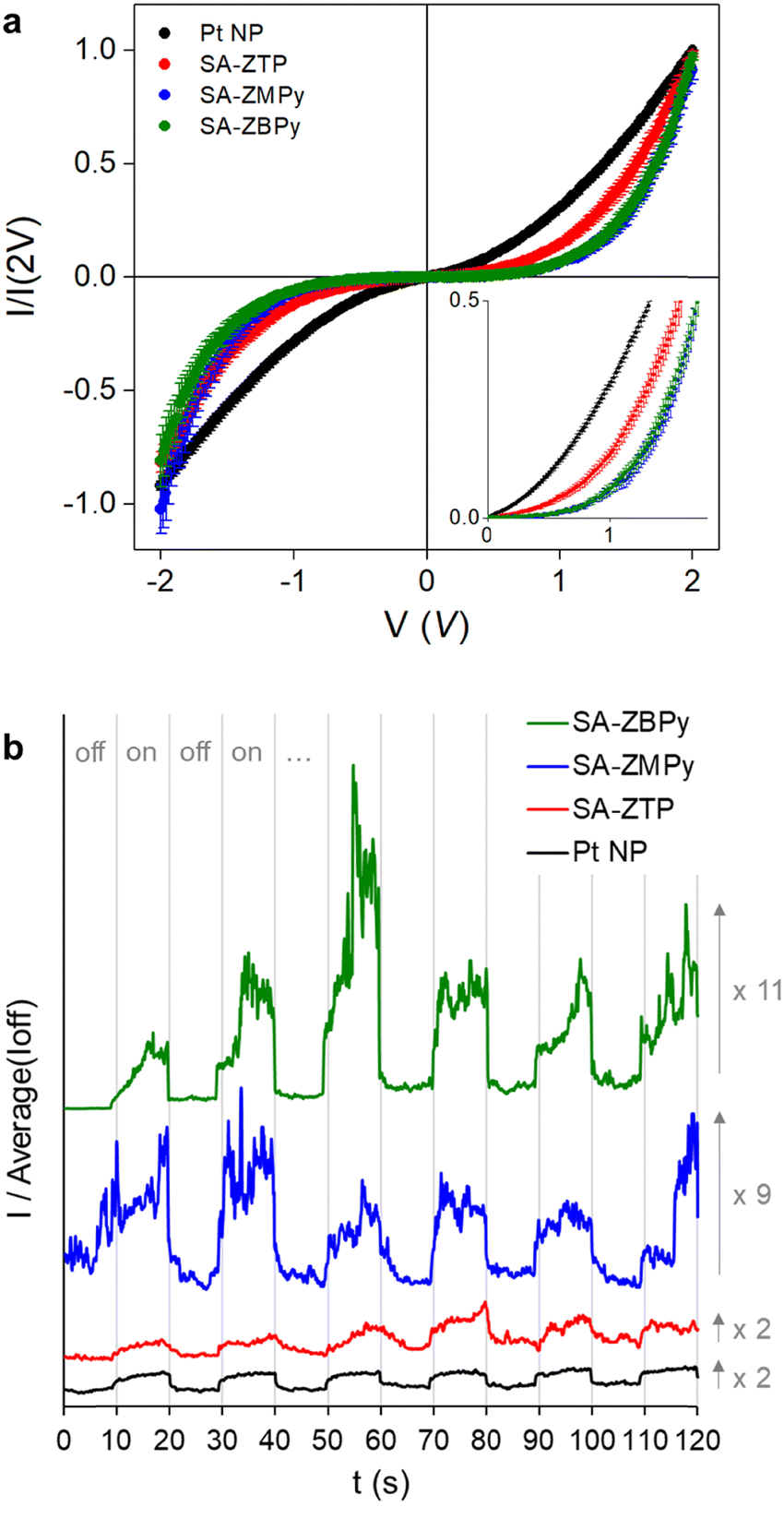 | ||
| Fig. 5 Charge transport measurements at the nanoscale, performed by conductive AFM on Pt NP, SA-ZTP, SA-ZMPy and SA-ZBPy, at room temperature. (a) I–V curves; the curves are normalized at 2 V; inset: magnification of the 0–1 V region. (b) Evolution of the current intensity under light irradiation at a voltage of 2 V; light is successively switched on and off. | ||
In summary, our approach lies at the interface between two fields that ultimately have little interaction: hybrid materials and coordination chemistry.3 We synthesized a new series of hybrid materials including ultra-small platinum nanoparticles and porphyrin derivatives. While unfunctionalized porphyrins interacted with the surface by weak interactions, the coordination bonds between the Pt surface and the pyridine moieties yielded self-assemblies of defined shapes and short-range ordering, as well as peculiar photophysical and electric properties. Particularly, energy transfer occurred in SA-ZMPy and SA-ZBPy systems and not in SA-ZTP. Coulomb blockade was more pronounced in the strongly interacting hybrid systems and conductivity enhancement appeared in SA-ZMPy and SA-ZBPy under light irradiation, whereas minor changes were observed with SA-ZTP. This work thus shows that the nature of the interactions between molecules and nanoparticles can be crucial to tune the photophysical properties of hybrid materials. Only a small amount of molecules, as low as 0.05 equivalents of ligand per Pt atom, was sufficient to significantly enhance the photoconductance properties of the hybrid materials, on condition that they were well-coordinated to the nanoparticles. In the future, coordination bond engineering should be seriously considered to tune the interactions between building blocks in hybrid materials in order to improve their photoresponse, a property which may be useful in fields as diverse as photoconductors, solar cells, photomemory devices, photocatalysts or neuromorphic electronics. For example, a challenging perspective to this work will be to control the light-induced spiking dynamics and to use it reliably for computations such as Boolean operations or image classification.45
Author contributions
SA, JB, GR and CK synthesized and characterized the functionalized porphyrins. NM, DB, AP, EC and ST worked on the Pt nanoparticle synthesis, self-assembly and characterization. AI performed the HR-STEM tomography imaging. DL performed the photoluminescence measurements. MT and JG performed the electrical measurements. CK and ST supervised the project. NM and ST wrote the manuscript. All authors discussed the results and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Sébastien Cher, Angélique Gillet, Francis Chouzenoux, Chloé Lhardy, Gaëlle Muraille and Donia Bouzouita for experimental support, and Thomas Blon for gold evaporation on substrates. Financial support from Agence Nationale de la Recherche (MOSC grant ANR-18-CE09-0007, DINAPO grant ANR-23-CE09-0006-02, and MOMA grant ANR-23-ERCC-0008-01) is acknowledged. This study has been partially supported through the EUR grant NanoX n° ANR-17-EURE-0009 in the framework of the Programme des Investissements d’Avenir. This work was also supported by the University Paul Sabatier (Toulouse) and CNRS. Technical assistance provided by the Institut de Chimie de Toulouse ICT-UAR 2599 (Université de Toulouse, CNRS, Toulouse, France) and by the Centre de Microcaractérisation Raimond Castaing (UAR 3623) is gratefully acknowledged. We acknowledge the use of instrumentation as well as the technical advice provided by the National Facility ELECMI ICTS node “ Laboratorio de Microscopías Avanzadas” at the University of Zaragoza.References
- S. H. Mir, L. A. Nagahara, T. Thundat, P. Mokarian-Tabari, H. Furukawa and A. Khosla, J. Electrochem. Soc., 2018, 165, B3137–B3156 CrossRef CAS.
- M. Arya, S. Heera, P. Meenu and K. G. Deepa, Chem. Phys. Mater., 2024, 3, 252–272 Search PubMed.
- P. Gomez-Romero, A. Pokhriyal, D. Rueda-García, L. N. Bengoa and R. M. González-Gil, Chem. Mater., 2024, 36, 8–27 CrossRef CAS.
- M. Sofos, J. Goldberger, D. A. Stone, J. E. Allen, Q. Ma, D. J. Herman, W.-W. Tsai, L. J. Lauhon and S. I. Stupp, Nat. Mater., 2009, 8, 68–75 CrossRef CAS.
- M. A. Mangold, M. Calame, M. Mayor and A. W. Holleitner, J. Am. Chem. Soc., 2011, 133, 12185–12191 CrossRef CAS.
- A. André, C. Theurer, J. Lauth, S. Maiti, M. Hodas, M. S. Khoshkhoo, S. Kinge, A. J. Meixner, F. Schreiber, L. D. A. Siebbeles, K. Braun and M. Scheele, Chem. Commun., 2017, 53, 1700–1703 RSC.
- J.-S. Lee, M. V. Kovalenko, J. Huang, D. S. Chung and D. V. Talapin, Nat. Nanotechnol., 2011, 6, 348–352 CrossRef CAS.
- J. Jiang, W. Xu, Z. Sun, L. Fu, S. Zhang, B. Qin, T. Fan, G. Li, S. Chen, S. Yang, W. Ge, B. Shen and N. Tang, Small, 2024, 20, 2306068 CrossRef CAS.
- A. Cao, S. Li, H. Chen, M. Deng, X. Xu, L. Shang, Y. Li, A. Cui and Z. Hu, Mater. Horiz., 2023, 10, 5099–5109 RSC.
- X. Chen, B. Chen, B. Jiang, T. Gao, G. Shang, S.-T. Han, C.-C. Kuo, V. A. L. Roy and Y. Zhou, Adv. Funct. Mater., 2023, 33, 2208807 CrossRef CAS.
- Z. Feng, S. Yuan, J. Zou, Z. Wu, X. Li, W. Guo, S. Tan, H. Wang, Y. Hao, H. Ruan, Z. Lin, Z. Xu, Y. Zhu, G. Wei and Y. Dai, Nanoscale Horiz., 2024, 9, 1813–1822 RSC.
- T. Zhang, C. Fan, L. Hu, F. Zhuge, X. Pan and Z. Ye, ACS Nano, 2024, 18, 16236–16247 CrossRef CAS PubMed.
- G. Vats, B. Hodges, A. J. Ferguson, L. M. Wheeler and J. L. Blackburn, Adv. Mater., 2023, 35, 2205459 CrossRef CAS.
- J.-M. Nam, J. S. Owen and D. V. Talapin, Acc. Chem. Res., 2023, 56, 2265–2266 CrossRef CAS.
- J. Liao, S. Blok, S. J. van der Molen, S. Diefenbach, A. W. Holleitner, C. Schönenberger, A. Vladyka and M. Calame, Chem. Soc. Rev., 2015, 44, 999–1014 RSC.
- T. Kister, J. H. M. Maurer, L. González-García and T. Kraus, ACS Appl. Mater. Interfaces, 2018, 10, 6079–6083 CrossRef CAS.
- Y. Min, H. Nasrallah, D. Poinsot, P. Lecante, Y. Tison, H. Martinez, P. Roblin, A. Falqui, R. Poteau, I. del Rosal, I. C. Gerber, J.-C. Hierso, M. R. Axet and P. Serp, Chem. Mater., 2020, 32, 2365–2378 CrossRef CAS.
- H. Nakanishi, K. J. M. Bishop, B. Kowalczyk, A. Nitzan, E. A. Weiss, K. V. Tretiakov, M. M. Apodaca, R. Klajn, J. F. Stoddart and B. A. Grzybowski, Nature, 2009, 460, 371–375 CrossRef CAS.
- D. E. Westmoreland, R. López-Arteaga and E. A. Weiss, J. Am. Chem. Soc., 2020, 142, 2690–2696 CrossRef CAS.
- S. Tricard, O. Said-Aizpuru, D. Bouzouita, S. Usmani, A. Gillet, M. Tassé, R. Poteau, G. Viau, P. Demont, J. Carrey and B. Chaudret, Mater. Horiz., 2017, 4, 487–492 RSC.
- A. Gillet, S. Cher, M. Tassé, T. Blon, S. Alves, G. Izzet, B. Chaudret, A. Proust, P. Demont, F. Volatron and S. Tricard, Nanoscale Horiz., 2021, 6, 271–276 RSC.
- S. Usmani, M. Mikolasek, A. Gillet, J. Sanchez Costa, M. Rigoulet, B. Chaudret, A. Bousseksou, B. Lassalle-Kaiser, P. Demont, G. Molnár, L. Salmon, J. Carrey and S. Tricard, Nanoscale, 2020, 12, 8180–8187 RSC.
- G. Manai, H. Houimel, M. Rigoulet, A. Gillet, P.-F. Fazzini, A. Ibarra, S. Balor, P. Roblin, J. Esvan, Y. Coppel, B. Chaudret, C. Bonduelle and S. Tricard, Nat. Commun., 2020, 11, 2051 CrossRef CAS PubMed.
- L. Merle, G. Manai, A. Pham, S. Lecommandoux, P. Demont, C. Bonduelle, S. Tricard, A. Mlayah and J. Grisolia, J. Phys. Chem. C, 2021, 125, 22643–22649 CrossRef CAS.
- X. Feng, L. Liu, Y. Honsho, A. Saeki, S. Seki, S. Irle, Y. Dong, A. Nagai and D. Jiang, Angew. Chem., Int. Ed., 2012, 51, 2618–2622 CrossRef CAS.
- S. M. B. Aly, M. Eita, J. I. Khan, E. Alarousu and O. F. Mohammed, J. Phys. Chem. C, 2014, 118, 12154–12161 CrossRef CAS.
- O. Penon, M. J. Marín, D. A. Russell and L. Pérez-García, J. Colloid Interface Sci., 2017, 496, 100–110 CrossRef CAS.
- S. Lian, M. S. Kodaimati and E. A. Weiss, ACS Nano, 2018, 12, 568–575 CrossRef CAS.
- Y. Liu, L. Wang, H. Feng, X. Ren, J. Ji, F. Bai and H. Fan, Nano Lett., 2019, 19, 2614–2619 CrossRef CAS.
- S. Gomez, L. Erades, K. Philippot, B. Chaudret, V. Collière, O. Balmes and J.-O. Bovin, Chem. Commun., 2001, 1474–1475 RSC.
- K. Kurotobi and A. Osuka, Org. Lett., 2005, 7, 1055–1058 CrossRef CAS.
- H. Wu, F. Yang, X.-L. Lv, B. Wang, Y.-Z. Zhang, M.-J. Zhao and J.-R. Li, J. Mater. Chem. A, 2017, 5, 14525–14529 RSC.
- S. Abid, Y. Gisbert, M. Kojima, N. Saffon-Merceron, J. Cuny, C. Kammerer and G. Rapenne, Chem. Sci., 2021, 12, 4709–4721 RSC.
- C. Dablemont, P. Lang, C. Mangeney, J.-Y. Piquemal, V. Petkov, F. Herbst and G. Viau, Langmuir, 2008, 24, 5832–5841 CrossRef CAS.
- M. Asha Jhonsi, A. Kathiravan and R. Renganathan, J. Lumin., 2009, 129, 854–860 CrossRef CAS.
- G. Muraille, S. Tricard, E. A. Baquero, B. Chekroun, D. Lagarde, X. Marie, B. Chaudret, C. Nayral and F. Delpech, J. Lumin., 2020, 217, 116778 CrossRef CAS.
- D. Genovese, M. Cingolani, E. Rampazzo, L. Prodi and N. Zaccheroni, Chem. Soc. Rev., 2021, 50, 8414–8427 RSC.
- H. Sahoo, J. Photochem. Photobiol., C, 2011, 12, 20–30 CrossRef CAS.
- M. Taniguchi, J. S. Lindsey, D. F. Bocian and D. Holten, J. Photochem. Photobiol., C, 2021, 46, 100401 CrossRef CAS.
- R. P. Tan, J. Carrey, C. Desvaux, L.-M. Lacroix, P. Renaud, B. Chaudret and M. Respaud, Phys. Rev. B, 2009, 79, 174428 CrossRef.
- P. Yang, I. Arfaoui, T. Cren, N. Goubet and M.-P. Pileni, Phys. Rev. B, 2012, 86, 075409 CrossRef.
- J. Lin and D. Shi, Appl. Phys. Rev., 2021, 8, 011302 CAS.
- M. D. Pike, S. K. Bose, J. B. Mallinson, S. K. Acharya, S. Shirai, E. Galli, S. J. Weddell, P. J. Bones, M. D. Arnold and S. A. Brown, Nano Lett., 2020, 20, 3935–3942 CrossRef CAS PubMed.
- L. V. Govor, G. H. Bauer, T. Lüdtke, R. J. Haug and J. Parisi, Phys. Lett. A, 2011, 375, 4041–4044 CrossRef CAS.
- S. J. Studholme, Z. E. Heywood, J. B. Mallinson, J. K. Steel, P. J. Bones, M. D. Arnold and S. A. Brown, Nano Lett., 2023, 23, 10594–10599 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh01327a |
| This journal is © The Royal Society of Chemistry 2025 |