
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment and clinical potential of 18F-PSiMA for prostate cancer PET imaging†
Lexi
Gower-Fry‡
 a,
Justin J.
Bailey‡§
b,
Melinda
Wuest
b,
Susan
Pike
b,
Alexey
Kostikov
cd,
Andreas
Dorian¶
a,
Carmen
Wängler
ef,
Frank
Wuest
ab and
Ralf
Schirrmacher
*ab
a,
Justin J.
Bailey‡§
b,
Melinda
Wuest
b,
Susan
Pike
b,
Alexey
Kostikov
cd,
Andreas
Dorian¶
a,
Carmen
Wängler
ef,
Frank
Wuest
ab and
Ralf
Schirrmacher
*ab
aDepartment of Chemistry, University of Alberta, Edmonton, Alberta, Canada. E-mail: schirrma@ualberta.ca
bDepartment of Oncology, University of Alberta, Edmonton, Alberta, Canada
cDepartment of Neurology and Neurosurgery, McGill University, Montreal, Quebec, Canada
dDepartment of Chemistry, McGill University, Montreal, Quebec, Canada
eBiomedical Chemistry, Clinic of Radiology and Nuclear Medicine, Medical Faculty Mannheim, Heidelberg University, Germany
fResearch Campus M2OLIE, Medical Faculty Mannheim, Heidelberg University, Germany
First published on 9th May 2025
Abstract
Prostate-specific membrane antigen (PSMA) is a key target for diagnosing prostate cancer through positron emission tomography (PET). While 68Ga-labeled PSMA compounds are widely used, 18F-labeled PSMA inhibitors have gained traction for clinical tumor imaging. We previously investigated PSMA-targeting compounds based on the Lys-urea-Glu motif, incorporating a silicon fluoride-acceptor (SiFA) and chemical auxiliaries to enhance in vivo biodistribution. This led to the development of 18F-PSiMA, a SiFA-based radiotracer with an optimized linker exhibiting favorable PSMA potency (IC50 = 154 ± 47 nM in LNCaP cells). 18F-PSiMA radiosynthesis with low to high concentrations of 18F and precursor achieved molar activities (Am) of 10.9–82.5 GBq μmol−1 and showed a 24–38% increase in tumor uptake in LNCaP tumors (SUV60min 1.56 ± 0.18; 7.23 ± 0.75% ID per g at lower Am and SUV60min 1.90 ± 0.29; 9.62 ± 1.29% ID per g at higher Am) compared to our previous lead, 18F-SiFA-Asp2-PEG3-PSMA. PSMA specificity was confirmed by a 20 ± 10% reduction in SUV60min upon co-injection with DCFPyl. These promising in vitro and in vivo results support further clinical translation of 18F-PSiMA for prostate cancer PET imaging.
Introduction
The detection of recurrent and metastatic prostate cancer is important for disease staging and planning an effective treatment regimen. Recent developments and combinations of different molecular imaging modalities such as positron emission tomography (PET) and magnetic resonance imaging (MRI) have led to an improved diagnosis and prognostic outcome for prostate cancer patients.1,2 Radiotracers targeting prostate-specific membrane antigen (PSMA), a glycoprotein that is highly overexpressed in prostate cancer, have significantly enhanced the diagnosis of this disease across different disease stages.3 The staging of high-risk prostate cancer, particularly in assessing biochemical recurrence in castration-resistant prostate cancer (CRPC), has greatly benefited from a wide range of clinically used PET and single-photon emission computed tomography (SPECT) radiotracers that target PSMA.4Recently, it has been clinically established that PSMA imaging is most beneficial to patients diagnosed early with intermediate to very-high-risk prostate cancer as well as biochemical recurrence and CRPC.3,4 Molecular imaging of PSMA using radiotracers bearing the structural Lys-urea-Glu motif provide the radiopharmacological tools for tumor staging and dosimetry for radionuclide-based prostate cancer therapy.5–8 Over the past decade an impressive variety of PSMA-binding tracers have been introduced into the clinic, most of which were either labeled with gallium-68 (68Ga, t1/2 = 68 min), a typically generator-produced radionuclide, or fluorine-18 (18F, t1/2 = 110 min), a cyclotron-based nuclide. All these compounds are small molecule inhibitors binding to the extracellular domain of PSMA. The synthesis of a diverse range of structurally similar compounds based on the Lys-urea-Glu motif has led to significant clinical advancements, resulting in FDA approvals for 68Ga-PSMA-11, 18F-DCFPyl, and most recently, 18F-rhPSMA-7.3.9–11 The latter is a radiohybrid radiotracer which utilizes the silicon fluoride-acceptor (SiFA) technology for 18F-radiolabeling, while also containing a DOTAGA chelator for labeling with 68Ga or therapeutic radiometals. A range of promising PSMA-targeting radiotracers, including 18F-PSMA-1007, 68Ga-PSMA-16 and 68Ga-PSMA-I&T, are currently in clinical trials, expanding the scope of diagnostic agents for PSMA imaging.5,12,13 Recently disclosed 18F-labeled tracers such as 18F-CTT1057, 18F-JK-PSMA-7, 18F-FSU-880 and 18F-AlF-PSMA-11 suggest a shift from 68Ga-labeled tracers to 18F-fluorinated radiopharmaceuticals, driven by the superior imaging resolution of 18F-based radiopharmaceuticals and the high cost and limited availability of approved 68Ge/68Ga-generator systems.14–17
The SiFA labeling concept, which utilizes isotopic exchange (IE) of nascent F-19 with [18F]fluoride, represents an important addition to the expanding array of 18F-labeling techniques. This method has been validated for human molecular imaging applications, as demonstrated by the introduction of 18F-SiTATE, a somatostatin receptor-binding peptide labeled via the SiFA approach.18–25 This peptide is clinically used in the diagnosis of neuroendocrine tumors, meningiomas, and, more recently, in cases of lenticulostriatal ischemia.18–20,23,25–28 While advancing our line of SiFA-based PSMA radiotracers, we previously developed 18F-SiFA-Asp2-PEG3-PSMA (Fig. 1), our lead in the second generation of SiFA-PSMA radiotracers.29 The first generation of SiFA-PSMA radiotracers consisted of the SiFA moiety and Lys-urea-Glu motif, with or without a PEG linker.2918F-SiFA-Asp2-PEG3-PSMA (Fig. 1), with an IC50 of 125 nM, demonstrated the highest LNCaP tumor uptake among nine novel SiFA-bearing PSMA inhibitors when injected with a molar activity (Am) of up to 86 GBq μmol−1.29 Its uptake into LNCaP tumor bearing mice was favorable with an SUV60min of 1.18, comparable to that of 18F-PSMA-1007.5,29
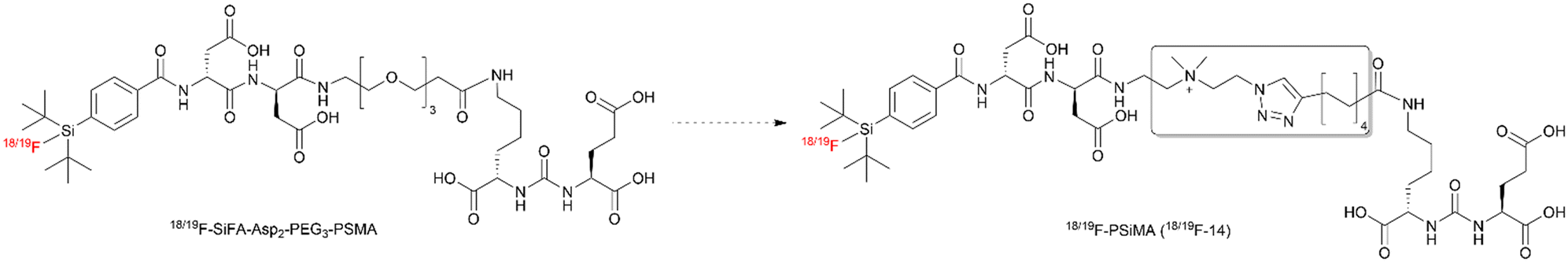 | ||
| Fig. 1 Structure of the previous second generation lead SiFA-PSMA radiotracer, 18/19F-SiFA-Asp2-PEG3-PSMA, compared to the novel third generation radiotracer, 18/19F-PSiMA (18/19F-14). | ||
In our previous investigations into structural auxiliaries to enhance SiFA-based imaging agents (e.g.18F-SiTATE), we identified several factors that influence biodistribution and non-specific binding. Key modifications, including the incorporation of aspartic acid, a PEG linker, and a positively charged ammonium group, were found to mitigate the inherently high lipophilicity of the SiFA functionality.27,29–31 This lipophilicity can negatively affect biodistribution, leading to elevated hepatobiliary clearance and non-specific binding.32 Notably, the addition of a quaternary ammonium group significantly influenced these parameters, reducing non-specific binding and improving overall biodistribution.
For the radiotracers 18F-SiTATE and 18F-SiFA-Asp2-PEG3-PSMA (Fig. 1), these modifications successfully redirected clearance towards a more renal clearance pathway.29,31 However, 18F-SiFA-Asp2-PEG3-PSMA still exhibited non-specific muscle uptake, which we hypothesized could be further reduced by decreasing the radiotracer's ability to passively diffuse through cell membranes.29 While PEG groups had minimal impact on tumor uptake compared to aspartic acid, the introduction of a quaternary ammonium salt showed promise in significantly enhancing the tumor-to-background ratio. A permanently positively charged motif was previously shown to decrease the lipophilicity of a SiFA-bearing molecule by a factor of 8, while retaining biological stability and high radiochemical yield (RCY) from the IE reaction with [18F]fluoride.30 The incorporation of the quaternary ammonium moiety alongside two aspartic acids, previously reported in 18F-SiTATE, resulted in higher tumor uptake, better tumor-to-background tissue ratios, and superior image quality compared to the gold standard, 68Ga-DOTATATE.21,27,31
Building on these findings, we have now synthesized 18F-PSiMA (18F-14, Fig. 1), our third generation radiotracer designed on a Lys-urea-Glu framework. This novel compound incorporates two aspartic acid groups, a quaternary ammonium group, and an alkyl linker, optimizing its structure for enhanced performance. To support its development, we established an efficient SiFA radiolabeling IE procedure, refined a robust purification protocol, and conducted comprehensive evaluations of 18F-PSiMA both in vitro and in vivo using the PSMA-expressing LNCaP prostate cancer model. These efforts underscore 18F-PSiMA's potential as a next-generation diagnostic tool in prostate cancer imaging.
Results and discussion
The 19F-PSiMA, 14 (Fig. 1), was synthesized following a protocol similar to previously reported methods, from three building blocks: the SiFA-Asp2 tag 3, a quaternary ammonium linker 7, and the Lys-urea-Glu targeting moiety 11 (Scheme 1).29 To construct the SiFA-Asp2 tag 3, an SiFA-NHS active ester 1 underwent an amidation reaction with t-butyl protected aspartic acid to produce SiFA-Asp1 intermediate 2 in 33% yield. A second aspartic acid was attached in a two-step method, first generating an activated ester using EDCI/NHS, followed by the addition of H-L-Asp(OtBu)-OH, to give the SiFA-Asp2 building block 3 in 52% yield after HPLC purification. | ||
| Scheme 1 Synthesis of PSiMA building blocks. aReagents and conditions: (a) H-L-Asp(OtBu)-OH, DIPEA, DMF, 33%; (b) step 1. NHS, EDCI, DMF, step 2. H-L-Asp(OtBu)-OH, DMF 26% over two steps; (c) NaN3, deionized H2O, 80 °C, 86%; (d) 2-(Boc-amino)ethyl bromide, MeCN; (e) 2 M HCl, Et2O, 71% over two steps; (f) step 1. P-Nitrophenyl chloroformate, DIPEA, CH2Cl2; step 2. H-Lys(Z)-OtBu, 86% over two steps; (g) H2, Pd/C, MeOH, 88%; (h) 10-undecynoic acid, HBTU, DIPEA, DMF, 70%. | ||
The quaternary ammonium linker 7 was synthesized with an azide and an amine moiety for orthogonal coupling to the SiFA-Asp23 and an alkyne-functionalized Lys-urea-Glu 11, respectively. Heating commercially available 2-chloro-N,N-dimethylethan-1-aminium chloride 4 in deionized water with sodium azide provided intermediate 5. Alkylation of 5 with N-Boc-2-bromoethyl-amine yielded 6, and subsequent deprotection using 2 M HCl in diethyl ether, gave the final quaternary ammonium building block 7, with a 71% yield, over two steps.
The PSMA binding motif, Lys-urea-Glu, was also retained in the structure of PSiMA. Synthesis of the Lys-urea-Glu PSMA binding motif 10 proceeded as previously reported via the cross-coupling of two commercially available tert-butyl- and CBZ-protected amino acids, H-Glu(OtBu)-OtBu 8 and H-Lys(Z)-OtBu, to produce intermediate 9.29,33,34 CBZ-deprotection using Pd/C hydrogenolysis provided the free amine 10 in 88% yield.
A hydrophobic linker adjacent to the PSMA-targeting motif was previously shown in literature to improve the binding affinity up to 60-fold due to its interactions with a lipophilic pocket a short distance away from the active site of PSMA.35 This also had a positive effect on the tumor uptake of PSMA radiotracers in LNCaP-tumor bearing mice.35 Therefore, a hydrophobic alkyl linker was utilized in the synthesis of 14. 10-undecynoic acid was converted to active ester with HBTU, then a coupling reaction was performed with the primary amine 10 to provide the Lys-urea-Glu building block 11 in 70% yield.
Synthesis of 14 was achieved through coupling of the SiFA-Asp23 carboxylic acid with the amine handle of the quaternary ammonium linker 7 (Scheme 2). To overcome solubility issues of 7, sonication and heat were utilized to assist the reaction. The SiFA-Asp2-QA intermediate 12 was successfully synthesized in 95% yield after HPLC purification. SiFA-Asp2-QA 12 was coupled to the Lys-urea-Glu alkyne via copper catalyzed alkyne–azide cycloaddition (CuAAC) to yield tert-butyl protected PSiMA 13. Without purification, the solvent was removed and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of TFA and CH2Cl2 was added for the final tert-butyl deprotection. After the deprotection, purification by HPLC gave the final product 14 in 20% yield, over two steps.
:1 mixture of TFA and CH2Cl2 was added for the final tert-butyl deprotection. After the deprotection, purification by HPLC gave the final product 14 in 20% yield, over two steps.
 | ||
| Scheme 2 Synthesis of 18F-PSiMA. aReagents and conditions: (i) 7, HBTU, DIPEA, DMF, 95%; (j) 11, CuSO4, sodium ascorbate, THF; (k) 1:1 TFA/CH2Cl2, 20% over two steps; (l) [18F]fluoride, K222, K2CO3, MeCN. | ||
The radiosynthesis of 18F-PSiMA was realized through 19F–18F IE of the SiFA moiety. To achieve this, the “four-drop method” was used to elute the [18F]fluoride from the QMA solid phase cartridge minimizing the amount of base in the reaction, obviating the use of oxalic acid to manage pH as with other SiFA labeling protocols.36 The final product, 18F-PSiMA, was obtained with an Am of 10.93 ± 3.70 GBq μmol−1 (n = 6), in 13 ± 8% radiochemical yield (RCY) (n = 6, uncorrected for radioactive decay). The total synthesis time was 90 min (from time of drying to formulation) and >99% radiochemical purity (RCP) was determined through quality control (QC) radio-HPLC.
The logD7.4 of 19F-PSiMA was determined as <−3, thus more hydrophilic than 18F-PSMA-1007 with −1.6, and similar to the previously reported second generation lead SiFA-PSMA radiotracer, 19F-SiFA-Asp2-PEG3-PSMA, with a logD7.4 of −3.03 as well as 19F,natGa-rhPSMA-7.3 logD7.4 of −3.2.29,37,38 A hydrolytic stability assay performed under physiological conditions (pH 7.4 and 37 °C) showed that 19F-PSiMA is sufficiently stable for in vivo application with very slow hydrolysis to the silanol (t1/2 of 177 ± 3 h).
Competitive displacement experiments in PSMA-expressing LNCaP cells revealed an IC50 value of 154 ± 47 nM (Fig. 2A, n = 6/2) which was similar to SiFA-Asp2-PEG3-PSMA (IC50 = 125 nM).2918F-PSiMA uptake into LNCaP cells resulted in 36 ± 8% radioactivity/mg protein (n = 6/2) after 120 min incubation time (Fig. 2B). An in vitro blocking study with inhibitor compound 2-PMPA resulted in 75% blocking of LNCaP cell uptake (Fig. 2C). Additionally, it was determined through a glycine wash of the LNCaP cells that ∼68% of the radiotracer is internalized (Fig. 2D, n = 6/1). Uptake into LNCaP prostate tumors in vivo was ∼24% higher compared to the previous analogue, 18F-SiFA-Asp2-PEG3-PSMA (Fig. 3A and D, SUV60min 1.56 ± 0.18 corresponding to 7.23 ± 0.75% ID per g (n = 3) versus 1.18 ± 0.12 and 5.64 ± 0.35% ID per g (n = 3) for 18F-SiFA-Asp2-PEG3-PSMA), despite having a significantly lower Am (10.9 GBq μmol−1vs. 86 GBq μmol−1).29 This tumor uptake was similar to that of 18F-PSMA-1007 with 8.0 ± 2.4% ID per g in mice, but lower than 18F-rhPSMA-7.3 with 18.3 ± 7.2% ID per g.5,38In vivo blocking studies using 300 μg of DCFPyL reduced tumor uptake by 20 ± 10% to a SUV60min value of 1.24 ± 0.06 (corresponding to 5.63 ± 0.25% ID per g, n = 3) (Fig. 3D), which is similar to the 32% blocking effect with 18F-SiFA-Asp2-PEG3-PSMA.29 Taken together, in vitro and in vivo blocking experiments have proven targeting efficacy of novel 18F-PSiMA despite some possible additional non-specific binding as experimentally observed.
 | ||
| Fig. 2 A) Inhibition-response curve against 18F-PSMA-1007. Data are shown as mean ± SEM from n = 6–9 data points from 1–2 experiments. B) In vitro LNCaP cell uptake of 18F-PSiMA over 120 min. Data are shown as mean ± SEM from n = 6 datapoints from two experiments. C) Blocking of in vitro18F-PSiMA cell uptake with PSMA inhibitor 2-PMPA (1 μM). Data are shown as mean ± SEM from n = 6 data points from one experiment. D) 18F-PSiMA internalization into LNCaP cells. Data are shown as mean ± SEM from n = 6 data points from one experiment. | ||
 | ||
| Fig. 3 Representative PET images as maximum intensity projections (MIP) of novel 18F-PSiMA (A and B) and 18F-PSMA-1007 (C) in LNCaP-tumor bearing mice at 60 min post injection. Corresponding time–activity curves (TACs) for radiotracer uptake into LNCaP tumor and muscle tissue (D–F) the salivary glands (G–I) and kidneys (J–L) in the absence and presence of 300 μg blocking DCFPyL. Data are shown as mean ± SEM from n = 3, n = 4 experiments. | ||
For direct comparison with a leading clinical PSMA-targeting tracer, 18F-PSMA-1007 was obtained from the routine clinical production done by the Edmonton Radiopharmaceutical Centre (ERC). The novel SiFA-PSMA compound 18F-PSiMA had a longer blood circulation time compared to clinical 18F-PSMA-1007, as well as a delayed non-target tissue and background clearance. However, it is not taken up by the salivary glands, resulting in a more prostate-specific binding to PSMA (Fig. 3G and I). This prostate-specific target binding is also supported by an around 4-fold lower kidney uptake (Fig. 3J and L), as mouse kidney tissue also express PSMA receptors.39 Blocking with DCFPyL confirmed PSMA binding of 18F-PSiMA and 18F-PSMA-1007 in PSMA expressing mouse kidney tissue (Fig. 3J and L), while blocking PSMA in salivary glands was detected with 18F-PSMA-1007 only (Fig. 3G and I). In vivo metabolism analysis revealed a 67–78% plasma availability and 7–16% protein binding of 18F-PSiMA over 5 to 60 min post injection (Fig. 4). No radioactive metabolites were detectable in blood plasma or urine over the entire 60 min post injection (Fig. 4). Taken together, 18F-PSiMA represents a metabolically stable radiotracer in vivo.
 | ||
| Fig. 4 A) Blood cell compartment distribution of 18F-PSiMA. Data are shown as mean ± SEM from n = 3 experiments. B) In vivo metabolic stability of 18F-PSiMA in normal control mouse. Representative HPLC chromatograms from blood plasma & urine samples. | ||
To increase Am, reaction parameters were optimized by reducing the precursor amount from 20 nmol to 5 nmol and increasing the starting activity of [18F]fluoride from 1–1.5 GBq to 4.1 GBq. The reaction volume was reduced from 200 μL to 50 μL, and the reaction vial was preheated to 80 °C to facilitate faster reaction initiation. These modifications resulted in an Am of 82.54 GBq μmol−1, with the resulting non-decay corrected RCY of 7.9% being slightly lower than the previous formulations of 13%.
In vivo PET imaging in LNCaP tumor-bearing mice, a well established PSMA-expressing prostate cancer model,29,39 demonstrated a 38% higher tumor uptake with the high Am18F-PSiMA tracer (SUV60min 1.90 ± 0.29; 9.62 ± 1.29% ID per g (n = 4)) (Fig. 3B and E) compared to the lower Am formulation (10.93 GBq μmol−1). Interestingly, no radioactivity was observed in the bladder with the higher Am tracer, with all radioactivity localized to the kidneys. This altered biodistribution suggests a potential pharmacological blocking of the renal uptake pathways at lower Am, leading to increased kidney clearance of the radioactivity. While the precise mechanism remains unclear, this observation aligns with previous findings on PSMA-targeting radiotracers exhibiting kidney-specific binding due to PSMA receptor expression in renal tissue.
Given the large amount of radioactivity required to achieve a higher Am, further studies using high Am18F-PSiMA should be conducted following translation to an automated synthesis platform.
Overall, the novel 18F-PSiMA radiotracer demonstrates superior tumor uptake compared to previous analogues revealing it as a promising candidate for a potential clinical translation. Optimization of the radiosynthetic procedure and automation would further increase its potential as a clinical radiopharmaceutical.
Notably, 18F-PSiMA offers an advantage over 18F-PSMA-1007 due to its negligible salivary gland uptake and lower kidney tissue uptake. Additionally, the prolonged blood circulation time of 18F-PSiMA may lead to enhanced delivery of the radiotracer to the target site reaching an SUV60min of 2.69 ± 0.12 in the blood pool versus 0.37 ± 0.04 for 18F-PSMA-1007 (both n = 3; Fig. S1†). This represents a significant difference between both investigated 18F radiotracers.
Moreover, further investigation into the non-specific binding of SiFA-PSMA compounds is warranted and will be investigated in the future.
Conclusions
18F-PSiMA, our third generation SiFA-PSMA radiotracer, which incorporates two aspartic acids, a hydrophilic quaternary ammonium cation, and an alkyl linker, demonstrates a 24–38% increase in in vivo tumor uptake in LNCaP tumors compared to the second generation lead SiFA-PSMA radiotracer, 18F-SiFA-Asp2-PEG3-PSMA.The significantly increased tumor uptake of 18F-PSiMA was accompanied by a negative side effect of enhanced non-specific binding which could be reduced at higher Am. This was particularly evident in the blocking studies with DCFPyL, where the blocking effect was much smaller compared to 18F-PSMA-1007. Nevertheless, 18F-PSiMA exhibited negligible salivary gland uptake and a much lower kidney tissue uptake, offering a distinct advantage over clinically used 18F-PSMA-1007. This also indicates a potential for further optimization and development of a radiotherapeutic derivative of 18F-PSiMA. With additional optimization of the radiosynthesis and further investigation into the nature of the non-specific binding, tumor uptake may be further enhanced while reducing non-specific binding, offering a new foundation for the development of SiFA-containing PSMA-targeting radiotracers.
Experimental
Materials and methods
Commercial reagents and solvents were purchased and used without further purification. 18F-PSMA-1007 was supplied by the Edmonton Radiopharmaceutical Centre (ERC) from routine clinical production. NMR spectra were recorded on a 700 Hz Agilent VNMRS four-channel, dual receiver spectrometer with a cryo-probe and 500 MHz Agilent/Varian VNMRS two-channel spectrometer with a 13C/1H dual cold probe. NMR spectra were calibrated in relation to the deuterated solvent signals. Multiplicities are indicated as s (singlet), d (doublet), dd (doublet of doublets), t (triplet), and m (multiplet). Analytical and semi-preparative HPLC (high-performance liquid chromatography) was conducted on an Agilent, Gilson, and Shimadzu with UV wavelength 254 nm and a radiodetector. HRMS (high-resolution mass spectrometry) was obtained using an Agilent Technologies 6220 oaTOF instrument. Purity of final compounds used for radiosynthesis, cell and animal studies were confirmed to be over 95% using HPLC.Chemical synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 139.0, 134.3, 134.2, 134.1, 126.1, 82.3, 82.2, 49.7, 37, 36.7, 28, 27.9, 27.2, 20.2, 20.1; 19F NMR (376 MHz, CDCl3): δ −188.64; HRMS (ESI/Q-TOF) m/z: [M − H]− calcd for C31H49FN2O8Si 623.3169, found 623.3156.
:2 → 1:1) to obtain 9 as a clear oil in 86% yield (4.35 g, 6.99 mmol). Rf (hexanes/EtOAc, 4:6) = 0.66. 1H NMR (500 MHz, CDCl3) δ 7.4–7.3 (m, 5H, Ar), 5.2–5.0 (m, 4H, CH2Ph, 2× NH), 4.4–4.3 (m, 2H, 2× CH), 3.2–3.1 (m, 2H, CH2), 2.28 (m, 2H, CH2), 2.05 (m, 1H, CH2), 1.6–1.5 (m, 3H, CH2), 1.44 (s, 18H, 2× OtBu), 1.42 (s, 9H, OtBu), 1.4–1.3 (m, 2H, CH2); 13C NMR (125 MHz, CDCl3) δ 172.4, 172.4, and 172.3 (3× COEster), 160.0 (COUrea), 157.0 (NHCO2), 136.8 (ArC–C), 128.5 (2× ArC–H), 128.1 (ArC–H), 128.0 (2× ArC–H), 82.9 (C(CH3)3), 82.8 (C(CH3)3), 81.8 (C(CH3)3), 66.6 (CH2Ph), 54.5 and 54.1 (CLys/Gluα), 40.8 (CLysε), 33.1, 32.5, 28.9, 28.4 (4× CH2), 28.7 (C(CH3)3), 28.3 (C(CH3)3), 28.2 (C(CH3)3), 23.6 (CLysγ); HRMS (ESI) m/z: [M + Na]+ calcd. For C32H51N3 644.3518, found 644.3516.
:1:1 CH2Cl2/MeOH/EtOAc + 1% TEA) to obtain 10 as a pink oil in 88% yield (291 mg, 0.60 mmol). Rf (8:1:1 CH2Cl2/MeOH/EtOAc + 1% TEA) = 0.3. 1H NMR (500 MHz, CD3OD) δ 4.2 (m, 2H, CHLys, CHGlu), 2.85 (m, 3H, CHLys, CHGlu), 2.35 (m, 2H, CHGlu), 2.05 (m, 1H, CH2Glu), 1.8 (m, 2H, CH2Glu, CH2Lys), 1.63 (m, 3H, CH2Lys), 1.45 (m, 27H, 3× tBu), 1.25 (m, 2H, CH2Lys); 13C NMR (125 MHz, CD3OD) δ 173.8 (COEster), 173.7 (COEster), 173.6 (COEster), 160.0 (COUrea), 82.9 (C(CH3)3), 82.8 (C(CH3)3), 81.8 (C(CH3)3), 54.5 and 54.1 (CLys/Gluα), 40.8 (CLysε), 33.1, 32.5, 28.9 (3× CH2), 28.7 (C(CH3)3), 28.3 (C(CH3)3), 28.2 (C(CH3)3), 23.6 (CLysγ); HRMS (ESI/Q-TOF) m/z: [M + H]+ calcd for C24H45N3O7 488.333, found 488.3333.
:1 → 3:7) to obtain 11 as a white solid in 70% yield (252 g, 0.39 mmol). Rf (EtOAc; 100%) = 0.8; 1H NMR (498 MHz, CDCl3) δ 5.79 (t, 1H, NHAmide), 5.12 (d, 2H, 2 × NHUrea), 4.31 (m, 2H, 2× CHLys/Gluα), 3.23 (m, 2H, CH2Lysε), 2.31 (m, 2H, CH2Gluγ), 2.17 (m, 4H, 2× CH2Alkyl), 2.08 (m, 1H, CH2Gluβ), 1.93 (t, 1H, CHAlkyne), 1.80 (m, 3H, CH2Glu/Lysβ), 1.62 (m, 3H, CH2Lysγ,δ), 1.50–1.69 (m, 4H, CH2Lysδ/Alkyl), 1.47 (s, 9H, OtBu), 1.47 (s, 9H, OtBu), 1.45 (s, 9H, OtBu), 1.30–1.41 (m, 9H, CH2Alkyl); 13C NMR (125 MHz, CDCl3) δ 173.4, 172.4, 172.4, 172.3, 156.6 (5× CO), 84.8 (CAlkyne), 82.1 (C(CH3)3), 81.8 (C(CH3)3), 80.6 (C(CH3)3), 68.1 (CHAlkyne), 53.3 and 53.0 (CLys/Gluα), 38.9, 36.8, 32.6, 31.6, 29.3, 29.2, 29.0, 28.9, 28.7, 28.5 and 28.3 (11× CH2), 28.1 (C(CH3)3), 28.0 (2× C(CH3)3), 25.8 (CH2Alkyl), 22.2 (CH2Lys), 18.4 (CH2Alkyl); HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C35H61N3O8Na 674.4351, found 674.4348.
O), 139.1 (ArC–Si, 2JC,F = 13.7 Hz), 135.9 (ArC–C), 135.3 (ArC–H, 3JC,F = 4.1 Hz), 127.7 (ArC–H), 82.7 and 82.6 (2× OC(CH3)3), 64.4 and 64.2 (2× NCH2), 52.5 and 52.4 (2× N(CH3)) 51.6 and 49.5 (2× CHAspα), 45.9 (N3CH2), 37.6 and 37.2 (2× CH2Aspβ), 34.9 (NHCH2), 28.4 and 28.3 (2× OC(CH3)3), 27.7 (2× SiC(CH3)3), 21.0 (2× SiC(CH3)3, 2JC,F = 12.1 Hz); HRMS (ESI/Q-TOF) m/z: [M+] calcd for C37H63FN7O7Si 764.4537, found 764.4534.
:1 CH2Cl2/TFA (4 mL), without purification. The reaction mixture was stirred for 24 h at room temperature. The crude mixture was concentrated then purified via HPLC (C18(2) column: Luna 5 μm 100 Å 250 mm × 10 mm; flow: 2.5 mL min−1; solvent A: H2O, solvent B: MeCN + 0.1% TFA, method: isocratic 45% B; tR = 16 min) to obtain 14 as a white powder, after lyophilization, in 20% yield (17.1 mg, 0.015 mmol). 1H NMR (500 MHz, CD3OD) δ 7.92 (s, 1H, 7.90, CHTriazole), 7.87 (d, 2H, J = 8.0 Hz, Ar), 7.73 (d, 2H, J = 8.0 Hz, Ar), 4.99 (t, 2H, J = 6.5 Hz, CH2Triazole), 4.87 (t, 1H, J = 6.5 Hz, CHAspα), 4.67 (t, 1H, J = 6.0 Hz, CHAspα), 4.30 (dd, 1H, J = 3, 5.5, CHGluα), 4.24 (dd, 1H, J = 3.5, 4.5, CHLysα), 3.70 (m, 2H, CH2), 3.61 (m, 2H, CH2), 3.18 (s, 6H, 2× NCH3), 3.16 (m, 2H, CH2), 3.03 (dd, 1H, J = 7, 9.5 Hz, CH2), 2.85 (m, 3H, CH2), 2.70 (t, 2H, J = 7.5 Hz, CH2), 2.41 (m, 2H, CH2), 2.15 (m, 3H, CH2), 1.86 (m, 2H, CH2), 1.67 (m, 3H, CH2), 1.61–1.39 (m, 6H, CH2), 1.32 (m, 8H, CH2), 1.06 (s, 18H, 2× SiC(CH3)3); 13C NMR (126 MHz, CD3OD) δ 176.6, 176.5, 176.3, 175.9, 175.3, 174.5, 173.7, 173.4, 170.5, 160.1 (10× CO), 150.2 (CTriazole), 139.8 (ArC–Si, 2JC,F = 13.4 Hz), 136.1 (ArC–C), 135.2 (ArC–H, 3JC,F = 4.2 Hz), 127.6 (ArC–H), 124.1 (CHTriazole), 64.5, 63.7 (2× NCH2), 54.1, 53.6 (2× CH2Aspα), 52.5 (2× N(CH3)), 52.3, 51.5 (2× CHLys/Gluα), 49.7 (CH2) 44.7 (CH2Triazole), 40.0 (CH2Lysε), 37.1, 36.0 (2× CH2Asp), 34.9 (CH2), 33.2 (CHLysβ), 31.1 (CH2Gluγ), 30.3, 30.2, 30.1, 30.1, 30.0, 29.9 (6× CH2), 28.9 (CH2Gluβ), 27.7, 27.0 (2× CH2), 26.2 (2× SiC(CH3)3), 24.0 (CH2), 21.0 (2× SiC(CH3)3, 2JC,F = 12.1 Hz); 19F NMR (376 MHz, CD3OD): δ −189.46, −76.9 (TFA); HRMS (ESI/Q-TOF) m/z: [M*]+ calcd for C52H84FN10O15Si 1135.5865, found 1135.5860.
000 rpm × 5 min). Precipitation of proteins in the supernatant was achieved by the addition of 2 volume parts of MeOH, and the samples were centrifuged again (13000 rpm for 5 min) to obtain the protein and plasma fraction. Fractions of blood cells, proteins, and plasma were measured using a HIDEX automated γ-counter (Hidex Oy, Turku, Finland) to determine radioactivity per sample. The clear plasma supernatant was injected into a Shimadzu HPLC system. The samples were analyzed by radioHPLC (C18(2) column: Luna 10 μm 100 Å 250 mm × 4.6 mm; flow: 1.0 mL min−1; solvent A: H2O + 0.2% TFA, solvent B: MeCN, method: 0–3 min 10% B, 10 min 30% B, 17 min 50% B, 23 min 70% B, 27–30 min 90% B).
:1) to reduce the viscosity, while the aliquot of an aqueous phase was diluted with PBS (1:19) to decrease the concentration. The samples were analyzed by HPLC to measure concentrations of the analyte and the logD of 19F-PSiMA was calculated as log(Coctanol/Cwater) factoring the ratio of solvents and dilutions of each phase. The measurement was repeated in triplicate.
O), 139.0, 134.3, 134.2, 134.1, 126.1, 82.3, 82.2, 49.7, 37, 36.7, 28, 27.9, 27.2, 20.2, 20.1; 19F NMR (376 MHz, CDCl3): δ −188.64; HRMS (ESI/Q-TOF) m/z: [M − H]− calcd for C31H49FN2O8Si 623.3169, found 623.3156.
:2 → 1:1) to obtain 9 as a clear oil in 86% yield (4.35 g, 6.99 mmol). Rf (hexanes/EtOAc, 4:6) = 0.66. 1H NMR (500 MHz, CDCl3) δ 7.4–7.3 (m, 5H, Ar), 5.2–5.0 (m, 4H, CH2Ph, 2× NH), 4.4–4.3 (m, 2H, 2× CH), 3.2–3.1 (m, 2H, CH2), 2.28 (m, 2H, CH2), 2.05 (m, 1H, CH2), 1.6–1.5 (m, 3H, CH2), 1.44 (s, 18H, 2× OtBu), 1.42 (s, 9H, OtBu), 1.4–1.3 (m, 2H, CH2); 13C NMR (125 MHz, CDCl3) δ 172.4, 172.4, and 172.3 (3× COEster), 160.0 (COUrea), 157.0 (NHCO2), 136.8 (ArC–C), 128.5 (2× ArC–H), 128.1 (ArC–H), 128.0 (2× ArC–H), 82.9 (C(CH3)3), 82.8 (C(CH3)3), 81.8 (C(CH3)3), 66.6 (CH2Ph), 54.5 and 54.1 (CLys/Gluα), 40.8 (CLysε), 33.1, 32.5, 28.9, 28.4 (4× CH2), 28.7 (C(CH3)3), 28.3 (C(CH3)3), 28.2 (C(CH3)3), 23.6 (CLysγ); HRMS (ESI) m/z: [M + Na]+ calcd. For C32H51N3 644.3518, found 644.3516.
:1:1 CH2Cl2/MeOH/EtOAc + 1% TEA) to obtain 10 as a pink oil in 88% yield (291 mg, 0.60 mmol). Rf (8:1:1 CH2Cl2/MeOH/EtOAc + 1% TEA) = 0.3. 1H NMR (500 MHz, CD3OD) δ 4.2 (m, 2H, CHLys, CHGlu), 2.85 (m, 3H, CHLys, CHGlu), 2.35 (m, 2H, CHGlu), 2.05 (m, 1H, CH2Glu), 1.8 (m, 2H, CH2Glu, CH2Lys), 1.63 (m, 3H, CH2Lys), 1.45 (m, 27H, 3× tBu), 1.25 (m, 2H, CH2Lys); 13C NMR (125 MHz, CD3OD) δ 173.8 (COEster), 173.7 (COEster), 173.6 (COEster), 160.0 (COUrea), 82.9 (C(CH3)3), 82.8 (C(CH3)3), 81.8 (C(CH3)3), 54.5 and 54.1 (CLys/Gluα), 40.8 (CLysε), 33.1, 32.5, 28.9 (3× CH2), 28.7 (C(CH3)3), 28.3 (C(CH3)3), 28.2 (C(CH3)3), 23.6 (CLysγ); HRMS (ESI/Q-TOF) m/z: [M + H]+ calcd for C24H45N3O7 488.333, found 488.3333.
:1 → 3:7) to obtain 11 as a white solid in 70% yield (252 g, 0.39 mmol). Rf (EtOAc; 100%) = 0.8; 1H NMR (498 MHz, CDCl3) δ 5.79 (t, 1H, NHAmide), 5.12 (d, 2H, 2 × NHUrea), 4.31 (m, 2H, 2× CHLys/Gluα), 3.23 (m, 2H, CH2Lysε), 2.31 (m, 2H, CH2Gluγ), 2.17 (m, 4H, 2× CH2Alkyl), 2.08 (m, 1H, CH2Gluβ), 1.93 (t, 1H, CHAlkyne), 1.80 (m, 3H, CH2Glu/Lysβ), 1.62 (m, 3H, CH2Lysγ,δ), 1.50–1.69 (m, 4H, CH2Lysδ/Alkyl), 1.47 (s, 9H, OtBu), 1.47 (s, 9H, OtBu), 1.45 (s, 9H, OtBu), 1.30–1.41 (m, 9H, CH2Alkyl); 13C NMR (125 MHz, CDCl3) δ 173.4, 172.4, 172.4, 172.3, 156.6 (5× CO), 84.8 (CAlkyne), 82.1 (C(CH3)3), 81.8 (C(CH3)3), 80.6 (C(CH3)3), 68.1 (CHAlkyne), 53.3 and 53.0 (CLys/Gluα), 38.9, 36.8, 32.6, 31.6, 29.3, 29.2, 29.0, 28.9, 28.7, 28.5 and 28.3 (11× CH2), 28.1 (C(CH3)3), 28.0 (2× C(CH3)3), 25.8 (CH2Alkyl), 22.2 (CH2Lys), 18.4 (CH2Alkyl); HRMS (ESI/Q-TOF) m/z: [M + Na]+ calcd for C35H61N3O8Na 674.4351, found 674.4348.
O), 139.1 (ArC–Si, 2JC,F = 13.7 Hz), 135.9 (ArC–C), 135.3 (ArC–H, 3JC,F = 4.1 Hz), 127.7 (ArC–H), 82.7 and 82.6 (2× OC(CH3)3), 64.4 and 64.2 (2× NCH2), 52.5 and 52.4 (2× N(CH3)) 51.6 and 49.5 (2× CHAspα), 45.9 (N3CH2), 37.6 and 37.2 (2× CH2Aspβ), 34.9 (NHCH2), 28.4 and 28.3 (2× OC(CH3)3), 27.7 (2× SiC(CH3)3), 21.0 (2× SiC(CH3)3, 2JC,F = 12.1 Hz); HRMS (ESI/Q-TOF) m/z: [M+] calcd for C37H63FN7O7Si 764.4537, found 764.4534.
:1 CH2Cl2/TFA (4 mL), without purification. The reaction mixture was stirred for 24 h at room temperature. The crude mixture was concentrated then purified via HPLC (C18(2) column: Luna 5 μm 100 Å 250 mm × 10 mm; flow: 2.5 mL min−1; solvent A: H2O, solvent B: MeCN + 0.1% TFA, method: isocratic 45% B; tR = 16 min) to obtain 14 as a white powder, after lyophilization, in 20% yield (17.1 mg, 0.015 mmol). 1H NMR (500 MHz, CD3OD) δ 7.92 (s, 1H, 7.90, CHTriazole), 7.87 (d, 2H, J = 8.0 Hz, Ar), 7.73 (d, 2H, J = 8.0 Hz, Ar), 4.99 (t, 2H, J = 6.5 Hz, CH2Triazole), 4.87 (t, 1H, J = 6.5 Hz, CHAspα), 4.67 (t, 1H, J = 6.0 Hz, CHAspα), 4.30 (dd, 1H, J = 3, 5.5, CHGluα), 4.24 (dd, 1H, J = 3.5, 4.5, CHLysα), 3.70 (m, 2H, CH2), 3.61 (m, 2H, CH2), 3.18 (s, 6H, 2× NCH3), 3.16 (m, 2H, CH2), 3.03 (dd, 1H, J = 7, 9.5 Hz, CH2), 2.85 (m, 3H, CH2), 2.70 (t, 2H, J = 7.5 Hz, CH2), 2.41 (m, 2H, CH2), 2.15 (m, 3H, CH2), 1.86 (m, 2H, CH2), 1.67 (m, 3H, CH2), 1.61–1.39 (m, 6H, CH2), 1.32 (m, 8H, CH2), 1.06 (s, 18H, 2× SiC(CH3)3); 13C NMR (126 MHz, CD3OD) δ 176.6, 176.5, 176.3, 175.9, 175.3, 174.5, 173.7, 173.4, 170.5, 160.1 (10× CO), 150.2 (CTriazole), 139.8 (ArC–Si, 2JC,F = 13.4 Hz), 136.1 (ArC–C), 135.2 (ArC–H, 3JC,F = 4.2 Hz), 127.6 (ArC–H), 124.1 (CHTriazole), 64.5, 63.7 (2× NCH2), 54.1, 53.6 (2× CH2Aspα), 52.5 (2× N(CH3)), 52.3, 51.5 (2× CHLys/Gluα), 49.7 (CH2) 44.7 (CH2Triazole), 40.0 (CH2Lysε), 37.1, 36.0 (2× CH2Asp), 34.9 (CH2), 33.2 (CHLysβ), 31.1 (CH2Gluγ), 30.3, 30.2, 30.1, 30.1, 30.0, 29.9 (6× CH2), 28.9 (CH2Gluβ), 27.7, 27.0 (2× CH2), 26.2 (2× SiC(CH3)3), 24.0 (CH2), 21.0 (2× SiC(CH3)3, 2JC,F = 12.1 Hz); 19F NMR (376 MHz, CD3OD): δ −189.46, −76.9 (TFA); HRMS (ESI/Q-TOF) m/z: [M*]+ calcd for C52H84FN10O15Si 1135.5865, found 1135.5860.
000 rpm × 5 min). Precipitation of proteins in the supernatant was achieved by the addition of 2 volume parts of MeOH, and the samples were centrifuged again (13000 rpm for 5 min) to obtain the protein and plasma fraction. Fractions of blood cells, proteins, and plasma were measured using a HIDEX automated γ-counter (Hidex Oy, Turku, Finland) to determine radioactivity per sample. The clear plasma supernatant was injected into a Shimadzu HPLC system. The samples were analyzed by radioHPLC (C18(2) column: Luna 10 μm 100 Å 250 mm × 4.6 mm; flow: 1.0 mL min−1; solvent A: H2O + 0.2% TFA, solvent B: MeCN, method: 0–3 min 10% B, 10 min 30% B, 17 min 50% B, 23 min 70% B, 27–30 min 90% B).
:1) to reduce the viscosity, while the aliquot of an aqueous phase was diluted with PBS (1:19) to decrease the concentration. The samples were analyzed by HPLC to measure concentrations of the analyte and the logD of 19F-PSiMA was calculated as log(Coctanol/Cwater) factoring the ratio of solvents and dilutions of each phase. The measurement was repeated in triplicate.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Lexi Gower-Fry: investigation, data curation, formal analysis, methodology, validation, visualization, writing. Justin J. Bailey: conceptualization, methodology, validation, investigation, formal analysis, writing. Melinda Wuest: investigation, formal analysis, writing, visualization. Susan Pike: investigation. Alexey Kostikov: investigation, writing. Andreas Dorian: investigation. Carmen Wängler: conceptualization, writing. Frank Wuest, Ralf Schirrmacher: conceptualization, funding acquisition, writing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Natural Science and Engineering Research Council of Canada (individual operating grant to RS) and the New Frontiers in Research Fund (grant NFRFT-2022-00269, to RS). This manuscript is dedicated to Prof. Dr. Klaus Jurkschat in profound recognition of his invaluable contributions to the advancement of SiFA radiochemistry. The authors would like to thank members of Dr. Frank Wuest's research group; Dr. Jenilee Woodfield, Jennifer Dufour, and Cody Bergman, for their assistance with the radiosynthesis, cell culture and in vitro cell experiments. The authors would also like to thank the Edmonton Radiopharmaceutical Centre (ERC) for 18F radionuclide production and for providing 18F-PSMA-1007 as well as the Vivarium at the Cross Cancer Institute for supporting the animal studies. In addition, the authors would like to thank members of Dr. Ralf Schirrmacher's group, specifically Yinglan Pu, Dr. Carolin Jaworski and Travis Kronemann for their assistance with the organic synthesis. Finally, we would like to thank the Alberta Cancer Foundation (ACF) for their financial support of this work.Notes and references
- L. Evangelista, F. Zattoni, G. Cassarino, P. Artioli, D. Cecchin, F. Dal Moro and P. Zucchetta, Eur. J. Nucl. Med. Mol. Imaging, 2021, 48, 859–873 CrossRef PubMed.
- M. A. Hoffmann, H. J. Wieler, C. Baues, N. J. Kuntz, I. Richardsen and M. Schreckenberger, Urology, 2019, 130, 1–12 CrossRef PubMed.
- S. Houshmand, C. Lawhn-Heath and S. Behr, Abdom. Radiol., 2023, 48, 3610–3623 CrossRef PubMed.
- M. Weber, C. Kurek, F. Barbato, M. Eiber, T. Maurer, M. Nader, B. Hadaschik, V. Grünwald, K. Herrmann, A. Wetter and W. P. Fendler, J. Nucl. Med., 2021, 62, 88–91 CrossRef CAS PubMed.
- J. Cardinale, M. Schäfer, M. Benešová, U. Bauder-Wüst, K. Leotta, M. Eder, O. C. Neels, U. Haberkorn, F. L. Giesel and K. Kopka, J. Nucl. Med., 2017, 58, 425–431 CrossRef CAS PubMed.
- Y. Chen, M. Pullambhatla, C. A. Foss, Y. Byun, S. Nimmagadda, S. Senthamizhchelvan, G. Sgouros, R. C. Mease and M. G. Pomper, Clin. Cancer Res., 2011, 17, 7645–7653 CrossRef CAS PubMed.
- M. Eiber, W. P. Fendler, S. P. Rowe, J. Calais, M. S. Hofman, T. Maurer, S. M. Schwarzenboeck, C. Kratowchil, K. Herrmann and F. L. Giesel, J. Nucl. Med., 2017, 58, 67S–76S CrossRef CAS PubMed.
- Z. Zhang, Z. Zhu, D. Yang, W. Fan, J. Wang, X. Li, X. Chen, Q. Wang and X. Song, Oncol. Lett., 2016, 12, 1001–1006 CrossRef CAS PubMed.
- U. Hennrich and M. Eder, Pharmaceuticals, 2021, 14, 713 CrossRef CAS PubMed.
- S. P. Rowe, K. J. Macura, E. Mena, A. L. Blackford, R. Nadal, E. S. Antonarakis, M. Eisenberger, M. Carducci, H. Fan, R. F. Dannals, Y. Chen, R. C. Mease, Z. Szabo, M. G. Pomper and S. Y. Cho, Mol. Imaging Biol., 2016, 18, 411–419 CrossRef CAS PubMed.
- M. Kroenke, L. Schweiger, T. Horn, B. Haller, K. Schwamborn, A. Wurzer, T. Maurer, H.-J. Wester, M. Eiber and I. Rauscher, J. Nucl. Med., 2022, 63, 1809–1814 CrossRef CAS PubMed.
- M. A. Green, G. D. Hutchins, C. D. Bahler, M. Tann, C. J. Mathias, W. Territo, J. Sims, H. Polson, D. Alexoff, W. C. Eckelman, H. F. Kung and J. W. Fletcher, Mol. Imaging Biol., 2020, 22, 752–763 CrossRef CAS PubMed.
- W. Cytawa, A. K. Seitz, S. Kircher, K. Fukushima, J. Tran-Gia, A. Schirbel, T. Bandurski, P. Lass, M. Krebs, W. Połom, M. Matuszewski, H.-J. Wester, A. K. Buck, H. Kübler and C. Lapa, Eur. J. Nucl. Med. Mol. Imaging, 2020, 47, 168–177 CrossRef CAS PubMed.
- S. C. Behr, R. Aggarwal, H. F. Vanbrocklin, R. R. Flavell, K. Gao, E. J. Small, J. Blecha, S. Jivan, T. A. Hope, J. P. Simko, J. Kurhanewicz, S. M. Noworolski, N. J. Korn, R. De Los Santos, M. R. Cooperberg, P. R. Carroll, H. G. Nguyen, K. L. Greene, B. Langton-Webster, C. E. Berkman and Y. Seo, J. Nucl. Med., 2019, 60, 910–916 CrossRef CAS PubMed.
- F. Dietlein, M. Hohberg, C. Kobe, B. D. Zlatopolskiy, P. Krapf, H. Endepols, P. Täger, J. Hammes, A. Heidenreich, B. Neumaier, A. Drzezga and M. Dietlein, J. Nucl. Med., 2020, 61, 202–209 CrossRef CAS PubMed.
- T. Saga, Y. Nakamoto, T. Ishimori, T. Inoue, Y. Shimizu, H. Kimura, S. Akamatsu, T. Goto, H. Watanabe, K. Kitaguchi, M. Watanabe, M. Ono, H. Saji, O. Ogawa and K. Togashi, Cancer Sci., 2019, 110, 742–750 CrossRef CAS PubMed.
- X. Li, M. Yu, J. Yang, D. Li, R. Li, J. Mao, C. Zuo, Z. Liang, Q. Li and C. Cheng, EJNMMI Rep., 2024, 8(1), 28 CrossRef PubMed.
- C. Wängler, L. Beyer, P. Bartenstein, B. Wängler, R. Schirrmacher and S. Lindner, EJNMMI Radiopharm. Chem., 2022, 7, 22 CrossRef PubMed.
- M. Unterrainer, S. C. Kunte, L. M. Unterrainer, A. Holzgreve, A. Delker, S. Lindner, L. Beyer, M. Brendel, W. G. Kunz, M. Winkelmann, C. C. Cyran, J. Ricke, K. Jurkschat, C. Wängler, B. Wängler, R. Schirrmacher, C. Belka, M. Niyazi, J.-C. Tonn, P. Bartenstein and N. L. Albert, Eur. J. Nucl. Med. Mol. Imaging, 2023, 50, 3390–3399 CrossRef CAS PubMed.
- R. S. Eschbach, M. Hofmann, L. Späth, G. T. Sheikh, A. Delker, S. Lindner, K. Jurkschat, C. Wängler, B. Wängler, R. Schirrmacher, R. Tiling, M. Brendel, V. Wenter, F. J. Dekorsy, M. J. Zacherl, A. Todica, H. Ilhan, F. Grawe, C. C. Cyran, M. Unterrainer, J. Rübenthaler, T. Knösel, T. Paul, S. Boeck, C. B. Westphalen, C. Spitzweg, C. J. Auernhammer, P. Bartenstein, L. M. Unterrainer and L. Beyer, Front. Oncol., 2023, 13, 992316 CrossRef CAS PubMed.
- C. Wängler, S. Niedermoser, J. Chin, K. Orchowski, E. Schirrmacher, K. Jurkschat, L. Iovkova-Berends, A. P. Kostikov, R. Schirrmacher and B. Wängler, Nat. Protoc., 2012, 7, 1946–1955 CrossRef PubMed.
- S. Lindner, C. Wängler, J. J. Bailey, K. Jurkschat, P. Bartenstein, B. Wängler and R. Schirrmacher, Nat. Protoc., 2020, 15, 3827–3843 CrossRef CAS PubMed.
- H. Ilhan, S. Lindner, A. Todica, C. C. Cyran, R. Tiling, C. J. Auernhammer, C. Spitzweg, S. Boeck, M. Unterrainer, F. J. Gildehaus, G. Böning, K. Jurkschat, C. Wängler, B. Wängler, R. Schirrmacher and P. Bartenstein, Eur. J. Nucl. Med. Mol. Imaging, 2020, 47, 870–880 CrossRef CAS PubMed.
- S. Lindner, M. Simmet, F. Gildehaus, K. Jurkschat, C. Wängler, B. Wängler, P. Bartenstein, R. Schirrmacher and H. Ilhan, Nucl. Med. Biol., 2020, 88–89, 86–95 CrossRef CAS PubMed.
- M. Unterrainer, S. Lindner, L. Beyer, F. Gildehaus, A. Todica, L. Mittlmeier, K. Jurkschat, C. Wängler, B. Wängler, R. Schirrmacher, J. Tonn, N. Albert, P. Bartenstein and H. Ilhan, Clin. Nucl. Med., 2021, 46(8), 667–668 CrossRef PubMed.
- L. Beyer, A. Gosewisch, S. Lindner, F. Völter, L. M. Mittlmeier, R. Tiling, M. Brendel, C. C. Cyran, M. Unterrainer, J. Rübenthaler, C. J. Auernhammer, C. Spitzweg, G. Böning, F. J. Gildehaus, K. Jurkschat, C. Wängler, B. Wängler, R. Schirrmacher, V. Wenter, A. Todica, P. Bartenstein and H. Ilhan, Eur. J. Nucl. Med. Mol. Imaging, 2021, 48, 3571–3581 CrossRef CAS PubMed.
- S. Niedermoser, J. Chin, C. Wängler, A. Kostikov, V. Bernard-Gauthier, N. Vogler, J.-P. Soucy, A. J. Mcewan, R. Schirrmacher and B. Wängler, J. Nucl. Med., 2015, 56, 1100–1105 CrossRef CAS PubMed.
- S. C. Kunte, L. M. Unterrainer, W. G. Kunz, M. Winkelmann, S. Lindner, K. Jurkschat, C. Wängler, B. Wängler, R. Schirrmacher, P. Bartenstein, C. Belka, C. Schichor, N. L. Albert and M. Unterrainer, Clin. Nucl. Med., 2024, 49(12), e689–e690 Search PubMed.
- J. Bailey, M. Wuest, M. Wagner, A. Bhardwaj, C. Wängler, B. Wängler, J. Valliant, R. Schirrmacher and F. Wuest, J. Med. Chem., 2021, 64(21), 15671–15689 CrossRef CAS PubMed.
- A. Kostikov, L. Iovkova, J. Chin, E. Schirrmacher, B. Wängler, C. Wängler, K. Jurkschat, G. Cosa and R. Schirrmacher, J. Fluorine Chem., 2011, 132(1), 27–34 Search PubMed.
- S. Litau, S. Niedermoser, N. Vogler, M. Roscher, R. Schirrmacher, G. Fricker, B. Wängler and C. Wängler, Bioconjugate Chem., 2015, 26(12), 2350–2359 CrossRef CAS PubMed.
- C. Wängler, A. Kostikov, J. Zhu, J. Chin, B. Wängler and R. Schirrmacher, Appl. Sci., 2012, 2, 277–302 CrossRef.
- A. Kozikowski, F. Nan, P. Conti, J. Zhang, E. Ramadan, T. Bzdega, B. Wroblewska, J. Neale, S. Pshenichkin and J. Wroblewski, J. Med. Chem., 2001, 44(3), 298–301 CrossRef CAS PubMed.
- A. Kozikowski, J. Zhang, F. Nan, P. Petukhov, E. Grajkowska, J. Wroblewski, T. Yamamoto, T. Bzdega, B. Wroblewska and J. Neale, J. Med. Chem., 2004, 47(7), 1729–1738 CrossRef CAS PubMed.
- M. Wirtz, A. Schmidt, M. Schottelius, S. Robu, T. Günther, M. Schwaiger and H.-J. Wester, EJNMMI Res., 2018, 8(1), 84 CrossRef PubMed.
- D. Connolly, J. Bailey, H. Ilhan, P. Bartenstein, C. Wängler, B. Wängler, M. Wuest, F. Wuest and R. Schirrmacher, J. Visualized Exp., 2020, 155, e60623 Search PubMed.
- S. Robu, A. Schmidt, M. Eiber, M. Schottelius, T. Günther, B. H. Yousefi, M. Schwaiger and H.-J. Wester, EJNMMI Res., 2018, 8(1), 30 CrossRef PubMed.
- A. Wurzer, M. Parzinger, M. Konrad, R. Beck, T. Günther, V. Felber, S. Färber, D. Di Carlo and H.-J. Wester, EJNMMI Res., 2020, 10(1), 149 Search PubMed.
- V. Bouvet, M. Wuest, J. J. Bailey, C. Bergman, N. Janzen, J. F. Valliant and F. Wuest, Mol. Imaging Biol., 2017, 19, 923–932 CrossRef CAS PubMed.
- Y. Yuan, W. Wu, S. Xu and B. Liu, Chem. Commun., 2017, 53, 5287–5290 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5md00275c |
| ‡ These authors contributed equally. |
| § Current address: TRIUMF, Vancouver, BC, Canada. |
| ¶ Current address: 48 Hour Discovery, Edmonton, AB, Canada. |
| This journal is © The Royal Society of Chemistry 2025 |