
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the in vitro anticancer potential of bis(imino)acenaphthene–N-heterocyclic carbene transition metal complexes revealed TrxR inhibition and triggering of immunogenic cell death (ICD) for allyl palladates†
Chiara
Donati
a,
Ishfaq Ibni
Hashim
b,
Nestor Bracho
Pozsoni
b,
Laurens
Bourda
 b,
Kristof
Van Hecke
b,
Catherine S. J.
Cazin
b,
Fabiano
Visentin
c,
Steven P.
Nolan
*b,
Valentina
Gandin
*a and
Thomas
Scattolin
*d
b,
Kristof
Van Hecke
b,
Catherine S. J.
Cazin
b,
Fabiano
Visentin
c,
Steven P.
Nolan
*b,
Valentina
Gandin
*a and
Thomas
Scattolin
*d
aDipartimento di Scienze del Farmaco, Università degli Studi di Padova, via Marzolo 5, 35131 Padova, Italy. E-mail: valentina.gandin@unipd.it
bDepartment of Chemistry and Centre for Sustainable Chemistry, Ghent University, Krijgslaan 281,S-3, 9000 Ghent, Belgium. E-mail: steven.nolan@ugent.be
cDipartimento di Scienze Molecolari e Nanosistemi, Università Ca' Foscari, Campus Scientifico, Via Torino 155, 30174 Venezia-Mestre, Italy
dDipartimento di Scienze Chimiche, Università degli Studi di Padova, via Marzolo 1, 35131 Padova, Italy. E-mail: thomas.scattolin@unipd.it
First published on 19th March 2025
Abstract
Immunogenic cell death (ICD) is a regulated form of cell death that activates an immune response through the release of danger-associated molecular patterns (DAMPs), including calreticulin, ATP, and HMGB1. Gold complexes are known to induce ICD, but the ICD-inducing potential of palladium complexes remains largely unexplored. We report the first examples of palladium compounds capable of inducing ICD, specifically allyl palladates bearing bis(imino)acenaphthene–NHC (BIAN–NHC) ligands. Cytotoxicity tests on human cancer cell lines revealed that allyl palladates outperform their cinnamyl analogues and gold(I)/copper(I) BIAN–NHC complexes. Notably, [BIAN–IMes·H][PdCl2(allyl)] 2a showed excellent TrxR inhibition, reducing activity by 67% and surpassing auranofin. This inhibition strongly correlates with ICD induction, as evidenced by enhanced DAMP marker expression, including superior ATP and HMGB1 release compared to doxorubicin. These findings establish allyl palladates as a novel class of ICD inducers with dual anticancer activity and immune activation potential.
Introduction
Immunogenic cell death (ICD) is a form of regulated cell death characterized by the release of danger-associated molecular patterns (DAMPs) and the activation of an immune response against the dying cells.1–5 This recently discovered pathway involves a sequence of events that lead to the activation of the immune system: i) DAMPs release: the dying cancer cells release DAMPs, such as calreticulin, ATP, and HMGB1 that act as signals to the immune system;6 ii) calreticulin exposure: calreticulin translocates to the cell surface, serving as an “eat-me” signal that promotes the phagocytosis of dying cells by dendritic cells and macrophages;7,8 iii) ATP secretion: ATP is released into the extracellular space, acting as a “find-me” signal to attract immune cells to the site of cell death;9 and iv) HMGB1 release: HMGB1 is released from the nucleus and binds to receptors of dendritic cells, enhancing the presentation of tumour antigens to T cells and triggering an adaptive immune response.10When the ICD is triggered by the use of metallodrugs it presents several advantages: i) dual action: metallodrugs can directly kill cancer cells and simultaneously activate the immune system, providing a two-pronged approach to cancer therapy;11 ii) synergy with immunotherapy: metallodrugs can enhance the efficacy of existing immunotherapies, such as immune checkpoint inhibitors, by increasing the immunogenicity of tumour cells;12 iii) overcoming resistance: by inducing ICD, metallodrugs can potentially overcome resistance mechanisms that often limit the effectiveness of traditional chemotherapies.1–5,13
Recent studies have shown that some metal-based anticancer agents are able to induce ICD through different mechanisms.14 For example, oxaliplatin has been shown to induce ICD by causing the release of DAMPs and activating the immune response.15,16 Its mechanism involves the induction of endoplasmic reticulum (ER) stress and the subsequent exposure of calreticulin on the cell surface. On the contrary, some ruthenium,17,18 iridium19 and copper20 complexes have shown potential in inducing ICD by generating reactive oxygen species (ROS) and disrupting cellular homeostasis. Finally, in the case of some gold complexes, inhibition of TrxR leads to oxidative stress and the release of DAMPs, triggering ICD and an immune response against cancer cells.21
Focusing on this last mechanism, targeting thioredoxin reductase (TrxR) is emerging as a promising strategy to induce ICD and harness the immune system to fight against cancer. It should be remembered that thioredoxin reductase (TrxR) is a pivotal enzyme in the redox regulation of cells, playing a critical role in maintaining the redox balance by reducing thioredoxin (Trx), which in turn regulates numerous cellular processes, including DNA synthesis and repair, and defence against oxidative stress.22 Overexpression of TrxR is often observed in cancer cells, contributing to their survival, proliferation, and resistance to chemotherapy.23
Gold-based anticancer agents have shown significant potential as anticancer agents due to their ability to inhibit TrxR.24–27 These complexes can bind to the selenocysteine residue at the active site of TrxR, leading to its inhibition. This event disrupts the redox homeostasis within the cancer cells, promoting oxidative stress and, ultimately, leading to cell death.
In contrast to gold, the ability of palladium complexes to inhibit TrxR is almost unexplored. Palladium compounds with antitumor properties are primarily known to target DNA28–31 or, in some cases, mitochondria.32–35 The molecular targets have been much less studied, with only a few examples of palladium compounds acting as inhibitors of key enzymes.36–38
In this work, we describe the first examples of organopalladium compounds capable of promoting ICD, which appears to be triggered by an efficient inhibition of TrxR. Specifically, the compounds described in this contribution belong to the category of allyl palladates. These species exhibit a unique interaction between the proton of an azolium salt and two chlorides of the Pd-allyl (or cinnamyl) fragment. We have recently demonstrated that these species are key intermediates in the preparation of [Pd(NHC)Cl(allyl/cinnamyl)] precatalysts,39 and can be obtained simply by grinding an azolium salt and the [PdCl(allyl/cinnamyl)]2 dimer with a mortar and pestle.40 The promising antitumor activity of allyl palladates bearing classical imidazol(in)ium salts against various cancer cell lines, also confirmed on patient-derived organoids, has prompted us to further investigate this class of unusual compounds.40 Moreover, we focus on bis(imino)acenaphthene–NHC (BIAN–NHC) salts, which are expected to confer a promising antitumor activity to the corresponding allyl palladates, due to their high steric hindrance. Indeed, in one of our previous contributions the best antitumor activity was achieved with the most sterically hindered imidazolium salt used (IPr*·HCl). Furthermore, bis(imino)acenaphthene–NHC (BIAN–NHC) salts and their carbene complexes have been extensively studied in recent years for their promising catalytic and anticancer properties, which appear to be attributable to the bis(imino)acenaphthene scaffold.41–46
In this contribution, we describe not only the preparation of allyl/cinnamyl palladates bearing BIAN–NHC salts but also the corresponding carbene complexes with coinage metals such as copper and gold. One of our objectives is to demonstrate the comparable or even superior anticancer activity of allyl palladates compared to the corresponding copper and gold complexes, which are known to exhibit important biological properties, including antitumor and antibacterial activities.
Results and discussion
Solvent-free synthesis of allyl palladates from acenaphthene-based azolium salts
From the synthetic route of allyl and indenyl palladates using conventional imidazolium and imidazolium salts,40 we hypothesized that this class of organopalladium compounds could also be synthesized with more extended π-systems such as acenaphthene-based azolium salts. Gratifyingly, employing the same solvent-free method, bis(imino)acenaphthene–NHC (BIAN–NHC) salts 1a–b were ground together with [Pd(allyl)(μ-Cl)]2 or [Pd(cinnamyl)(μ-Cl)]2 precursors using a simple mortar and pestle. The target complexes 2a–b and 3a–b were obtained quantitatively and fully characterized by NMR spectroscopy and elemental analyses (Scheme 1).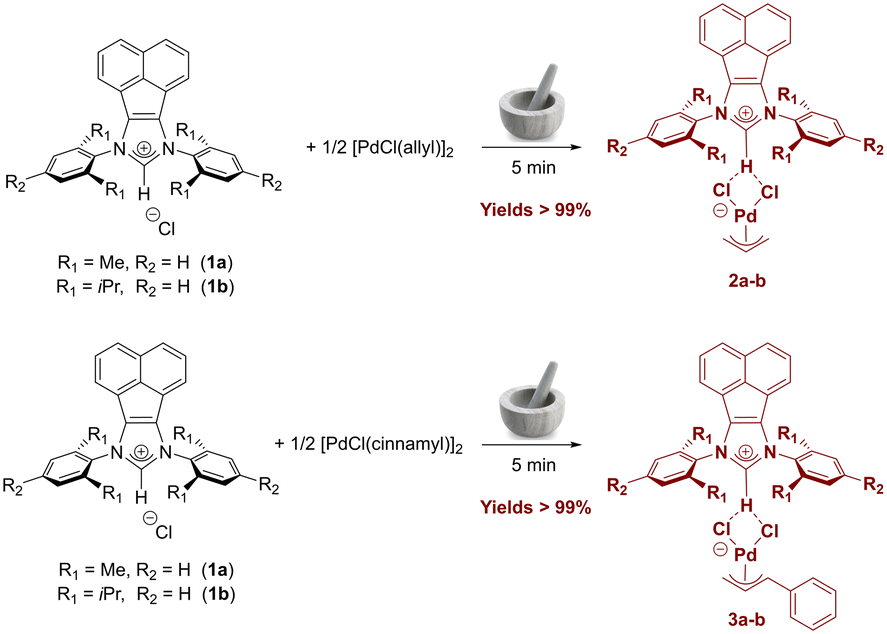 | ||
| Scheme 1 Mechanochemical synthesis of BIAN–NHC based allyl palladate complexes 2a–b and 3a–b. | ||
In the 1H NMR spectra, the shift of all aromatic signals of the bis(imino)acenaphthene fragment compared to the starting BIAN–NHC salts 1a–b can be clearly observed. Regarding the cinnamyl fragment, four distinct signals are present in the spectra of complexes 3a–b at ca. 5.6, 4.4, 3.8, and 2.8 ppm, in addition to those of the phenyl substituent. In allyl-supported complexes 2a–b, three sets of characteristic signals can be observed, which are attributable to the syn protons (doublet at ca. 3.8 ppm, J = 7 Hz), the anti protons (doublet at 2.6–2.9 ppm, J = 12 Hz), and the central allyl proton (multiplet at ca. 5 ppm). Both signals from the allyl and cinnamyl fragments are significantly shifted compared to the [Pd(allyl)(μ-Cl)]2 and [Pd(cinnamyl)(μ-Cl)]2 precursors, respectively. The upfield shift of the imidazolium proton NCHN signal (Δδ = 0.2–0.5 ppm) compared to the starting imidazolium salt further indicates interaction between the imidazolium moiety and the palladium-allyl fragment.
Furthermore, in the case of complex 2b, the atom connectivity in the product was unequivocally confirmed by single crystal X-ray diffraction analysis (Fig. 1). Suitable crystals were grown by slow vapor diffusion of diethyl ether into a CHCl3 solution.
 | ||
| Fig. 1 X-ray molecular structure of 2b is presented, showing thermal displacement ellipsoids at the 50% probability level with CHCl3 (solvent molecule) and hydrogen atoms (except for the NCHN one) omitted for clarity. CCDC: 2415263. | ||
Unified synthetic procedure to acenaphthene-based NHC copper(I) and gold(I) complexes
One of the objectives of this work was to compare the potential anticancer activity of the allyl/cinnamyl palladates described above with that of the more classical carbene derivatives of coinage metals. Specifically, we have developed a simple general method for the preparation of both gold(I) and copper(I) complexes bearing BIAN–NHC ligands derived from 1a–b azolium salts, as well as from an extremely bulky azolium, 1c (BIAN–IPr#·HCl). To this end, CuCl and [AuCl(dms)] (dms = dimethylsulfide) precursors were reacted with various azolium salts in the presence of potassium carbonate as a weak and inexpensive base (Scheme 2). All reactions proceeded under extremely mild and aerobic conditions (40–60 °C, 1.5–20 h) using technical grade acetone (green acetone) as solvent. As expected, the metalation–deprotonation (MD) process occurs more slowly when the extremely bulky acenaphthene-based azolium salt 1c is employed. Complexes 4a–c and 5a–c were obtained in high yields (91–95%) and purity through a simple purification procedure. Complexes 4a–b and 5a–b, whose synthesis has already been reported in the literature,47 albeit with lower yields and conducted under an inert atmosphere and with more toxic and anhydrous solvents (e.g., dichloromethane), display NMR spectra comparable to those previously reported. In contrast, the BIAN–IPr# derivatives 4c and 5c have never been reported and were fully characterized by NMR spectroscopy and elemental analyses. In the 1H NMR spectra, the disappearance of the imidazole proton NCHN because of the base-assisted metalation process is consistently observed, along with a shift in the aromatic signals compared to the starting azolium salt. Moreover, in the 13C NMR spectra, the typical signal of the carbene carbon (ca. 180–185 ppm) is observed, thus confirming the deprotonation–metalation process.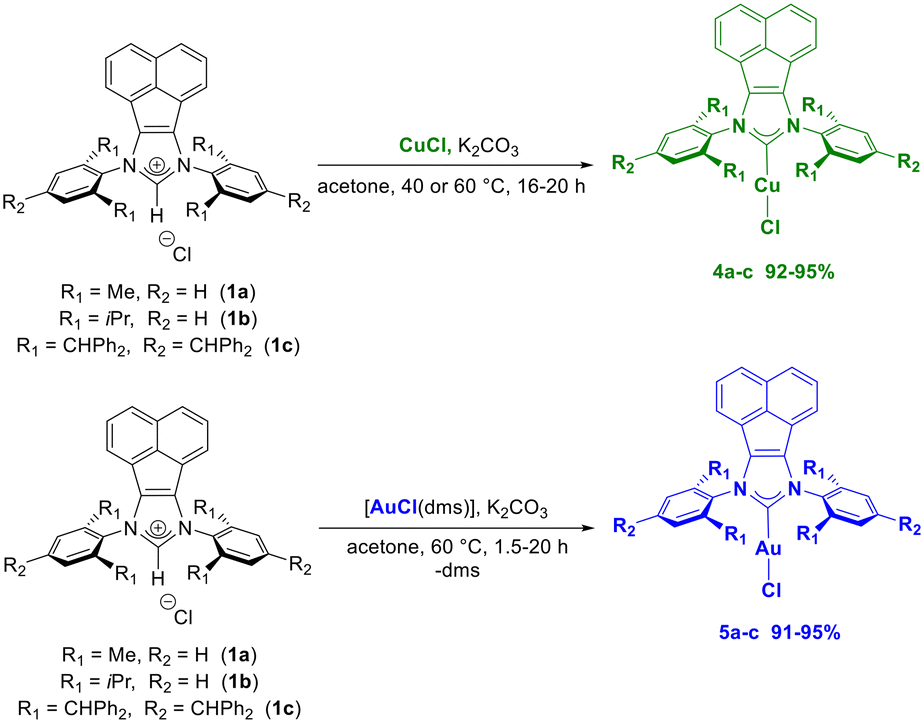 | ||
| Scheme 2 Simple synthetic route to BIAN–NHC copper(I) and gold(I) complexes 4a–c and 5a–c. | ||
Antiproliferative activity towards cancer and normal cell lines
The in vitro anticancer potential of the newly developed complexes was evaluated on the in-house panel of human cancer cell lines including HCT-15 (colorectal), 2008 (ovarian), PSN-1 (pancreatic), MDA-MB-231 (breast) and NTERA-2 (embryonal) cancer cells. For comparison purposes, the efficacy of cisplatin, the milestone of metal-based drugs, was assessed under the same experimental conditions. The cytotoxicity parameters, expressed in terms of IC50 and obtained after 72 h of drug exposure by MTT assay, are reported in Table 1.| Complex | IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|---|
| NTERA-2 | MDA-MB-231 | PSN-1 | HCT-15 | 2008 | C13* | HEK-293 (S.I.) | CHO (S.I.) | |
| a Stock solutions in DMSO for all complexes; stock solutions in 0.9% NaCl for cisplatin. | ||||||||
| Cisplatin | 15 ± 3 | 22 ± 4 | 12 ± 3 | 14 ± 2 | 2 ± 1 | 25 ± 2 (11.7) | 14 ± 3 (1.1) | 29 ± 3 (2.3) |
| 2a | 5 ± 2 | 7 ± 1 | 5 ± 2 | 3 ± 1 | 2.7 ± 0.6 | 1.8 ± 0.4 (0.7) | 1.8 ± 0.1 (0.4) | 3.7 ± 0.1 (0.8) |
| 2b | 0.4 ± 0.2 | 4 ± 1 | 5 ± 1 | 10.4 ± 0.6 | 5.5 ± 0.3 | 3.5 ± 0.7 (0.6) | 1.7 ± 0.8 (0.3) | 9 ± 1 (1.7) |
| 3a | 1.1 ± 0.8 | 6 ± 4 | 5 ± 2 | 10.3 ± 0.2 | 4 ± 1 | 2.6 ± 0.4 (0.7) | 1.1 ± 0.8 (0.2) | 5 ± 1 (0.8) |
| 3b | 2 ± 1 | 4 ± 2 | 7.2 ± 0.3 | 10.6 ± 0.8 | 7.2 ± 0.8 | 5.5 ± 0.4 (0.8) | 2.3 ± 0.6 (0.4) | 8 ± 2 (1.3) |
| 4a | 4 ± 1 | 5 ± 2 | 4 ± 3 | 9 ± 6 | 3 ± 2 | 4 ± 1 (1.2) | 1.1 ± 0.5 (0.2) | 1.3 ± 0.3 (0.3) |
| 4b | 4 ± 2 | 4 ± 2 | 7 ± 2 | 5.9 ± 0.5 | 7 ± 1 | 7.7 ± 0.7 (1.1) | 3.2 ± 0.4 (0.6) | 1.5 ± 0.1 (0.3) |
| 4c | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 5a | 11 ± 4 | 19 ± 5 | 14 ± 4 | 17 ± 3 | 8 ± 1 | 8 ± 1 (1.0) | 36 ± 6 (2.6) | 28 ± 1 (2.0) |
| 5b | 40 ± 8 | 33 ± 9 | 15.0 ± 0.1 | 22 ± 1 | 16 ± 1 | 18 ± 2 (1.2) | 31 ± 7 (1.2) | >50 |
| 5c | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
Preliminarily, we evaluated the stability of the synthesized complexes in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 D2O/DMSO-d6 solution through NMR spectroscopy. After 48 hours, there were no notable changes detected in the spectra, indicating the complexes maintained their structural integrity. The same result was obtained by recording UV-vis spectra in different phosphate buffer solutions (pH = 6, 7.2 and 8) with a 10% of DMSO (see ESI†).
:1 D2O/DMSO-d6 solution through NMR spectroscopy. After 48 hours, there were no notable changes detected in the spectra, indicating the complexes maintained their structural integrity. The same result was obtained by recording UV-vis spectra in different phosphate buffer solutions (pH = 6, 7.2 and 8) with a 10% of DMSO (see ESI†).
Among all tested complexes, gold derivatives were those characterised by the lowest in vitro antitumor potential, with complex [(BIAN–IPr#)AuCl] (5c) being completely ineffective against all tested cancer cell lines. Gold derivative [(BIAN–IMes)AuCl] (5a) showed, on average, the same cytotoxicity profile as cisplatin, whereas [(BIAN–IPr)AuCl] (5b) elicited average IC50 values about 2-fold higher than those detected with the metal-based reference drug.
Conversely, except for derivative [(BIAN–IPr#)CuCl] (4c), all copper and palladate derivatives showed remarkable cytotoxicity against tested cancer cell lines, with average IC50 values in the low-micromolar range. Allyl palladate complexes, [BIAN–IMes·H][PdCl2(allyl)] (2a) and [BIAN–IPr·H][PdCl2(allyl)] (2b), were on average much more effective than the corresponding cinnamyl derivatives, [BIAN–IMes·H][PdCl2(cin)] (3a) and [BIAN–IPr·H][PdCl2(cin)] (3b). Noteworthy, against NTERA-2 embryonal and MDA-MB-231 breast cancer cells, both characterized by low sensitivity to cisplatin, palladate complexes were up to 36 and 5.8 times more active than the reference metallodrug cisplatin, respectively.
Considering these very promising results and keeping in mind that drug resistance represents a critical issue in anticancer therapy, we also evaluated the ability of the tested complexes to bypass acquired drug resistance. The in vitro antitumor effect of gold, copper and palladium-based complexes was hence assessed on human ovarian cancer cells selected for their resistance to cisplatin, namely C13* cancer cells. These cells were generated from 2008 cells through monthly selection with low doses of cisplatin.48 The most important molecular mechanisms involved in drug resistance in C13* cancer cells involves high glutathione and TrxR cellular levels, reduced cellular drug uptake, and/or enhanced DNA damage repair.49In vitro antitumor effect in both sensitive 2008 and resistant C13* cells was detected after 72 h treatment using the MTT test. Cross-resistance profiles were evaluated by means of the resistance factor (RF), defined as the ratio between IC50 values for the resistant cells and those derived from the sensitive ones (Table 1).
All tested complexes proved to be similarly effective against both cisplatin-sensitive and resistant cell lines, with RFs ranging from 9.7 to 16.7 times lower than that of cisplatin, thus attesting to their ability to overcome acquired cisplatin resistance and ruling out the occurrence of cross-resistance phenomena.
The antiproliferative activity of the newly developed Au, Cu and Pd complexes was also evaluated against two non-tumor cell lines, Chinese hamster ovary (CHO) and the human embryonic kidney (HEK-293) cells, with the aim to preliminarily assess their cell selectivity. As highlighted by the selectivity index values (SI = the quotient of the average IC50 toward normal cells divided by the average IC50 for the malignant cells), only gold compound [(BIAN–IMes)AuCl] (5a) proved to be much more effective in affecting cancer cells with respect to non-cancer cells, whereas for all the other complexes no preferential cytotoxicity against cancer cells could be detected.
Antiproliferative activity towards 3D spheroids
Even if the 2D cell cultures are the most employed model for in vitro drug screening due to the low cost, simplicity, and reliability, 2D methods are unable to properly reproduce the characteristics of in vivo solid tumors. In contrast, 3D cell cultures being more efficient in closely mimicking the heterogeneity and complexity of the tumor microenvironment, are more predictive for in vivo outcomes.50 On these bases, the cytotoxicity of palladate derivatives was also tested in 3D cell cultures of human colon ovarian (2008) and embryonal (NTERA-2) cancers. Cancer cell spheroids were treated for 72 h with the palladate compounds, and cell viability was assessed by means of a modified acid phosphatase (APH) assay (Table 2).| Complex | 2008 | NTERA-2 |
|---|---|---|
| a Stock solutions in DMSO for all complexes; stock solutions in 0.9% NaCl for cisplatin. | ||
| Cisplatin | 13.9 ± 0.8 | 50 ± 5 |
| [BIAN–IMes·H][PdCl2(allyl)] (2a) | 24.3 ± 0.9 | 36 ± 5 |
| [BIAN–IPr·H][PdCl2(allyl)] (2b) | 27 ± 7 | 17 ± 2 |
| [BIAN–IMes·H][PdCl2(cin)] (3a) | 45 ± 5 | 31.5 ± 0.7 |
| [BIAN–IPr·H][PdCl2(cin)] (3b) | 31 ± 8 | 48 ± 2 |
The IC50 values obtained in this set of experiments were slightly higher than those calculated in 2D models, due to the greater resistance to drug treatment generally displayed by cells in 3D cultures.
In 3D spheroids, the presence of an extracellular matrix and cell–cell interactions creates a more complex microenvironment that limits drug penetration. Additionally, the hypoxic core of spheroids reduces cell proliferation, making cells less susceptible to drugs that target rapidly dividing cells. Furthermore, differences in gene expression and signaling pathways in 3D models contribute to enhanced drug resistance mechanisms, leading to lower overall cytotoxicity compared to traditional 2D monolayer cultures.
Nevertheless, the results clearly confirm that allyl complexes [BIAN–IMes·H][PdCl2(allyl)] 2a and [BIAN–IPr·H][PdCl2(allyl)] 2b were much more effective than cinnamyl derivatives [BIAN–IMes·H][PdCl2(cin)] 3a and [BIAN–IPr·H][PdCl2(cin)] 3b. Moreover, all palladate complexes proved to be more potent than cisplatin against embryonal cancer NTERA-2 spheroids. In contrast, the gold–NHC complex 5a, which exhibited the highest selectivity index in 2D models, was found to be inactive (IC50 > 50 μM) towards 3D spheroids.
Inhibition of thioredoxin reductase (TrxR) enzymes
As previously highlighted by Arnér and co-workers, other noble metal-based compounds in addition to gold are extremely effective in targeting TrxR.51 Actually, even though the anticancer mechanism of several reported Pd compounds is generally believed to occur through DNA interaction, Pd electrophilic compounds also strongly react with nucleophiles other than DNA, such as the selenocysteine residue of selenoproteins. Recently, some terpyridine Pd(II) complexes were demonstrated to selectively inhibit isolated mammalian TrxR.52On this basis, we evaluated the ability to hamper TrxR in cell systems. Human ovarian 2008 cancer cells were treated for 24 h with equimolar concentrations (5 μM) of the tested derivatives 2a–b and 3a–b. TrxR activity was assayed by measuring the NADPH-dependent reduction of DTNB at 412 nm, as described in the Experimental section. Auranofin, a well-known metal-based TrxR inhibitor was used as a positive control under the same experimental conditions.
All Pd-based complexes were effective in hampering TrxR activity in 2008 cells (Fig. 2A). Compound [BIAN–IMes·H][PdCl2(allyl)] 2a was even more effective than auranofin in inhibiting cellular TrxR, being able to reduce cellular TrxR activity by about 67%. In general, TrxR inhibition data evidence a good correlation between the ability of the complexes to inhibit TrxR and their cytotoxicity in 2008 cells (see Table 1).
 | ||
| Fig. 2 TrxR inhibition (A) and ROS production (B). A. 2008 cells were incubated for 24 h with the tested compounds. Subsequently, cells were washed twice with PBS and lysed. TrxR activity was tested by measuring NADPH dependent reduction of DTNB at 412 nm. Error bars indicate SD; **p < 0.1. B. 2008 cells were preincubated in PBS/10 mM glucose medium for 20 min at 37 °C in the presence of 10 μM CM-H2DCFDA and then treated with the tested compounds at 5 μM. Antimycin (3 μM), a well-known inhibitor of mitochondrial complex III in the respiratory chain, was used as positive control. | ||
It is widely known that Trx system plays an essential role in cellular redox homeostasis, and inhibition of this redox regulatory system has been shown to determine cellular redox unbalance in terms of sulfhydryl redox status and cellular production of reactive oxygen species (ROS).53 We hence evaluated the ability of the tested complexes to increase the total basal production of ROS in 2008 cancer cells. As shown in Fig. 2B, all tested compounds exhibit a time-dependent increase in the cellular basal hydrogen peroxide production which was, however, substantially lower than that induced by antimycin, a well-known inhibitor of mitochondrial complex III in the respiratory chain.
Immunogenic cell-death (ICD) assay
Recent studies have shown a clear connection between TrxR inhibitors and the induction of ICD. As stated in the introduction, ICD is a unique type of cancer cell death that can induce adaptive immune responses, thus contributing to the elimination of residual tumor cells. ICD inducers are generally classified into type I and type II, with type II inducers generally more efficient than type I, being able to trigger ICD directly by ROS-mediated endoplasmic reticulum stress (ERS).2 On the other hand, compounds possessing the ability to hamper TrxR can induce the apoptosis of cancer cells by eliciting ROS-mediated ERS. Hence, one can speculate that compounds possessing the ability to inhibit TrxR can induce the apoptosis of cancer cells by eliciting ICD.To prove if our newly developed palladate complexes were effective in inducing ICD, we evaluated their ability to activate two specific ICD signals, named DAMPs (DAMPs = damage-associated molecular patterns). We evaluated the extracellular release of high-mobility group box 1 (HMGB1) and secretion of adenosine triphosphate (ATP) in 2008 cells treated for 24 h with tested complexes. The extracellular release of the HMGB1 protein was assayed using the ELISA kit (ThermoFisher).
As evident in Fig. 3, all palladate derivatives were able to significantly induce the release of both DAMP markers. Remarkably, compound [BIAN–IMes·H][PdCl2(allyl)] 2a was able to induce a HMGB1 release and an ATP excretion higher compared to those detected with the reference ICD inducer doxorubicin. Overall, comparing these data with those concerning TrxR inhibition, it is possible to underline that compound 2a which is the most effective in hampering TrxR activity it is the most effective in stimulating both DAMP markers.
 | ||
| Fig. 3 DAMPs on 2008 cells. 2008 cells were incubated for 24 h with the tested compounds (5 μM) or the ICD inducer doxorubicin (0.5 μM) as a positive control. A. HMGB1 excretion induced by tested compound was measured by HMGB1 ELISA kit. B. ATP secretion induced by tested compound was measured by ATP bioluminescence assay kit according to manufacturer's instructions. Error bars indicate S.D.; **p < 0.1. | ||
Conclusions
In summary, we have developed a unified protocol for synthesizing six gold(I) and copper(I) complexes featuring BIAN–NHC ligands, along with a straightforward solvent-free method to produce four novel allyl and cinnamyl palladates derived from BIAN–NHC salts. All compounds were thoroughly characterized using NMR spectroscopy and elemental analyses, proving their structures and purity. Remarkably, for the allyl palladate complex 2b, the connectivity between atoms was unequivocally confirmed through single-crystal X-ray diffraction.The synthesized compounds were evaluated for their cytotoxic effects on a panel of human cancer cell lines, including HCT-15 (colorectal), 2008 (ovarian), PSN-1 (pancreatic), MDA-MB-231 (breast), and NTERA-2 (embryonal) cancer cells. The results highlighted superior cytotoxicity of the copper complexes compared to their gold congeners. However, the allyl palladates emerged as the most promising class of compounds, demonstrating higher cytotoxicity than their cinnamyl analogues.
Notably, all allyl palladates effectively inhibited TrxR activity in 2008 ovarian cancer cells. Specifically, the compound [BIAN–IMes·H][PdCl2(allyl)] 2a outperformed auranofin in reducing cellular TrxR activity. Overall, the TrxR inhibition data revealed a strong correlation between the complexes' ability to inhibit TrxR and their cytotoxicity in 2008 cells. Furthermore, we have demonstrated that the allyl palladates can induce an immunogenic cell death (ICD) mechanism. To our knowledge, this represents the first class of palladium-based compounds identified as ICD inducers.
Remarkably, compound [BIAN–IMes·H][PdCl2(allyl)] 2a exhibited higher levels of HMGB1 release and ATP secretion compared to the reference ICD inducer doxorubicin.
When correlating these findings with the TrxR inhibition data, it becomes clear that compound 2a, which is the most effective in inhibiting TrxR activity, also shows the greatest efficacy in stimulating both DAMP markers. This suggests a strong link between TrxR inhibition and the ability of these compounds to function as ICD inducers. Encouraged by these promising results, we are currently conducting further in vitro and in vivo studies to explore their potential.
Experimental
Azolium salts 1a–c synthesized according to previously published procedures.54–56 The metal precursors CuCl, [AuCl(dms)], [Pd(allyl)(μ-Cl)]2 and [Pd(cinnamyl)(μ-Cl)]2, and other reagents were purchased and used as received without further purification, unless otherwise stated.1H, 13C{1H} NMR and bidimensional (HSQC, HMBC) spectra were recorded on a Bruker Advance 400 spectrometer at room temperature (298 K). Elemental analysis was carried out using an Elemental CHN “CUBO Micro Vario” analyzer. X-ray intensity data were collected at 100 K, on a Rigaku Oxford Diffraction Supernova Dual Source diffractometer equipped with an Atlas CCD detector.
Synthesis of [BIAN–IMes·H][PdCl2(allyl)] (2a)
In air, the azolium salt BIAN–IMes·HCl 1a (50.0 mg, 0.108 mmol) and the dimeric precursor [Pd(allyl)(μ-Cl)]2 (19.6 mg, 0.054 mmol) were added to a mortar. The two solids were mixed and grinded using a pestle for 5 min. A yellow powder was obtained in quantitative yield.1H NMR (300 MHz, CDCl3) δ: 10.80 (s, 1H, NCHN), 7.95 (d, J = 8.3 Hz, 2H, aryl-H), 7.55 (dd, J = 8.3, 7.0 Hz, 2H, aryl-H), 7.28 (m, 2H, aryl-H), 7.18 (s, 4H, aryl-H), 5.14 (m, 1H, central allyl-H), 3.78 (d, J = 6.7 Hz, 2H, syn allyl-H), 2.59 (d, J = 12.0 Hz, 2H, anti allyl-H), 2.44 (s, 6H, p-CH3(IMes)), 2.35 (s, 12H, o-CH3(IMes)).
13C NMR (101 MHz, CDCl3) δ: 142.9, 141.4 (CNCHN), 136.6, 134.7, 130.6, 130.4, 130.2, 130.1, 129.9, 128.3, 123.8, 122.8, 21.5 (p-CH3(IMes)), 18.2 (o-CH3(IMes)).
Elemental analysis calcd (%) for C35H38Cl2N2Pd: C, 63.31; H, 5.77; N, 4.22; found: C, 63.52; H, 5.59; N, 4.30.
Synthesis of [BIAN–IPr·H][PdCl2(allyl)] (2b)
In air, the azolium salt BIAN–IPr·HCl 1b (50 mg, 0.091 mmol) and the dimeric precursor [Pd(allyl)(μ-Cl)]2 (16.5 mg, 0.046 mmol) were added to a mortar. The two solids were mixed and grinded using a pestle for 5 min. A yellow powder was obtained in quantitative yield.
1H NMR (300 MHz, CDCl3) δ: 10.99 (s, 1H, NCHN), 8.01 (d, J = 8.1 Hz, 2H, aryl-H), 7.71–7.62 (m, 2H, aryl-H), 7.57 (dd, J = 8.4, 7.1 Hz, 2H, aryl-H), 7.46 (d, J = 7.9 Hz, 4H, aryl-H), 7.22 (d, J = 6.9 Hz, 2H, aryl-H), 5.15 (m, 1H, central allyl-H), 3.78 (d, J = 6.7 Hz, 2H, syn allyl-H), 2.91 (d, J = 12.0 Hz, 2H, anti allyl-H), 2.82–2.61 (m, 4H, ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) (CH3)dipp), 1.40 (d, J = 6.8 Hz, 12H, CH(CH3)dipp), 1.12 (d, J = 6.9 Hz, 12H, CH(CH3)dipp).
(CH3)dipp), 1.40 (d, J = 6.8 Hz, 12H, CH(CH3)dipp), 1.12 (d, J = 6.9 Hz, 12H, CH(CH3)dipp).
13C NMR (101 MHz, CDCl3) δ: 145.2, 137.7 (CNCHN), 132.2, 130.8, 130.4, 130.2, 129.7, 128.4, 125.1, 123.6, 123.1, 29.6 ((CH3)dipp), 24.6 (CH3(dipp)), 24.0 (CH3(dipp)).
Elemental analysis calcd (%) for C41H50Cl2N2Pd: C, 65.82; H, 6.74; N, 3.74; found: C, 65.65; H, 6.89; N, 3.64.
Synthesis of [BIAN–IMes·H][PdCl2(cinnamyl)] (3a)
In air, the azolium salt BIAN–IMes·HCl 1a (50.0 mg, 0.108 mmol) and the dimeric precursor [Pd(cinnamyl)(μ-Cl)]2 (27.9 mg, 0.054 mmol) were added to a mortar. The two solids were mixed and grinded using a pestle for 5 min. A pale-yellow powder was obtained in quantitative yield.1H NMR (300 MHz, CDCl3) δ: 11.01 (s, 1H, NCHN), 7.96 (d, J = 8.0 Hz, 2H, aryl-H), 7.55 (dd, J = 8.3, 7.0 Hz, 2H, aryl-H), 7.43 (d, J = 7.1 Hz, 2H, aryl-H), 7.27 (m, 5H, aryl-H), 7.15 (s, 4H, aryl-H), 5.59 (bs, 1H, CHcin), 4.32 (bs, 1H, CHcin), 3.82 (bs, 1H, CHcin), 2.78 (bs, 1H, CHcin), 2.43 (s, 6H, p-CH3(IMes)), 2.33 (s, 12H, o-CH3(IMes)).
13C NMR (101 MHz, CDCl3) δ: 143.2, 141.3 (CNCHN), 136.5, 134.7, 130.6, 130.3, 130.1, 130.0, 129.9, 128.2, 128.0, 123.9, 122.8, 21.5 (p-CH3(IMes)), 18.2 (o-CH3(IMes)).
Elemental analysis calcd (%) for C41H42Cl2N2Pd: C, 66.54; H, 5.72; N, 3.79; found: C, 66.39; H, 5.78; N, 3.90.
Synthesis of [BIAN–IPr·H][PdCl2(cinnamyl)] (3b)
In air, the azolium salt BIAN–IPr·HCl 1b (50 mg, 0.091 mmol) and the dimeric precursor [Pd(cinnamyl)(μ-Cl)]2 (23.5 mg, 0.046 mmol) were added to a mortar. The two solids were mixed and grinded using a pestle for 5 min. A yellow powder was obtained in quantitative yield.
1H NMR (300 MHz, CDCl3) δ: 11.80 (s, 1H, NCHN), 7.99 (d, J = 8.3 Hz, 2H, aryl-H), 7.69–7.62 (m, 2H, aryl-H), 7.56 (dd, J = 8.3, 7.0 Hz, 2H, aryl-H), 7.45 (d, J = 7.8 Hz, 6H, aryl-H), 7.24–7.16 (m, 5H, aryl-H), 5.66 (bs, 1H, CHcin), 4.48 (bs, 1H, CHcin), 3.85 (bs, 1H, CHcin), 2.89 (bs, 1H, CHcin), 2.74 (hept, J = 6.8 Hz, 4H, (CH3)dipp), 1.39 (d, J = 6.8 Hz, 12H, CH3(dipp)), 1.13 (d, J = 6.8 Hz, 12H, CH3(dipp)).
13C NMR (101 MHz, CDCl3) δ: 145.2, 144.1 (CNCHN), 137.6, 132.2, 130.8, 130.4, 130.2, 129.6, 128.9, 128.4, 128.1, 125.1, 123.5, 123.0, 29.6 ((CH3)dipp), 24.7 (CH3(dipp)), 23.9 (CH3(dipp)).
Elemental analysis calcd (%) for C46H50Cl2N2Pd: C, 68.36; H, 6.24; N, 3.47; found: C, 68.20; H, 6.33; N, 3.56.
Synthesis of [(BIAN–IMes)CuCl] (4a)
A 4 mL screw cap vial was charged with BIAN–IMes·HCl 1a (50 mg, 0.108 mmol), CuCl (11.0 mg, 0.108 mmol) and freshly grinded K2CO3 (44.6 mg, 0.323 mmol). Acetone (1.0 mL) was added to the mixture and left to stir in air at 40 °C for 16 h. Then solvent was removed in vacuo and to the residue THF (2.0 mL) was added. The mixture was filtered over a celite plug and eluted with THF (3.0 mL), the filtrate was concentrated, and addition of pentane (2 mL) resulted in the formation of precipitate. The precipitate was filtered and washed with pentane (3 × 2 mL), dried under vacuum yielding the desired product as orange solid (52.1 mg, 92%).1H NMR (300 MHz, CDCl3) δ: 7.79 (d, J = 8.8 Hz, 2H, aryl-H), 7.43 (dd, J = 8.3, 7.0 Hz, 2H, aryl-H), 7.10 (s, 4H, aryl-H), 7.05 (d, J = 6.6 Hz, 2H, aryl-H), 2.43 (s, 6H, p-CH3(Mes)), 2.23 (s, 12H, o-CH3(Mes)).
13C NMR (101 MHz, CDCl3) δ: 184.4 (Ccarbene), 139.8, 138.1, 134.6, 133.8, 130.8, 129.9, 129.8, 128.4, 127.8, 125.5, 121.0, 21.4 (p-CH3(Mes)), 18.1 (o-CH3(Mes)).
Elemental analysis calcd (%) for C31H28ClCuN2: C, 70.58; H, 5.35; N, 5.31; found: C, 70.90; H, 5.17; N, 5.43.
Synthesis of [(BIAN–IPr)CuCl] (4b)
A 4 mL screw cap vial was charged with BIAN–IPr·HCl 1b (50 mg, 0.091 mmol), CuCl (9.0 mg, 0.091 mmol) and freshly grinded K2CO3 (37.7 mg, 0.273 mmol). Acetone (1.0 mL) was added to the mixture and left to stir in air at 40 °C for 16 h. Then solvent was removed in vacuo and to the residue THF (2.0 mL) was added. The mixture was filtered over a celite plug and eluted with THF (3.0 mL), the filtrate was concentrated, and addition of pentane (2 mL) resulted in the formation of precipitate. The precipitate was filtered and washed with pentane (3 × 2 mL), dried under vacuum yielding the desired product as yellow solid (51.0 mg, 92%).
1H NMR (300 MHz, CDCl3) δ: 7.80 (d, J = 7.9 Hz, 2H, aryl-H), 7.60 (t, J = 7.8 Hz, 2H, aryl-H), 7.46–7.39 (m, 6H, aryl-H), 7.01 (d, J = 6.6 Hz, 2H, aryl-H), 2.85 (hept, J = 6.9 Hz, 4H, CH3(dipp)), 1.35 (d, J = 6.9 Hz, 12H, CH3(dipp)), 1.13 (d, J = 6.9 Hz, 12H, CH3(dipp)).
13C NMR (101 MHz, CDCl3) δ: 185.9 (Ccarbene), 145.7, 138.9, 133.1, 131.0, 130.0, 128.5, 127.9, 125.4, 124.7, 121.1, 29.1 (CHdipp), 25.0 (CH3(dipp)), 23.9 (CH3(dipp)).
Elemental analysis calcd (%) for C37H40ClCuN2: C, 72.65; H, 6.59; N, 4.58; found: C, 72.40; H, 6.81; N, 4.50.
Synthesis of [(BIAN–IPr#)CuCl] (4c)
A 4 mL screw cap vial was charged with BIAN–IPr#·HCl 1c (100 mg, 0.073 mmol), CuCl (8.0 mg, 0.080 mmol) and freshly grinded K2CO3 (30.1 mg, 0.218 mmol). Acetone (1.0 mL) was added to the mixture and left to stir at 60 °C for 20 h. Then volatiles were evacuated under reduced pressure and to the residue THF (5.0 mL) was added, microfiltered, filtrate concentrated. Then pentane (4 mL) was added resulting in the formation of precipitate, filtered and washed with pentane (2.0 × 2.0 mL), dried under vacuum yielding the desired product as yellow solid (99 mg, 95%).
1H NMR (300 MHz, CDCl3) δ: 7.53 (d, J = 8.3 Hz, 2H, aryl-H), 7.25–7.14 (m, 12H, aryl-H), 7.02–6.89 (m, 26H, aryl-H), 6.85 (d, J = 7.4 Hz, 8H, aryl-H), 6.78–6.66 (m, 12H, aryl-H), 6.61 (d, J = 6.7 Hz, 8H, aryl-H), 6.10 (d, J = 6.9 Hz, 2H, aryl-H), 5.47 (s, 2H, p-Ph2), 5.36 (s, 4H, o-Ph2).
13C NMR (101 MHz, CDCl3) δ: 185.8 (Ccarbene), 145.8, 143.3, 142.1, 141.8, 141.8, 139.0, 134.0, 131.0, 129.7, 129.7, 129.4, 129.3, 128.5, 128.0, 127.3, 126.5, 126.4, 124.2, 121.5, 56.4, 51.6.
Elemental analysis calcd (%) for C103H76ClCuN2: C, 85.87; H, 5.32; N, 1.94; found: C, 85.53; H, 5.48; N, 1.85.
Synthesis of [(BIAN–IMes)AuCl] (5a)
A 4 mL screw cap vial was charged with BIAN–IMes·HCl 1a (50 mg, 0.108 mmol), [Au(DMS)Cl] (31.6 mg, 0.108 mmol) and freshly grinded K2CO3 (44.6 mg, 0.323 mmol). Acetone (1.0 mL) was added to the mixture and left to stir at 60 °C for 1.5 h. Then volatiles were evacuated under reduced pressure and to the residue THF (5.0 mL) was added, microfiltered and eluted with THF. After that the filtrate was concentrated, and pentane (4 mL) was added resulting in the formation of precipitate, filtered and washed with pentane (2.0 × 2.0 mL), dried under vacuum yielding the desired product as yellow solid (65 mg, 91%).1H NMR (300 MHz, CDCl3) δ: 7.80 (dd, J = 8.3, 0.4 Hz, 2H, aryl-H), 7.43 (dd, J = 8.3, 7.0 Hz, 2H, aryl-H), 7.09 (s, 4H, aryl-H), 7.04 (d, J = 6.6 Hz, 2H, aryl-H), 2.42 (s, 6H, CH3(Mes)), 2.21 (s, 12H, CH3(Mes)).
13C NMR (101 MHz, CDCl3) δ: 177.6 (Ccarbene), 140.0, 137.3, 134.7, 133.5, 130.4, 129.9, 129.7, 128.6, 127.89, 125.41, 121.25 (s, CHMes), 21.41 (s, p-CH3(Mes)), 18.06 (s, o-CH3(Mes)).
Elemental analysis calcd (%) for C31H28AuClN2: C, 56.33; H, 4.27; N, 4.24; found: C, 56.01; H, 4.39; N, 4.31.
Synthesis of [(BIAN–IPr)AuCl] (5b)
A 4 mL screw cap vial was charged with BIAN–IPr·HCl 1b (50 mg, 0.091 mmol), [Au(DMS)Cl] (29.2 mg, 0.091 mmol) and freshly grinded K2CO3 (37.8 mg, 0.273 mmol). Acetone (1.0 mL) was added to the mixture and left to stir in air at 60 °C for 1.5 h. Then the solvent was removed in vacuo and to the residue THF (5.0 mL) was added. The mixture was filtered on a syringe membrane, the filtrate was concentrated, and addition of pentane (2 mL) resulted in the formation of precipitate. The precipitate was filtered and washed with pentane (3 × 3 mL), dried under vacuum yielding the desired product as yellow solid (62.4 mg, 92%).
1H NMR (300 MHz, CDCl3) δ: 7.82 (d, J = 8.3 Hz, 2H, aryl-H), 7.61 (t, J = 7.8 Hz, 2H, aryl-H), 7.47–7.44 (m, 2H, aryl-H), 7.41 (d, J = 7.8 Hz, 4H, aryl-H), 7.01 (d, J = 7.0 Hz, 2H, aryl-H), 2.83 (hept, J = 6.8 Hz, 4H, CH3(dipp)), 1.40 (d, J = 6.9 Hz, 12H, CH3(dipp)), 1.12 (d, J = 6.9 Hz, 12H, CH3(dipp)).
13C NMR (101 MHz, CDCl3) δ: 179.8 (Ccarbene), 145.8, 138.1, 132.9, 131.1, 130.5, 129.9, 128.8, 128.0, 125.5, 124.7, 121.4, 29.2 (CHdipp), 24.7 (CH3(dipp)), 24.0 (CH3(dipp)).
Elemental analysis calcd (%) for C37H40AuClN2: C, 59.64; H, 5.41; N, 3.76; found: C, 59.87; H, 5.30; N, 3.89.
Synthesis of [(BIAN–IPr#)AuCl] (5c)
A 4 mL screw cap vial was charged with BIAN–IPr#·HCl 1c (50 mg, 0.036 mmol), [Au(DMS)Cl] (11.7 mg, 0.039 mmol) and freshly grinded K2CO3 (15.1 mg, 0.109 mmol). Acetone (1.0 mL) was added to the mixture and left to stir at 60 °C for 20 h. Then volatiles were evacuated under reduced pressure and to the residue THF (5.0 mL) was added, microfiltered, filtrate concentrated. Then pentane (4 mL) was added resulting in the formation of precipitate, filtered, and washed with pentane (2.0 × 2.0 mL), dried under vacuum yielding the desired product as yellow solid (54 mg, 95%).
1H NMR (300 MHz, CDCl3) δ: 7.53 (d, J = 8.3 Hz, 2H, aryl-H), 7.25–7.21 (m, 6H, aryl-H), 7.20–7.16 (m, 4H, aryl-H), 7.03–6.89 (m, 36H, aryl-H), 6.75–6.65 (m, 12H, aryl-H), 6.63–6.58 (m, 8H, aryl-H), 6.07 (d, J = 6.9 Hz, 2H, aryl-H), 5.48 (s, 2H, p-Ph2), 5.42 (s, 4H, o-Ph2).
13C NMR (101 MHz, CDCl3) δ: 179.7 (Ccarbene), 145.9, 143.2, 142.0, 141.8, 141.7, 138.3, 133.5, 131.1, 129.6, 129.5, 129.4, 129.2, 128.5, 128.4, 128.0, 127.5, 126.5, 126.5, 126.4, 124.1, 121.8, 56.4, 51.6.
Elemental analysis calcd (%) for C103H76AuClN2: C, 78.59; H, 4.87; N, 1.78; found: C, 78.36; H, 4.98; N, 1.63.
Experiments with cultured human cancer cells
Tested complexes were dissolved in DMSO just before the experiment, and a calculated amount of drug solution was added to the cell growth medium to a final solvent concentration of 0.5%, which had no detectable effects on cell viability. Cisplatin was dissolved in 0.9% sodium chloride solution. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), p-nitrophenyl phosphate, cisplatin, auranofin and doxorubicin were obtained from Sigma Chemical Co, St. Louis, MO, USA.Cell cultures
Human breast (MDA-MB-231), embryonal (NTERA-2), colon (HCT-15), and pancreatic (PSN-1) carcinoma cell lines along with human embryonic kidney (HEK293) as well as Chinese ovary hamster (CHO) non-cancer cells were obtained by American Type Culture Collection (ATCC, Rockville, MD, USA). Human ovarian 2008 and C13* cancer cells were kindly provided by Prof. G. Marverti (Dept. of Biomedical Science of Modena University, Italy).Cell lines were maintained in the logarithmic phase at 37 °C in a 5% carbon dioxide atmosphere using the following culture media containing 10% fetal calf serum (EuroClone, Milan, Italy), antibiotics (50 units per mL penicillin and 50 μg mL−1 streptomycin), and 2 mM L-glutamine: (i) RPMI-1640 medium (EuroClone) for HCT-15, PSN-1, NTERA-2 and 2008 and C13* cells; (ii) DMEM (EuroClone) for MDA-MB-231, HEK293 cells; (iii) F-12 HAM's for CHO cells.
MTT assay
The growth inhibitory effect toward tumor cells was evaluated by means of MTT assay as previously described.57 IC50 values, the drug concentrations that reduce the mean absorbance at 570 nm to 50% of those in the untreated control wells, were calculated by the four-parameter logistic (4-PL) model.Spheroid cultures and acid phosphatase (APH) assay
Spheroid cultures were obtained by seeding 2.5 × 103 NTERA-2 or 2008 human cancer cells per well in a round-bottom non-treated tissue culture 96-well plate (Greiner Bio-one, Kremsmünster, Austria) in phenol red free F-12 HAMs medium (Sigma Chemical Co., St. Louis, MO, USA) containing 10% fetal calf serum and supplemented with 20% methyl cellulose stock solution. An APH modified assay was employed for evaluating cell viability in 3D spheroids. IC50 values (drug concentrations that reduce the mean absorbance at 405 nm 50% of those in the untreated control wells) were calculated by 4-PL model.Reactive oxygen species (ROS) production
The production of ROS was measured in 2008 cells (104 per well) grown for 24 h in a 96-well plate in RPMI medium without phenol red (Sigma Chemical Co.). Cells were then washed with PBS and loaded with 10 μM 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA) (Molecular Probes-Invitrogen, Eugene, OR) for 25 min, in the dark. Afterwards, cells were washed with PBS and incubated with increasing concentrations of tested compounds. Fluorescence increase was estimated utilizing the wavelengths of 485 nm (excitation) and 527 nm (emission) in an Infinite® 200 PRO (Tecan, Switzerland) plate reader. Antimycin (3 μM, Sigma Chemical Co), a potent inhibitor of complex III in the electron transport chain, and auranofin were used as positive controls.Inhibition of TrxR
2008 cells (1 × 106) were grown in 75 cm2 flasks at the confluence and treated for 24 h with the metal complexes at equimolar concentrations (5 μM). Cell monolayers were harvested, washed with PBS, and centrifuged. Each sample was lysed with RIPA buffer added immediately before the use of an antiprotease cocktail (Roche, Basel, Switzerland). Aliquots were employed for the determination of protein content by means of the BioRad assay (BioRad). The TrxR assay was performed in 0.2 M Na–K-phosphate buffer pH 7.4, containing 5 mM EDTA, 0.250 mM nicotinamide adenine dinucleotide phosphate (NADPH), and 75 nmol of TrxR1 (IMCO, Sweden). Tested complexes as well as auranofin were preincubated for 5 min at room temperature; the reaction started with 1 mM DTNB (5,5′-dithiobis(2-nitrobenzoic acid)), and the increase of absorbance was monitored at 412 nm over 5 min at 25 °C. Enzyme activity was calculated taking into account that mol of NADPH yields 2 mol of CNTP anion (carboxy-nitrothiophenol, reduced DTNB).DAMPs detection
2008 cells (1 × 106) were seeded on 25 cm2 flasks and treated for 24 h with equitoxic concentrations (15 μM) of tested compounds. Supernatants from the samples were transferred to the 96 well plates and analysed using the HMGB1 ELISA kit or the ATP bioluminescence assay kit (ThermoFisher) according to manufacturer's instructions. Absorbance or luminescence were measured using the Infinite® 200 PRO (Tecan, Switzerland) plate reader. Data were normalized to the untreated control samples.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Conflicts of interest
“There are no conflicts to declare”.Acknowledgements
The FWO is gratefully acknowledged for support of this work (G0A6823N). Umicore AG is thanked for continued support through gifts of materials. VC was financially supported by Ministero della Salute – Ricerca Corrente.Notes and references
- L. Galluzzi, A. Buqué, O. Kepp, L. Zitvogel and G. Kroemer, Nat. Rev. Immunol., 2016, 17, 97–111 CrossRef PubMed.
- A. D. Garg, A. M. Dudek-Peric, E. Romano and P. Agostinis, Int. J. Dev. Biol., 2015, 59, 131–140 CAS.
- G. Kroemer, L. Galluzzi, O. Kepp and L. Zitvogel, Annu. Rev. Immunol., 2013, 31, 51–72 CAS.
- J. Fucikova, O. Kepp, L. Kasikova, G. Petroni, T. Yamazaki, P. Liu, L. Zhao, R. Spisek, G. Kroemer and L. Galluzzi, Cell Death Dis., 2020, 11, 1013 CAS.
- J. Zhou, G. Wang, Y. Chen, H. Wang, Y. Hua and Z. Cai, J. Cell. Mol. Med., 2019, 23, 4854–4865 Search PubMed.
- D. V. Krysko, A. D. Garg, A. Kaczmarek, O. Krysko, P. Agostinis and P. Vandenabeele, Nat. Rev. Cancer, 2012, 12, 860–875 CAS.
- L. Zitvogel, O. Kepp, L. Senovilla, L. Menger, N. Chaput and G. Kroemer, Clin. Cancer Res., 2010, 16, 3100–3104 CAS.
- M. Obeid, A. Tesniere, F. Ghiringhelli, G. M. Fimia, L. Apetoh, J.-L. Perfettini, M. Castedo, G. Mignot, T. Panaretakis, N. Casares, D. Métivier, N. Larochette, P. Van Endert, F. Ciccosanti, M. Piacentini, L. Zitvogel and G. Kroemer, Nat. Med., 2006, 13, 54–61 Search PubMed.
- I. Martins, Y. Wang, M. Michaud, Y. Ma, A. Q. Sukkurwala, S. Shen, O. Kepp, D. Métivier, L. Galluzzi, J. Perfettini, L. Zitvogel and G. Kroemer, Cell Death Differ., 2013, 21, 79–91 CrossRef PubMed.
- T. Yamazaki, D. Hannani, V. Poirier-Colame, S. Ladoire, C. Locher, A. Sistigu, N. Prada, S. Adjemian, J. P. P. P. Catani, M. Freudenberg, C. Galanos, F. André, G. Kroemer and L. Zitvogel, Cell Death Differ., 2013, 21, 69–78 CrossRef PubMed.
- S. Sen, M. Won, M. S. Levine, Y. Noh, A. C. Sedgwick, J. S. Kim, J. L. Sessler and J. F. Arambula, Chem. Soc. Rev., 2022, 51, 1212–1233 RSC.
- B. Englinger, C. Pirker, P. Heffeter, A. Terenzi, C. R. Kowol, B. K. Keppler and W. Berger, Chem. Rev., 2018, 119, 1519–1624 CrossRef PubMed.
- D. Longley and P. Johnston, Molecular mechanisms of drug resistance, J. Pathol., 2005, 205, 275–292 CrossRef CAS PubMed.
- L. Zhang, N. Montesdeoca, J. Karges and H. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202300662 CrossRef CAS PubMed.
- H. Zhu, Y. Shan, K. Ge, J. Lu, W. Kong and C. Jia, Cell. Oncol., 2020, 43, 1203–1214 CrossRef CAS PubMed.
- A. Tesniere, F. Schlemmer, V. Boige, O. Kepp, I. Martins, F. Ghiringhelli, L. Aymeric, M. Michaud, L. Apetoh, L. Barault, J. Mendiboure, J. Pignon, V. Jooste, P. Van Endert, M. Ducreux, L. Zitvogel, F. Piard and G. Kroemer, Oncogene, 2009, 29, 482–491 CrossRef PubMed.
- Z. Xu, M. Xu, X. Wu, S. Guo, Z. Tian, D. Zhu, J. Yang, J. Fu, X. Li, G. Song, Z. Liu and X. Song, ChemMedChem, 2023, 18, e202300131 CrossRef CAS PubMed.
- L. M. Lifshits, J. A. Roque III, P. Konda, S. Monro, H. D. Cole, D. Von Dohlen, S. Kim, G. Deep, R. P. Thummel, C. G. Cameron, S. Gujar and S. A. McFarland, Chem. Sci., 2020, 11, 11740–11762 RSC.
- H. Chao, L. Wang, J. Karges, F. Wei, L. Xie, Z. Chen, G. Gasser and L. Ji, Chem. Sci., 2023, 14, 1461–1471 RSC.
- Y. Wang, Y. Ding, D. Yao, H. Dong, C. Ji, J. Wu, Y. Hu and A. Yuan, Small, 2021, 17, 2006231 CrossRef CAS PubMed.
- S. Sen, S. Hufnagel, E. Y. Maier, I. Aguilar, J. Selvakumar, J. E. DeVore, V. M. Lynch, K. Arumugam, Z. Cui, J. L. Sessler and J. F. Arambula, J. Am. Chem. Soc., 2020, 142, 20536–20541 CrossRef CAS PubMed.
- R. Gencheva and E. S. J. Arnér, Annu. Rev. Pharmacol. Toxicol., 2022, 62, 177–196 CrossRef CAS PubMed.
- H. Ghareeb and N. Metanis, Chem. – Eur. J., 2020, 26, 10175–10184 CrossRef CAS PubMed.
- A. Bindoli, M. P. Rigobello, G. Scutari, C. Gabbiani, A. Casini and L. Messori, Coord. Chem. Rev., 2009, 253, 1692–1707 CrossRef CAS.
- C. Gabbiani, G. Mastrobuoni, F. Sorrentino, B. Dani, M. P. Rigobello, A. Bindoli, M. A. Cinellu, G. Pieraccini, L. Messori and A. Casini, MedChemComm, 2011, 2, 50–54 RSC.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 CrossRef CAS PubMed.
- T. Scattolin, G. Tonon, E. Botter, S. G. Guillet, N. V. Tzouras and S. P. Nolan, Chem. – Eur. J., 2023, 29, e202301961 CrossRef CAS PubMed.
- M. K. Amir, S. Z. Khan, F. Hayat, A. Hassan, I. S. Butler and N. Zia-Ur-Rehman, Inorg. Chim. Acta, 2016, 451, 31–40 CrossRef CAS.
- M. Heydari, M. E. Moghadam, A. Tarlani and H. Farhangian, Appl. Biochem. Biotechnol., 2016, 182, 110–127 Search PubMed.
- T. Scattolin, I. Caligiuri, N. Mouawad, M. E. Boustani, N. Demitri, F. Rizzolio and F. Visentin, Eur. J. Med. Chem., 2019, 179, 325–334 CAS.
- G. Tonon, M. Mauceri, E. Cavarzerani, R. Piccolo, C. Santo, N. Demitri, L. Orian, P. A. Nogara, J. B. T. Rocha, V. Canzonieri, F. Rizzolio, F. Visentin and T. Scattolin, Dalton Trans., 2024, 53, 8463–8477 CAS.
- T. Scattolin, E. Bortolamiol, F. Visentin, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio and A. Togni, Chem. – Eur. J., 2020, 26, 11868–11876 CAS.
- T. Scattolin, E. Bortolamiol, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio, L. Cavallo, B. Dereli, M. V. Mane, S. P. Nolan and F. Visentin, Chem. Commun., 2020, 56, 12238–12241 CAS.
- T. Scattolin, V. A. Voloshkin, F. Visentin and S. P. Nolan, Cell Rep. Phys. Sci., 2021, 2, 100446 CAS.
- A. R. Kapdi and I. J. S. Fairlamb, Chem. Soc. Rev., 2014, 43, 4751–4777 CAS.
- L. Szűčová, Z. Trávníček, M. Zatloukal and I. Popa, Bioorg. Med. Chem., 2006, 14, 479–491 Search PubMed.
- C. G. Oliveira, I. Romero-Canelón, M. M. Silva, J. P. C. Coverdale, P. I. S. Maia, A. A. Batista, S. Castelli, A. Desideri, P. J. Sadler and V. M. Deflon, Dalton Trans., 2019, 48, 16509–16517 CAS.
- T. Scattolin, E. Cavarzerani, D. Alessi, M. Mauceri, E. Botter, G. Tonon, I. Caligiuri, O. Repetto, U. Kamensek, S. K. Brezar, M. Dalla Pozza, S. Palazzolo, M. Cemazar, V. Canzonieri, N. Demitri, S. P. Nolan, G. Gasser, F. Visentin and F. Rizzolio, Dalton Trans., 2025, 54, 4685–4696 CAS.
- C. M. Zinser, F. Nahra, M. Brill, R. E. Meadows, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and C. S. J. Cazin, Chem. Commun., 2017, 53, 7990–7993 CAS.
- T. Scattolin, I. Pessotto, E. Cavarzerani, V. Canzonieri, L. Orian, N. Demitri, C. Schmidt, A. Casini, E. Bortolamiol, F. Visentin, F. Rizzolio and S. P. Nolan, Eur. J. Inorg. Chem., 2022, e202200103 CAS.
- C. Chen, F. Liu and M. Szostak, Chem. – Eur. J., 2021, 27, 4478–4499 CAS.
- J. Zhang, T. Li, X. Li, G. Zhang, S. Fang, W. Yan, X. Li, X. Yang, Y. Ma and M. Szostak, Chem. Commun., 2022, 58, 7404–7407 CAS.
- M. Farooq, N. Taha, R. Butorac, D. Evans, A. Elzatahry, E. Elsayed, M. Wadaan, S. Al-Deyab and A. Cowley, Int. J. Mol. Sci., 2015, 16, 24718–24731 CrossRef CAS PubMed.
- P. Baczewska, K. Śniady, W. Kośnik and M. Michalak, Catalysts, 2021, 11, 972 CrossRef CAS.
- T. Scattolin, A. A. Logvinov, N. V. Tzouras, C. S. J. Cazin and S. P. Nolan, Organometallics, 2023, 42, 2692–2730 CrossRef CAS.
- I. I. Hashim, N. V. Tzouras, W. Janssens, T. Scattolin, L. Bourda, S. Bhandary, K. Van Hecke, S. P. Nolan and C. S. J. Cazin, Chem. – Eur. J., 2022, 28, e202201224 CrossRef PubMed.
- H. Zhu, Y. Shen, Q. Deng, J. Chen and T. Tu, ACS Catal., 2017, 7, 4655–4659 CAS.
- K. Zinkewich-Péotti and P. A. Andrews, Cancer Res., 1992, 52, 1902–1906 Search PubMed.
- C. Marzano, V. Gandin, A. Folda, G. Scutari, A. Bindoli and M. P. Rigobello, Free Radical Biol. Med., 2006, 42, 872–881 Search PubMed.
- L. A. Kunz-Schughart, J. P. Freyer, F. Hofstaedter and R. Ebner, SLAS Discov., 2004, 9, 273–285 CAS.
- S. Prast-Nielsen, M. Cebula, I. Pader and E. S. J. Arnér, Free Radical Biol. Med., 2010, 49, 1765–1778 CAS.
- V. V. L. Müller, P. V. Simpson, K. Peng, U. Basu, D. Moreth, C. Nagel, S. Türck, L. Oehninger, I. Ott and U. Schatzschneider, Inorg. Chem., 2023, 62, 16203–16214 Search PubMed.
- V. Scalcon, A. Bindoli and M. P. Rigobello, Free Radical Biol. Med., 2018, 127, 62–79 CAS.
- H. Türkmen, O. Şahin, O. Büyükgüngör and B. Çetinkaya, Silver, Eur. J. Inorg. Chem., 2006, 2006, 4915–4921 Search PubMed.
- K. V. Vasudevan, R. R. Butorac, C. D. Abernethy and A. H. Cowley, Dalton Trans., 2010, 39, 7401 CAS.
- G. Utecht-Jarzyńska, S. Jarzyński, M. M. Rahman, G. Meng, R. Lalancette, R. Szostak and M. Szostak, Organometallics, 2024, 43, 2305–2313 Search PubMed.
- C. Saviozzi, L. Biancalana, T. Funaioli, M. Bortoluzzi, M. De Franco, M. Guelfi, V. Gandin and F. Marchetti, Inorg. Chem., 2024, 63, 1054–1067 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2415263. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5md00039d |
| This journal is © The Royal Society of Chemistry 2025 |