
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToward understanding biomolecular materials comprising intrinsically disordered proteins via simulation and experiment
Bin
Wang†
 a,
Tianren
Zhang†
ab,
Sirui
Shen
a,
Darrin J.
Pochan
a,
Jeffery G.
Saven
*b and
Kristi L.
Kiick
*ac
a,
Tianren
Zhang†
ab,
Sirui
Shen
a,
Darrin J.
Pochan
a,
Jeffery G.
Saven
*b and
Kristi L.
Kiick
*ac
aDepartment of Materials Science and Engineering, University of Delaware, Newark, DE, USA. E-mail: kiick@udel.edu
bDepartment of Chemistry, University of Pennsylvania, Philadelphia, PA, USA. E-mail: saven@sas.upenn.edu
cDepartment of Biomedical Engineering, University of Delaware, Newark, DE, USA
First published on 25th April 2025
Abstract
Intrinsically disordered proteins (IDPs) yield solutions with tunable phase transition behavior and have been widely applied in designing stimuli-responsive materials. Understanding interactions between amino acid residues of the IDP sequence is critical to designing new IDP-based materials with selective phase behavior, assembly, and mechanical properties. The lack of defined structure for this class of proteins complicates accurate prediction of their molecular-scale behavior. In this review, recent progress is presented in the development and application of simulation methods to describe the behavior of IDPs. Results for elastin-like polypeptides (ELPs) and resilin-like polypeptides (RLPs) are highlighted, focusing on studies that compare simulation results with experimental findings.
Design, System, ApplicationIntrinsically disordered proteins (IDPs) yield solutions with tunable phase transition behavior and have been widely applied in designing stimuli-responsive materials. Understanding interactions between amino acid residues of the IDP sequence is critical to designing new IDP-based materials with selective phase behavior, assembly, and mechanical properties. The lack of defined structure for this class of proteins complicates accurate prediction of their molecular-scale behavior. In this review, two IDPs, elastin-like polypeptides (ELPs) and resilin-like polypeptides (RLPs), and recent development of simulation methods for understanding and predicting important properties of these IDPs such as conformational features, polymer properties, and phase transition behavior are highlighted. The comparisons between simulation and experimental results are included to reveal the advances and current limitations in IDP simulations. Several examples of successful simulation of peptide systems integrating IDPs and ordered domains demonstrate the potential to design advanced biomaterials with desired and versatile properties through the assistance of simulation. |
Introduction
Protein structure is closely related to solution behavior and function.1–3 As a result, structure has served over the past few decades as the primary basis for the design and realization of de novo protein-based materials with targeted assembly, responsiveness, and stability. More recent and rapidly developing material characterization methods and genomic tools have successfully provided approaches to create and characterize protein compositions and structures. For example, the development of mass spectrometry and amino acid analysis allows the precise measurement of protein molecular weight and has been widely used to characterize the sequence and post-translational modification of proteins;4,5 circular dichroism and IR spectrometry provide insights into protein secondary structure;6,7 and (cryogenic) electron microscopy has been successfully applied in the reconstruction of protein three-dimensional (3D) structure and the direct observation of folded proteins.8,9 In addition to these developments in characterization methods, gene and protein databases, accompanied by bioinformatic tools, have also facilitated the identification of specific proteins from a wide range of organisms for further elaboration in the development of new biological materials based on structured proteins.10On the other hand, intrinsically disordered proteins (IDPs) are characterized by the presence of intrinsically disordered regions that lack well-defined three-dimensional (3D) structures.11 The disordered nature of IDPs has been shown to be important in biological processes including transcription, translation, signal transduction, and cell-cycle control.12 The abilities of IDPs to undergo tunable, stimuli-responsive assembly as well as promote liquid–liquid phase separation (LLPS) have been successfully applied in designing stimuli-responsive materials including drug delivery carriers, hydrogels, and nanoparticles.13 IDPs can also be versatile building blocks for new materials and have been exploited to guide supramolecular self-assembly and materials properties via the architecture and sequence of fused IDPs.14
Elastin-like peptides (ELPs) are derived from the hydrophobic domain of tropoelastin, a key extracellular matrix protein responsible for the elasticity of many tissues. ELPs are one of the most widely explored and earliest-studied IDPs. ELPs exhibit unique thermoresponsive behavior, making them highly versatile in various biotechnological applications, particularly in drug delivery15,16 and tissue engineering.16,17 Derived from the repetitive amino acid sequences found in elastin (e.g., [VPGXG]n, X is guest residue which can be any amino acid except proline, n is the number of repeat units), ELPs undergo reversible phase transitions in response to changes in temperature, typically characterized by the lower critical solution temperature (LCST).18 This tunable thermoresponsiveness has supported the development of dynamic systems for targeted delivery and controlled assembly. Recent advances in the design and functionalization of ELPs have focused on tailoring their molecular architectures, such as varying chain length,19,20 incorporating guest residues,21,22 and conjugating them with other functional domains.23–25 These modifications aim to enhance ELP properties, including stability and responsiveness.26 By understanding molecular mechanisms governing ELP behavior, researchers can better design ELP-based systems with tunable properties for a broad range of applications.
Resilin is another IDP that serves energy storage functions in insects27 and has been used both as a model for understanding IDP behavior and as a candidate for biomaterials, drug delivery vehicles, and actuators.28–30 Crosslinked resilin forms a thermal stable rubber-like material with low stiffness, high extensibility, high resilience, and long fatigue times. The protein is prevalent in the mechanically active exoskeleton of insects where it serves as a high-frequency responsive elastomer.28,31 Inspired by resilin, resilin-like polypeptides (RLPs) have been designed and recombinantly synthesized in E. coli, based on the putative pro-resilin sequence from the first exon of the Drosophila melanogaster CG 15920 gene.32 More recently, resilins from various insects, including mosquitos, fleas, and buffalo flies, have been produced recombinantly and used in hydrogels, stimuli-responsive materials, and as a bioactive component in hybrid materials.28,30,33,34 By exploiting protein engineering techniques, bioactive and structural domains have been successfully fused with RLPs. RLP-based polypeptides with tunable mechanical properties, versatile assembly into nano- and micro-structures, and high biocompatibility have been produced for biomedical and materials applications.35–39
Various IDP-based materials, especially ELP/RLP-based materials, have been designed and applied as smart biomaterial platforms, leveraging their reversible phase transition behavior, tunable phase transition temperatures, and high biocompatibility.13 Opportunities remain in fine-tuning their physicochemical properties and on-demand phase separation, which require a better understanding of how sequence can be used to control these properties. Compared to other polymeric materials, however, the large numbers of potential sequences involving a wide range of amino acids provide not only diversity in tuning properties but also difficulty in predicting resulting behavior.40 In addition, the variety of short- and long-range interactions possible between amino acid residues (e.g., hydrophobic interactions, electrostatic interactions, π–π interactions, among others), coupled with the absence of well-defined structures in IDPs, hinder the precise characterization of residue interactions within IDP-based assemblies.8,41,42
To understand their association behavior and address key interactions in IDPs, a variety of computational methods have been developed to aid in the understanding of the complex assembly behavior of these polypeptide-based materials.43–45 Molecular dynamics (MD) simulations and related computational methods have been extensively utilized as a complement to experimental studies to better understand LLPS, self-assembly, and intermolecular interactions in IDP-based materials.46–49 Depending on the length and time scales of the modeled systems, both all-atom simulations and coarse-grained (CG) models have been widely employed. All-atom simulations leverage force fields, such as CHARMM,50,51 AMBER,52–54 or OPLS,22,55 and advanced sampling techniques, such as replica exchange molecular dynamics (REMD),56,57 metadynamics, or tempering-based methods,58 to overcome the limitations of standard MD in exploring rugged energy landscapes. Notably, several studies have combined advanced sampling techniques to improve the exploration of IDP conformational landscapes. For instance, hybrid methods such as replica exchange molecular dynamics integrated with metadynamics (REMD-MetaD) have been applied to overcome free energy barriers more effectively to enhance the sampling of free energy landscapes in peptide folding studies.59 Similarly, bias-exchange metadynamics, where multiple replicas, each biased along a different collective variable, periodically exchange configurations, has been successfully applied to capture both global and local conformational dynamics in protein folding.60 Those simulations and various sampling techniques provide molecular-level insights into structure, energetics, interactions, dynamics, and conformational fluctuations, and these techniques have been insightful in studying IDP-based materials, enabling high-resolution analysis of molecular features and the complex dynamics of these systems.47,53,54,61–67
While all-atom simulation provides a powerful way of understanding IDP and related materials, the study of LLPS and complex large-scale aggregates has been limited by the significant computational demands of all-atom simulation. To overcome these limitations, CG models, with their reduced numbers of degrees of freedom, have been developed for simulating systems at larger length and time scales but with lower molecular resolution. Various CG models,68–70 ranging from simplified polymer models to the carefully parameterized Martini CG models, have been successfully applied to biomolecular systems.57,71–77 Polymer models, such as bead-spring models, lattice models and DPD methods, are particularly useful for capturing the overall characteristics of biopolymers, such as chain conformation and assembly of nano- and micro-scale structures, while reducing computational cost and focusing on key interactions.55,78–89 However, the reduction in computational cost provided by these models comes at the expense of detailed molecular accuracy. For IDPs, assembly and LLPS is driven by complex noncovalent and often weak interactions between multiple amino acids, which are often too intricate for simple polymer models to accurately capture.53,90–95 To address this limitation, hydrophobicity scale (HPS) models that parameterize amino acid interactions without explicit solvent have been developed.57,96–99 These models allow for a more refined representation of the specific interactions that govern assembly and phase separation associated with IDPs. While HPS models reduce computational expense relative to all-atom simulations, they do so at the cost of lower molecular detail, potentially overlooking solvent-mediated effects and finer interaction nuances critical to IDP behavior. In addition, the Martini CG model, which employs a coarse-grained explicit solvent, has been modified to study IDP-based materials.100–103 These approaches allow for modeling of the specific interactions that govern assembly and phase separation associated with IDPs. Nonetheless, both Martini models and HPS models enhance computational feasibility and efficiency, although they inherently introduce approximations that can affect the accuracy of the simulated thermodynamics and kinetics, and thus require rigorous calibration and validation.
Furthermore, hybrid simulations combining all-atom and CG models have emerged as an effective approach to studying IDP-based materials, taking advantage of the detailed accuracy of all-atom representation and the computational efficiency of CG models. For instance, the formation and dynamics of the LLPS of IDPs has been studied at the CG level, while key interaction motifs or regions responsible for the phase-separation process can be modeled at the all-atom level for detailed structural insights.53,54,104 However, hybrid simulations present their own challenges, such as maintaining a consistent interface between all-atom and CG representations, addressing parameterization differences, and striking the right balance between computational efficiency and the required level of detail. The integration of these approaches requires careful optimization to ensure that the benefits of broader sampling and reduced computational expense do not compromise the structural and dynamical accuracy of key interaction motifs.
Although IDP sequences have been studied experimentally for decades in a range of different materials, simulation models for IDPs have only been more recently developed for characterizing their association, conformational properties, and interactions. Below, some of the recent progress in the simulation of specific IDPs and their conjugates with ordered assembly-promoting sequences (e.g., collagen-like peptides and coiled-coil peptides) is presented. The emphasis is on how these methods can aid both in interpreting experimental observations from a molecular perspective and in understanding features of IDP-based materials, including transition temperatures, physical properties, residue interactions, and assembled structures.
Simulations of elastin-like (poly)peptides (ELPs)
Since the thermal responsiveness behavior of ELPs was discovered, multiple factors including chain length and guest residues (X in [VPGXG]n motif) have been widely tuned to design ELPs with versatile properties.105,106 McDaniel et al. first developed an experimental model to predict ELP transition temperature based on their chain length and composition; however, this experimental model only applied to ELPs with Ala and Val guest residues.107 MD simulation with an all-atom CHARMM force field for ELP were conducted by Rousseau et al.,108 and although these approaches captured the conformational transition of the octapeptide GVG[VPGVG], the models were unable to simulate phase separation or to be directly compared with experimental studies. Since these early studies, molecular modeling and MD simulations, ranging from CG to all-atom simulations, have been developed and applied to understand transition temperatures and liquid–liquid phase separation (LLPS) associated with ELPs and other intrinsically disordered proteins (IDPs).109,110More recent simulations of ELPs have focused on both simulating the molecular properties near the phase transition and guest–residue interactions. Hydrophobic interactions have been regarded as the main driving force of the phase transition, and researchers have shown the difference between multi-chain and mono-chain simulations.111,112 In substantial studies by Barrat et al., the investigation focused on molecular properties near the lower critical solution temperature (LCST) of ELPs using all-atom simulations of the sequence GVG[VPGVG]3;113 here the primary objective was to understand how intra- and inter-peptide interactions evolve below, near, and above the LCST. The role of peptide concentration on the structural and dynamic properties of aqueous solutions of short ELPs was explored over a temperature range near the LCST. Utilizing the AMBER99SB-ILDN114 force field for peptides and the TIP4P-D water model,115 ELP behavior was simulated in single-chain, two-peptide, and multichain systems. Because of the limited length of the peptide sequence, only a slight reduction in polymer size at the LCST was observed at the single-chain level; nevertheless, Markov state modeling116 revealed clear two-state behavior. In the two-peptide system, consistent with LCST behavior, the potential mean force (PMF) between the chains shifted with temperature. In multichain systems, the PMF was employed to characterize the interactions; the peptides experience a weak attraction between the chains at high temperature, as shown in Fig. 1a. By analyzing the potential of the mean force of the chains, it was shown that the peptides adopted more extended conformations compared to single-chain systems, where the peptides behaved more like ideal chains. This extension in the multichain systems was attributed to the presence of central valine residues that drive inter-chain contact formation. The study suggested that substituting these valine residues with more hydrophilic or weakly interacting amino acids could potentially suppress contact formation, which provided more detailed insight into residue interaction in ELP and associated LLPS and similar interactions have been observed by the Shaw group and the Pomes group.112,114 Overall, the simulations provided valuable data for ELP design and deeper insights into the mechanisms governing ELP behavior near the LCST.
 | ||
| Fig. 1 a) Potential of mean force (PMF) w(Δr) as a function of distance Δr for a pair of ELP chains as a function of temperature. The positive w(Δr) indicates more probable distances peptides and the negative w(Δr) indicates less probable distances between peptides. Temporal evolution of b) nonbonded interaction energy within the 27-polypeptide cluster, c) interaction energy between 27 polypeptides and water, d) total number of interchain peptide–peptide H-bonds in the system, and e) average number of intrachain H-bonds per peptide chain. a) Reprinted with permission.113 Copyright 2023, American Chemical Society. b–e) Reprinted with permission.117 Copyright 2021, American Chemical Society. | ||
Longer ELP sequences have also been successfully simulated by Yingling et al. in all-atom simulations to investigate the structural and aggregation behavior of ELP coacervates.111,117–119 The sequences [VGPVG]18 and [VPGVG]18 were simulated in explicit water at 350 K to capture hysteresis effects caused by differences in amino acid patterning.117 As shown in Fig. 1b and c, in the initial stages, peptide–peptide interactions intensify while peptide–water interactions diminish, leading to conformational changes, followed by aggregation of the peptides without formation of a defined hydrophobic core. While hydrogen bonds were indicated as the primary driving force in these interactions (Fig. 1d and e), factors such as peptide surface hydrophobicity, torsion angles, and secondary structural motif propensities were also shown to contribute to both the LCST and the observed hysteresis.117,119 By comparing the thermal behavior of [VGPVG]18 and [VPGVG]18, deeper insights into LCST phenomena were obtained. The consistency of the simulation results with experimental data further validated the simulation findings, bolstering confidence in the ability to understand LCST behavior based on peptide sequence and structure.
All-atom simulations were also utilized in other reports to study the relationship between LCST and the degree of polymerization in ELPs, spanning from three to over 30 repeat units in single-chain systems, while CG simulations examined chains with up to 150 repeat units.64–66,120 These all-atom and CG simulations both demonstrated that the relationship between the inverse transition temperatures of ELPs and their lengths follows power law exponents, aligning well with experimental findings. Moreover, the all-atom simulation study suggested that the transition behavior of [VPGVG]n, can be explained by a combination of the thermal disruption of the water network surrounding the polypeptide, the peptide's solvent-accessible surface area, and the peptide's hydrophobicity.64,65 Together these studies provide valuable insights into the coacervation of ELPs, particularly highlighting the influence of ELP length, which can be readily controlled in fine-tuning the behavior of ELP-based systems.
Although all atom-level MD simulations have provided molecularly detailed representations of ELP properties, these simulations become impractical with increasing ELP chain length and numbers of chains. As a result, CG models have been introduced to simulate ELPs. Muraoka et al. explored the formation of coacervates and the internal structural and dynamic properties of ELPs using CG simulations with a Martini3 CG model.121 ELPs were modeled with a variable number n of [VPGVG]n repeat units, employing an explicit, CG solvent model.122 From simulations of the self-assembly of two distinct ELPs having molecular weights of 2.1 kDa and 8.6 kDa, a 6-fold difference in the inter-chain contact surface area was observed using differences in solvent-accessible surface area,123 as shown in Fig. 2. These findings indicate that increasing both the concentration and chain length of ELPs enhances coacervate formation. Complementing the simulations, experiments conducted at higher ELP concentrations corroborated the simulation results, confirming the observed trends in concentration and length effects. While CG models offer significant computational advantages, they also introduce approximations that can compromise structural accuracy and the detailed representation of molecular interactions. For instance, the Martini CG model does not capture secondary structures well and thus requires initial definition of the secondary structures, which could introduce artifacts in the simulations. On the molecular level, due to its CG strategies, hydrogen bonds were not included.46,70 This trade-off is critical when studying systems where these fine-grain interactions can play a pivotal role. Thus, although CG simulations provide valuable insights into the overall assembly behavior of ELPs, care must be taken in interpreting results, particularly when extrapolating to molecular-level details.
 | ||
| Fig. 2 a) Mapping of the poly[VPGVG] chemical structure to the coarse-grained model in b), where particle types used for the Martini 3.0 mapping are indicated. Different colors correspond to different particle types. Contact surface area of (c) 2.1 kDa and (d) 8.6 kDa models. The contact surface area of 8.6 kDa sample is 5–6 times larger than the 2.1 kDa sample. The low contact surface area in (c) quantifies association and indicates the absence of an enhanced local polymer density. The increasing contact surface area in (d) reveals the formation of densely associated polymers, consistent with formation of the coacervate phase. Reprinted with permission.121 Copyright 2021, American Chemical Society. | ||
Simulations of resilin-like (poly)peptide interactions
The interactions of RLPs have been more complex than those of ELPs due to the myriad types of interactions present, including those involving charged and aromatic residues. The upper critical solution temperature (UCST) behavior of RLPs has been largely attributed to π–π and cation–π interactions.28,90,124,125 By combining simulations and experiments, Rekhi et al. further elucidated the roles of specific residues in the LLPS and resulting physical properties (e.g., viscosity) of condensates formed by the artificial IDPs (A-IDPs), such as [GRGDSPYS]25 and its variants that exhibit (UCST) phase behavior.53 In the simulations, the systems were initially constructed using a hydrophobicity-based (physics-based) CG model (HPS) within simulation boxes of slab geometry. These systems were then mapped onto all-atom structures, as shown in Fig. 3a, with the simulations conducted over microsecond-long timescales as in previous computational studies.54,57 For the wild-type (WT) A-IDP, [GRGDSPYS]25, in addition to the commonly observed interactions (e.g., between Tyr and Arg, Tyr with itself, and electrostatic interactions between Arg and Asp), interactions between Ser and Gly residues were also identified. The detailed average residue pairwise contacts, estimated from atomistic simulations of the WT condensed phase, are illustrated in Fig. 3a. By substituting Arg with Gln and Asp with Asn [G![[Q with combining low line]](https://www.rsc.org/images/entities/char_0051_0332.gif) G
G![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) SPYS] to eliminate the zwitterionic pair, most amino acid interactions remained similar to those in the WT sequence. Although the overall number of contacts was reduced, this similarity to the WT emphasized the crucial role of interactions among uncharged residues in driving LLPS. Moreover, when comparing Arg [G
SPYS] to eliminate the zwitterionic pair, most amino acid interactions remained similar to those in the WT sequence. Although the overall number of contacts was reduced, this similarity to the WT emphasized the crucial role of interactions among uncharged residues in driving LLPS. Moreover, when comparing Arg [G![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif) GSPYS] with Lys [G
GSPYS] with Lys [G![[K with combining low line]](https://www.rsc.org/images/entities/char_004b_0332.gif) GSPYS], a decrease in all contacts involving Lys was observed, suggesting that the interactions involving Arg with other residues may be critical for polymer association and LLPS. The phase separation observed in the polycationic Arg variant implies that short-range interactions encoded within the polypeptide sequence are able to compensate for long-range electrostatic repulsion resulting from the high net charge. Last, the sequences with the repeat units GRGSP
GSPYS], a decrease in all contacts involving Lys was observed, suggesting that the interactions involving Arg with other residues may be critical for polymer association and LLPS. The phase separation observed in the polycationic Arg variant implies that short-range interactions encoded within the polypeptide sequence are able to compensate for long-range electrostatic repulsion resulting from the high net charge. Last, the sequences with the repeat units GRGSP![[Y with combining low line]](https://www.rsc.org/images/entities/char_0059_0332.gif) S and GRGSP
S and GRGSP![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) S were simulated to isolate the contribution of Tyr. The pairwise contact analysis revealed a loss of all contacts at the amino acid position after the Y to A substitution, not just the loss of Y–Y interactions between polymers, further demonstrating that interactions of Tyr with non-aromatic residues are also essential for driving association and LLPS in these A-IDPs.
S were simulated to isolate the contribution of Tyr. The pairwise contact analysis revealed a loss of all contacts at the amino acid position after the Y to A substitution, not just the loss of Y–Y interactions between polymers, further demonstrating that interactions of Tyr with non-aromatic residues are also essential for driving association and LLPS in these A-IDPs.
 | ||
| Fig. 3 a) (I): Representative rendering of the atomistic simulation of the condensed phase of [GRGDSPYS]25. (II): Snapshot from atomistic simulations of [GRGDSPYS]25, highlighting representative examples of the wide variety of contacts driving phase separation of WT. (III): Non normalized (top) and normalized (bottom) average residue pairwise contacts (top), separated into backbone–backbone (bb–bb), backbone–side chain (bb–sc) and side chain–side chain (sc–sc). The inset show the average contacts formed by Tyr:Ser Tyr:Tyr Arg:Asp Arg:Tyr Arg:Arg Arg:Ser. b) Fluorescence microscopy image of 0.5 μm yellow–green fluorescent polystyrene beads embedded in IDP coacervates. Scale bar, 5 μm. Inset: Representative trajectory from two-dimensional particle tracking showing Brownian motion of the beads (the length of the inset box is 0.02 μm). c) State diagram showing saturation concentration, Csat, at 18 °C and viscosities for the variants. Reprinted with permission.53 Copyright 2024, Springer Nature Limited. | ||
To design IDP-containing materials for applications, it is critical to understand materials properties such as mechanical strength and nanostructure after phase separation. While most simulations cannot quantitatively predict such experimental properties, efforts have been made to connect measured material properties with simulation to obtain qualitative insight. In the above work, Rekhi et al. correlated the viscosity from experiment with the critical concentration identified in simulation;53 the viscosity was estimated by mean squared displacement (MSD) of fluorescent tracer beads embedded in the IDP coacervates (Fig. 3b). These studies found that viscosity decreased with the saturation concentration of the peptide sequences (Fig. 3c). Detailed analysis of the different sequences indicated that changes in side-chain interactions generally correlated with the variation of viscosities of different sequences, highlighting that while a nonlinear relationship existed between propensity for phase separation and coacervate viscosity, the independent modulation of phase behavior and material properties is feasible.
All-atom simulations using parallel tempering in the well-tempered ensemble (PT-WTE) were also applied to study short resilin-like polypeptides ((RLP-X) [GGRPSDSXGAPGGGN]3) based on the full consensus repeat from D. melanogaster, with aromatic (X = Y, F, W) and aliphatic (X = A, L, V) guest residues at the X position (Fig. 4a).37 For the aromatic guest residues, π–π and cation–π interactions were observed, with RLP-W forming these interactions twice as frequently as RLP-Y or RLP-F; RLP-Y exhibited a higher number of cation–π contacts. These observations involving aromatic residues were consistent with previous experimental studies and were believed to be the primary driving forces for phase separation.90,91,126,127 Moreover, RLP-W and RLP-Y formed twice as many hydrogen bonds involving donor/acceptor side chain atoms. As a result, RLP-W showed the highest propensity for LLPS, whereas RLP-F exhibited the lowest among the aromatic residues. In contrast, the aliphatic residues displayed overall less frequent contacts than the aromatic variants, potentially making them less likely to undergo LLPS. The detailed residue pairwise-contact maps for RLP-W, as well as the hydrogen bonds, π–π, and π–cation interactions for all the variants, are provided in Fig. 4a.
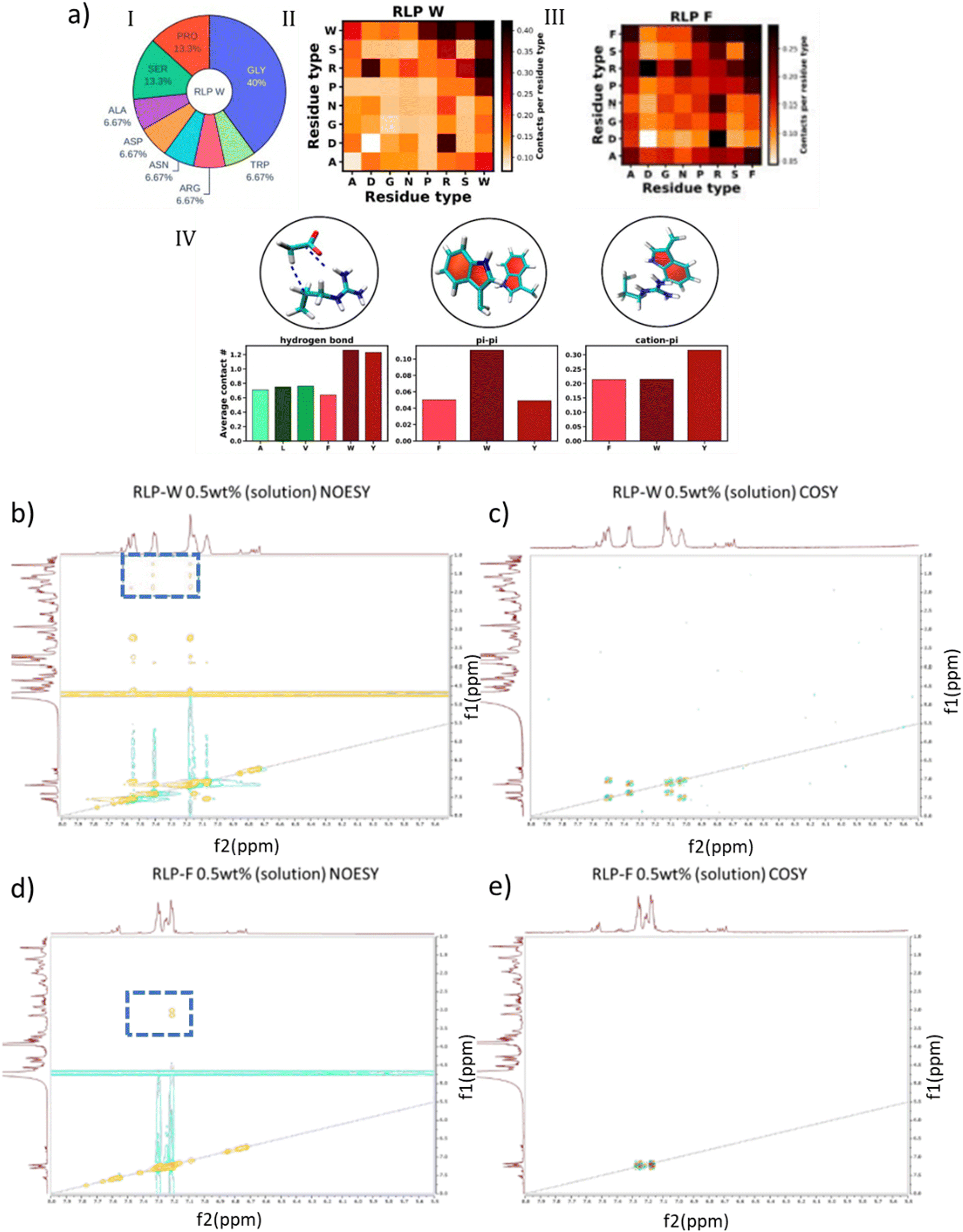 | ||
| Fig. 4 a) (I) Amino acid composition of the 45-residue long RLP-W peptide that was simulated using an all-atom model. (II) Total number of intramolecular van der Waals contacts formed between each residue type pair reflecting the abundance of each pair type in the RLP-W sequence. (III) Contact propensity in (II) normalized to the number of possible interaction pairs of a given type in the RLP-W sequence to obtain insights into their intrinsic interaction strengths. (IV) Number of intramolecular van der Waals contacts formed by residue type X (A, L, V, F, W, or Y) further analyzed in terms of important known modes, hydrogen bond, π–π, and cation–π interactions. 2D b) NOESY and c) COSY NMR spectra of RLP-W. Only the signals from 1 to 5 ppm (Y-axis) in the NOESY spectra represent interactions through space. 2D d) NOESY and e) COSY NMR spectra of RLP-F. Only the signals from 3 to 5 ppm (Y-axis) in the NOESY spectra represent interactions through space. Reprinted with permission.37 Copyright 2024, American Chemical Society. | ||
Experimental verification of the different interactions in the RLP-X constructs was confirmed experimentally by Garcia et al.37via NMR methods, double quantum-filtered correlation spectroscopy (DQF-COSY) and nuclear Overhauser effect spectroscopy (NOESY). NOESY-unique signals (which appeared in NOESY but not in COSY), captured through-space interactions of specific amino acid residues in the RLP-X chains. As shown in Fig. 4b and c, the NOESY-unique signals of RLP-W suggested high spatial proximity of Trp–Trp and Trp–Arg side chains (aromatic Trp protons: δ 7.00–7.60 ppm, Hα tryptophan: δ 4.60 ppm H arginine δ 3.23, 1.86, 1.58, and 1.21 ppm, highlighted in blue boxes), which was consistent with simulation results (Fig. 4a). However, with phenylalanine guest residues, the most common contacts observed in NOESY-unique signals (Fig. 4d and e) only involved Phe aromatic protons (δ 7.15–7.40 ppm) and Hα and Hβ from Phe (δ 3.00, 3.15, and 4.56 ppm; highlighted in blue boxes), which corroborated the prevalence of only Phe–Phe interactions, consistent with simulation (Fig. 4a). In addition, an increased UCST transition temperature of RLP-W compared to RLP-F was consistent with the increased interaction strength for RLP-W.
In other reports, Bevan and coworkers applied MD simulations to investigate the roles of the SYGAP sequence motif and its less common variants SYSAP and TYGAP in influencing the elastomeric properties of D. melanogaster resilin containing Gly-rich repetitive motifs comprising the amino acids PSSSYGAPGGGNGGR.128 Their findings revealed that the SYGAP motif forms a bent structure with greater extensibility than the other variations, primarily due to the conformational freedom of glycine and the presence of hydrogen bonding within the motif. These structural features may significantly impact the elastic behavior of resilin and potentially other elastomeric proteins.
Simulations of other IDPs
CG models are widely used to study association and LLPS involving cytoplasmic IDPs, which control a variety of cellular processes including transcription and aggregation.96–98,129–131 One such extensively studied model is the HPS model. An improved model, HPS-Urry, used the Urry hydropathy scale, and parameters were optimized so as to recover experimental radius of gyration Rg values from 42 different IDP sequences. The model has demonstrated excellent agreement between simulated and experimental coexistence densities of both DEAD-box helicase 4 (DDX4) and low complexity domain of RNA-binding protein FUS (FUS LC).57,96,97 Additionally, the Mpipi model, developed by Collepardo-Guevara and colleagues, was parameterized using a combination of atomistic simulations and bioinformatics data. In this model, atomistic PMF calculations and bioinformatics data were integrated to parameterize short-ranged pairwise interactions and long-range charge–charge interactions, achieving a balanced representation of π–π and non-π-based contacts. This model was able to capture key interactions, such as π–π and cation–π interactions, and its predictions aligned well with experimental radii of gyration (Rg) with a Pearson correlation coefficient (r) = 0.92. In addition, estimated critical temperatures Tc for selected 17 IDPs were also in quantitative agreement with the Tc determined from in vitro phase diagrams, with r = 0.97.98 As a comparison, for the same IDPs, using the HPS-Urry model, weaker correlation of computation and experiment was observed for both the Rg (r = 0.87) and the critical temperature Tc (r = 0.91). These simulation methods significantly enhance our understanding of LLPS in IDPs at the molecular level and provide valuable insights into the biochemical functions of IDPs.A more recent study by Pesce et al. has revealed the possibility of designing IDPs de novo,132 starting with naturally occurring IDP sequences, including α-synuclein (αSyn), the low complexity domain from hnRNPA1 (A1-LCD), the prion-like domain of FUS (FUS-PLD), and the R-/G-rich domain of the P granule protein LAF-1 (LAF-1-RGG). New sequences based on the naturally occurring IDP sequences were identified through a Markov chain Monte Carlo scheme to sample sequence variants. The sequences were simulated by MD simulations with the CG CALVADOS force field97 to generate structural parameters such as radius of gyration and saturation concentration. Based on the simulation, 15 variants were selected, five of which could be successfully expressed recombinantly. Characterization via differential interference contrast (DIC) microscopy confirmed the formation of coacervates for the purified variants (Fig. 5a). The saturation concentration (Fig. 5b) and radius of gyration (Fig. 5c) measured experimentally aligned well for all but one variant V5, showing the possibility of the de novo design of artificial IDPs.
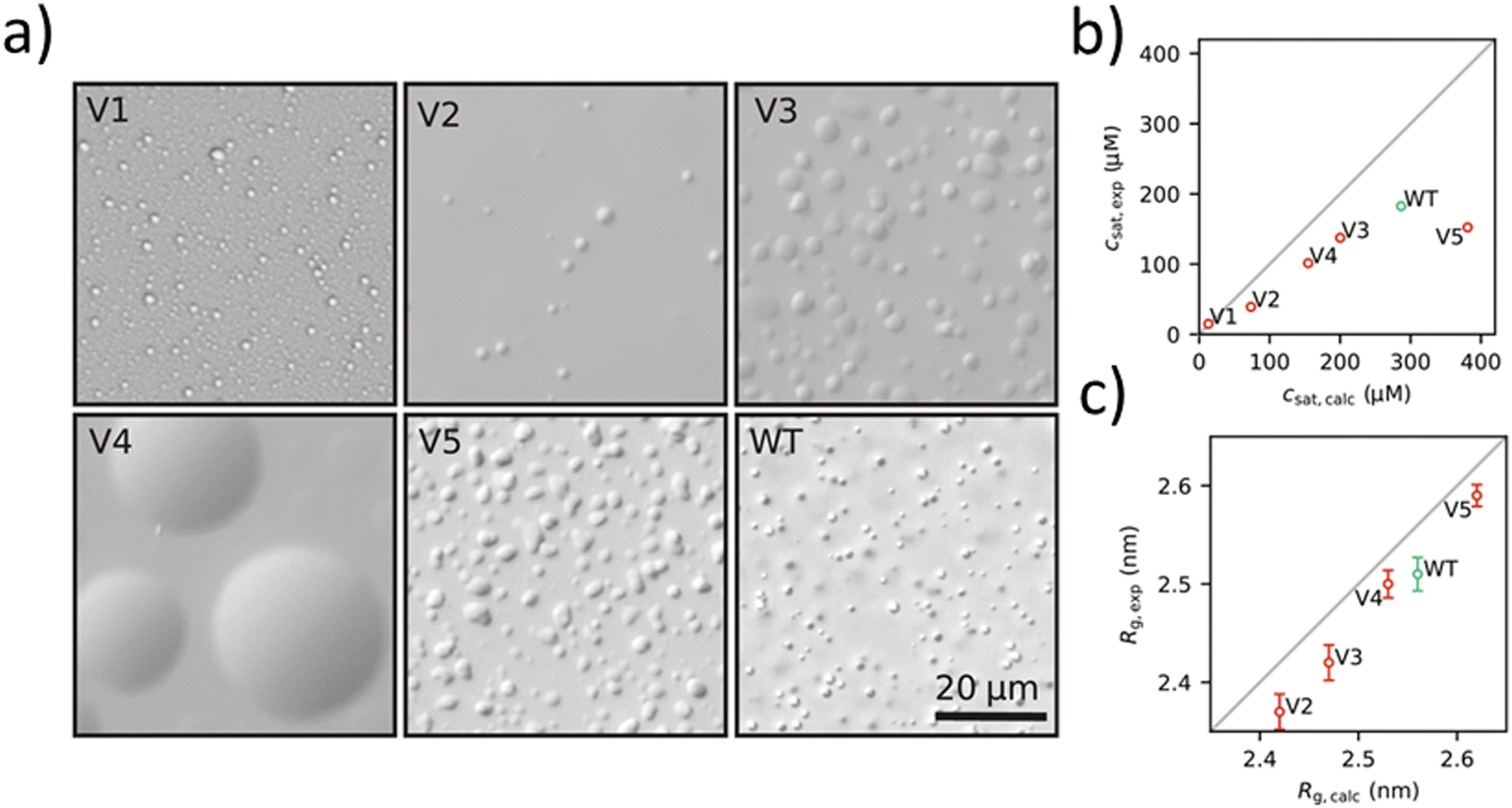 | ||
| Fig. 5 a) DIC microscopy WT A1-LCD and the five variants showing phase separation behavior of artificial IDPs. b) Comparison of experimental and calculated values of Csat at 298 K. c) Comparison of experimental and calculated values of Rg for WT A1-LCD and V2 to V5. Reprinted under a Creative Commons Attribution Non-Commercial License 4.0 (CC BY-NC).132 Copyright 2024, the authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. | ||
Simulation of IDPs conjugated with ordered domains
The simulation of IDPs has successfully highlighted specific residue interactions important in the phase behavior of these proteins and has also captured chain conformation changes near their transition temperatures. New opportunities for controlling association and materials properties have also been observed by fusing IDP domains (ELPs and RLPs) with ordered assembly-promoting domains including peptide sequences and lipid tails.14,133,134 Additional simulation and experimental studies have explored IDP conjugation to ordered domains, providing intriguing opportunities to further tune interactions in the control of tuning of phase transition behavior,22,134 hydrogel structure and hysteresis,111,135 and the assembly of functional nanoscale structures.38 The nonequilibrium, hysteretic phase separation behavior of IDPs has also been successfully applied and incorporated as a parameter in materials design.14Roberts et al. showed that the inclusion of polyalanine helical sequences (An or (KAAA)n) in sequences containing in ELP sequence repeats [VPG(Ax/V1−x)G]n caused thermal hysteresis as monitored by turbidity; differences in heating/cooling transition temperatures increased with increasing polyalanine ratio and decreasing charge.135 A CG bead-spring model was employed and showed an aggregate of ELP domains above the transition temperature. However, the exchange of alanine-rich regions with neighboring clusters entangled them into a network which prevented the re-solubilization of ELP domains upon cooling. A more detailed all-atom level MD simulation of this hysteretic phase behavior was conducted by Quiroz et al.,136 in which it was found that the hysteresis can be promoted by reversing the ELP sequence, e.g. using [VPGVG]n rather than [GVGPV]n (Fig. 6a). In the simulations, minor changes in secondary-structure propensities, primarily in the unstructured motif, were observed after sequence reversal (Fig. 6b). However, the differences in radius of gyration (ΔRg) were close to 0 after sequence reversal (indicating increased backbone rigidity and a less compact conformation of reversal sequences compared to original sequences at high temperature), and in those cases the new sequence tended to show large hysteresis experimentally (Fig. 6c).
 | ||
| Fig. 6 a) Temperature-dependent optical turbidity to probe the phase behavior of three IDP pairs, I) (VPGVG)30 and (GVGPA)30, II) (VPAGVG)25 and (GVGAPV)25, and III) (APVGLG)40 and (VPAGLG)40. Each pair consists of two IDPPs with identical number of repeats of motif sequences that are interrelated by sequence reversal. b) Fraction of unstructured motifs residues for single IDPP chains and c) differential between values at high and low temperatures of radius of gyrations (ΔRg, black) and absolute peptide–water interaction (ΔEpw, blue) energy of 18-mer, single IDPP chains at low (290 to 310 K) and high (350 to 390 K) temperatures for three IDPP pairs in a). d) Fraction of value changes (f) in interchain interaction quantities after cooling two chain “phase separated” systems to 290 K for 25 ns. f = (end value − initial value)/(initial value). e) Snapshots from two-chain simulations for an IDPP with marked hysteresis (VGAPVG)18 (shown in Fig. 6a II) and for an IDPP with negligible hysteresis (LGAPVG)18 (shown in Fig. 6a III). a–d) Reprinted under a Creative Commons Attribution Non-Commercial License 4.0 (CC BY-NC).136 Copyright 2019, the authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. e) Reprinted with permission.111 Copyright 2014, American Chemical Society. | ||
The significant decrease in peptide–water interaction energy (ΔEpw) was also regarded as a predictor of hysteresis behavior of [VGPVG]18 and [VGAPVG]18, indicating that minor changes in hydrophobic hydration can be a main driving force for fine tuning IDP LCST-associated phase separation (Fig. 6c).111 In a two-chain model, the hysteresis was characterized by the increase in fractional difference of interaction energy, number of interchain hydrogen bonds, number of interchain hydrophobic contacts, and (absolute) hydrophobic interaction energy (Fig. 6d); the visualization of the differences between IDPs with strong hysteresis ([VGAPVG]18) and weak hysteresis ([LGAPV]18) is shown in Fig. 6e. These studies illustrate the ability to predict the hysteretic phase separation of IDP via MD simulation, which should be applicable to a variety of related ELP- and IDP-based materials.
In collaborative studies by the Jayaraman and Kiick laboratories on short ELPs conjugated to trimer-forming collagen-like peptides (CLPs), a series of all-atom and CG simulations were employed, complemented by experimental work. In all-atom simulations, the LCST transition temperature, Tt, of ELP oligomers with the sequence [VPGFG]6 was characterized.55 The LLPS behavior of ELP oligomers anchored to fixed points was also simulated as a model of the ELP conjugation to the triple-helix-forming CLP; the results qualitatively aligned with previous experimental observations, describing the thermoresponsive assembly of ELP–CLP, by Luo and Kiick.137 In the CG simulations of ELPs conjugated to CLPs, where the CLPs were modeled as rigid blocks, it was observed that the increased local density of ELPs made them more prone to aggregation at lower polymer–polymer attraction energies compared to when the ELPs were untethered. Furthermore, similar studies examined the influence of various guest residues (X) on the Tt in short ELPs [VPGXG]4.138 ELPs containing Trp (WWWW) were more prone to form β-turn structures at lower temperatures compared to Phe variants (FFFF). The bulkier aromatic side chain of Trp likely contributes to increased local stiffness in the Trp variants, which could be another factor for the significantly lower Tt (Tt < 278 K) compared to that observed for the Phe variants (Tt < 353 K), in addition to differences in their hydrophobicity. The simulations provided results closely matching experimental findings when the effects of local stiffness and the stronger attractive interactions among Trp residues compared to Phe residues were incorporated into the CG model. These types of CG models were rooted in polymer physics and were primarily designed to capture the key interactions between ELPs and CLPs, while minimizing the influence of other interactions. This approach allowed researchers to focus on the main driving forces behind the system's behavior. However, due to the simplified nature of the model, the results were typically limited to qualitative comparisons with experimental data.
In subsequent work, the effect of Tyr substitutions and their placement in the ELP ‘parent’ sequences [VPGFG]6 was explored and was shown to influence Tt.139 Simulations revealed that the Tyr guest residues enhanced intra- and inter-peptide hydrogen bonding, which promoted the aggregation of ELPs and lowered Tt. Moreover, placing Tyr away from the tethering point to the CLPs resulted in a lower Tt compared to placements closer to the tethering point, as a greater number of π–π stacking interactions were possible at low temperatures, owing to the increased conformational flexibility of the ELP chains further from the tethering point.
CG simulations using the Martini force field have also been applied to understand the self-assembly of block copolypeptides in which RLPs [GGRPSDSWGAPGGGN]1 are conjugated to designed coiled-coil bundle-forming peptides (BFP) that form tetrameric, α-helical, antiparallel bundles.38,39,140,141 These simulations successfully captured the UCST behavior of RLP–BFP–RLP conjugates and revealed 1D self-assembly mediated by RLP interactions at low temperatures, consistent with experimental observations as shown in Fig. 7a–c.38 Further experimental studies explored the self-assembly of hybrid polypeptides with BFP on the termini (BFP–(RLP-L)6–BFP) where Trp was replaced by Leu in the RLP sequence.39 Upon fusion with BFP peptides, the RLPs retained their thermoresponsive phase behavior, and cryogenic transmission electron microscopy (Fig. 7d) along with CG simulations (Fig. 7e and f) revealed that BFP–(RLP-L)6–BFP adopted a closed-loop dimer structure (Fig. 7f), assembling into 1D nanofibers that further formed into 2D ribbon-like or sheet-like structures. These findings suggest broader opportunities for fusing stimuli-responsive IDPs with assembled peptide domains to prepare hybrid materials that adopt properties from each domain.
 | ||
| Fig. 7 Cryo TEM reveals that genetic fusion of RLPs with 1 repeat unit a) or 3 repeat unit b) to BFP mediates nanofibrillar assembly of coiled coil bundles. c) Rendering of Martini CG of RLP–BFP–RLP assembly with alignment of BFP bundles in the nanofibrils (RLP indicated in blue and BFP indicated in orange). The length of RLP was one repeat unit in the simulations. d) CryoTEM BFP–(RLP-L)6–BFP constructs assembled into micrometer-sized individual nanofibrils, scale bar = 500 nm. e) and f) Two CG BFP–(RLP-L)6–BFP conjugates model that self-assemble into e) chain nanostructures or f) closed loop configuration. Left is the Martini CG representation of each self-assembled nanostructure, and right is a snapshot of the aggregation morphologies achieved from the simulations. a–c) Reprinted with permission.38 Copyright 2023 Wiley-VCH GmbH. d–f) Reprinted with permission.39 Copyright 2024, American Chemical Society. | ||
Conclusions and perspectives
IDPs are extensively used in biomaterials due to their stimuli-responsiveness, biocompatibility, and tunable physiochemical properties. IDP-based materials have been developed for applications in tissue regeneration, biomanufacturing, and controlled drug delivery, where the precise control of IDP interactions and the decoupling of parameters such as transition temperatures and length are critical.13,39 Simulation approaches provide promising tools to both understand IDP-associated processes and to design IDPs with targeted properties. Several approaches, including all-atom simulation and CG modeling, have successfully captured experimental behavior involving short-range interactions (typically less than 1 nm), such as hydrophobic interactions, van der Waals forces and electrostatic attractions between specific amino acid residues. These approaches also capture single-chain properties, including the tendency of certain amino acids to come into close proximity, such as those forming π–π interactions, as well as global structural features like the radius of gyration.53,132 Such insight supports the potential for predicting specific and sequence-dependent properties of IDP-based materials. In addition, simulation approaches have been successfully employed to study the phase transition behavior of IDPs at different spatiotemporal scales; conformational changes and aggregation processes near important transitions have been successfully captured. Residue interactions in all-atom level simulations suggest important interactions that drive the phase transition process. CG modeling provides insights into the nanostructures formed upon association, especially when IDPs have been fused to other domains. Although most simulations have been applied to understand experimentally observed properties of IDP systems, several approaches have successfully predicted behavior de novo with all-atom models.61,132 Already, a deeper understanding of residue interactions at the molecular level has advanced the design of new IDPs.However, the precise prediction of the behavior of IDP systems, such as transition temperature, saturation concentration, and mechanical properties, still requires more investigation. A variety of factors limit the detailed quantitative comparison of simulation and experiment regarding IDP properties. These include the necessarily large systems (micrometer-size in experiment vs. nanometer-size in simulation) and long timescales (second-scale in experiment vs. nanosecond-scale in simulation) associated with liquid–liquid phase separation, the quantitative alignment of experimental and simulation conditions, appropriate parameterization of important intermolecular interactions (including accurately modeling hydrogen bonding, hydrophobic effects, electrostatics, and solvent-mediated interactions), and the subtlety of methods associated with simulating systems comprising multiple phases and their transitions. Appropriate parameterization needs to consider residue-specific interaction potentials, temperature-dependent effects, solvation dynamics, and the influence of ionic strength; new parameters could also include detailed representations of post-translational modifications, crowding effects, and explicit solvent molecules.
Most all-atom level simulations focus on single-chain systems or small oligomers due to the complexity of the model, and even with a simplified CG model, simulation sizes are still limited to nanometer dimensions while IDP protein-rich phases in experiments are micrometer-size coacervates. While CG approaches can potentially simulate the phase separation processes on a larger scale, they have to date been only widely applied in explaining general features of IDP assembly, mainly due to the lack of accuracy resulting from the necessarily simplified residue interactions in the CG models. Optimized force fields address the residue interactions in IDPs more accurately, but developing and implementing these in CG modeling still requires further investigation in the context of larger-scale simulations. Specific improvements include the incorporation of more accurate potentials that reflect hydrogen bonds, accurately capture secondary structures, and represent solvation energetics more effectively. Enhancing the resolution of side-chain representations and introducing sequence-specific interaction rules derived from experimental or high-resolution all-atom data may significantly improve CG model fidelity. Additionally, hybrid modeling approaches combining atomistic and coarse-grained representations could be explored to better capture critical interactions in larger-scale systems.
Data-based approaches such as machine learning have been extensively used in the simulation and prediction of polymeric materials properties including polymer swelling,142 polymer-surface interactions,143 and assembled membrane properties.144 Similar data-derived methods will provide interesting opportunities for the simulation and prediction of IDP properties as training datasets become more available and more extensive for this class of polypeptides. Classification of diverse IDP sequences, recognition of key parameters that influence IDP properties, and significant additional contributions to the current IDP datasets will be required for successful implementation of data-based simulation of IDPs. In summary, continued synergistic improvement in the coordinated application of simulation and experiment will be critical for to advance the prediction of materials properties and the successful de-novo design of artificial IDPs.
Data availability
All data needed to evaluate the conclusions in the review are presented in the review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work of the authors and previous research from the authors' laboratories was primarily supported by National Science Foundation through the University of Delaware Materials Research Science and Engineering Center, DMR-2011824. Partial support for the work was also provided by the NSF (DMR-1609544; DMR-0907478; DMR-2004796; DMR-0239744; DMR-2004890; CBET-1159466; CBET-2023668; CBET-1605130; CBET-1703402); and the National Institutes of Health (P20 RR017716; RO1 DC011377; P20 GM104316, P20 GM103446, P20 GM139760). The authors also acknowledge support from the Department of Energy, Office of Basic Energy Sciences, Biomolecular Materials Program under grant numbers DE-SC0019355 and DE-SC0019282, TG-CHE110041 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #1548562, #2138259, #2138286, #2138307, #2137603, and #2138296. The contents of this manuscript are solely the responsibility of the authors and do not necessarily reflect the views of the funding agencies.References
- B. Kuhlman and P. Bradley, Advances in protein structure prediction and design, Nat. Rev. Mol. Cell Biol., 2019, 20(11), 681–697 CrossRef CAS PubMed.
- S. J. Piper, R. M. Johnson, D. Wootten and P. M. Sexton, Membranes under the Magnetic Lens: A Dive into the Diverse World of Membrane Protein Structures Using Cryo-EM, Chem. Rev., 2022, 122(17), 13989–14017 CrossRef CAS PubMed.
- O. K. Parfenova, V. G. Kukes and D. V. Grishin, Follistatin-Like Proteins: Structure, Functions and Biomedical Importance, Biomedicines, 2021, 9(8), 999 CrossRef CAS PubMed.
- L. Piersimoni, P. L. Kastritis, C. Arlt and A. Sinz, Cross-Linking Mass Spectrometry for Investigating Protein Conformations and Protein–Protein Interactions—A Method for All Seasons, Chem. Rev., 2022, 122(8), 7500–7531 CrossRef CAS PubMed.
- W. Xu, C. Zhong, C. Zou, B. Wang and N. Zhang, Analytical methods for amino acid determination in organisms, Amino Acids, 2020, 52(8), 1071–1088 CrossRef CAS PubMed.
- A. J. Miles, R. W. Janes and B. A. Wallace, Tools and methods for circular dichroism spectroscopy of proteins: a tutorial review, Chem. Soc. Rev., 2021, 50(15), 8400–8413 RSC.
- A. Sadat and I. J. Joye, Peak Fitting Applied to Fourier Transform Infrared and Raman Spectroscopic Analysis of Proteins, Appl. Sci., 2020, 10(17), 5918 CrossRef CAS.
- K. M. Yip, N. Fischer, E. Paknia, A. Chari and H. Stark, Atomic-resolution protein structure determination by cryo-EM, Nature, 2020, 587(7832), 157–161 CrossRef CAS PubMed.
- X. Yao, X. Fan and N. Yan, Cryo-EM analysis of a membrane protein embedded in the liposome, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(31), 18497–18503 CrossRef CAS PubMed.
- The UniProt Consortium, UniProt: the universal protein knowledgebase in 2021, Nucleic Acids Res., 2021, 49(D1), D480–D489 CrossRef PubMed.
- R. van der Lee, M. Buljan, B. Lang, R. J. Weatheritt, G. W. Daughdrill and A. K. Dunker, et al. Classification of Intrinsically Disordered Regions and Proteins, Chem. Rev., 2014, 114(13), 6589–6631 CrossRef CAS PubMed.
- M. M. Babu, The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease, Biochem. Soc. Trans., 2016, 44(5), 1185–1200 CrossRef CAS PubMed.
- B. Wang, S. S. Patkar and K. L. Kiick, Application of Thermoresponsive Intrinsically Disordered Protein Polymers in Nanostructured and Microstructured Materials, Macromol. Biosci., 2021, 21(9), 2100129 CrossRef CAS PubMed.
- S. S. Patkar, B. Wang, A. M. Mosquera and K. L. Kiick, Genetically Fusing Order-Promoting and Thermoresponsive Building Blocks to Design Hybrid Biomaterials, Chem. – Eur. J., 2024, 30(30), e202400582 CrossRef CAS PubMed.
- Q. Li, X. Yang and X. Xia, et al. Affibody-Functionalized Elastin-like Peptide–Drug Conjugate Nanomicelle for Targeted Ovarian Cancer Therapy[J], Biomacromolecules, 2024, 25(10), 6474–6484 CrossRef CAS PubMed.
- D. Jiang, Y. Yang, X. Yang, B. Wang, W. Fan and Y. Liu, et al. The application of elastin-like peptides in cancer, tissue engineering and ocular disease, OpenNano, 2023, 9, 100113 CrossRef CAS.
- A. K. Varanko, J. C. Su and A. Chilkoti, Elastin-Like Polypeptides for Biomedical Applications, Annu. Rev. Biomed. Eng., 2020, 22(1), 343–369 CrossRef CAS PubMed.
- D. W. Urry, M. M. Long, B. A. Cox, T. Ohnishi, L. W. Mitchell and M. Jacobs, The synthetic polypentapeptide of elastin coacervates and forms filamentous aggregates, Biochim. Biophys. Acta, Protein Struct., 1974, 371(2), 597–602 CrossRef CAS PubMed.
- T. A. T. Lee, A. Cooper, R. P. Apkarian and V. P. Conticello, Thermo-Reversible Self-Assembly of Nanoparticles Derived from Elastin-Mimetic Polypeptides, Adv. Mater., 2000, 12(15), 1105–1110 CrossRef CAS.
- M. R. Dreher, A. J. Simnick, K. Fischer, R. J. Smith, A. Patel and M. Schmidt, et al. Temperature Triggered Self-Assembly of Polypeptides into Multivalent Spherical Micelles, J. Am. Chem. Soc., 2008, 130(2), 687–694 CrossRef CAS PubMed.
- P. A. Taylor, H. Huang, K. L. Kiick and A. Jayaraman, Placement of tyrosine residues as a design element for tuning the phase transition of elastin-peptide-containing conjugates: experiments and simulations, Mol. Syst. Des. Eng., 2020, 5(7), 1239–1254 RSC.
- A. Prhashanna, P. A. Taylor, J. Qin, K. L. Kiick and A. Jayaraman, Effect of Peptide Sequence on the LCST-Like Transition of Elastin-Like Peptides and Elastin-Like Peptide–Collagen-Like Peptide Conjugates: Simulations and Experiments, Biomacromolecules, 2019, 20(3), 1178–1189 CrossRef CAS PubMed.
- L. A. Navarro, J. J. Ryan, M. Dzuricky, M. Gradzielski, A. Chilkoti and S. Zauscher, Microphase Separation of Resilin-like and Elastin-like Diblock Copolypeptides in Concentrated Solutions, Biomacromolecules, 2021, 22(9), 3827–3838 CrossRef CAS PubMed.
- W. M. Park and J. A. Champion, Thermally Triggered Self-Assembly of Folded Proteins into Vesicles, J. Am. Chem. Soc., 2014, 136(52), 17906–17909 CrossRef CAS PubMed.
- T. Luo and K. L. Kiick, Noncovalent Modulation of the Inverse Temperature Transition and Self-Assembly of Elastin-b-Collagen-like Peptide Bioconjugates, J. Am. Chem. Soc., 2015, 137(49), 15362–15365 CrossRef CAS PubMed.
- D. R. Dautel, W. T. Heller and J. A. Champion, Protein Vesicles with pH-Responsive Disassembly, Biomacromolecules, 2022, 23(9), 3678–3687 CrossRef CAS PubMed.
- T. Weis-Fogh, A Rubber-Like Protein in Insect Cuticle, J. Exp. Biol., 1960, 37(4), 889–907 CrossRef CAS.
- R. Balu, N. K. Dutta, A. K. Dutta and N. R. Choudhury, Resilin-mimetics as a smart biomaterial platform for biomedical applications, Nat. Commun., 2021, 12(1), 149 CrossRef CAS PubMed.
- C. Gonzalez-Obeso, E. Jane Hartzell, R. A. Scheel and D. L. Kaplan, Delivering on the promise of recombinant silk-inspired proteins for drug delivery, Adv. Drug Delivery Rev., 2023, 192, 114622 CrossRef CAS PubMed.
- S. C. Huang, Y. J. Zhu, X. Y. Huang, X. X. Xia and Z. G. Qian, Programmable adhesion and morphing of protein hydrogels for underwater robots, Nat. Commun., 2024, 15(1), 195 CrossRef CAS PubMed.
- L. Li and K. L. Kiick, Resilin-Based Materials for Biomedical Applications, ACS Macro Lett., 2013, 2(8), 635–640 CrossRef CAS PubMed.
- C. M. Elvin, A. G. Carr, M. G. Huson, J. M. Maxwell, R. D. Pearson and T. Vuocolo, et al. Synthesis and properties of crosslinked recombinant pro-resilin, Nature, 2005, 437(7061), 999–1002 CrossRef CAS PubMed.
- R. E. Lyons, D. C. C. Wong, M. Kim, N. Lekieffre, M. G. Huson and T. Vuocolo, et al. Molecular and functional characterisation of resilin across three insect orders, Insect Biochem. Mol. Biol., 2011, 41(11), 881–890 CrossRef CAS PubMed.
- A. Basheer, S. Shahid, M. J. Kang, J. H. Lee, J. S. Lee and D. W. Lim, Switchable Self-Assembly of Elastin- and Resilin-Based Block Copolypeptides with Converse Phase Transition Behaviors, ACS Appl. Mater. Interfaces, 2021, 13(21), 24385–24400 CrossRef CAS PubMed.
- W. Ahn, J. H. Lee, S. R. Kim, J. Lee and E. J. Lee, Designed protein- and peptide-based hydrogels for biomedical sciences, J. Mater. Chem. B, 2021, 9(8), 1919–1940 RSC.
- P. Weber, M. Dzuricky, J. Min, I. Jenkins and A. Chilkoti, Concentration-Independent Multivalent Targeting of Cancer Cells by Genetically Encoded Core-Crosslinked Elastin/Resilin-like Polypeptide Micelles, Biomacromolecules, 2021, 22(10), 4347–4356 CrossRef CAS PubMed.
- C. G. Garcia, S. S. Patkar, N. Jovic, J. Mittal and K. L. Kiick, Alteration of Microstructure in Biopolymeric Hydrogels via Compositional Modification of Resilin-Like Polypeptides, ACS Biomater. Sci. Eng., 2021, 7(9), 4244–4257 CrossRef PubMed.
- S. S. Patkar, Y. Tang, A. M. Bisram, T. Zhang, J. G. Saven and D. J. Pochan, et al. Genetic Fusion of Thermoresponsive Polypeptides with UCST-type Behavior Mediates 1D Assembly of Coiled-Coil Bundlemers, Angew. Chem., Int. Ed., 2023, 62(25), 1–7 CrossRef PubMed.
- S. S. Patkar, Y. Tang, T. Zhang, A. M. Bisram, J. G. Saven and D. J. Pochan, et al. Genetically Fused Resilin-like Polypeptide–Coiled Coil Bundlemer Conjugates Exhibit Tunable Multistimuli-Responsiveness and Undergo Nanofibrillar Assembly, Biomacromolecules, 2024, 25(4), 2449–2461 CrossRef CAS PubMed.
- F. T. Doole, C. P. Camp and M. Kim, Tailoring the formation and stability of self-assembled structures from precisely engineered intrinsically disordered protein polymers: A comprehensive review, Giant, 2023, 14, 100158 CrossRef CAS.
- B. Gao, L. Chi, Y. Zhu, X. Shi, P. Tu and B. Li, et al. An Introduction to Next Generation Sequencing Bioinformatic Analysis in Gut Microbiome Studies, Biomolecules, 2021, 11(4), 530 CrossRef CAS PubMed.
- S. Mei, F. Li, A. Leier, T. T. Marquez-Lago, K. Giam and N. P. Croft, et al. A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction, Briefings Bioinf., 2020, 21(4), 1119–1135 CrossRef CAS PubMed.
- A. H. Elcock, D. Sept and J. A. McCammon, Computer Simulation of Protein−Protein Interactions, J. Phys. Chem. B, 2001, 105(8), 1504–1518 CrossRef CAS.
- V. Daggett, Protein Folding−Simulation, Chem. Rev., 2006, 106(5), 1898–1916 CrossRef CAS PubMed.
- M. AlQuraishi, Machine learning in protein structure prediction, Curr. Opin. Chem. Biol., 2021, 65, 1–8 CrossRef CAS PubMed.
- S. Shi, L. Zhao and Z. Y. Lu, Coarse-Grained Modeling of Liquid–Liquid Phase Separation in Cells: Challenges and Opportunities, J. Phys. Chem. Lett., 2024, 15(28), 7280–7287 CrossRef CAS PubMed.
- M. Paloni, R. Bailly, L. Ciandrini and A. Barducci, Unraveling Molecular Interactions in Liquid-Liquid Phase Separation of Disordered Proteins by Atomistic Simulations, J. Phys. Chem. B, 2020, 124(41), 9009–9016 CrossRef CAS PubMed.
- K. Shmilovich, R. A. Mansbach, H. Sidky, O. E. Dunne, S. S. Panda and J. D. Tovar, et al. Discovery of Self-Assembling π-Conjugated Peptides by Active Learning-Directed Coarse-Grained Molecular Simulation, J. Phys. Chem. B, 2020, 124(19), 3873–3891 CrossRef CAS PubMed.
- S. Sarma, T. R. Sudarshan, V. Nguyen, A. S. Robang, X. Xiao and J. V. Le, et al. Design of parallel β-sheet nanofibrils using Monte Carlo search, coarse-grained simulations, and experimental testing, Protein Sci., 2024, 33(8), e5102 CrossRef CAS PubMed.
- H. Liu, D. Song, H. Lu, R. Luo and H. F. Chen, Intrinsically disordered protein-specific force field CHARMM36IDPSFF, Chem. Biol. Drug Des., 2018, 92(4), 1722–1735 CrossRef CAS PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig and B. L. De Groot, et al. CHARMM36m: An improved force field for folded and intrinsically disordered proteins, Nat. Methods, 2016, 14(1), 71–73 CrossRef PubMed.
- R. B. Best, W. Zheng and J. Mittal, Balanced protein-water interactions improve properties of disordered proteins and non-specific protein association, J. Chem. Theory Comput., 2014, 10(11), 5113–5124 CrossRef CAS PubMed.
- S. Rekhi, C. G. Garcia, M. Barai, A. Rizuan, B. S. Schuster and K. L. Kiick, et al. Expanding the molecular language of protein liquid–liquid phase separation, Nat. Chem., 2024, 16(7), 1113–1124 CrossRef CAS PubMed.
- W. Zheng, G. L. Dignon, N. Jovic, X. Xu, R. M. Regy and N. L. Fawzi, et al. Molecular details of protein condensates probed by microsecond long atomistic simulations, J. Phys. Chem. B, 2020, 124(51), 11671–11679 CrossRef CAS PubMed.
- J. E. Condon, T. B. Martin and A. Jayaraman, Effect of conjugation on phase transitions in thermoresponsive polymers: An atomistic and coarse-grained simulation study, Soft Matter, 2017, 13(16), 2907–2918 RSC.
- Y. Sugita and Y. Okamoto, Replica-exchange molecular dynamics method for protein folding, Chem. Phys. Lett., 1999, 314(1–2), 141–151 CrossRef CAS.
- R. M. Regy, J. Thompson, Y. C. Kim and J. Mittal, Improved coarse-grained model for studying sequence dependent phase separation of disordered proteins, Protein Sci., 2021, 30(7), 1371–1379 CrossRef CAS PubMed.
- X. Gong, Y. Zhang and J. Chen, Advanced sampling methods for multiscale simulation of disordered proteins and dynamic interactions[J], Biomolecules, 2021, 11(10), 1416 CrossRef CAS PubMed.
- G. Bussi, F. L. Gervasio, A. Laio and M. Parrinello, Free-Energy Landscape for β Hairpin Folding from Combined Parallel Tempering and Metadynamics, J. Am. Chem. Soc., 2006, 128(41), 13435–13441 CrossRef CAS PubMed.
- S. Piana and A. Laio, A Bias-Exchange Approach to Protein Folding, J. Phys. Chem. B, 2007, 111(17), 4553–4559 CrossRef CAS PubMed.
- N. Galvanetto, M. T. Ivanović, A. Chowdhury, A. Sottini, M. F. Nüesch and D. Nettels, et al. Extreme dynamics in a biomolecular condensate, Nature, 2023, 619(7971), 876–883 CrossRef CAS PubMed.
- S. L. Perry, L. Leon, K. Q. Hoffmann, M. J. Kade, D. Priftis and K. A. Black, et al. Chirality-selected phase behaviour in ionic polypeptide complexes, Nat. Commun., 2015, 6(1), 6052 CrossRef CAS PubMed.
- K. Q. Hoffmann, S. L. Perry, L. Leon, D. Priftis, M. Tirrell and J. J. De Pablo, A molecular view of the role of chirality in charge-driven polypeptide complexation, Soft Matter, 2015, 11(8), 1525–1538 RSC.
- B. Zhao, N. K. Li, Y. G. Yingling and C. K. Hall, LCST Behavior is Manifested in a Single Molecule: Elastin-Like polypeptide (VPGVG)n, Biomacromolecules, 2016, 17(1), 111–118 CrossRef CAS PubMed.
- N. K. Li, F. G. Quiroz, C. K. Hall, A. Chilkoti and Y. G. Yingling, Molecular description of the lcst behavior of an elastin-like polypeptide, Biomacromolecules, 2014, 15(10), 3522–3530 CrossRef CAS PubMed.
- A. Tarakanova, W. Huang, A. S. Weiss, D. L. Kaplan and M. J. Buehler, Computational smart polymer design based on elastin protein mutability, Biomaterials, 2017, 127, 49–60 CrossRef CAS PubMed.
- W. Zheng, A. Borgia, K. Buholzer, A. Grishaev, B. Schuler and R. B. Best, Probing the Action of Chemical Denaturant on an Intrinsically Disordered Protein by Simulation and Experiment, J. Am. Chem. Soc., 2016, 138(36), 11702–11713 CrossRef CAS PubMed.
- H. Wu, P. G. Wolynes and G. A. Papoian, AWSEM-IDP: A Coarse-Grained Force Field for Intrinsically Disordered Proteins, J. Phys. Chem. B, 2018, 122(49), 11115–11125 CrossRef CAS PubMed.
- A. P. Latham and B. Zhang, Improving Coarse-Grained Protein Force Fields with Small-Angle X-ray Scattering Data, J. Phys. Chem. B, 2019, 123(5), 1026–1034 CrossRef CAS PubMed.
- W. G. Noid, R. J. Szukalo, K. M. Kidder and M. C. Lesniewski, Rigorous Progress in Coarse-Graining, Annu. Rev. Phys. Chem., 2024, 75(1), 21–45 CrossRef CAS PubMed.
- A. C. Murthy, G. L. Dignon, Y. Kan, G. H. Zerze, S. H. Parekh and J. Mittal, et al. Molecular interactions underlying liquid−liquid phase separation of the FUS low-complexity domain, Nat. Struct. Mol. Biol., 2019, 26(7), 637–648 CrossRef CAS PubMed.
- G. L. Dignon, W. Zheng, Y. C. Kim, R. B. Best and J. Mittal, Sequence determinants of protein phase behavior from a coarse-grained model, PLoS Comput. Biol., 2018, 14(1), e1005941 CrossRef PubMed.
- G. Krainer, T. J. Welsh, J. A. Joseph, J. R. Espinosa, S. Wittmann and E. de Csilléry, et al. Reentrant liquid condensate phase of proteins is stabilized by hydrophobic and non-ionic interactions, Nat. Commun., 2021, 12(1), 1085 CrossRef CAS PubMed.
- A. E. Conicella, G. L. Dignon, G. H. Zerze, H. B. Schmidt, A. M. D'Ordine and Y. C. Kim, et al. TDP-43 α-helical structure tunes liquid–liquid phase separation and function, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(11), 5883–5894 CrossRef CAS PubMed.
- J. McCarty, K. T. Delaney, S. P. O. Danielsen, G. H. Fredrickson and J. E. Shea, Complete Phase Diagram for Liquid–Liquid Phase Separation of Intrinsically Disordered Proteins, J. Phys. Chem. Lett., 2019, 10(8), 1644–1652 CrossRef CAS PubMed.
- Z. A. Levine and J. E. Shea, Simulations of disordered proteins and systems with conformational heterogeneity, Curr. Opin. Struct. Biol., 2017, 43, 95–103 CrossRef CAS PubMed.
- U. Baul, D. Chakraborty, M. L. Mugnai, J. E. Straub and D. Thirumalai, Sequence Effects on Size, Shape, and Structural Heterogeneity in Intrinsically Disordered Proteins, J. Phys. Chem. B, 2019, 123(16), 3462–3474 CrossRef CAS PubMed.
- L. W. Chang, T. K. Lytle, M. Radhakrishna, J. J. Madinya, J. Vélez and C. E. Sing, et al. Sequence and entropy-based control of complex coacervates, Nat. Commun., 2017, 8(1), 1–7 CrossRef PubMed.
- C. Li, X. Fu, W. Zhong and J. Liu, Dissipative Particle Dynamics Simulations of a Protein-Directed Self-Assembly of Nanoparticles, ACS Omega, 2019, 4(6), 10216–10224 CrossRef CAS PubMed.
- M. L. Mansfield and J. F. Douglas, Shape characteristics of equilibrium and non-equilibrium fractal clusters, J. Chem. Phys., 2013, 139(4), 044901 CrossRef PubMed.
- Y. Jung, C. Jeon, J. Kim, H. Jeong, S. Jun and B. Y. Ha, Ring polymers as model bacterial chromosomes: confinement, chain topology, single chain statistics, and how they interact, Soft Matter, 2012, 8(7), 2095–2102 RSC.
- T. Zhang, K. I. Winey and R. A. Riggleman, Conformation and dynamics of ring polymers under symmetric thin film confinement, J. Chem. Phys., 2020, 153, 184905 CrossRef CAS PubMed.
- J. C. Shillcock, C. Lagisquet, J. Alexandre, L. Vuillon and J. H. Ipsen, Model biomolecular condensates have heterogeneous structure quantitatively dependent on the interaction profile of their constituent macromolecules, Soft Matter, 2022, 18(35), 6674–6693 RSC.
- M. Streek, F. Schmid, T. T. Duong and A. Ros, Mechanisms of DNA separation in entropic trap arrays: A Brownian dynamics simulation, J. Biotechnol., 2004, 112(1–2), 79–89 CrossRef CAS PubMed.
- K. Kremer and G. S. Grest, Dynamics of entangled linear polymer melts: A molecular-dynamics simulation, J. Chem. Phys., 1990, 92(8), 5057–5086 CrossRef CAS.
- Y. Zhao, R. Cortes-Huerto, K. Kremer and J. F. Rudzinski, Investigating the Conformational Ensembles of Intrinsically Disordered Proteins with a Simple Physics-Based Model, J. Phys. Chem. B, 2020, 124(20), 4097–4113 CrossRef CAS PubMed.
- A. M. Rumyantsev, A. Johner, M. V. Tirrell and J. J. De Pablo, Unifying Weak and Strong Charge Correlations within the Random Phase Approximation: Polyampholytes of Various Sequences, Macromolecules, 2022, 55(14), 6260–6274 CrossRef CAS.
- G. S. Freeman, D. M. Hinckley, J. P. Lequieu, J. K. Whitmer and J. J. De Pablo, Coarse-grained modeling of DNA curvature, J. Chem. Phys., 2014, 141, 165103 CrossRef PubMed.
- J. M. Choi, F. Dar, R. V. Pappu and O. Keskin, LASSI: A Lattice Model for Simulating Phase Transitions of Multivalent Proteins, PLoS Comput. Biol., 2019, 15(10), e1007028 CrossRef CAS PubMed.
- M. Dzuricky, B. A. Rogers, A. Shahid, P. S. Cremer and A. Chilkoti, De novo engineering of intracellular condensates using artificial disordered proteins, Nat. Chem., 2020, 12(9), 814–825 CrossRef CAS PubMed.
- J. Wang, J. M. Choi, A. S. Holehouse, H. O. Lee, X. Zhang and M. Jahnel, et al. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins, Cell, 2018, 174(3), 688–699.e16 CrossRef CAS PubMed.
- A. Bremer, M. Farag, W. M. Borcherds, I. Peran, E. W. Martin and R. V. Pappu, et al. Deciphering how naturally occurring sequence features impact the phase behaviours of disordered prion-like domains, Nat. Chem., 2022, 14(2), 196–207 CrossRef CAS PubMed.
- I. Alshareedah, W. M. Borcherds, S. R. Cohen, A. Singh, A. E. Posey and M. Farag, et al. Sequence-specific interactions determine viscoelasticity and ageing dynamics of protein condensates, Nat. Phys., 2024, 20(9), 1482–1491 Search PubMed.
- J. M. Choi, A. S. Holehouse and R. V. Pappu, Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions, Annu. Rev. Biophys., 2020, 49(1), 107–133 Search PubMed.
- Y. Dai, L. You and A. Chilkoti, Engineering synthetic biomolecular condensates, Nat. Rev. Bioeng., 2023, 1(7), 466–480 Search PubMed.
- R. M. Regy, W. Zheng and J. Mittal, Using a sequence-specific coarse-grained model for studying protein liquid–liquid phase separation, in Physiology & behavior, 2021, pp. 1–17 Search PubMed.
- G. Tesei, T. K. Schulze and R. Crehuet, et al. Accurate model of liquid–liquid phase behavior of intrinsically disordered proteins from optimization of single-chain properties[J], Proc. Natl. Acad. Sci. U. S. A., 2021, 118(44), e2111696118 CrossRef CAS PubMed.
- J. A. Joseph, A. Reinhardt, A. Aguirre, P. Y. Chew, K. O. Russell, J. R. Espinosa, A. Garaizar and R. Collepardo-Guevara, Physics-driven coarse-grained model for biomolecular phase separation with near-quantitative accuracy, Nat. Comput. Sci., 2021, 1(11), 732–743 CrossRef PubMed.
- J. Jung, C. Tan and Y. Sugita, GENESIS CGDYN: large-scale coarse-grained MD simulation with dynamic load balancing for heterogeneous biomolecular systems[J], Nat. Commun., 2024, 15(1), 3370 CrossRef CAS PubMed.
- F. E. Thomasen, F. Pesce, M. A. Roesgaard, G. Tesei and K. Lindorff-Larsen, Improving Martini 3 for Disordered and Multidomain Proteins, J. Chem. Theory Comput., 2022, 18(4), 2033–2041 CrossRef CAS PubMed.
- P. C. T. Souza, R. Alessandri, J. Barnoud, S. Thallmair, I. Faustino and F. Grünewald, et al. Martini 3: a general purpose force field for coarse-grained molecular dynamics, Nat. Methods, 2021, 18(4), 382–388 CrossRef CAS PubMed.
- F. E. Thomasen, T. Skaalum and A. Kumar, et al. Rescaling protein-protein interactions improves Martini 3 for flexible proteins in solution[J], Nat. Commun., 2024, 15(1), 6645 CrossRef CAS PubMed.
- Z. Benayad, S. von Bülow, L. S. Stelzl and G. Hummer, Simulation of FUS Protein Condensates with an Adapted Coarse-Grained Model, J. Chem. Theory Comput., 2021, 17(1), 525–537 CrossRef CAS PubMed.
- B. S. Schuster, G. L. Dignon and W. S. Tang, et al. Identifying sequence perturbations to an intrinsically disordered protein that determine its phase-separation behavior[J], Proc. Natl. Acad. Sci. U. S. A., 2020, 117(21), 11421–11431 CrossRef CAS PubMed.
- D. E. Meyer and A. Chilkoti, Quantification of the Effects of Chain Length and Concentration on the Thermal Behavior of Elastin-like Polypeptides, Biomacromolecules, 2004, 5(3), 846–851 CrossRef CAS PubMed.
- F. Aladini, C. Araman and C. F. W. Becker, Chemical synthesis and characterization of elastin-like polypeptides (ELPs) with variable guest residues, J. Pept. Sci., 2016, 22(5), 334–342 CrossRef CAS PubMed.
- J. R. McDaniel, D. C. Radford and A. Chilkoti, A Unified Model for De Novo Design of Elastin-like Polypeptides with Tunable Inverse Transition Temperatures, Biomacromolecules, 2013, 14(8), 2866–2872 CrossRef CAS PubMed.
- R. Rousseau, E. Schreiner, A. Kohlmeyer and D. Marx, Temperature-Dependent Conformational Transitions and Hydrogen-Bond Dynamics of the Elastin-Like Octapeptide GVG(VPGVG): A Molecular-Dynamics Study, Biophys. J., 2004, 86(3), 1393–1407 CrossRef CAS PubMed.
- S. H. Chong, P. Chatterjee and S. Ham, Computer Simulations of Intrinsically Disordered Proteins, Annu. Rev. Phys. Chem., 2017, 68, 117–134 CrossRef CAS PubMed.
- K. J. Bari and D. D. Prakashchand, Fundamental Challenges and Outlook in Simulating Liquid–Liquid Phase Separation of Intrinsically Disordered Proteins, J. Phys. Chem. Lett., 2021, 12(6), 1644–1656 CrossRef CAS PubMed.
- N. K. Li, F. G. Quiroz, C. K. Hall, A. Chilkoti and Y. G. Yingling, Molecular Description of the LCST Behavior of an Elastin-Like Polypeptide, Biomacromolecules, 2014, 15(10), 3522–3530 CrossRef CAS PubMed.
- S. Rauscher and R. Pomès, The liquid structure of elastin, eLife, 2017, 6, e26526 CrossRef PubMed.
- T. I. Morozova, N. A. García, O. Matsarskaia, F. Roosen-Runge and J. L. Barrat, Structural and Dynamical Properties of Elastin-Like Peptides near Their Lower Critical Solution Temperature, Biomacromolecules, 2023, 24(4), 1912–1923 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis and R. O. Dror, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field, Proteins:Struct., Funct., Bioinf., 2010, 78(8), 1950–1958 CrossRef CAS PubMed.
- S. Piana, A. G. Donchev, P. Robustelli and D. E. Shaw, Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States, J. Phys. Chem. B, 2015, 119(16), 5113–5123 CrossRef CAS PubMed.
- C. Wehmeyer, M. K. Scherer, T. Hempel, B. E. Husic, S. Olsson and F. Noé, Introduction to Markov state modeling with the PyEMMA software [Article v1.0], Living Journal of Computational Molecular Science, 2019, 1(1), 5965 CrossRef.
- N. K. Li, Y. Xie and Y. G. Yingling, Insights into Structure and Aggregation Behavior of Elastin-like Polypeptide Coacervates: All-Atom Molecular Dynamics Simulations, J. Phys. Chem. B, 2021, 125(30), 8627–8635 CrossRef CAS PubMed.
- B. Zhao, N. K. Li, Y. G. Yingling and C. K. Hall, LCST Behavior is Manifested in a Single Molecule: Elastin-Like polypeptide (VPGVG)n, Biomacromolecules, 2016, 17(1), 111–118 CrossRef CAS PubMed.
- N. K. Li, S. Roberts, F. G. Quiroz, A. Chilkoti and Y. G. Yingling, Sequence Directionality Dramatically Affects LCST Behavior of Elastin-Like Polypeptides, Biomacromolecules, 2018, 19(7), 2496–2505 CrossRef CAS PubMed.
- U. Baul, M. Bley and J. Dzubiella, Thermal Compaction of Disordered and Elastin-like Polypeptides: A Temperature-Dependent, Sequence-Specific Coarse-Grained Simulation Model, Biomacromolecules, 2020, 21(9), 3523–3538 CrossRef CAS PubMed.
- T. Mabuchi, J. Kijima, Y. Yamashita, E. Miura and T. Muraoka, Coacervate Formation of Elastin-like Polypeptides in Explicit Aqueous Solution Using Coarse-Grained Molecular Dynamics Simulations, Macromolecules, 2023, 56(3), 794–805 CrossRef CAS.
- H. Zhang, V. Juraskova and F. Duarte, Modelling chemical processes in explicit solvents with machine learning potentials, Nat. Commun., 2024, 15(1), 6114 CrossRef CAS PubMed.
- J. Ribeiro, C. Ríos-Vera, F. Melo and A. Schüller, Calculation of accurate interatomic contact surface areas for the quantitative analysis of non-bonded molecular interactions, Bioinformatics, 2019, 35(18), 3499–3501 CrossRef CAS PubMed.
- H. X. Zhou, D. Kota, S. Qin and R. Prasad, Fundamental Aspects of Phase-Separated Biomolecular Condensates, Chem. Rev., 2024, 124(13), 8550–8595 CrossRef CAS PubMed.
- F. G. Quiroz and A. Chilkoti, Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers, Nat. Mater., 2015, 14(11), 1164–1171 CrossRef CAS PubMed.
- R. M. C. Vernon, P. A. Chong, B. Tsang, T. H. Kim, A. Bah and P. Farber, et al. Pi-Pi contacts are an overlooked protein feature relevant to phase separation, eLife, 2018, 7, 1–48 CrossRef PubMed.
- J. P. Brady, P. J. Farber, A. Sekhar, Y. H. Lin, R. Huang and A. Bah, et al. Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(39), E8194–E8203 CrossRef CAS PubMed.
- M. S. K. Khandaker, D. M. Dudek, E. P. Beers, D. A. Dillard and D. R. Bevan, Molecular modeling of the elastomeric properties of repeating units and building blocks of resilin, a disordered elastic protein, J. Mech. Behav. Biomed. Mater., 2016, 61, 110–121 CrossRef CAS PubMed.
- L. H. Kapcha and P. J. Rossky, A simple atomic-level hydrophobicity scale reveals protein interfacial structure, J. Mol. Biol., 2014, 426(2), 484–498 CrossRef CAS PubMed.
- T. Dannenhoffer-Lafage and R. B. Best, A Data-Driven Hydrophobicity Scale for Predicting Liquid-Liquid Phase Separation of Proteins, J. Phys. Chem. B, 2021, 125(16), 4046–4056 CrossRef CAS PubMed.
- G. L. Dignon, W. Zheng, R. B. Best, Y. C. Kim and J. Mittal, Relation between single-molecule properties and phase behavior of intrinsically disordered proteins, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(40), 9929–9934 CrossRef CAS PubMed.
- F. Pesce, A. Bremer, G. Tesei, J. B. Hopkins, C. R. Grace and T. Mittag, et al. Design of intrinsically disordered protein variants with diverse structural properties, Sci. Adv., 2024, 10(35), eadm9926 CrossRef CAS PubMed.
- J. Ji, M. S. Hossain, E. N. Krueger, Z. Zhang, S. Nangia and B. Carpentier, et al. Lipidation Alters the Structure and Hydration of Myristoylated Intrinsically Disordered Proteins, Biomacromolecules, 2023, 24(3), 1244–1257 CrossRef CAS PubMed.
- Z. Zhang, J. Ji, M. D. S. Hossain, B. Bailey, S. Nangia and D. Mozhdehi, Lipidation alters the phase-separation of resilin-like polypeptides, Soft Matter, 2024, 20(19), 4007–4014 RSC.
- S. Roberts, T. S. Harmon, J. L. Schaal, V. Miao, K. (Jonathan) Li and A. Hunt, et al. Injectable tissue integrating networks from recombinant polypeptides with tunable order, Nat. Mater., 2018, 17(12), 1154–1163 CrossRef CAS PubMed.
- F. G. Quiroz, N. K. Li, S. Roberts, P. Weber, M. Dzuricky and I. Weitzhandler, et al. Intrinsically disordered proteins access a range of hysteretic phase separation behaviors, Sci. Adv., 2019, 5(10), eaax5177 CrossRef CAS PubMed.
- T. Luo, L. He, P. Theato and K. L. Kiick, Thermoresponsive self-assembly of nanostructures from a collagen-like peptide-containing diblock copolymera, Macromol. Biosci., 2015, 15(1), 111–123 CrossRef CAS PubMed.
- A. Prhashanna, P. A. Taylor, J. Qin, K. L. Kiick and A. Jayaraman, Effect of Peptide Sequence on the LCST-Like Transition of Elastin-Like Peptides and Elastin-Like Peptide-Collagen-Like Peptide Conjugates: Simulations and Experiments, Biomacromolecules, 2019, 20(3), 1178–1189 CrossRef CAS PubMed.
- P. A. Taylor, H. Huang, K. L. Kiick and A. Jayaraman, Placement of tyrosine residues as a design element for tuning the phase transition of elastin-peptide-containing conjugates: Experiments and simulations, Mol. Syst. Des. Eng., 2020, 5(7), 1239–1254 RSC.
- D. Wu, N. Sinha, J. Lee, B. P. Sutherland, N. I. Halaszynski and Y. Tian, et al. Polymers with controlled assembly and rigidity made with click-functional peptide bundles, Nature, 2019, 574(7780), 658–662 CrossRef CAS PubMed.
- H. V. Zhang, F. Polzer and M. J. Haider, et al. Computationally designed peptides for self-assembly of nanostructured lattices[J], Sci. Adv., 2016, 2(9), e1600307 CrossRef PubMed.
- Q. Xu and J. Jiang, Machine Learning for Polymer Swelling in Liquids, ACS Appl. Polym. Mater., 2020, 2(8), 3576–3586 CrossRef CAS.
- J. Shi, M. J. Quevillon, P. H. A. Valença and J. K. Whitmer, Predicting Adhesive Free Energies of Polymer–Surface Interactions with Machine Learning, ACS Appl. Mater. Interfaces, 2022, 14(32), 37161–37169 CrossRef CAS PubMed.
- S. Glass, M. Schmidt, P. Merten, A. Abdul Latif, K. Fischer and A. Schulze, et al. Design of Modified Polymer Membranes Using Machine Learning, ACS Appl. Mater. Interfaces, 2024, 16(16), 20990–21000 CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |