
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeta-cyclodextrin formulation of a disulfide-bond disrupting agent for improved systemic exposure†
Zaafir M.
Dulloo
 a,
Ion
Ghiviriga
a,
Mary E.
Law
b,
Sarvesh K.
Verma
c,
Abhisheak
Sharma
c,
Brian K.
Law
*b and
Ronald K.
Castellano
*a
a,
Ion
Ghiviriga
a,
Mary E.
Law
b,
Sarvesh K.
Verma
c,
Abhisheak
Sharma
c,
Brian K.
Law
*b and
Ronald K.
Castellano
*a
aDepartment of Chemistry, University of Florida, PO BOX 117200, Gainesville, FL 32611-7200, USA. E-mail: castellano@chem.ufl.edu
bDepartment of Pharmacology & Therapeutics, University of Florida, PO BOX 100267, Gainesville, FL 32610-0267, USA. E-mail: bklaw@ufl.edu
cDepartment of Pharmaceutics, University of Florida, PO BOX 100495, Gainesville, FL 32610-0495, USA
First published on 21st May 2025
Abstract
Disulfide-bond disrupting agents (DDAs) are a class of cyclic thiosulfonates that have been shown to kill human epidermal growth factor receptor (HER) family-overexpressing breast cancer (BC) cells selectively and with no adverse side effects. Previous structure–activity relationships suggested a strong correlation between DDA lipophilicity and potency. In this study, we present the use of cyclodextrins (CDs) as molecular excipients to address the possible solubilizing drawback of increasingly lipophilic DDAs in oral administrations. The formulation of tcyDTDO, a potent second-generation DDA, with beta-cyclodextrin (BCD) and 2-hydroxypropyl-beta-cyclodextrin (HPB) was investigated. The choice of BCD as an optimal host over other CDs was guided by two in silico methods, namely: (1) host cavity volume estimations using a computational modeling approach and (2) binding energy (BE) calculations from simulations of different complexation geometries. A solid-state inclusion complex (IC) between tcyDTDO and BCD was prepared by kneading. Characterization by ATR-FTIR revealed positioning of tcyDTDO inside the cavity of BCD. Phase-solubility plots were constructed using NMR spectroscopy to measure the concentrations of host and guest in solution; a powerful technique that has yet to be exploited in the context of host–guest chemistry. The AL-type plots obtained pointed to the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with both BCD and HPB. BCD formed a stronger complex with tcyDTDO (Ka of 4090 M−1) although the solubility of tcyDTDO was enhanced by only 3-fold from an intrinsic solubility of 1.58 mM. Contrastingly, HPB displayed a lower affinity for tcyDTDO (Ka of 81 M−1) but resulted in a remarkable 90-fold increase in solubility with tcyDTDO concentrations approaching 150 mM. Encapsulation of tcyDTDO in both cases did not hinder its anti-cancer activity as they retained cytotoxicity against MDA-MB-468 (EGFR+) BC cells in vitro. More striking was the superior pharmacokinetic profile and systemic exposure of tcyDTDO observed in male Sprague-Dawley rats when formulated with BCD as indicated by an area under the concentration vs. time curve (AUC0–24) of 3150 ± 381 ng h mL−1. This work suggests a correlation between Ka and in vivo pharmacokinetics of DDAs following their complexation with CDs and provides an ameliorated approach for their oral administration in future animal studies.
:1 complexes with both BCD and HPB. BCD formed a stronger complex with tcyDTDO (Ka of 4090 M−1) although the solubility of tcyDTDO was enhanced by only 3-fold from an intrinsic solubility of 1.58 mM. Contrastingly, HPB displayed a lower affinity for tcyDTDO (Ka of 81 M−1) but resulted in a remarkable 90-fold increase in solubility with tcyDTDO concentrations approaching 150 mM. Encapsulation of tcyDTDO in both cases did not hinder its anti-cancer activity as they retained cytotoxicity against MDA-MB-468 (EGFR+) BC cells in vitro. More striking was the superior pharmacokinetic profile and systemic exposure of tcyDTDO observed in male Sprague-Dawley rats when formulated with BCD as indicated by an area under the concentration vs. time curve (AUC0–24) of 3150 ± 381 ng h mL−1. This work suggests a correlation between Ka and in vivo pharmacokinetics of DDAs following their complexation with CDs and provides an ameliorated approach for their oral administration in future animal studies.
Introduction
Cancer persists as a major health problem on the global front. According to the World Health Organization (WHO), cancer was responsible for an estimated 9.7 million deaths worldwide in 2022.1 Global predictions from the WHO foresee an increase of 10 million new cancer cases by 2040,1 which would likely result in an uptick in cancer fatalities. Breast cancer (BC) is an important contributor to cancer morbidity and mortality. BC remains the most widespread cancer in women with 2.3 million diagnoses and 670000 victims globally in 2022.2 In the United States (US), BC prevalence is just as critical. National statistics compiled by the American Cancer Society (ACS) in 2025 foresee BC as the most common cancer diagnosed in American women making up an estimated 32% of all cancer cases in this demographic, excluding skin cancers.3 The same report ranks BC as the second leading cause of cancer death in women after lung cancer. The ACS also notes a slight regression in the rate of decline in BC mortality (from 2% per year to 1% per year) accompanied by a steady increase in incidence since the mid-2000s,3 some numbers which accentuate the urgency of this public health issue.
BC is a heterogenous disease divided into four main molecular subtypes based on the levels of expression of estrogen and progesterone receptors and the human epidermal growth factor receptor-2 (HER2).4 BC heterogeneity is associated with different levels of aggressiveness and patient outcomes. HER2-enriched and triple-negative (TN) BCs have the worst prognoses.5,6 The HER2 receptor is overexpressed in roughly 15–25% of all BCs and is considered an attractive therapeutic target.7 For instance, current anti-BC drugs such as monoclonal antibodies and small molecule tyrosine kinase inhibitors inhibit HER2 through various mechanisms.8 TNBC, which accounts for approximately 10–15% of all BCs, is characterized by the absence of hormone receptors and HER2 overexpression.9 Due to this singularity in its molecular composition, TNBC does not present an obvious biomarker to guide TNBC-targeted therapies. However, it has been reported that the epidermal growth factor receptor (EGFR or HER1), the defining member of the EGFR family, is frequently overexpressed in TNBC and non-small cell lung cancer (NSCLC).10,11 Consequently, EGFR is a potential chemotherapeutic target that has inspired the emergence of new treatment strategies and anti-EGFR agents.12 However, these new therapeutic regimens primarily address NSCLC such that TNBC-specific treatments remain scarce and require further research and development.12,13 Another challenge associated with the treatment of BCs is the intrinsic and/or acquired resistance of tumors to existing drugs.14,15 Resistance results in reduced or short-lived therapeutic actions which eventually lead to poorer outcomes.
Amid the need for new treatment strategies, we have been developing a class of sulfur-containing compounds called disulfide-bond disrupting agents (DDAs). DDAs possess the unique ability to selectively kill EGFR-overexpressing (EGFR+) and HER2+ BC cells in vitro and suppress tumor growth in vivo with no adverse side effects.16,17 From lead optimization efforts starting with DTDO, depicted in Fig. 1, we identified the bicyclic derivative, tcyDTDO, as the most potent second-generation DDA candidate.18 The potency of tcyDTDO was further enhanced by structural modifications at strategic positions to generate more cytotoxic third-generation candidates such as dMtcyDTDO and dFtcyDTDO featuring methoxy and fluorine substituents, respectively.19 A condensed structure–activity relationship campaign established by our group is outlined in Fig. 1 and presents select DDAs across three generations. The improvement in cytotoxicity against the MDA-MB-468 (EGFR+) BC cell line, deduced from the smaller IC50 values, was partly attributed to elevated lipophilicity measured in clogP.19 The hypothesis pertaining to this cytotoxicity–lipophilicity interplay was supported by the complete inactivity of some DDAs from previous generations that contained acetoxy groups presumably enzymatically hydrolyzed into polar hydroxyl groups. More recently, this hypothesis was supported by the lack of activity of dHtcyDTDO.19
 | ||
| Fig. 1 Structure–activity relationships of some DDAs including (IC50) and [clogP] values. The potency of DTDO was improved by fusing a cyclohexane ring to its structure to generate tcyDTDO. Decorating the scaffold of tcyDTDO at strategic positions with methoxy and/or fluorine groups further improved its potency. Contrastingly, dHtcyDTDO, bearing two hydroxyl groups, was biologically inactive. This led to the realization of a cytotoxicity–lipophilicity interplay regarding the DDAs. | ||
According to the biopharmaceutics classification system (BCS), two key parameters that govern the extent of drug absorption and ultimately drug bioavailability, are drug dissolution rate, reflected in aqueous solubility, and permeability.20 From the theoretical basis established by Amidon and colleagues in 1995, oral drug candidates can be sorted into four different classes (I through IV) of varying high/low solubility and permeability characteristics, and an ideal drug candidate (class I) with most favorable pharmacokinetics exhibits both high aqueous solubility and high gastrointestinal permeability.20 If the increased lipophilicity of a compound can be beneficial in improving its cell membrane permeation and subsequent absorption from the gastrointestinal tract,21 the accompanied reduced aqueous solubility is disadvantageous as it can compromise the compound's overall pharmacokinetics. This issue may potentially arise in the next generations of DDAs especially if future synthetic efforts, guided by our previous SAR campaign, aim to further enhance DDA lipophilicity thereby creating compounds that fall under class II of the BCS scheme.20 Such compounds, characterized by high permeability and low solubility, would require higher dosages to compensate for their lower bioavailability or alternate administration routes. Elevated drug dosages are associated with potential complications.22
An effective solution to improving the aqueous solubility of lipophilic compounds is through a variety of formulation strategies. One such strategy involves complexation with solubilizing agents.23 Among the most well-substantiated supramolecular excipients used for this purpose are cyclodextrins (CDs), a set of macrocycles composed of D-glucose building blocks held together by α-1,4-glycosidic bonds.24 The three naturally-occurring CDs are alpha-, beta- and gamma-CD, or ACD, BCD and GCD, consisting of 6, 7 and 8 repeating glucose units, respectively.24 The ability of CDs to act as supramolecular hosts is a consequence of their distinct molecular shape, depicted in Fig. 2, which is comparable to a truncated cone. The narrow rim is constituted of primary OH groups whereas the wide rim of secondary OH groups. These outward pointing polar functional groups make the rims hydrophilic. CDs are, however, amphiphilic in nature due to the inward pointing methine hydrogens conversely forming a hydrophobic cavity.24 This molecular structure allows CDs to encapsulate structurally diverse hydrophobic guests in aqueous solutions to form inclusion complexes (ICs), a concept evoked for the first time by the German Chemist, Friedrich Cramer, in 1949.25 The degree of inclusion is dictated by both molecular shape and size complementarity between host and guest and can result in partial or complete encapsulation. In a host–guest supramolecular system, a dynamic equilibrium exists between complexed and non-complexed (or “free”) forms and is defined by an association constant, Ka, which reflects on the strength of the IC.
 | ||
| Fig. 2 3-D representation of the molecular structure of CD. CDs adopt the shape of truncated cones of varying dimensions based on the number of glucopyranose units (6, 7, or 8). CDs are amphiphilic due to the presence of both hydrophilic and hydrophobic sites. | ||
Aside from improving aqueous solubility, complexation with CDs offers additional benefits such as bioavailability and metabolic stability enhancements, controlled release, toxicity reduction and taste- and odor-masking.26 Moreover, ACD, BCD, and GCD are readily available from numerous chemical vendors. All three native CDs exhibit well-substantiated favorable safety profiles and no pathological evidence of systemic toxicity following oral administration.27–30 As a result, ACD, BCD, and GCD are categorized as “generally recognized as safe” or “GRAS” by the US Food and Drug Administration (FDA). An executive summary regarding the clinical safety of CDs as excipients via several administration routes is compiled by the European Medicines Agency.31 Since their discovery in the late 19th century by the French Chemist Antoine Villiers,25 natural CDs have been modified structurally to provide derivatives with altered physicochemical properties fine-tuned for certain applications or with complexing capabilities tailored to the inclusion of distinct guests.32 Given the confluence of advantages associated with this spectacular host family, both unmodified and modified CDs have found applications in several industries including agriculture, cosmetics, food, and pharmaceutical. From the approval of the first CD-formulated drug (prostaglandin E2 with BCD) in Japan in 1976 to the first US-approved drug-CD oral solution (itraconazole with HPB) in 1997, approximately 130 drugs formulated with CDs have been marketed worldwide as of 2022.33
The purpose of this work is to study the complexation of a second-generation DDA, tcyDTDO, with BCD and the derivative 2-hydroxypropyl-beta-cyclodextrin (HPB). We present herein the preparation of a tcyDTDO–BCD complex by kneading and its characterization by ATR-FTIR. The two tcyDTDO–CD complexes are investigated to assess the effects of CD formulation on the physicochemical and biological properties of tcyDTDO such as its aqueous solubility, in vitro cytotoxicity against the MDA-MB-468 (EGFR+) BC cell line, and in vivo pharmacokinetics in male Sprague-Dawley rats.
Results and discussion
Optimal host determination
To determine if ACD, BCD, or GCD would be the most favorable host for tcyDTDO, and other bicyclic DDAs, two in silico methods were used to simulate the kinetic and thermodynamic parameters of host–guest complexation. The first method was a direct comparison of the volume occupied by some bicyclic DDAs and the space available inside the cavities of the three CDs. DDA molecular structures were built in Hyperchem 8.0.6 using the “model build” function. Initial ACD, BCD, and GCD structures were retrieved from the Cambridge Crystallographic Data Center where they are deposited as BAJJAX (1105430), WEWTOJ (762697), and CYDXPL (1134600) respectively, using ConQuest 2.0.4 and edited with Mercury 4.3.0. All structures were minimized at the semi-empirical parametric method 3 (PM3) level.The minimized structures were imported into SwissPDB 4.1.0 where the desired volumes were generated as illustrated in Fig. 3. Both Hyperchem 8.0.6 and SwissPDB 4.1.0 are available free of charge from the internet. The volumes of tcyDTDO, dMtcyDTDO, and dFtcyDTDO were 180 Å3, 186 Å3, and 229 Å3, respectively, as shown in Table 1. Estimated cavity volumes of ACD, BCD, and GCD were generated using a simple and effective computational method inspired by the work of Grabicki et al. in which benzenes and coronenes were used to cap a series of pyrenylenes during the mapping of their internal cavities by a probe sphere to prevent the latter from exiting prematurely.34 Employing a similar strategy, described step-by-step in the ESI,† the estimated cavity volumes of ACD, BCD, and GCD were calculated as 175 Å3, 261 Å3, and 429 Å3 respectively, and are in good agreement with literature-reported CD cavity volumes (Table 1).24 Given the limitations associated with the calculation of cavity sizes,35,36 the method employed here should be well appreciated for its simplicity, accessibility, and accuracy. However, this method does not report on cavity shape, and shape complementarity is another important contributor to favorable host–guest complexation next to cavity size.
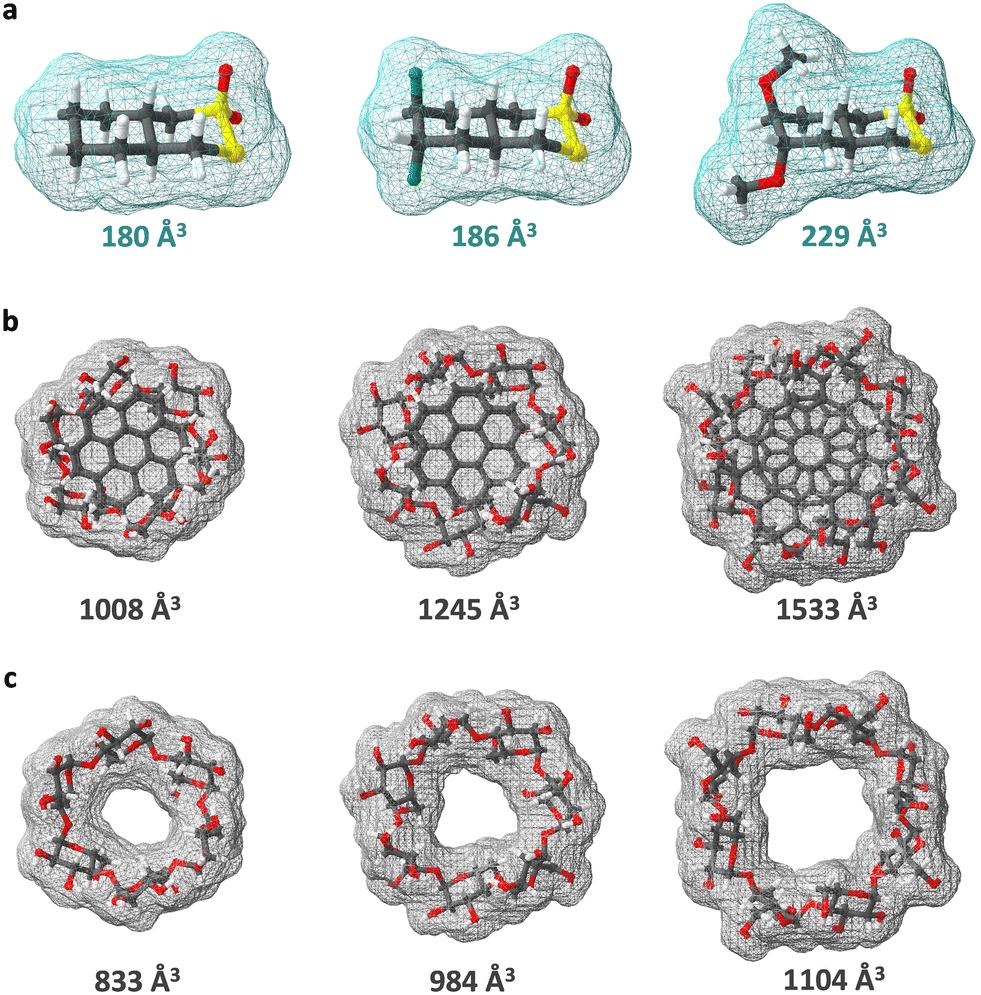 | ||
| Fig. 3 Van der Waals surfaces based on fixed aromatic radii generated using SwissPDB 4.1.0. (a) TcyDTDO, dFtcyDTDO, and dMtcyDTDO (left to right). (b) Alpha-CD (ACD), beta-CD (BCD), and gamma-CD (GCD) with coronene and/or circumcoronene positioned near the rims to form closed surfaces (left to right). (c) ACD, BCD, and GCD (left to right). Cavity volumes of the CDs were obtained by subtracting (c) from (b). | ||
| Cavity volume (Å3) | ACD | BCD | GCD |
|---|---|---|---|
| a Previously reported CD cavity volumes can be found here.24 b Determined with the combined use of Hyperchem 8.0.6 and SwissPDB 4.1.0 following a method described in the ESI.† c PCs of some bicyclic DDAs calculated by dividing the volume of DDA by the estimated cavity volume of the appropriate CD. | |||
| Literaturea | 174 | 262 | 427 |
| This workb | 175 | 261 | 429 |
| Packing coefficient (PC)c | |||
|---|---|---|---|
| tcyDTDO | 1.03 | 0.69 | 0.42 |
| dFtcyDTDO | 1.06 | 0.71 | 0.43 |
| dMtcyDTDO | 1.31 | 0.88 | 0.53 |
From a study performed by Mecozzi and Rebek to investigate the physical basis behind host–guest recognition came a rule that predicts an ideal host–guest pair.37 The rule is based on the packing coefficient (PC) of the guest in the well-defined internal cavity of a host where PC is described as the volume fraction of the host's cavity occupied by the guest. The rule assumes full encapsulation of the guest by the host. For a variety of organic liquids and certain adamantane derivatives, Mecozzi and Rebek estimated a narrow range of PCs of 0.55 ± 0.09 for which encapsulation of the guests was optimal in various molecular capsules.37 From the PCs reported in Table 1, it can be inferred that the cavity of ACD is too small to fully encapsulate any of the three bicyclic DDAs (tcyDTDO, dMtcyDTDO, and dFtcyDTDO). Therefore, complexation between ACD and the concerned DDAs may be partial or in a non-1:1 fashion. For BCD, all the calculated PCs fall outside of the 0.55 ± 0.09 range with tcyDTDO being the closest at satisfying the rule with a PC of 0.69. Although this PC exceeds the proposed range, the largely non-polar cyclohexane-based structure of tcyDTDO can contribute some extra stabilization enthalpy through van der Waals interactions with the hydrophobic internal cavity of BCD. This additional stabilization may be enough to compensate for the tighter packing of tcyDTDO and any associated entropy loss. Indeed, favorable non-covalent interactions in supramolecular systems can accommodate for PCs of up to 70%.37 Lastly, for GCD, dMtcyDTDO is predicted to be an ideal guest with a PC of 0.53. From the calculated PCs, both BCD and GCD were nominated as potential hosts for the encapsulation of tcyDTDO.
Since volume considerations alone are not necessarily enough to evaluate the complexation capabilities of the CDs, complexation was simulated by running sequential geometry optimizations as a second method. In this approach, inspired by the work of Liu and Guo,38 Hyperchem 8.0.6 was used to simulate the complexation between tcyDTDO and ACD, BCD, and GCD to locate the global minima on the potential energy surfaces of the resulting complexes. The simulation was set up as shown in Fig. 4 with the thiosulfonate-end of tcyDTDO approaching the wide rim of the various CDs. To simulate complexation, tcyDTDO was moved along an axis (z-axis) that passes through the cavity of each CD from −10 Å to +10 Å with the origin taken as the center of the CD cavity. With each 1 Å step from the starting point to the end point, the supramolecular system was geometry optimized at the PM3 level. The binding energy (BE) of the system is estimated as the difference between the energy of the complex and the sum of energies of the free host and guest. All BEs obtained were negative, except for ACD, which indicates favorable stabilizing interactions and complexation. BE was plotted against distance along the z-axis and shows the highest magnitude of −29.2 kcal mol−1 recorded between tcyDTDO and BCD. The equation used to calculate BE accompanied by a plot of BE vs. distance along the z-axis are provided in the ESI.† This study proposes BCD as a better host for tcyDTDO compared to GCD which displayed lower, and hence less favorable, BEs. The unfavorable encapsulation of tcyDTDO by ACD was expected since the estimated surface volume of tcyDTDO is larger than the estimated cavity volume of ACD as highlighted earlier.
 | ||
| Fig. 4 Inclusion complexation as simulated using Hyperchem 8.0.6. This figure features tcyDTDO and BCD as an example. Simulations were carried out with AMBER99 and a dielectric of 78.4 to mimic water. | ||
Dynamic complexation is driven mainly by the classical hydrophobic effect and has two components: (1) full or partial encapsulation of the hydrophobic guest into the CD cavity and (2) dehydration of the hydrophobic guest molecule.39 Both processes can be thermodynamically favorable. The encapsulation of the guest is prompted by the formation of non-covalent interactions between host–guest and results in a decrease in enthalpy. As understood for the hydrophobic effect, the solvation of a hydrophobic molecule disrupts the bulk water medium and initiates the formation of ordered clathrate-like solvent cages. In this model, water molecules possess stronger and more rigid hydrogen bonds. Complexation of the guest by the host, and its subsequent removal from surrounding water molecules, or “dehydration”, restores the bulk water medium and results in an increase in entropy.40 Contrastingly, select cases where complexation follows a non-classical model have also been reported.41,42
Although explicit solvent and entropic effects were not part of the calculations, the trend in BE observed speaks reliably to host–guest complementarity and was independent of the orientations of the guest or host. The same trend was obtained when either extremity (thiosulfonate or cyclohexane) of tcyDTDO was used to approach either rim (narrow or wide) of the CDs. From the two in silico methods, BCD was determined as the optimal host for tcyDTDO. Parent tcyDTDO is the only bicyclic DDA that will be investigated as a model guest in the remaining sections of this paper due to its facile synthesis. The knowledge gained with tcyDTDO can be applied to more synthetically precious and more potent DDAs but can also be extended to other classes of small molecules.
Complexation characterization with ATR-FTIR spectroscopy
ATR-FTIR is a fast and non-destructive method that has been extensively used in the literature to characterize host–guest complexation in the solid state where changes such as wavenumber shifts and broadening and/or attenuation of infrared (IR) bands are direct indications of interactions between a host and a guest at the molecular level.43 These changes can also reveal which specific part (or parts) of a guest is participating in the IC formation.43 Solid inclusion complexes (KN) were prepared by kneading equimolar amounts of tcyDTDO and BCD with a mortar and pestle. Deionized water was added in portions to maintain a paste-like consistency while the mixture was thoroughly blended for 20–30 minutes. The resulting complex was placed in an oven at 60 °C overnight to remove any excess water. The ATR-FTIR spectra of neat tcyDTDO, neat BCD, and the KN complex were recorded on a Perkin Elmer Spectrum 1™ instrument equipped with a zinc selenide crystal tip.Fig. 5a is a superimposition of a section of the IR spectra of neat tcyDTDO and complexed tcyDTDO (KN). Neat tcyDTDO, which is composed mainly of C and H atoms, exhibited strong characteristic absorption bands at 713 cm−1, 773 cm−1, and 888 cm−1 attributed to S–O stretches,44 and at 1125 cm−1, 1253 cm−1, and 1294 cm−1 which are additional S–O stretches typically present in sulfone-containing compounds as surveyed by Schreiber,45 and at 2857 cm−1 and 2920 cm−1 corresponding to C–H stretches. Given that decalin, a saturated hydrocarbon consisting of two fused cyclohexane rings, does not exhibit many strong absorption bands in the lower frequency region,46 it can be assumed that most of the bands observed in the IR spectrum of tcyDTDO between 500 cm−1 and 1300 cm−1 belong to the cyclic thiosulfonate moiety. In KN, attenuation of the characteristic bands of tcyDTDO was observed, and some of the most pronounced changes are highlighted in Fig. 5a. The differences can be explained by a decrease in the ability of the bonds of tcyDTDO to bend and to stretch upon encapsulation by BCD.43 The reduced intensity of the bands was accompanied by slight changes in frequency. For instance, the S–O stretch at 773 cm−1 shifted to 776 cm−1 while characteristic sulfone bands shifted from 1125 cm−1 to 1132 cm−1 and 1294 cm−1 to 1309 cm−1. These shifts to higher wavenumber, although small, reflect on the increase in electron density around tcyDTDO resulting from its positioning within the electron-rich cavity of BCD.24,47
 | ||
| Fig. 5 (a) Superimposed ATR-FTIR spectra of neat tcyDTDO and kneaded tcyDTDO–BCD complex. The more pronounced differences are highlighted. (b) Superimposed ATR-FTIR spectra of neat BCD and kneaded tcyDTDO–BCD complex. The IR spectrum of the complex bears more resemblance to the IR spectrum of neat BCD. The combined results from (a) and (b) point to the successful encapsulation of tcyDTDO by BCD. | ||
The IR spectra of neat BCD and KN are overlaid in Fig. 5b. Neat BCD displayed prominent absorption bands around 3310 cm−1 (O–H stretching), 2930 cm−1 (C–H stretching in the pyranoid ring), 1645 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1153 cm−1 (asymmetric stretching of the glycosidic C–O–C bridge), and 1025 cm−1 (CH2–OH vibration).47 The spectrum of KN bears more resemblance to the spectrum of neat BCD than neat tcyDTDO which is another indication that tcyDTDO is located inside the cavity of BCD. Overall, the combined changes point strongly to the successful encapsulation of tcyDTDO by BCD and the formation of an IC. Additional IR overlays supporting this conclusion are included in the ESI.†
O stretching), 1153 cm−1 (asymmetric stretching of the glycosidic C–O–C bridge), and 1025 cm−1 (CH2–OH vibration).47 The spectrum of KN bears more resemblance to the spectrum of neat BCD than neat tcyDTDO which is another indication that tcyDTDO is located inside the cavity of BCD. Overall, the combined changes point strongly to the successful encapsulation of tcyDTDO by BCD and the formation of an IC. Additional IR overlays supporting this conclusion are included in the ESI.†
Phase-solubility studies
Phase-solubility plots are useful in analyzing and understanding host–guest complexation events as they report on the solubilizing capacity of the host, the complexation stoichiometry, and the strength of the association between host–guest defined by an association constant, Ka. To construct the phase-solubility plots in Fig. 6a and b, the principles delineated by Higuchi and Connors were adopted with some modifications.48 To measure the concentrations of a substrate and an excipient in solution, a recently developed method was applied which utilizes nuclear magnetic resonance (NMR) spectroscopy with water suppression to measure the aqueous solubility of organic compounds.49 Deionized water, with a calculated concentration of 55.3 M−1 at 25.0 °C, is set as a reference and the concentrations of both the substrate and the excipient are determined using the PULCON relationship.50 This creative approach was proven to have the same accuracy, precision, and range as the gold-standard “shake-flask” method with several added benefits.49 Additionally, its validity was confirmed in the same study by comparing the experimental aqueous solubilities of a variety of organic compounds, both solids and liquids, to literature-reported ones.49 This work is the first reported instance, to the best of our knowledge, where this technique is applied in the construction of phase-solubility plots. | ||
| Fig. 6 A L-Type phase-solubility plots of tcyDTDO with BCD and the more soluble derivative 2-hydroxypropyl-beta-cyclodextrin (HPB). (a) Ka between tcyDTDO and BCD was calculated as 4080 M−1 and a 3-fold increase in the solubility of tcyDTDO was achieved. (b) Ka between tcyDTDO and HPB was calculated as 81 M−1 and an increase in the solubility of tcyDTDO of almost 100-fold was achieved. | ||
From the above method, an AL-type solubility profile was obtained denoted by a linear increase in the water-solubility of tcyDTDO with increasing concentrations of BCD (Fig. 6a). According to Higuchi and Connors, an AL-type profile suggests the formation of soluble complexes between substrate and excipient resulting in an increase in the concentration of the substrate in solution.48 To collect the last data point in Fig. 6a, excess BCD was added to a saturated solution of tcyDTDO in an NMR tube. The mixture was subjected to prolonged agitation in a water bath kept constant at 25.0 °C. Solids consisting of neat undissolved tcyDTDO and BCD were allowed to settle at the bottom of the tube for over 48 hours after which measurements of concentrations were recorded by NMR. This data point represents the highest achievable concentrations of tcyDTDO (5.75 mM) and BCD (4.43 mM) in solution at 25.0 °C before the formation of supersaturated solutions. A similar approach was adopted to determine the intrinsic solubility, S0, of tcyDTDO in water (1.58 mM). This number was used as the first data points in Fig. 6a and b.
The slope of the trendline in Fig. 6a was 0.866, or less than unity, which indicates a 1:1 complexation stoichiometry between tcyDTDO and BCD.48 Although the intrinsic solubility of tcyDTDO in water at 25.0 °C can be extrapolated as 1.62 mM from the plot, the value of 1.58 mM determined experimentally was used instead in the calculation of Ka. With a slope of 0.866 and an intrinsic solubility of 1.58 mM, Ka was calculated as 4090 M−1 revealing a high affinity between tcyDTDO and BCD. All Ka calculations are provided in the ESI.† Upon complexation with BCD, the water-solubility of tcyDTDO was improved by 3.5-fold, a modest enhancement as a direct consequence of the poor intrinsic solubility of BCD in water. Indeed, out of the three naturally occurring CDs, BCD is the least soluble (18.5 mg mL−1) compared to ACD (145 mg mL−1) and GCD (232 mg mL−1) at 25.0 °C.24 In BCD, intramolecular hydrogen-bonds between the neighboring C2–OH and C3–OH of each glucopyranose monomer, labeled in Fig. 2 as part of the introduction, results in a complete hydrogen bond network or “belt” which confers additional rigidity to BCD and prevents intermolecular hydrogen bonds with water.24
To further enhance the solubility of tcyDTDO upon complexation, BCD was replaced with a derivative with better solubilizing ability, namely 2-hydroxypropyl-beta-cyclodextrin (HPB) in which the intramolecular hydrogen bond “belt” is incomplete because of the lack of hydrogens at randomly substituted positions. The solubility of HPB is advertised as roughly 45 g per 100 mL of water by Millipore Sigma®. The AL-type profile of Fig. 6b indicates that a 1:1 complex was formed between tcyDTDO and HPB. Although the calculated Ka of 81 M−1 is two orders of magnitude lower than in the previous case, indicating a much weaker association, the solubility of tcyDTDO was significantly enhanced by almost 100-fold as expected. At the supersaturation point, the highest achievable concentrations of tcyDTDO and HPB are 147 mM and 1325 mM in solution, respectively, at 25.0 °C. The lower affinity of HPB for tcyDTDO can be justified by sterics caused by the number, position, and bulkiness of the substituents hindering the entry of tcyDTDO into the cavity of HPB, a general phenomenon noted by Pattarino and co-workers.51
Biological evaluation of tcyDTDO–CD complexes
Stock solutions of the complexes for biological evaluation were prepared using the traditional “shake-flask” method. The required amounts of tcyDTDO, BCD, and HPB were determined from the phase-solubility plots in the previous section. Briefly, solid CD was added in successive portions to a saturated solution of tcyDTDO in deionized water. After every portion, the mixture was agitated gently with a stir bar at room temperature until all the CD was added. In cases where a slight excess of tcyDTDO or CD was used, creating a suspension, the resulting solution was passed through a 0.45 μm syringe filter to remove any solids. Final stock solutions of the formulations contained tcyDTDO (5.75 mM)/BCD (4.43 mM) and tcyDTDO (147 mM)/HPB (1325 mM) from which the desired concentrations of tcyDTDO for biological studies were achieved by serial dilutions. Solutions of neat BCD and neat HPB were also prepared as negative controls.MTT cell viability assay
From the MTT assay in Fig. 7, treatment of the MDA-MB-468 (EGFR+) BC cell line with neat BCD and neat HPB, up to concentrations as high as 100 μM, did not have any significant adverse effect on cell viability. This observation is in accordance with the low toxicity profiles of BCD and HPB.52 Contrastingly, treatment of cells with both formulations resulted in cancer cell death. The observed in vitro cytotoxicity associated with the formulations suggests that tcyDTDO retains its anti-cancer activity following complexation with BCD and HPB. By applying a sigmoidal fit in Origin 8.5 to the MTT assay data, IC50 values were generated and compared. From Table 2, neat tcyDTDO displayed the highest potency (IC50 of 1.01 μM) followed by the tcyDTDO–BCD complex (IC50 of 2.74 μM) and the tcyDTDO–HPB complex (IC50 of 4.40 μM). CD encapsulation has been shown to mitigate the in vitro cytotoxicity of some bioactive compounds.53 Therefore, we expected the complexes to have lower potency than neat tcyDTDO. | ||
| Fig. 7 MTT viability assays of MDA-MB-468 (EGFR+) cells treated with the indicated compounds or formulations at the specified concentrations. Each data point is the mean of 10 replicates ± standard deviation. | ||
| Pharmacokinetic parameter | tcyDTDO–BCD | tcyDTDO–HPB | tcyDTDO–NaCMC |
|---|---|---|---|
| C max: maximum concentration of tcyDTDO; Tmax: time taken to reach Cmax; AUC0–24: area under the concentration–time curve over a 24 hour period (systemic exposure). IC50 values were calculated by applying a sigmoidal fit in Origin 8.5 to the MTT assay data provided in the ESI.† | |||
| T max (minutes) | 4.8 | 4.8 | 4.8 |
| C max (ng mL−1) | 6590.7 ± 899.0 | 1246.5 ± 43.7 | 3081.4 ± 278.6 |
| AUC0–24 (ng h mL−1) | 3153.1 ± 381.3 | 1135.1 ± 117.1 | 2066.6 ± 177.5 |
| IC50 (μM) | 2.74 | 4.40 | Not tested |
Since the Ka of tcyDTDO with BCD is two orders of magnitude higher than with HPB (4090 M−1vs. 81 M−1), we anticipated that in the case of BCD, a lower concentration of “free” tcyDTDO would be available resulting in a lower potency. It is a reasonable assumption that a stronger association with BCD can impact the release kinetics of tcyDTDO and mask its biological activity to a larger extent than HPB. Nonetheless, the IC50 values observed with both complexes are comparable suggesting that Ka does not have a significant influence on cytotoxicity in vitro but it might be of more significance in vivo where dilution is, in a sense, “infinite” after administration. Aihara and colleagues compared the difference between the in vitro and in vivo oral absorption of poorly soluble drugs formulated with CDs and noted some discrepancies in the behavior of the complexes.54 Similar inconsistencies in in vitro–in vivo correlations pertaining to potency, or other parameters, are expected and, if present, may be attributed to the large difference in Ka values.
Oral pharmacokinetics in Sprague-Dawley rats
Pharmacokinetic parameters of tcyDTDO were assessed in male Sprague-Dawley rats by oral gavage. Given the low intrinsic solubility of tcyDTDO in water, the corresponding volume of tcyDTDO solution that is required to attain the desired dose of 9 mg kg−1 exceeds the recommended dosing volume for oral administration in rats.55 Therefore, a standard sodium carboxymethyl cellulose (NaCMC) suspension was employed for dosing of tcyDTDO in water.The data from Table 2, derived from Fig. 8, shows that tcyDTDO exhibited a fast absorption 5 minutes after administration across all three systems. The maximum concentration (Cmax) of tcyDTDO measured in plasma samples after this time was highest for the BCD formulation at 6590 ± 899 ng mL−1. This concentration of tcyDTDO exceeds the levels recorded with HPB and NaCMC by 5-fold and 2-fold, respectively. The area under the concentration–time curve (AUC0–24) represents the total amount of tcyDTDO that reached systemic circulation over a period of 24 hours after oral administration and hence reports on the systemic exposure of tcyDTDO.56 AUC0–24 was highest for the BCD formulation at 3150 ± 381 ng h mL−1. This is almost a 3-fold increase in systemic exposure compared to the HPB formulation and a 1.5-fold increase compared to neat tcyDTDO. From similar reports in the literature, formulation with BCD and its different derivatives typically improves Cmax by roughly 2-fold and AUC0–t by 1.5- to 4-fold which is consistent with what we observed.57–60
 | ||
| Fig. 8 Plasma concentration–time profile of tcyDTDO in male Sprague-Dawley rats following oral administration of the three indicated systems at an equivalent tcyDTDO dose of 9 mg kg−1. Each data point is the mean of 4 replicates ± standard deviation. | ||
The Cmax attained with all three formulations were below the toxicity threshold that we have established previously.16,17 Despite the strong association between tcyDTDO and BCD, the Cmax was highest for the BCD formulation and was recorded only 5 minutes after administration. The largest differences in the average concentration of tcyDTDO were observed between the two CD formulations for up to 2 hours after administration. When comparing BCD-formulated tcyDTDO to neat tcyDTDO, although the Cmax attained with BCD was highest after 5 minutes, there was no significant difference in tcyDTDO concentration for the remainder of the experiment. Statistical analysis and supporting P-values are provided in the ESI.† Hence, by comparing the two formulations, it appears that a Ka surpassing 4000 M−1 does not hamper the release of tcyDTDO in vivo but instead enhances its overall pharmacokinetics.
DDAs are susceptible to oxidative metabolic degradation by cytochrome P450 (CYPs) at the least oxidized sulfur adjacent to the sulfone which can be further oxidized. From our previous findings, the oxidation states of the sulfurs play a pivotal role in governing DDA cytotoxicity.19 Consequently, complexation with CDs can shield DDAs from CYPs and enhance their metabolic stability. In this regard, a more tightly-bound complex can presumably offer elevated protection against oxidative metabolic degradation explaining the superior pharmacokinetic profile of the BCD formulation. From the combined pharmacokinetic parameters, we infer that BCD is the best excipient for the formulation of tcyDTDO and that a Ka of roughly 4000 M−1 leads to an improved pharmacokinetic profile in small animals. In similar studies, complexes with Ka approximating 5000 M−1 performed better in male Wistar rats compared to complexes with Ka nearing 40000 M−1.61 Our findings, taken together with previously reported ones, suggest that a range of Ka likely exists for the optimal pharmacokinetic behavior of a CD-formulated drug in vivo.
Lastly, neat tcyDTDO outperforming the HPB formulated-tcyDTDO during the first hour following administration can be rationalized as follows. First, although NaCMC was primarily used to suspend tcyDTDO in water for administration purposes, it remains a popular formulating agent, albeit with vastly different properties and for different applications than CDs.62 Second, given the high water-solubility of the HPB complex, to achieve a practical dosing volume, the latter was diluted by 10-fold with a saturated solution of HPB in water. A saturated solution of HPB was chosen for dilution, instead of water or plasma alone, to minimize alterations in the complexation event. However, a large excess of HPB is already present in the formulation at the supersaturation point; a proportion which is further skewed by dilution with HPB such that the propensity of finding “free” tcyDTDO is, we assume, greatly reduced regardless of the weaker Ka between tcyDTDO and HPB. This theory may explain the lower Cmax observed with the HPB formulation despite its highest solubility. A situation that may be resolved by using larger animals than Sprague-Dawley rats to bypass the need for sample dilution or by increasing the dose of tcyDTDO above 9 mg kg−1.
Conclusions
Shown from the present study, CDs can be utilized as molecular excipients for the second-generation bicyclic DDA, tcyDTDO, to improve its water-solubility and pharmacokinetic profile. Two in silico methods, both straightforward and accessible, have been presented to study host–guest complementarity. The two methods, one based on space considerations and the other on BEs, nominated BCD as the optimal host for tcyDTDO. Solid-state IC, prepared by kneading equimolar tcyDTDO and BCD, was characterized by ATR-FTIR for successful complexation. To the best of our knowledge, this work is the first to implement a newly developed NMR spectroscopy technique with water suppression to construct phase-solubility plots. The plots with AL profiles and slopes less than unity indicated the formation of 1:1 ICs between tcyDTDO and both CDs. The complexes exhibited contrasting physicochemical properties. For instance, BCD formed a much stronger complex with tcyDTDO compared to HPB likely due to steric considerations introduced by the HPB substituents. However, these same substituents greatly improved the solubility of the HPB-formulated tcyDTDO. A 100-fold increase in the solubility of tcyDTDO was achieved with HPB compared to a 3-fold increase with BCD. Both complexes retained their cytotoxicity against MDA-MB-468 (EGFR+) BC cells as demonstrated by MTT assays. Lastly, formulation with BCD resulted in a superior pharmacokinetic profile and increased systemic exposure of tcyDTDO in male Sprague-Dawley rats compared to HPB-formulated tcyDTDO. The findings herein will be extended to the formulation of next-generation DDAs with CDs for their oral administration in small animals for future biological evaluations.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Zaafir M. Dulloo and Ronald K. Castellano conceptualized the project. Zaafir M. Dulloo synthesized tcyDTDO, performed in silico studies, and prepared and characterized the ICs under the supervision of Ronald K. Castellano. Ion Ghiviriga carried out phase-solubility studies. Mary E. Law and Brian K. Law performed MTT assays. Sarvesh K. Verma conducted animal studies under the supervision of Abhisheak Sharma. Zaafir M. Dulloo wrote the manuscript, prepared the figures, tables, and graphical abstract, and assembled the article which was collectively reviewed and edited.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded in part by the following grants to Ronald K. Castellano and Brian K. Law: NIH/NCI R21 CA252400, NIH/NCI R21 CA277485, Florida Department of Health, James & Esther King Cancer Research Program grants 23K06 and 22K04, Florida Department of Health, Bankhead-Coley Research Program grant 23B03, and a grant from the Florida Breast Cancer Foundation. We acknowledge Cole Stearns for his help with the processing of graphics.References
- World Health Organization, International Agency for Research on Cancer, Press Release No. 345: Global cancer burden growing, amidst mounting need for services, 2024.
- World Health Organization, Breast Cancer, 2025.
- R. L. Siegel, T. B. Kratzer, A. N. Giaquinto, H. Sung and A. Jemal, Ca-Cancer J. Clin., 2025, 75, 10–45 CrossRef PubMed.
- E. Orrantia-Borunda, P. Anchondo-Nuñez, L. E. Acuña-Aguilar, F. O. Gómez-Valles and C. A. Ramírez-Valdespino, Breast Cancer, Exon Publications, Brisbane, 2022 Search PubMed.
- D. J. Slamon, G. M. Clark, S. G. Wong, W. J. Levin, A. Ullrich and W. L. McGuire, Science, 1987, 235, 177–182 CrossRef CAS PubMed.
- X. Li, J. Yang, L. Peng, A. A. Sahin, L. Huo, K. C. Ward, R. O'Regan, M. A. Torres and J. L. Meisel, Breast Cancer Res. Treat., 2017, 161, 279–287 CrossRef PubMed.
- H. J. Burstein, N. Engl. J. Med., 2005, 353, 1652–1654 CrossRef CAS PubMed.
- S. M. Swain, M. Shastry and E. Hamilton, Nat. Rev. Drug Discovery, 2023, 22, 101–126 CrossRef CAS PubMed.
- W. D. Foulkes, I. E. Smith and J. S. Reis-Filho, N. Engl. J. Med., 2010, 363, 1938–1948 CrossRef CAS PubMed.
- Y. Ohsaki, S. Tanno, Y. Fujita, E. Toyoshima, S. Fujiuchi, Y. Nishigaki, S. Ishida, A. Nagase, N. Miyokawa, S. Hirata and K. Kikuchi, Oncol. Rep., 2000, 7, 603–607 CAS.
- T. O. Nielsen, F. D. Hsu, K. Jensen, M. Cheang, G. Karaca, Z. Hu, T. Hernandez-Boussard, C. Livasy, D. Cowan, L. Dressler, L. A. Akslen, J. Ragaz, A. M. Gown, C. B. Gilks, M. Van De Rijn and C. M. Perou, Clin. Cancer Res., 2004, 10, 5367–5374 CrossRef CAS PubMed.
- I. Ahmad and H. M. Patel, Eur. J. Med. Chem., 2025, 284, 117178–117215 CrossRef CAS PubMed.
- T. Zubair and D. Bandyopadhyay, Int. J. Mol. Sci., 2023, 24, 2651–2670 CrossRef CAS PubMed.
- S. Moulder, Womens Health, 2010, 6, 821–830 Search PubMed.
- C. Marquette and L. Nabell, Curr. Treat. Options Oncol., 2012, 13, 263–275 CrossRef PubMed.
- R. B. Ferreira, M. E. Law, S. C. Jahn, B. J. Davis, C. D. Heldermon, M. Reinhard, R. K. Castellano and B. K. Law, OncoTargets Ther., 2015, 6, 10445–10459 CrossRef PubMed.
- M. E. Law, B. J. Davis, A. F. Ghilardi, E. Yaaghubi, Z. M. Dulloo, M. Wang, O. A. Guryanova, C. D. Heldermon, S. C. Jahn, R. K. Castellano and B. K. Law, Front. Pharmacol., 2022, 12, 792600 CrossRef PubMed.
- M. Wang, R. B. Ferreira, M. E. Law, B. J. Davis, E. Yaaghubi, A. F. Ghilardi, A. Sharma, B. A. Avery, E. Rodriguez, C. W. Chiang, S. Narayan, C. D. Heldermon, R. K. Castellano and B. K. Law, Oncogene, 2019, 38, 4264–4282 CrossRef CAS PubMed.
- A. F. Ghilardi, E. Yaaghubi, R. B. Ferreira, M. E. Law, Y. Yang, B. J. Davis, C. M. Schilson, I. Ghiviriga, A. E. Roitberg, B. K. Law and R. K. Castellano, ChemMedChem, 2022, 17, e202200165 CrossRef CAS PubMed.
- G. L. Amidon, H. Lennernäs, V. P. Shah and J. R. Crison, Pharm. Res., 1995, 12, 413–420 CrossRef CAS PubMed.
- J. A. Arnott and S. L. Planey, Expert Opin. Drug Discovery, 2012, 7, 863–875 CrossRef CAS PubMed.
- C. Le Tourneau, J. J. Lee and L. L. Siu, J. Natl. Cancer Inst., 2009, 101, 708–720 CrossRef CAS PubMed.
- B. Xie, Y. Liu, X. Li, P. Yang and W. He, Acta Pharm. Sin. B, 2024, 14, 4683–4716 CrossRef CAS PubMed.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1754 CrossRef CAS PubMed.
- N. Morin-Crini, S. Fourmentin, É. Fenyvesi, E. Lichtfouse, G. Torri, M. Fourmentin and G. Crini, Environ. Chem. Lett., 2021, 19, 2581–2617 CrossRef CAS.
- O. E. Nicolaescu, I. Belu, A. G. Mocanu, V. C. Manda, G. Rău, A. S. Pîrvu, C. Ionescu, F. Ciulu-Costinescu, M. Popescu and M. V. Ciocîlteu, Pharmaceutics, 2025, 17, 288–324 CrossRef PubMed.
- B. A. R. Lina and A. Bär, Regul. Toxicol. Pharmacol., 2004, 39, 27–33 CrossRef PubMed.
- B. A. R. Lina and A. Bär, Regul. Toxicol. Pharmacol., 2004, 39, 14–26 CrossRef PubMed.
- M. E. Bellringer, T. G. Smith, R. Read, C. Gopinath and P. Olivier, Food Chem. Toxicol., 1995, 33, 367–376 CrossRef CAS PubMed.
- I. C. Munro, P. M. Newberne, V. R. Young and A. Bär, Regul. Toxicol. Pharmacol., 2004, 39, 3–13 CrossRef PubMed.
- European Medicines Agency, Committee for Human Medicinal Products (CHMP), Cyclodextrins used as excipients, 2017.
- L. Szente and J. Szejtli, Adv. Drug Delivery Rev., 1999, 36, 17–28 CrossRef CAS PubMed.
- I. Puskás, L. Szente, L. Szőcs and É. Fenyvesi, Period. Polytech., Chem. Eng., 2023, 67, 11–17 CrossRef.
- N. Grabicki, K. Nguyen, S. Weidner and O. Dumele, Angew. Chem., Int. Ed., 2021, 60, 14909–14914 CrossRef CAS PubMed.
- B. C. Hamann, K. D. Shimizu and J. Rebek, Angew. Chem., Int. Ed. Engl., 1996, 35, 1326–1329 CrossRef CAS.
- C. N. Eid and D. J. Cram, J. Chem. Educ., 1993, 70, 349–351 CrossRef CAS.
- S. Mecozzi and J. Rebek, Chem. – Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- L. Liu and Q.-X. Guo, J. Inclusion Phenom. Macrocyclic Chem., 2004, 50, 95–103 CrossRef CAS.
- M. V. Rekharsky and Y. Inoue, Chem. Rev., 1998, 98, 1875–1917 CrossRef CAS PubMed.
- F. Franks, Faraday Symp. Chem. Soc., 1982, 17, 7–10 RSC.
- P. Setny, R. Baron and J. A. McCammon, J. Chem. Theory Comput., 2010, 6, 2866–2871 CrossRef CAS PubMed.
- S. B. Ferguson, E. M. Seward, F. Diederich, E. M. Sanford, A. Chou, P. Inocencio-Szweda and C. B. Knobler, J. Org. Chem., 1988, 53, 5593–5595 CrossRef CAS.
- R. Stancanelli, R. Ficarra, C. Cannavà, M. Guardo, M. L. Calabrò, P. Ficarra, R. Ottanà, R. Maccari, V. Crupi, D. Majolino and V. Venuti, J. Pharm. Biomed. Anal., 2008, 47, 704–709 CrossRef CAS PubMed.
- R. Venkataraghavan and T. R. Kasturi, Can. J. Chem., 1964, 42, 36–42 CrossRef.
- K. C. Schreiber, Anal. Chem., 1949, 21, 1168–1172 CrossRef CAS.
- B. G. Ershov, N. M. Panich, G. L. Bykov, A. L. Kustov, V. G. Krasovskiy and L. M. Kustov, Molecules, 2021, 26, 5565–5578 CrossRef CAS PubMed.
- A. Saifi, J. P. Joseph, A. P. Singh, A. Pal and K. Kumar, ACS Omega, 2021, 6, 4776–4782 CrossRef CAS PubMed.
- T. Higuchi and K. A. Connors, Adv. Anal. Chem. Instrum., 1965, 4, 117–212 CAS.
- I. Ghiviriga, Anal. Chem., 2023, 95, 2706–2712 CrossRef CAS PubMed.
- G. Wider and L. Dreier, J. Am. Chem. Soc., 2006, 128, 2571–2576 CrossRef CAS PubMed.
- F. Pattarino, L. Giovannelli, G. Giovenzana, M. Rinaldi and M. Trotta, J. Drug Deliv. Sci. Technol., 2005, 15, 465–468 CrossRef CAS.
- S. Gould and R. C. Scott, Food Chem. Toxicol., 2005, 43, 1451–1459 CrossRef CAS PubMed.
- U. M. Hanumegowda, Y. Wu and S. P. Adams, J. Pharm. Pharmacol, 2014, 2, 5–9 Search PubMed.
- R. Aihara, R. Messerschmid, M. Mizoguchi, K. Wada, K. Minami, H. Higashino, T. Takagi, M. Kataoka and S. Yamashita, Int. J. Pharm., 2021, 600, 120494–120501 CrossRef CAS.
- A. P. Brown, N. Dinger and B. S. Levine, Contemp. Top. Lab. Anim. Sci., 2000, 39, 17–21 CAS.
- A. Rescigno, A. Marzo and J. S. Beck, J. Pharmacokinet. Biopharm., 1991, 19, 473–482 CrossRef CAS PubMed.
- M. Wang, J. Jiang, Y. Cai, M. Zhao, Q. Wu, Y. Cui and C. Zhao, Biomed. Chromatogr., 2018, 32, e4364 CrossRef PubMed.
- S. M. Omar, F. Ibrahim and A. Ismail, Saudi Pharm. J., 2020, 28, 349–361 CrossRef CAS PubMed.
- Y. Zeng, Y. Lv, M. Hu, F. Guo and C. Zhang, Xenobiotica, 2022, 52, 718–728 CrossRef CAS PubMed.
- V. R. Maddula and R. S. R. Dachuru, AAPS PharmSciTech, 2023, 24, 115–132 CrossRef CAS PubMed.
- Y. Kubota, M. Fukuda, M. Muroguchi and K. Koizumi, Biol. Pharm. Bull., 1996, 19, 1068–1072 CrossRef CAS PubMed.
- M. S. Rahman, M. S. Hasan, A. S. Nitai, S. Nam, A. K. Karmakar, M. S. Ahsan, M. J. A. Shiddiky and M. B. Ahmed, Polymers, 2021, 13, 1345–1393 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Method to determine host cavity volume, binding energy equation and plot, inclusion complex preparation by kneading, ATR-FTIR overlays, phase-solubility plots, association constants, MTT cell viability assays, and pharmacokinetic studies in Sprague-Dawley rats performed in compliance with the Guidelines for Care and Use of Laboratory Animals of the University of Florida (UF) and approved by the UF Institutional Animal Care and Use Committee (IACUC). See DOI: https://doi.org/10.1039/d5md00334b |
| This journal is © The Royal Society of Chemistry 2025 |