
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards catalytic fluoroquinolones: from metal-catalyzed to metal-free DNA cleavage†
Moshe N.
Goldmeier‡
a,
Alina
Khononov‡
a,
Tomasz
Pieńko‡
a,
Valery
Belakhov
a,
Feng-Chun
Yen
b,
Limor
Baruch
b,
Marcelle
Machluf
b and
Timor
Baasov
 *a
*a
aEdith and Joseph Fischer Enzyme Inhibitors Laboratory, Schulich Faculty of Chemistry, Technion – Israel Institute of Technology, Haifa 3200003, Israel. E-mail: chtimor@technion.ac.il
bFaculty of Biotechnology and Food Engineering, Technion – Israel Institute of Technology, Haifa 3200003, Israel
First published on 5th March 2025
Abstract
A library of eight new fluoroquinolone–nuclease conjugates containing a guanidinoethyl or aminoethyl auxiliary pendant on the 1,4,7-triazacyclononane (TACN) moiety was designed and synthesized to investigate their potential as catalytic antibiotics. The Cu(II) complexes of the designer structures showed significant in vitro hydrolytic and oxidative DNA cleavage activity and good antibacterial activity against both Gram-negative and Gram-positive bacteria. The observed activity of all the Cu(II)–TACN–ciprofloxacin complexes was strongly inhibited in the presence of Cu(II)-chelating agents, thereby demonstrating “vulnerability” under physiological conditions. However, selected TACN–ciprofloxacin conjugates in their metal-free form efficiently cleaved plasmid DNA under physiological conditions. The lead compound 1 showed good DNase activity which was retained in the presence of strong metal chelators and exhibited excellent antibacterial activity against both Gram-negative and Gram-positive bacteria. Density functional theory calculations combined with quantum mechanics/molecular mechanics simulations suggest a general base–general acid mechanism for the hydrolytic DNA cleavage mechanism by compound 1.
Introduction
The rapid increase in antimicrobial resistance poses a significant threat to public health, with warnings about entering a post-antibiotic era already issued.1,2 This has sparked renewed efforts to formulate novel antibacterial strategies that can combat existing bacterial resistance and slow down the development of new resistance in the future. Currently, the most promising approaches include combination therapies, modification of existing antibiotics and efficacy enhancement by metabolic stimulation or employing more efficient delivery systems. In addition to these conventional approaches, alternative strategies based on bacteriophages, anti-biofilm agents, probiotics, vaccines, and antibodies have also been proposed.3,4In recent years, several new antibiotics, such as delafloxacin or plazomicin, have been approved for use in the clinic to deal with multidrug-resistant bacteria.5 However, it has been well-documented that once a new antibiotic is introduced into the clinic, whether it is a novel chemical entity acting on a distinct bacterial target or a semisynthetic derivative that counters the resistance to its parent drug, it is only a matter of time until new resistance will yet again emerge.6 Therefore, new generations of classical antibiotics that allow us to survive another round of bacterial combat do not necessarily affect the roots of resistance development.
A promising strategy to overcome antibacterial resistance is the development of catalytic antibiotics, which are meant to act as small-molecule-based therapeutic agents that catalytically inactivate specific bacterial targets.7 This approach, akin to the Michaelis–Menten enzyme model, involves noncovalent binding to the target followed by its chemical modification. Catalytic antibiotics promote multiple turnovers of a catalytic cycle, thus potentially improving antibacterial activity, reducing toxicity due to lower dosage requirements, and delaying new resistance development.8
Recent studies have highlighted multifunctional antibacterial metallopeptides that modify nucleic acids, proteins, or phospholipids via reactive oxygen species (ROS) generated by an amino terminal copper and nickel (ATCUN) binding motif. Research by Cowan9,10 and Angeles-Boza and co-workers11,12 has shown that Cu(II)–ATCUN conjugates with antimicrobial peptides significantly improve antibacterial activity compared to the parent peptides. These conjugates can also act synergistically with conventional antibiotics to reduce biofilm formation13 which is challenging to eradicate.14 However, the observed improvements in minimal inhibitory concentration (MIC) were not as significant as expected from catalytic metallodrugs, indicating a need for more complex design strategies to fully realize their clinical potential.
To address these challenges, we explored repurposing known classical antibiotics as selective binding motifs to facilitate the cleavage of critical chemical bonds in bacterial targets, leading to their immediate deactivation. Initially, we applied this concept to aminoglycoside antibiotics15 and more recently to the fluoroquinolone scaffold.8,16
Fluoroquinolones are highly effective, broad-spectrum antibiotics that are widely prescribed worldwide. They exhibit concentration-dependent bactericidal activity by inhibiting DNA gyrase and topoisomerase IV, essential enzymes for DNA replication and transcription. However, resistance to fluoroquinolones is limiting their use, often due to specific mutations in DNA gyrase and topoisomerase IV. Consequently, new bacterial topoisomerase type IIA inhibitors have been developed to offer alternative binding modes or mechanisms of action.17,18
Within the concept of catalytic fluoroquinolones, we synthesized ciprofloxacin–nuclease conjugates (see first-generation cyclen-based conjugates, Fig. 1) designed to catalytically cleave a specific phosphodiester bond at the site of the fluoroquinolone–topoisomerase–DNA ternary complex. Under oxidative conditions, Cu(II)-complexes of these conjugates fragmented supercoiled plasmid DNA into linear DNA in the presence of DNA gyrase, demonstrating a proof of concept in vitro. The lead compound also showed enhanced antibacterial activity under oxidative conditions compared to the metal-free ligand.8
 | ||
| Fig. 1 Designed structures of cyclen-based first-generation8 and second-generation16 conjugates of ciprofloxacin, along with the new TACN-based, third-generation conjugates (1–8) and the reference TACN-warheads (9 and 10). | ||
However, the hydrolytic nuclease activity of the first-generation Cu(II)–cyclen–ciprofloxacin complexes was inhibited under normal cellular conditions due to Cu(II)–aqua ligand exchange with endogenous ligands such as glutamate, spermidine or ATP. This “vulnerability” is a significant issue in vivo for metallodrugs working via a hydrolytic mechanism. To address this, we designed second-generation ciprofloxacin–nuclease conjugates with a dynamic intramolecular cap to protect the metal center (Fig. 1).16 We have shown that installing the amine pendant at the Co(III)–cyclen warhead preserved in vitro DNase activity of the conjugates under physiological conditions. Due to the low pKa of the amine arm (below 4), the Co(III)–cyclen complex remained capped in solution. At the DNA interface, the complex was uncapped as it formed strong electrostatic interactions with the DNA phosphates. However, the high positive charge of the Co(III) ion and ammonium pendant (+4 in total) could significantly slow down dissociation from the DNA and thereby limit catalytic performance.16
Consequently, we were motivated to solve the vulnerability issue of the Cu(II) derivatives that bear less positive charge, thus potentially enhancing catalytic turnover. For the second-generation cyclen complexes of Cu(II), the pKa of the aminoethyl or guanidinoethyl pendants was around 9; therefore, the Cu(II) complexes remained uncapped and thus unprotected under physiological conditions.16 To lower the pKa of the Cu(II) complexes, we substituted cyclen with 1,4,7-triazacyclononane (TACN) as it has been shown that the pKa of the guanidinoethyl pendant in the Cu(II)–TACN complex is 5.6,19 thus favoring the capped state at physiological pH.
With this premise, we assembled a library of third-generation conjugates of ciprofloxacin and Cu(II)–TACN with guanidinoethyl or aminoethyl pendants (compounds 1–8, Fig. 1), along with their corresponding warheads as reference compounds (compounds 9 and 10, Fig. 1). We found that the new designer compounds demonstrated excellent in vitro hydrolytic and oxidative DNA cleavage activity and good antibacterial activity against both Gram-negative and Gram-positive bacteria, with improvements regarding the respective cyclen analogues. Surprisingly however, their nuclease activity was inhibited under “normal” cellular conditions indicating that the metal vulnerability issue remained unsolved. Nevertheless, we were pleased to discover a new lead compound based on the TACN–guanidinoethyl warhead that cleaved DNA hydrolytically in a “metal-free” mechanism under physiological conditions and exhibited significant antibacterial activity against both Gram-negative and Gram-positive bacteria. By using a variety of in silico tools, we suggest the potential molecular mechanism of DNA cleavage and discuss the benefits of metal-independent nuclease activity for future development of catalytic antibiotics.
Results and discussion
Synthesis of 1–8 and their Cu(II) complexes
Compounds 1–8 were synthesized in four or five chemical steps from terminal bromide derivatives 11a–d (ref. 16) and common intermediate 12 (TFA salt form) incorporating a TACN ring appended to a two-carbon linked protected amine (Scheme 1). It is notable that derivative 12 was prepared from the known bis-Boc-protected derivative19 of 12 by treatment with TFA, as detailed in the Experimental section. | ||
| Scheme 1 Synthesis of compounds 1–4 (A) and compounds 5–8 (B). Reagents and conditions: (a) CH3CN, K2CO3, −15 °C; (b) CH3CN, Cs2CO3, 60 °C; (c) dry DCM, Et3N, Boc2O; (d) EtOH, CH3NH2, K2CO3, 0 °C–rt, 21% over 3 steps (13a), 35% over 3 steps (13b), 30% over 3 steps (13c), 40% over 3 steps (13d); (e) CH3CN, N,N-di-Boc-1H-pyrazole-1-carboxamide, K2CO3, rt, 72% (14a), 75% (14b), 53% (14c), 49% (14d); (f) 6 M HCl(aq), 90 °C, 37% (1), 41% (2), 34% (3), 45% (4), 48% (5), 45% (6), 42% (7), 47% (8). | ||
First, the coupling of primary bromides 11a–d with 12 under basic conditions (Cs2CO3), followed by Boc-protection with Boc2O, provided the corresponding phthalimide derivatives, which after treatment with methylamine (in the presence of K2CO3 (ref. 20)) afforded the corresponding amines 13a–d, which also contained a methyl amide instead of a methyl ester in the ciprofloxacin moiety. The amines were then treated with N,N-di-Boc-1H-pyrazole-1-carboxamide in the presence of a base (K2CO3) to afford the corresponding protected guanidine derivatives 14a–d.
One-step deprotection using 6 M HCl at 90 °C, followed by purification of the crude products (using an LH-20 size-exclusion column), provided the desired compounds 1–4 as hydrochloride salts (Scheme 1A), with guanidinium pendants on the TACN warhead. Alternatively, one-step deprotection of amine derivatives 13a–d using 6 M HCl at 90 °C provided the desired compounds 5–8 as hydrochloride salts (Scheme 1B), with amine pendants. The final 1–8 structures were characterized by combined 1D and 2D nuclear magnetic resonance (NMR) and high-resolution mass spectrometry (HRMS) techniques. Finally, the reference compound 9 was prepared according to a published procedure,19 and the second reference compound 10 was prepared from the known bis-Boc-protected derivative19 of 10 by treatment with TFA, as detailed in the Experimental section.
The new set of ligands 1–8 as hydrochloride salts were treated with a stoichiometric amount of CuCl2, followed by pH adjustment (using 1 M NaOH(aq)) to facilitate metal-to-ligand coordination. The complexes were characterized by UV-vis and HRMS (see the Experimental section for details).
DNA cleavage assays
Biologically relevant nuclease activity tests of metal-free compounds 1–8 and their Cu(II) complexes were performed as previously reported by our group.8,16 Briefly, agarose gel electrophoresis (1% agarose) and ethidium bromide staining were used to monitor the conversion of supercoiled (form I or F-I) pHOT-1 plasmid DNA into its nicked form (form II or F-II) or into multiply nicked DNA; no linear form (form III or F-III) was observed. As reported previously,8,16 due to the presence of multiple positive charges in the ligands, it is necessary to use an ion-exchange based microscale procedure for removal of the ligands prior to electrophoresis to ensure the migration of DNA in the electric field. In all DNA cleavage assays, we used compound A (Chart 1) from our first-generation cyclen–ciprofloxacin conjugates8 as the positive control. | ||
| Chart 1 Compound A from our first-generation cyclen–ciprofloxacin conjugates,8 which was used in this study as the positive control. | ||
 | ||
| Fig. 2 Comparative concentration-dependent cleavage of (+) supercoiled pHOT-1 plasmid (0.014 μg μL−1) in HEPES buffer (50 mM, pH 7.4) over 5 h. Compounds include A–Cu(II) as a positive control. | ||
For the amine series, however, three of the Cu(II) complexes, 5–Cu(II), 6–Cu(II) and 8–Cu(II), show significantly enhanced DNase activity compared to the parent complex 10–Cu(II) (see Fig. 1 and 2), whilst 7–Cu(II) shows no significant activity (see Fig. S1†).
Collectively, these data indicate that (i) DNA binding and (ii) DNase activity are highly dependent on the linker length, both for the guanidine and amine series. However, the intercalating cipro scaffold apparently mediates enhanced DNase activity for the amine series, but not for the guanidine series. It is reasonable to suggest that the conformational restrictions imposed upon the bulky guanidine pendant by scaffold-intercalation offset any potential DNase enhancement mediated by the “proximity effect” (or its increased effective molarity21). Indeed, we have previously reported16 a similar discrepancy between the guanidine and amine pendants for the cyclen-based analogues (i.e., Cu(II) complexes of second-generation derivatives, Fig. 1), for which the guanidine pendant (but not the amine pendant) shows a strong inhibitory effect on DNase activity.16
Finally, to investigate whether the amine or guanidine pendants provide effective protection (electronic and/or steric) against Cu(II)–aqua ligand exchange,8 we investigated the DNase activity of 1–Cu(II), 5–Cu(II) and 6–Cu(II) in the presence of Cu(II)-chelating Tris buffer. Unfortunately, as depicted in Fig. S2,† all DNase activity was lost under these conditions, which demonstrates that the Cu(II) complexes are vulnerable to ligand exchange. We also performed potentiometric titration (pH 2–12.5) in combination with UV-vis spectroscopy to probe whether the pendants coordinate to the metal. Specifically, we investigated 3–Cu(II) (guanidine series) and 7–Cu(II) (amine series). In both these cases, the UV-vis spectra showed no significant perturbation in the investigated pH range until the pKa of the pendant was reached, i.e., around 12 for 3–Cu(II) and around 10 for 7–Cu(II). These data indicated that the guanidine and amine pendants do not interact with the metal centre at physiological pH.
As depicted in Fig. S3,†1–Cu(II) shows no significant activity at micromolar concentrations (>40 μM). In stark contrast, the cyclen-based analogues (second-generation derivatives with guanidine pendants, Fig. 1) all show complete conversion of form I to form II at 5 μM.8 This distinction however is not unexpected, as Cowan and co-workers22 have reported that unlike Cu(II)–cyclen, Cu(II)–TACN has no significant DNA cleavage activity in the presence of ascorbic acid under aerobic conditions.
Interestingly however, 5–Cu(II) and 6–Cu(II) do exhibit significant DNase activity under these conditions, with complete conversion of form I to multiply nicked DNA (as evidenced by a DNA smear) at 20 μM and 80 μM, respectively (Fig. S3†). Although these activities are much less pronounced than their respective cyclen-based analogues (showing complete conversion at 2.5 μM and 0.5 μM, respectively16), these data nevertheless highlight the critical impact of the amine vs. guanidine pendant on the oxidative DNA cleavage activity of these complexes.
 | ||
| Fig. 3 Comparative concentration-dependent cleavage of (+) supercoiled pHOT-1 plasmid (0.014 μg μL−1) in HEPES buffer (50 mM, pH 7.4) over 5 h. Compounds include A–Cu(II) as a positive control. | ||
For the amine series, compound 5 exhibits significantly enhanced DNase activity compared to the parent compound 10 (see Fig. 3). In contrast, compounds 7 and 8 both exhibit very strong DNA binding, whilst compound 6 shows no significant DNase activity (see Fig. S4†).
In summary, these data suggest that (i) DNA binding and (ii) DNA cleavage activity are highly dependent on the linker length, for both the guanidine and amine series. Furthermore, compound 1 is shown to be the most effective metal-free nuclease across both series (see Fig. 3).
To investigate whether the mechanism of cleavage for compound 1 is metal-mediated, a scavenging assay was performed in 1 mM EDTA. As illustrated in Fig. 4, whilst 3 loses all DNase activity in the presence of EDTA (and therefore serves as a negative control), compound 1 retains significant activity. Furthermore, in contrast to 1–Cu(II) which loses all DNase activity in the presence of metal-chelating Tris buffer (see Fig. S2†), 1 retains full DNase activity under the same conditions (see Fig. 5).
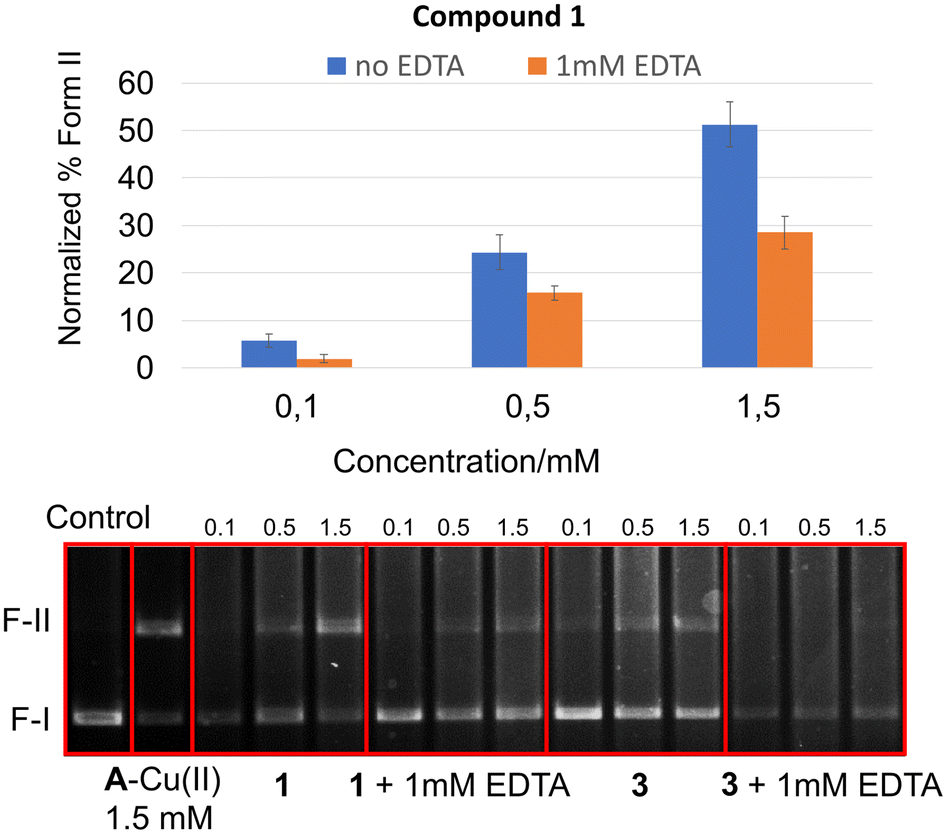 | ||
| Fig. 4 Comparative concentration-dependent cleavage of (+) supercoiled pHOT-1 plasmid (0.014 μg μL−1) in HEPES buffer (50 mM, pH 7.4) over 5 h, with or without 1 mM EDTA. Compounds include A–Cu(II) as a positive control. | ||
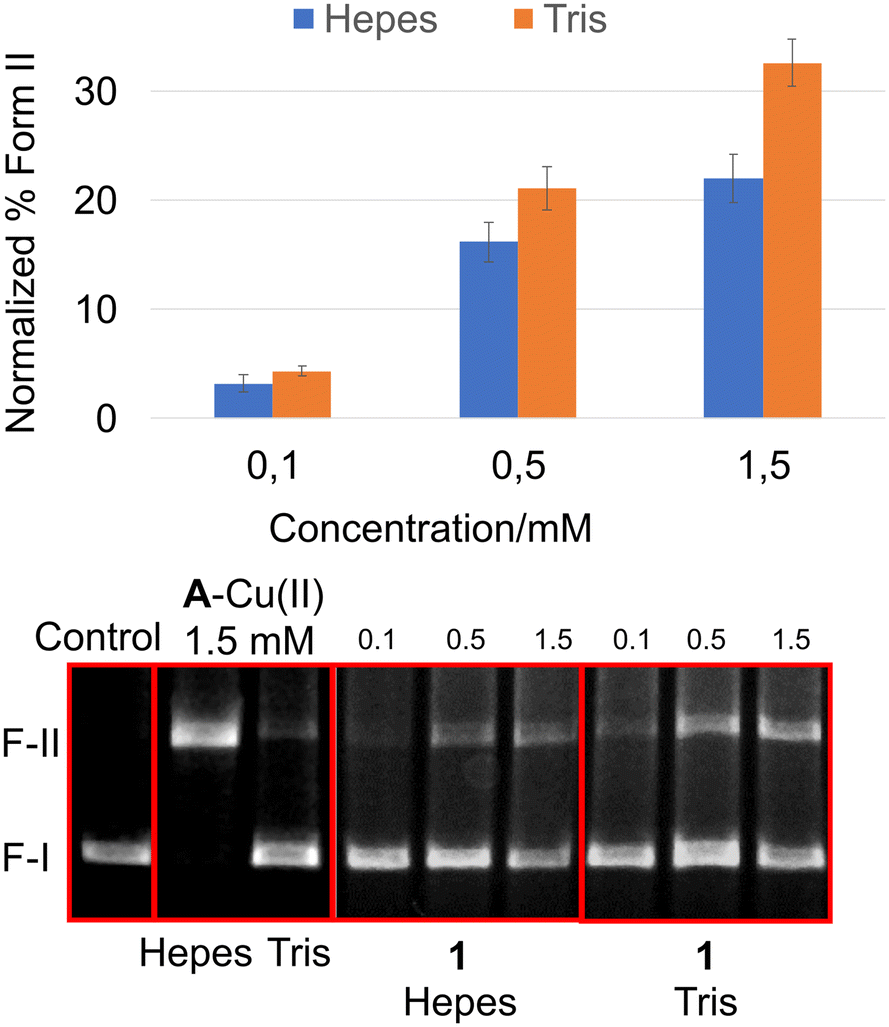 | ||
| Fig. 5 Comparative concentration-dependent cleavage of (+) supercoiled pHOT-1 plasmid (0.014 μg μL−1) in HEPES or Tris buffer (50 mM, pH 7.4) over 5 h. Compounds include A–Cu(II) as a positive control. | ||
Collectively, these data strongly suggest that the combination of the DNA-intercalating ciprofloxacin scaffold and the macrocyclic polyamine TACN facilitates metal-free, hydrolytic DNA cleavage. Indeed, several TACN–anthraquinone intercalator conjugates have been reported to exclusively exhibit metal-free DNA cleavage activity.23,24 Those compounds retained their activity in the presence of metal chelators and ROS scavengers, thereby excluding DNA cleavage via metals, autooxidation and ROS formation. It is reasonable to speculate that an intercalator moiety (i.e., ciprofloxacin or anthraquinone) and an optimal spacer enable TACN to be proximal to the DNA backbone without becoming strongly bound to it.
Antibacterial activity and cytotoxicity
We tested compounds 1–8 and their Cu(II) complexes for their comparative antibacterial activity by determining MIC values against two Gram-negative E. coli strains (R477-100 and 25922) and two Gram-positive strains, Staphylococcus epidermidis and Bacillus subtilis (Table 1).| Bacteria/compound | ||||
|---|---|---|---|---|
| Gram-negative | Gram-positive | |||
| Ciprofloxacin | 0.05 | 0.05 | 0.19 | 0.02 |
| a Escherichia coli R477-100. b Escherichia coli 25922. c Staphylococcus epidermidis. d Bacillus subtilis 6633. | ||||
| 1 | 6 | 3 | 3 | 3 |
| 1–Cu(II) | 6 | 3 | 3 | 3 |
| 2 | 12 | 12 | 12 | 12 |
| 2–Cu(II) | 12 | 6 | 12 | 6 |
| 3 | 3 | 3 | 3 | 3 |
| 3–Cu(II) | 6 | 6 | 6 | 6 |
| 4 | 12 | 12 | 12 | 6 |
| 4–Cu(II) | 12 | 12 | 12 | 6 |
| 5 | 12 | 6 | 6 | 3 |
| 5–Cu(II) | 12 | 12 | 6 | 6 |
| 6 | 12 | 6 | 12 | 6 |
| 6–Cu(II) | 12 | 12 | 6 | 6 |
| 7 | 12 | 6 | 3 | 3 |
| 7–Cu(II) | 6 | 6 | 6 | 3 |
| 8 | 12 | 12 | 3 | 3 |
| 8–Cu(II) | 12 | 12 | 6 | 3 |
All the new derivatives of ciprofloxacin exhibit significant antibacterial activity against Gram-negative and Gram-positive bacteria. The two lead compounds against the two E. coli strains tested (Gram-negative) are 1 and 3 (3–6 μg mL−1), which show 2- to 4-fold improvement in comparison with the other compounds. However, against the two Gram-positive strains tested, 1, 3, 7 and 8 all exhibit similarly good antibacterial activity (3 μg mL−1).
Furthermore, all the Cu(II) complexes of 1–8 exhibit similar antibacterial activity compared to the metal-free ligands. Of particular note in this regard is 5–Cu(II), which exhibits strong DNase activity compared to the metal-free ligand (i.e., full conversion of form I to form II at 0.1 mM vs. no DNase activity whatsoever for 5 – see Fig. 1vs.2) but no improvement in antibacterial activity in any of the tested strains. Thus, the MIC data corroborate the notion that these Cu(II) complexes are indeed vulnerable to ligand exchange. However, the new compounds have significantly reduced antibacterial activity relative to the parent ciprofloxacin (Table 1). A very similar reduction in antibacterial potency of ciprofloxacin derivatives was also observed when ciprofloxacin was conjugated with other highly positively charged molecules like cyclen8,16 and aminoglycosides to yield hybrid antibiotics.25 It was suggested that the cellular permeability of these ciprofloxacin conjugates is significantly lower than that of the parent drug or the conjugates have increased offsite binding. Both phenomena could be attributed to the high polarity of either cyclen,8,16 TACN–guanidine/TACN–amine (current work), or aminoglycosides,25 covalently linked to ciprofloxacin.
Given the fact that 1 shows (i) significant, metal-free, hydrolytic DNase activity (see Fig. 4 and 5) and (ii) exhibits strong antibacterial activity against both Gram-negative and Gram-positive bacteria (Table 1), it would be intriguing to investigate the possibility that 1 is a dual-acting antibacterial agent with the potential to slow down resistance development.
To gain a better understanding of the safety of the lead compounds 1, 2, 5 and 8, we determined their comparative cell toxicity to ciprofloxacin by measuring the half-maximal lethal concentrations values (LC50) in baby hamster kidney (BHK) and in human embryonic kidney (HEK293-FT) cells (Table 2). Among the tested compounds, the parent drug ciprofloxacin is the most cytotoxic compound with LC50 values of 0.2 and 0.11 mM in BHK and HEK293-FT, respectively. Compound 2 is the most toxic compound among the synthetic derivatives with an LC50 value of 0.3 mM for both cell lines. Nevertheless, it is still 1.5- and 3-fold less cytotoxic than ciprofloxacin. In BHK cells, the LC50 of compound 8 is 0.74 mM, making it the least cytotoxic compound among all. Finally, the lead compound 1 has a cytotoxicity of 0.5 mM for both cell lines, 2.6- and 4.4- fold less toxic than ciprofloxacin.
| Cells/compounda | BHK | HEK293-FT |
|---|---|---|
| a Cell toxicity was measured in baby hamster kidney (BHK) and human embryonic kidney (HEK293-FT) cells and calculated as a ratio between the number of living cells in cultures grown with and without the presence of the tested compounds. | ||
| Ciprofloxacin | 0.20 | 0.11 |
| 1 | 0.51 | 0.48 |
| 2 | 0.31 | 0.34 |
| 5 | 0.42 | 0.71 |
| 8 | 0.74 | 0.46 |
Molecular mechanism of metal-free DNA cleavage
Intrigued by the metal-free DNase activity of compound 1, we investigated its molecular mechanism by combining several computational methods including quantum chemical calculations based on density functional theory (DFT), molecular docking, classical molecular dynamics (MD), and hybrid quantum mechanics/molecular mechanics (QM/MM) simulations (see Computational methods in the ESI†).Initially, we focused on studying the structure of the TACN–guanidine warhead in solution. It is of note that the two tertiary amines of the warhead are chiral yielding four possible configurations (RR, RS, SR, SS). We sampled and energy-scored all possible protonation states of the warhead (see Fig. S5 and Table S1†) using GFN2-xTB26 in CREST 2.12 (ref. 27) and CENSO 1.2.0 (ref. 28) followed by DFT calculations in Orca 6.0 (ref. 29) coupled with the COSMO-RS implicit solvation model in OpenCOSMO-RS.30 The free-energy calculations showed that in all the protonation states considered, the most energetically stable are stereoisomers with either (R, R) or (S, S) configurations. For the first protonation, we found that the most basic amine within the warhead is the guanidine pendant, for which we estimated a pKa of 14.48, which agrees with its exceptionally high pKa in arginine (13.8).31 The second protonation favorably occurs at the secondary amine, with the pKa calculated to be 12.20. The triprotonation is preferred at the tertiary amine connected with the linker, with the pKa calculated to be 5.77. The second and the third deprotonation constants correspond to those observed for disubstituted TACN derivatives, such as Hnoapy32 (pK1 = 12.15 and pK2 = 5.78) or H2noda33 (pK1 = 11.82 and pK2 = 6.70), supporting the in silico predictions. For the quadruple protonated state, we estimated a pKa of 3.34. The pKa results indicate that under physiological conditions, the TACN–guanidine warhead is predominantly diprotonated.
To study specific interactions of the diprotonated TACN–guanidine warhead with water, we performed classical MD simulations in explicit solvent using the AMBER force field.34 We found that the warhead can recruit a single water molecule from the bulk, bridging the guanidine pendant and TACN amine, stabilizing two positively charged amines in proximity (Fig. 6A). The capability of the TACN–guanidine warhead to bind to a single water molecule has brought us to the hypothesis that this water could be used as a nucleophile for attack on the DNA phosphate in an analogous manner to metal-based catalysis of DNA cleavage.35 To test this hypothesis, we used the DFT-optimized geometries of the complexes of the TACN–guanidine warhead with water observed in the MD simulations, in both (R, R) and (S, S) configurations (Fig. 6A) to perform fixed-anchor docking36 of compound 1 to the selected binding sites of DNA (PDB code: 2XKK37) in many conformations which we generated in the Hamiltonian replica-exchange MD (H-REMD)38 simulations (see Fig. S6 and S7†).
 | ||
| Fig. 6 A: DFT-optimized structures of water complexes of the TACN warhead with the guanidinoethyl pendant in two favorable stereoisomers observed in the MD simulations. For computational efficiency, the ciprofloxacin-linker part is represented by an ethyl group. B: The most probable binding mode of compound 1 during DNA cleavage, derived from the QM/MM simulation and visualized using VMD.39 The O–P–O angle and key interactions (distances in Å) for catalysis between compound 1 and DNA are depicted. C: General acid–general base mechanism of DNA cleavage by compound 1. | ||
Only among the docked structures with the (S, S) configuration of the chiral centers in the warhead, we found poses in which the TACN–guanidine warhead oriented the water molecule in the in-line geometry for nucleophilic attack on the DNA phosphate (with ≥150° angle between the oxygen atom of water Ow, the phosphorus atom P, and the oxygen atom of the DNA phosphate O5′ and the distance Ow–P of ≤4.0 Å). The selected structures were further simulated using the classical MD and AMBER force field40–42 (see Fig. S8†). The simulations revealed instability of the complex of the catalytic warhead and DNA on the pico–nanosecond timescale. We observed that using three-point charge representation of water (e.g. OPC3 (ref. 43)) destabilized the complex even more than four-point charge models (e.g. OPC (ref. 40)). The instability issue persisted even after refinement of the AMBER parameters44,45 of compound 1 (see Fig. S9†), leading us to the notion that the AMBER force field might be an oversimplified model to study the interactions between compound 1 and DNA prior to the hydrolysis reaction. Therefore, we tested different semi-empirical potentials in the QM/MM approach.46
Only one setup, combining the third-order density-functional tight-binding (DFTB3) method corrected with D4 three-body dispersion47,48 and using the OPC water model, provided stability of the DNA–warhead complex on the timescale of hundreds of picoseconds (see Fig. S10†). During the QM/MM simulations, the nearly in-line geometry of water approaching the DNA phosphate was stable (with Ow–P ≈ 3.78 Å and Ow–P–O5′ ≈ 163°) (Fig. 6B and S10†). The hydrogen atoms of the water molecule interacted preferably with the TACN tertiary amine connected to the linker (the N3 atom in Fig. 6B) (with Hw–N3 ≈ 2.91 Å) (Fig. 6B and S10†). The in-line orientation of water within the pre-reaction complex is stabilized by two interactions between the DNA and the warhead: i) between the protonated secondary amine of TACN and the adjacent DNA phosphate (N2–OP1 ≈ 2.85 Å), and ii) between the guanidine pendant and the cleaved phosphate (N1–OP2 ≈ 3.02 Å) (Fig. 6B and S10†).
The MD and QM/MM simulations suggested that accurate description of electrostatic and dispersion interactions within the DNA–warhead complex at the DFT level is mandatory to study the next steps of the mechanism of cleavage. Nevertheless, collectively, our calculations suggest that compound 1 cleaves DNA in a general base–general acid mechanism (Fig. 6C). Since the TACN tertiary amine conjugated with the linker (the N3 atom in Fig. 6B) is mostly deprotonated at physiological pH (pKa = 5.77) and favorably interacts with the water molecule in the in-line geometry at the DNA interface, it can serve as a general base, activating the water molecule for attack on the DNA phosphate. Meanwhile, the close interaction of the protonated guanidine arm with the cleaved phosphate group indicates that it may function as a general acid, neutralizing the negative charge of the phosphate and activating the leaving group. Because of the high pKa of guanidine, its role in shuttling protons on the phosphorane intermediate formed during the hydrolysis has been under debate.49 In this context, we can rationalize the enhanced DNase activity of compound 1 in Tris buffer (relative to HEPES buffer, Fig. 5), in which a primary amine with a pKa of 8.6 (ref. 50) could serve as a more potent proton donor for the leaving group than guanidine.
Perspective
To date, metal-free artificial DNases have been commonly built as bis-guanidinium receptors based on the scaffold of phenyl,51 calix[4]arenes,52 Dervan-type polyamides53 and cyclic peptides54 decorated with moieties bearing hydroxyl groups, imitating DNA topoisomerase I which uses tyrosine as a nucleophile and catalyzes a transphosphorylation reaction.55 An alternative strategy for designing non-metallic nucleases has involved cyclic polyamines like cyclen and TACN. DNase capabilities have been observed in various compounds: N-dimethylated cyclen,56 bis-cyclen connected with urea,57 a cyclen conjugate with peptide nucleic acid58 and TACN combined with anthraquinone.23 Additionally, modifications involving guanidine or amine pendants, and hydroxyethyl were made to TACN to perform transphosphorylation, leading to DNase activity at low micromolar concentrations.24,59 The transphosphorylation mechanism effectively cuts DNA, yet compounds operating in this manner were thought to become covalently trapped in DNA, preventing subsequent catalytic cycles.To the best of our knowledge, our study is the first report of an antibiotic molecule bearing a small organic fragment that cleaves DNA with no metal involvement in a general base–general acid mechanism by recruiting a water molecule from bulk. Unlike the transphosphorylation mechanism applied in most artificial metal-free DNases, our proposed DNA hydrolysis mechanism allows the compound to be released after the reaction and perform multiple catalytic turnovers.
Furthermore, metal-free DNA cleavage avoids the problems associated with metals, such as vulnerability under physiological conditions or the associated toxicity due to the ROS formation. With a purely hydrolytic mechanism and strong dependence of the nuclease activity on its structure, it is also feasible to improve site selectivity of cleavage and limit any adverse effects.
It is of note that since ciprofloxacin conjugates of TACN with amine or guanidine pendants were initially designed for metal-catalyzed DNA cleavage, their efficiency in non-metallic catalysis may be limited by the imperfect structure. The QM/MM simulations revealed that compound 1 can cleave DNA only when ciprofloxacin is arranged in an orientation parallel to the DNA bases, whereas, the H-REMD simulations of the ciprofloxacin–linker conjugate suggested that it preferred perpendicular orientation to the DNA bases (Fig. S7†). Thus, it can be speculated that, depending on the DNA sequence context, compound 1 can be trapped in catalytically inactive conformations, significantly limiting its global nuclease efficiency. Optimization of the linker connecting the binding and catalytic domains seems crucial for increasing the rate of the hydrolysis reaction. In addition, higher nuclease activity could be provided by modifying the linker attachment site in the catalytic warhead to increase general base pKa as well as modifying the structure of the guanidine moiety to be a stronger general acid.60
Despite those shortcomings, we believe that the presented concept of metal-free DNA cleavage by TACN with a guanidinoethyl pendant can be applied to other antibiotics targeting bacterial nucleic acids, such as aminoglycosides, and paves a new path in the future development of catalytic antibiotics.
Conclusions
In this study, we explored the potential to solve the issue of metal vulnerability for the Cu(II) complexes of the second-generation nuclease-conjugated ciprofloxacin by replacing the cyclen moiety with TACN to effectively enhance the amine or guanidine cap formation at the metal center.Firstly, the DNA cleavage assays underscore the critical role of the linker length in determining the DNase activity and DNA binding affinity of both guanidine and amine series compounds. For the guanidine series, only 1–Cu(II) exhibited significant DNase activity comparable to the parent complex 9–Cu(II), while 2–Cu(II) showed strong DNA binding but poor catalytic efficiency. In contrast, for the amine series, 5–Cu(II), 6–Cu(II), and 8–Cu(II) demonstrated enhanced DNase activity compared to the parent compound 10–Cu(II), with 7–Cu(II) showing no significant activity. These findings indicate that while the guanidine pendant may exacerbate DNase activity of the Cu(II) complexes due to conformational restrictions, the amine pendant can enhance DNase activity. Furthermore, investigations using Cu(II)-chelating agents like Tris buffer revealed that all tested Cu(II) complexes lost their DNase activity, suggesting vulnerability to ligand exchange.
Surprisingly, the metal-free compounds also displayed significant variability in their DNase activity. Compound 1 emerged as the most effective metal-free nuclease, retaining significant activity even in the presence of EDTA, indicating a metal-free, hydrolytic mechanism of DNA cleavage. This suggests that compound 1 could act as a dual-acting antibacterial agent with both DNA cleavage and antibacterial properties due to inhibiting topoisomerase II enzymes, offering potential to slow down resistance development.
The antibacterial activity assays further reinforced the potential of these compounds. Compounds 1 and 3 were particularly effective against Gram-negative E. coli strains, showing 2- to 4-fold improvement over other compounds, while compounds 1, 3, 7, and 8 exhibited strong activity against Gram-positive strains. Notably, the Cu(II) complexes showed similar antibacterial activity to the metal-free ligands, with no significant enhancement, corroborating the susceptibility of these complexes to ligand exchange.
DFT calculations combined with MD and QM/MM simulations provided deeper insights into the DNA cleavage mechanism of compound 1. The simulations revealed that the DNA sequence context may influence the binding mode of compound 1, which can affect its cleavage capability. Importantly, the structural analysis of the TACN–guanidine warhead in solution indicated that the warhead could recruit a water molecule, which may act as a nucleophile in DNA cleavage, supporting a general base–general acid mechanism of hydrolysis.
In conclusion, this study has identified compound 1 as a promising candidate for further development of antibacterial metal-free nuclease-active agents. The findings suggest a dual mechanism of action, potentially enhancing its therapeutic efficacy and reducing the likelihood of resistance development. Future research should focus on optimizing the DNase activity of these compounds under physiological conditions selectively within the ternary complex of topoisomerase IV or DNA gyrase, exploring their detailed mechanisms of action through advanced simulations and evaluating their efficacy in vivo. The insights gained from this study provide a solid foundation for the development of novel antibacterial agents with enhanced metal-free DNA cleavage capabilities.
Experimental
General techniques
1H and 13C NMR, DEPT, COSY, HMQC, and HMBC spectra were recorded on Bruker AvanceTM 600/500 spectrometers. Chemical shifts reported (in ppm) are relative to CHCl3 (δ = 7.26) with CDCl3 as the solvent, to HOD (δ = 4.79) or internal TMS with D2O as the solvent, and to CD3OH (δ = 3.31) or internal TMS with CD3OD as the solvent. Mass spectral analyses were performed on a Bruker Maxis Impact under electron spray ionization (ESI+) QTOFMS or on a Bruker Autoflex Speed under matrix-assisted laser desorption ionization (MALDI) TOFMS. Reactions were monitored by TLC on silica gel (Gel 60 F254, 0.25 mm, Merck), and spots were visualized using a UV lamp and iodine, or by charring with a yellow solution containing (NH4)6Mo7O24·4H2O (120 g) and (NH4)2Ce(NO3)6 (5 g) in 10% H2SO4 (800 mL). Flash column chromatography was performed on silica gel 60 (70–230 mesh). The EPR spectra were recorded on a Bruker EMX-10/12 X-band (ν = 9.3 GHz) digital EPR spectrometer. The spectra were recorded at a microwave power of 200 mW, 100 kHz, and magnetic field modulation of 3 G amplitude. The digital field resolution was 2048 points per spectrum. Spectral processing was performed with the Bruker WIN-EPR and SimFonia software. The UV-vis spectra were recorded on an Ultrospec 2100 pro spectrometer. All chemicals unless otherwise stated were obtained from commercial sources. The purity of the lead compound 1 was determined by using HPLC analysis, which indicated >95% purity (see the ESI†).Removal of ligands prior to electrophoresis
The procedure proved to be necessary when more than 10 μM compound (metal-free, Cu(II)) was employed, that is, (only) for the DNA cleavage assays that were performed in the absence of any adjuvants. An Amberlyst 15 (Sigma-Aldrich) ion-exchange resin was prepared according to the reported literature61 and then utilized for ligand removal as previously reported by our group.16DNA cleavage assays
DNA cleavage activity of 1–8 and their Cu(II) complexes toward supercoiled (+) pHOT-1 plasmid DNA (TopoGEN) was monitored by gel electrophoresis. In a typical experiment, plasmid DNA (400 ng, 0.014 μg mL−1) in HEPES buffer (50 mM, pH 7.4) was mixed with different concentrations of the compounds in the presence or absence of ascorbic acid (0.32 mM). All stock solutions of buffers and of the compounds were prepared using HPLC grade water (ChromAR). Molecular biology reagent grade water (Sigma) was added up to a total reaction volume of 30 μL before incubation for a given time. For the ascorbic acid assays, the reaction was quenched immediately after incubation with EDTA (54 mM). For analysis, 10 μL of loading buffer (40% sucrose, 100 mM Tris-HCl (pH 7.5), 1 mM EDTA, 0.5 mg mL−1 bromophenol blue) was added to the incubated solution, and the mixture was loaded onto an agarose (SeaKemLE) gel [1% in TAE × 1 buffer (40 mM Tris, 20 mM acetic acid, 1 mM EDTA)]. Electrophoresis was carried out at 60 V for 2 h. The gels were then stained with 1 μg mL−1 ethidium bromide for 30 min and de-stained in TAE × 1 buffer. DNA bands were visualized with medium-range ultraviolet light using a Bio-RadGel Doc XR+ imaging system. The quantity of different DNA forms (I or II) was estimated using ImageJ62 software.Antibacterial and cytotoxicity tests
Comparative antibacterial activities were determined by measuring the MIC values using the double-microdilution method according to the Clinical and Laboratory Standards Institute63 (CLSI). A Luria–Bertani growth medium and polypropylene 96-well plates (Thermo) were used. All the experiments were performed in triplicate, and analogous results were obtained in three different experiments.For the cytotoxicity assays, HEK-293FT cells were seeded overnight in 96-well plates (5000 cells per well) in a DMEM medium containing 10% FBS, 1% penicillin/streptomycin, 1% glutamine, and 1% pyruvate at 37 °C under 5% CO2. BHK cells were seeded overnight in 96-well plates (5000 cells per well) in a DMEM medium containing 10% FBS and 1% penicillin/streptomycin. Different concentrations of the tested compounds were added to the medium and incubated with the cells for 48 h. A cell proliferation assay (resazurin-based colorimetric assay, Sigma-Aldrich) was performed under 3 h incubation. The fluorescence values were then read by using a plate reader. Cell viability values were normalized to the untreated samples of each tested compound. The half-maximal lethal concentration (LC50) values were obtained by fitting concentration–response curves to the data of at least four biological repeats within each of 2 independent experiments using an online LC50 calculator (AAT Bioquest, https://www.aatbio.com/tools/lc50-calculator).
Compound 13a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH:MeNH2 (33% MeNH2 in EtOH) 30:4:1, which indicated that the reaction stopped progressing after 18 h. The reaction mixture was filtered under gravity and washed extensively with CH3CN and then the solvent was removed in vacuo.
:MeOH:MeNH2 (33% MeNH2 in EtOH) 30:4:1.3 and DCM:MeOH:Et3N 30:1:0.2. The reaction mixture was filtered under gravity and washed extensively with DCM and then the solvent was removed in vacuo. The crude product was then loaded onto a DCM-packed silica column as a DCM solution; the desired product was eluted using a DCM:MeOH:Et3N (30:1:0.1) solvent system. A post-column workup was then performed with DCM and water; the desired product was isolated in the organic phase, dried with MgSO4 and then evaporated to yield the pure compound (1.39 g, 1.63 mmol, 35% over two steps); 1H NMR (600 MHz, CDCl3) δH 8.47 (s, 1H, QH-2), 7.95–7.93 (d, 1H, QH-5), 7.77–7.76 (m, 2H, phthalimide Ar), 7.65–7.64 (m, 2H, phthalimide Ar), 7.28–7.18 (m, 5H, QH-8, linker Ar), 3.84 (s, 3H, OCH3), 3.69–3.67 (m, 2H, C
:MeOH:MeNH2 (33% MeNH2 in EtOH) 30:4:1, which indicated that the reaction stopped progressing after 18 h. The reaction mixture was filtered under gravity and washed extensively with CH3CN and then the solvent was removed in vacuo.
:MeOH:MeNH2 (33% MeNH2 in EtOH) 30:4:1.3 and DCM:MeOH:Et3N 30:1:0.2. The reaction mixture was filtered under gravity and washed extensively with DCM and then the solvent was removed in vacuo. The crude product was then loaded onto a DCM-packed silica column as a DCM solution; the desired product was eluted using a DCM:MeOH:Et3N (30:1:0.1) solvent system. A post-column workup was then performed with DCM and water; the desired product was isolated in the organic phase, dried with MgSO4 and then evaporated to yield the pure compound (1.39 g, 1.63 mmol, 35% over two steps); 1H NMR (600 MHz, CDCl3) δH 8.47 (s, 1H, QH-2), 7.95–7.93 (d, 1H, QH-5), 7.77–7.76 (m, 2H, phthalimide Ar), 7.65–7.64 (m, 2H, phthalimide Ar), 7.28–7.18 (m, 5H, QH-8, linker Ar), 3.84 (s, 3H, OCH3), 3.69–3.67 (m, 2H, C![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) -phthalimide), 3.52 (m, 2H, Ph-C-piperazine), 3.36–3.33 (m, 1H, cyclopropane CH), 3.22–3.16 (m, 8H, piperazine (4H), TACN (4H)), 2.87–2.74 (m, 6H, TACN (4H), CCH2-phthalimide (2H)), 2.61–2.58 (m, 10H, piperazine (4H), TACN (4H), TACN-C-Ph (2H)), 1.41–1.37 (m, 9H, Boc), 1.25–1.19 (m, 2H, cyclopropane CH2), 1.09–1.06 (m, 2H, cyclopropane CH2); 13C NMR (150 MHz, CDCl3) δC 173.11 (cipro C
-phthalimide), 3.52 (m, 2H, Ph-C-piperazine), 3.36–3.33 (m, 1H, cyclopropane CH), 3.22–3.16 (m, 8H, piperazine (4H), TACN (4H)), 2.87–2.74 (m, 6H, TACN (4H), CCH2-phthalimide (2H)), 2.61–2.58 (m, 10H, piperazine (4H), TACN (4H), TACN-C-Ph (2H)), 1.41–1.37 (m, 9H, Boc), 1.25–1.19 (m, 2H, cyclopropane CH2), 1.09–1.06 (m, 2H, cyclopropane CH2); 13C NMR (150 MHz, CDCl3) δC 173.11 (cipro C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 168.38 (phthalimide CO), 166.50 (cipro CO), 155.51 (Boc CO), 154.27 (cipro), 152.62 (cipro C–H), 148.36 (cipro), 144.63 (linker Ar), 138.02 (cipro), 134.03 (linker Ar), 133.96, 132.11 (C–H phthalimide Ar), 132.05, 131.84 (phthalimide Ar), 129.69 (C–H linker Ar), 129.30, 129.07 (C–H linker Ar), 123.37, 123.22 (C–H phthalimide Ar), 122.96 (cipro), 113.18 (cipro C–H), 110.03 (cipro), 104.79 (cipro C–H), 76.84 (Boc
O), 168.38 (phthalimide CO), 166.50 (cipro CO), 155.51 (Boc CO), 154.27 (cipro), 152.62 (cipro C–H), 148.36 (cipro), 144.63 (linker Ar), 138.02 (cipro), 134.03 (linker Ar), 133.96, 132.11 (C–H phthalimide Ar), 132.05, 131.84 (phthalimide Ar), 129.69 (C–H linker Ar), 129.30, 129.07 (C–H linker Ar), 123.37, 123.22 (C–H phthalimide Ar), 122.96 (cipro), 113.18 (cipro C–H), 110.03 (cipro), 104.79 (cipro C–H), 76.84 (Boc ![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) (CH3)3), 62.57 (Ph–H2–piperazine), 52.75 (piperazine), 52.08 (OH3), 49.89 (piperazine), 34.51 (cyclopropane H), 29.69, 28.56, 28.43, 28.35 (Boc C(H3)3), 8.14 (cyclopropane H2). MS (ESI+ QTOFMS) calculated for C47H57FN7O7 ([M + H]+) m/z 850.42; measured m/z 850.42.
–CO), 8.76 (s, 1H, QH-2), 7.96–7.94 (d, 1H, QH-5), 7–80–7.78 (d, 1H, QH-8), 7.66, 7.26–7.20 (m, 4H, linker Ar), 3.72–3.69 (m, 2H, Ph-C-piperazine), 3.65–3.61 (m, 1H, cyclopropane CH), 3.56–3.50 (m, 4H, CH2N), 3.47–3.39 (m, 5H, piperazine (4H), CH2N (1H)), 3.24–3.13 (m, 4H, C
(CH3)3), 62.57 (Ph–H2–piperazine), 52.75 (piperazine), 52.08 (OH3), 49.89 (piperazine), 34.51 (cyclopropane H), 29.69, 28.56, 28.43, 28.35 (Boc C(H3)3), 8.14 (cyclopropane H2). MS (ESI+ QTOFMS) calculated for C47H57FN7O7 ([M + H]+) m/z 850.42; measured m/z 850.42.
–CO), 8.76 (s, 1H, QH-2), 7.96–7.94 (d, 1H, QH-5), 7–80–7.78 (d, 1H, QH-8), 7.66, 7.26–7.20 (m, 4H, linker Ar), 3.72–3.69 (m, 2H, Ph-C-piperazine), 3.65–3.61 (m, 1H, cyclopropane CH), 3.56–3.50 (m, 4H, CH2N), 3.47–3.39 (m, 5H, piperazine (4H), CH2N (1H)), 3.24–3.13 (m, 4H, C![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) –NH–CO (3H), CH2N (1H)), 2.94–2.88 (m, 7H, CH2N), 2.80–2.78 (m, 3H, piperazine), 2.62, 2.54–2.49 (m, 6H, TACN–C–Ph (2H), CH2N (4H)), 1.41–1.39 (m, 9H, Boc), 1.31, 1.12–1.09 (m, 3H, cyclopropane CH2), 0.82–0.78 (m, 1H, cyclopropane CH2); 13C NMR (150 MHz, CDCl3) δC 1752.54 (cipro CO), 168.95 (cipro CO), 156.84 (Boc CO), 154.14 (cipro), 145.58 (cipro C–H), 145.02 (cipro), 140.76 (linker Ar), 137.52 (cipro), 134.96 (linker Ar), 130.54, 128.71 (C–H linker Ar), 126.57 (C–H linker Ar), 123.75 (cipro), 113.61 (cipro C–H), 110.35 (cipro), 108.98, 105.62 (cipro C–H), 79.06 (Boc (CH3)3), 64.75 (Ph–H2–piperazine), 56.05, 52.87, 51.93, 50.41 (H2N), 52.76 (piperazine), 51.84 (H2N), 50.03 (piperazine), 40.33, 38.05 (H2–NH2), 35.01 (cyclopropane H), 29.53–27.46 (TACN–CH2H2CH2–Ph, Boc C(H3)3), 22.86 (H3–NH–CO), 9.06 (cyclopropane H2). MS (ESI+ QTOFMS) calculated for C39H56FN8O4 ([M + H]+) m/z 719.43; measured m/z 719.43.
–NH–CO (3H), CH2N (1H)), 2.94–2.88 (m, 7H, CH2N), 2.80–2.78 (m, 3H, piperazine), 2.62, 2.54–2.49 (m, 6H, TACN–C–Ph (2H), CH2N (4H)), 1.41–1.39 (m, 9H, Boc), 1.31, 1.12–1.09 (m, 3H, cyclopropane CH2), 0.82–0.78 (m, 1H, cyclopropane CH2); 13C NMR (150 MHz, CDCl3) δC 1752.54 (cipro CO), 168.95 (cipro CO), 156.84 (Boc CO), 154.14 (cipro), 145.58 (cipro C–H), 145.02 (cipro), 140.76 (linker Ar), 137.52 (cipro), 134.96 (linker Ar), 130.54, 128.71 (C–H linker Ar), 126.57 (C–H linker Ar), 123.75 (cipro), 113.61 (cipro C–H), 110.35 (cipro), 108.98, 105.62 (cipro C–H), 79.06 (Boc (CH3)3), 64.75 (Ph–H2–piperazine), 56.05, 52.87, 51.93, 50.41 (H2N), 52.76 (piperazine), 51.84 (H2N), 50.03 (piperazine), 40.33, 38.05 (H2–NH2), 35.01 (cyclopropane H), 29.53–27.46 (TACN–CH2H2CH2–Ph, Boc C(H3)3), 22.86 (H3–NH–CO), 9.06 (cyclopropane H2). MS (ESI+ QTOFMS) calculated for C39H56FN8O4 ([M + H]+) m/z 719.43; measured m/z 719.43.
General procedure for the synthesis of compounds 13b–d
:MeOH:MeNH2 (33% MeNH2 in EtOH) 30:4:1. The reaction mixture was filtered under gravity and washed extensively with CH3CN, and then the solvent was removed in vacuo.
:MeOH:MeNH2 (33% MeNH2 in EtOH) 30:4:1.3 and DCM:MeOH:Et3N 30:1:0.2. The reaction mixture was filtered under gravity and washed extensively with DCM, and then the solvent was removed in vacuo. The crude product was then loaded onto a DCM-packed silica column as a DCM solution; the desired product was eluted using a DCM:MeOH:Et3N (30:1:0.1) solvent system. A post-column workup was then performed with DCM and water; the desired product was isolated in the organic phase, dried with MgSO4 and then evaporated to yield the pure compound.
General procedure for the synthesis of compounds 14a–d
The compounds were prepared from the free-amine derivative using a procedure previously reported by our group.16 To an anhydrous CH3CN (50 mL g−113a–d) solution of the appropriate primary amine (13a–d) were added the reagents N,N-di-Boc-1H-pyrazole-1-carboxamide (1 equiv.) and anhydrous K2CO3 (2 equiv.) and the mixture was set stirring at r.t. under an argon atmosphere. TLC analysis (CHCl3/MeOH/Et3N 30:4.5:0.3) indicated complete conversion after overnight. The reaction mixture was filtered and washed extensively with CH3CN and DCM, and then the solvent was removed in vacuo. The dry residue was purified by silica chromatography using a MeOH/DCM/MeNH2 elution system, to yield pure compounds 14a–d. Chemical yields of 14a–d and their complete analytical data are given in the ESI.†
General procedure for the synthesis of compounds 1–8
The compounds were prepared using a procedure previously reported by our group.16 The appropriate derivative (13a–d or 14a–d) was dissolved in an aqueous solution of 6 M HCl (60 mL g−1 starting material) and set stirring at 90 °C under argon. The reaction was monitored by TLC [MeOH/DCM/MeNH2 (33% MeNH2 in EtOH)/H2O 3.3:3.3:2.3:1.1], mass spectrometry, and 1H NMR, which indicated complete conversion after 7 days. The crude solution was evaporated in vacuo, washed extensively with diethyl ether to remove any residual HCl, and then dried in vacuo. The dry residue was then purified using Sephadex LH20 and then lyophilized to yield pure compounds 1–8.
Compound 1
Following the general procedure, 14a (694 mg, 0.723 mmol) yielded 37% of compound 1 (225 mg, 0.273 mmol); 1H NMR (600 MHz, D2O pH 1) δH 8.46 (s, 1H, QH-2), 7.76 (d, 2H, linker Ar), 7.67 (d, 2H, linker Ar), 7.35 (d, 1H, QH-5), 7.27 (d, 1H, QH-8), 4.50 (s, 2H, Ph–C–piperazine), 3.86–3.76 (m, 4H, CH2N), 3.64–3.58 (m, 6H, cyclopropane CH (1H), CH2N (5H)), 3.44–3.20 (m, 12H, C–guanidine (2H), CH2N (10H)), 3.05 (m, 2H, CCH2–guanidine), 3.00–2.98 (m, 1H, CH2N), 2.81 (m, 2H, TACN–C–Ph), 1.34–1.33 (m, 2H, cyclopropane CH2), 1.07 (m, 2H, cyclopropane CH2); 13C NMR (150 MHz, D2O pH 1) δC 175.71 (cipro CO), 168.75 (cipro CO), 156.62 (guanidine C), 154.04 (cipro), 152.38, 148.17 (cipro C–H), 144.04 (cipro), 143.98 (Ar), 138.82 (Ar), 132.45 (C–H linker Ar), 131.33 (linker Ar), 130.31 (C–H linker Ar), 118.64 (cipro), 110.67 (cipro C–H), 110.52 (cipro C–H), 106.65 (cipro), 105.52, 60.54 (Ph–H2–piperazine), 59.71, 52.06, 51.39, 51.10, 49.29, 49.03, 46.64, 46.32, 45.46, 43.30, 40.19 (CH2N), 36.90 (H2–guanidine), 36.16 (cyclopropane H), 8.21 (cyclopropane H2), 7.49 (cyclopropane H2). HRMS (ESI+ QTOFMS) calculated for C34H47FN9O3 ([M + H]+) m/z 648.3786; measured m/z 648.3781.
Compound 2
Following the general procedure, 14b (522 mg, 0536 mmol) yielded 41% of compound 2 (182 mg, 0.218 mmol); 1H NMR (500 MHz, D2O pH 1) δH 8.48 (s, 1H, QH-2), 7.47–7.44 (d, 2H, linker Ar), 7.38–7.37 (m, 4H, linker Ar (2H), QH-5 (1H), QH-8 (1H)), 4.37 (s, 2H, Ph-C-piperazine), 3.84–3.74 (m, 2H, CH2N), 3.59–3.45 (m, 7H, cyclopropane CH (1H), CH2N (6H)), 3.34–3.18 (m, 12H, TACN–CCH2–Ph (2H), C–guanidine (2H), CH2N (8H)), 2.93–2.90 (m, 4H, CH2N), 2.85–2.83 (t, CCH2–guanidine, 2H), 2.48 (t, 2H, TACN–CH2C–Ph), 1.31–1.29 (m, 2H, cyclopropane CH2), 1.06–1.05 (m, 2H, cyclopropane CH2); 13C NMR (125 MHz, D2O) δC 175.92 (cipro CO), 168.94 (cipro CO), 156.74 (guanidine C), 154.30, 152.29 (cipro), 148.25 (cipro C–H), 145.02 (cipro), 143.98 (Ar), 138.94 (Ar), 131.82 (C–H linker Ar), 129.70 (C–H linker Ar), 126.64 (linker Ar), 118.89 (cipro), 110.80 (cipro C–H), 106.73 (cipro C–H), 105.67 (cipro), 60.03 (Ph–H2–piperazine), 57.80, 52.52, 50.89, 50.46, 48.10, 46.77, 46.40, 43.29, 41.94 (CH2N), 37.61 (H2–guanidine), 36.12 (cyclopropane H), 34.55, 30.55 (TACN–CH2H2–Ph), 24.48 (TACN–H2CH2–Ph), 8.19, 7.37 (cyclopropane H2). HRMS (ESI+ QTOFMS) calculated for C35H49FN9O3 ([M + H]+) m/z 662.3942; measured m/z 662.3942.
Compound 3
Following the general procedure, 14c (486 mg, 0.491 mmol) yielded 34% of compound 3 (143 mg, 0.167 mmol); 1H NMR (600 MHz, D2O pH 1) δH 8.46 (s, 1H, QH-2), 7.44 (d, 2H, linker Ar), 7.34–7.28 (m, 4H, linker Ar (2H), QH-5 (1H), QH-8 (1H)), 4.37 (s, 2H, Ph–C-piperazine), 3.81 (m, 2H, CH2N), 3.58–3.57 (m, 7H, cyclopropane CH (1H), CH2N (6H)), 3.32–3.22 (m, 12H, TACN–C(CH2)2–Ph (2H), C–guanidine (2H), CH2N (8H)), 2.96 (m, 4H, CH2N), 2.87–2.86 (t, CCH2–guanidine, 2H), 2.71–2.69 (t, 2H, TACN–(CH2)2C–Ph), 2.13–2.04 (m, 2H, TACN–CH2CCH2–Ph), 1.30 (m, 2H, cyclopropane CH2), 1.05 (m, 2H, cyclopropane CH2); 13C NMR (150 MHz, D2O pH 1) δC 175.83 (cipro CO), 168.86 (cipro CO), 156.72 (guanidine C), 153.26 (cipro), 148.22 (cipro C–H), 144.07 (cipro), 142.61 (Ar), 138.87 (Ar), 131.69 (C–H linker Ar), 129.36 (C–H linker Ar), 126.14 (linker Ar), 118.74 (cipro), 110.64 (cipro C–H), 106.67 (cipro C–H), 105.58 (cipro), 60.12 (Ph–H2–piperazine), 57.72, 52.03, 50.87, 50.70, 50.50, 47.56, 46.64, 46.36, 43.34, 41.12 (CH2N), 37.15 (H2–guanidine), 36.15 (cyclopropane H), 31.47 (TACN–(CH2)2H2–Ph), 25.26 (TACN–CH2H2CH2–Ph), 7.47 (cyclopropane H2). HRMS (ESI+ QTOFMS) calculated for C36H51FN9O3 ([M + H]+) m/z 676.4099; measured m/z 676.4100.
Compound 4
Following the general procedure, 14d (403 mg, 0.402 mmol) yielded 45% of compound 4 (156 mg, 0.181 mmol); 1H NMR (600 MHz, D2O pH 1) δH 8.41 (s, 1H, QH-2), 7.43–7.32 (m, 6H, linker Ar (4H), QH-5 (1H), QH-8 (1H)), 4.36 (s, 2H, Ph–C–piperazine), 3.83–3.53 (m, 9H, cyclopropane CH (1H), CH2N (8H)), 3.36–3.25 (m, 12H, TACN–C(CH2)3–Ph (2H), C–guanidine (2H), CH2N (8H)), 2.82 (m, 8H, CH2N (4H), CCH2–guanidine (2H), TACN–(CH2)3C–Ph (2H)), 1.73–1.61 (m, 4H, TACN–CH2(C)2CH2–Ph), 1.31 (m, 2H, cyclopropane CH2), 1.04–1.02 (m, 2H, cyclopropane CH2); 13C NMR (150 MHz, D2O pH 1) δC 175.68 (cipro CO), 168.70 (cipro CO), 156.78 (guanidine C), 148.07 (cipro), 147.04 (cipro C–H), 144.39 (cipro), 143.94 (Ar), 138.78 (Ar), 131.56 (C–H linker Ar), 129.35 (C–H linker Ar), 125.58 (linker Ar), 121.75 (cipro), 110.68 (cipro C–H), 106.60 (cipro C–H), 105.51 (cipro), 60.13 (Ph-H2–piperazine), 58.49, 54.76, 53.65, 53.10, 51.96, 51.85, 50.78, 48.07, 46.54, 46.37 (CH2N), 38.70 (H2–guanidine), 36.14 (cyclopropane H), 34.14 (TACN–(CH2)3H2–Ph), 27.41, 24.50–23.02 (TACN–CH2(H2)2CH2–Ph), 7.38 (cyclopropane H2). HRMS (ESI+ QTOFMS) calculated for C37H53FN9O3 ([M + H]+) m/z 690.4255; measured m/z 690.4253.
Compound 5
Following the general procedure, 13a (268 mg, 0.373 mmol) yielded 48% of product 5 (140 mg, 0.179 mmol); 1H NMR (600 MHz, D2O pH 1) δH 8.6 (s, 1H, QH-2), 7.63–7.54 (m, 4H, linker Ar), 7.41–7.39 (m, 2H, QH-8 (1H), QH-5 (1H)), 4.37 (s, 2H, Ph–C–piperazine), 3.80–3.78 (m, 2H, cyclopropane CH (1H), CH2N (1H)), 3.51–3.44 (m, 7H, CH2N), 3.33–3.28 (m, 7H, CH2N (7H)), 3.18–3.00 (m, 3H, CH2N), 2.91–2.86 (m, 6H, CH2N), 2.39 (t, 2H, TACN–C–Ph), 1.23–1.19 (m, 2H, cyclopropane CH2), 1.02 (m, 2H, cyclopropane CH2); 13C NMR (150 MHz, D2O pH 1) δC 174.95 (cipro CO), 167.82 (cipro CO), 153.02 (cipro), 147.34 (cipro C–H), 142.85 (cipro), 142.44 (Ar), 137.72 (Ar), 131.06 (C–H linker Ar), 127.08 (C–H linker Ar), 125.77 (linker Ar), 117.96 (cipro), 111.75 (cipro C–H), 106.89 (cipro C–H), 104.89 (cipro), 61.23 (Ph–H2–piperazine), 58.95, 51.17, 45.98, 44.78, 43.56, 42.89 (CH2N), 35.78 (cyclopropane H), 35.16 (H2–NH2), 31.15 (TACN–H2–Ph), 7.22 (cyclopropane H2). HRMS (ESI+ QTOFMS) calculated for C33H45FN7O3 ([M + H]+) m/z 606.3568; measured m/z 606.3564.
Compound 6
Following the general procedure, 13b (310 mg, 0.423 mmol) yielded 45% of compound 6 (154 mg, 0.194 mmol); 1H NMR (600 MHz, D2O pH 1) δH 8.39 (s, 1H, QH-2), 7.40–7.38 (d, 2H, linker Ar), 7.32–7.21 (m, 4H, linker Ar (2H), QH-8 (1H), QH-5 (1H)), 4.31 (s, 2H, Ph–C–piperazine), 3.74–3.69 (m, 2H, cyclopropane CH (1H), CH2N (1H)), 3.51–3.44 (m, 7H, CH2N), 3.26–3.17 (m, 9H, TACN–CCH2–Ph (2H), CH2N (7H)), 3.11–3.09 (m, 3H, CH2N), 2.93–2.91 (m, 6H, CH2N), 2.40 (m, 2H, TACN–CH2C–Ph), 1.23–1.20 (m, 2H, cyclopropane CH2), 1.00–0.97 (m, 2H, cyclopropane CH2); 13C NMR (150 MHz, D2O) δC 176.12 (cipro CO), 169.01 (cipro CO), 152.38 (cipro), 147.89 (cipro C–H), 144.97 (cipro), 138.72 (Ar), 131.05 (C–H linker Ar), 129.02 (C–H linker Ar), 125.24 (linker Ar), 118.54 (cipro), 110.65 (cipro C–H), 106.84 (cipro C–H), 106.01 (cipro), 61.18 (Ph–H2–piperazine), 58.48, 51.74, 50.56, 49.42, 47.48, 44.07, 42.59, 40.97 (CH2N), 36.08 (cyclopropane H), 34.50 (H2–NH2), 28.39 (TACN–(CH2)2H2–Ph), 24.91 (TACN–CH2H2CH2–Ph), 22.85 (TACN–H2CH2CH2–Ph), 7.86 (cyclopropane H2). HRMS (ESI+ QTOFMS) calculated for C34H47FN7O3 ([M + H]+) m/z 620.3724; measured m/z 620.3720.
Compound 7
Following the general procedure, 13c (331 mg, 0.443 mmol) yielded 42% of compound 7 (152 mg, 0.186 mmol); 1H NMR (600 MHz, D2O pH 1) δH 8.46 (s, 1H, QH-2), 7.44 (d, 2H, linker Ar), 7.35–7.34 (m, 3H, linker Ar (2H), QH-8 (1H)), 7.30–7.28 (d, 1H, QH-5), 4.38 (s, 2H, Ph–C–piperazine), 3.84–3.82 (m, 2H, cyclopropane CH (1H), CH2N (1H)), 3.60–3.57 (m, 7H, CH2N), 3.35–3.25 (m, 9H, TACN–C(CH2)2–Ph (2H), CH2N (7H)), 3.16–3.12 (m, 3H, CH2N), 2.97–2.94 (m, 6H, CH2N), 2.72–2.70 (t, 2H, TACN–(CH2)2C–Ph), 2.14–2.06 (m, 2H, TACN–CH2CCH2–Ph), 1.32–1.31 (m, 2H, cyclopropane CH2), 1.06 (m, 2H, cyclopropane CH2); 13C NMR (150 MHz, D2O pH 1) δC 175.80 (cipro CO), 168.84 (cipro CO), 153.28 (cipro), 148.21 (cipro C–H), 144.07 (cipro), 142.56 (Ar), 138.86 (Ar), 131.66 (C–H linker Ar), 129.36 (C–H linker Ar), 126.18 (linker Ar), 118.76 (cipro), 110.64 (cipro C–H), 106.66 (cipro C–H), 105.56 (cipro), 60.15 (Ph–H2–piperazine), 57.89, 50.91, 50.68, 50.28, 47.34, 46.38, 43.67, 43.19, 42.06, 41.27 (CH2N), 36.14 (cyclopropane H), 34.49 (H2–NH2), 31.45 (TACN–(CH2)2H2–Ph), 25.17 (TACN–CH2H2CH2–Ph), 7.47 (cyclopropane H2). HRMS (ESI+ QTOFMS) calculated for C35H49FN7O3 ([M + H]+) m/z 634.3881; measured m/z 634.3882.
Compound 8
Following the general procedure, 13d (240 mg, 0.316 mmol0) yielded 47% of product 8 (125 mg, 0.152 mmol); 1H NMR (500 MHz, D2O pH 1) δH 8.35 (s, 1H, QH-2), 7.43–7.34 (m, 6H, linker Ar (4H), QH-8 (1H), QH-5 (1H)), 4.37 (s, 2H, Ph–C–piperazine), 3.53–3.48 (m, 9H, CH2N (8H), cyclopropane CH (1H)), 3.30–3.25 (m, 9H, TACN–C(CH2)3–Ph (2H), CH2N (7H)), 3.18–3.15 (m, 6H, CH2N), 2.99–2.95 (m, 3H, CH2N), 2.72–2.66 (m, 2H, TACN–(CH2)3C–Ph), 1.75–1.64 (m, 4H, TACN–CH2(C)2CH2–Ph), 1.31 (m, 2H, cyclopropane CH2), 1.01 (m, 2H, cyclopropane CH2); 13C NMR (125 MHz, D2O) δC 175.52 (cipro CO), 168.58 (cipro CO), 152.53 (cipro), 147.92 (cipro C–H), 144.56 (cipro), 138.68 (Ar), 131.59 (C–H linker Ar), 129.37 (C–H linker Ar), 125.61 (linker Ar), 118.65 (cipro), 110.63 (cipro C–H), 106.50 (cipro C–H), 105.60 (cipro), 60.16 (Ph–H2–piperazine), 57.78, 51.29, 50.83, 50.73, 47.78, 46.79, 43.71, 43.33, 42.09, 41.64 (CH2N), 36.14 (cyclopropane H), 35.02, 34.20 (H2–NH2), 27.56 (TACN–(CH2)3H2–Ph), 25.68 (TACN–CH2H2CH2CH2–Ph), 23.62 (TACN–CH2CH2H2CH2–Ph), 7.53 (cyclopropane H2). HRMS (ESI+ QTOFMS) calculated for C36H51FN7O3 ([M + H]+) m/z 648.4037; measured m/z 648.3993.
Compound 12
To a stirred solution of N,N,N-bis-Boc-phthalimide-TACN19 (2.07 g, 4.12 mmol) in dry DCM (40 mL) was added neat TFA (42 mL) at room temperature under an argon atmosphere. The reaction was monitored by TLC using the following system: CHCl3:MeOH:MeNH2 (33% MeNH2 in EtOH) 24:4.5:1.5, which indicated complete conversion after 40 min. The reaction mixture was then set stirring in an ice bath, and diethyl ether was carefully added (150 mL), causing immediate precipitation of the desired product. The reaction mixture was then filtered using a Buchner apparatus and washed thoroughly with diethyl ether to yield a white solid (1.99 g, 3.75 mmol, 91%); 1H NMR (600 MHz, D2O) δH 7.78–7.63 (m, 4H, phthalimide Ar), 3.75 (t, 2H, C–phthalimide), 3.54 (s, 4H, TACN CN), 3.26 (t, TACN CN), 3.01 (t, 4H, TACN CN), 2.91 (t, 2H, CCH2–phthalimide); 13C NMR (150 MHz, D2O) δC 170.50 (phthalimide CO), 162.89 (TFA CO), 134.82 (phthalimide Ar), 131.18 (phthalimide Ar), 123.40 (phthalimide Ar), 116.30 (TFA F3), 51.80 (H2N), 47.33 (H2N), 43.68 (H2N), 42.06 (H2N), 33.83 (H2–phthalimide). MS (ESI+ QTOFMS) calculated for C16H23N4O2 ([M–H–2TFA]+) m/z 303.18; measured m/z 303.22.
Compound 10
The compound 1-(2-aminoethyl)-4,7-bis(tert-butoxycarbonyl)–TACN (390 mg, 1.06 mmol)19 was dissolved in dry DCM (40 mL) and cooled to 0 °C (ice bath). Then TFA (10 mL) was added and after 30 min, the reaction vessel was allowed to increase to r.t. The reaction was monitored by TLC using the following systems: CHCl3:MeOH:MeNH2 24:4.5:1.5 and CHCl3:MeOH 9:1, which indicated complete conversion after 1 h. Then, the reaction mixture was cooled, Et2O (450 mL) was added, and precipitation was observed. The precipitate was filtered through a Buchner apparatus and washed with Et2O. The final crystallized compound was dried in vacuo (152 mg, 0.880 mmol, 83%); 1H NMR (600 MHz, D2O) δH 3.45 (s, 4H, NHCRNH), 3.17–3.15 (m, 4H, NHCCH2N), 3.03–3.00 (t, 2H, RCNH2), 2.86–2.82 (m, 6H, NHCH2CN). 13C NMR (150 MHz, D2O) δC 51.01, 43.59, 42.01, 34.66. MS (ESI+ QTOFMS) calculated for C8H21N4 ([M–H–3TFA]+) m/z 172.11; measured m/z 172.25.
General procedure for the synthesis and characterization of Cu(II) complexes (1–8)
A stoichiometric amount of CuCl2 was added to an aqueous solution of the hydrochloride salt of the TACN derivative (1–8). The pH was then adjusted to 10 using 1 M NaOH(aq) and then stirring took place overnight at RT. UV-vis spectroscopy (λmax ≈ 600 nm) was used to monitor the progress of the complexation until completion. The reaction solution was pH adjusted to 7.4 and then microfiltered to obtain the stock for analysis and testing. The resulting Cu(II) complexes were characterized by mass spectrometry (see the ESI†). In addition, one representative member each from the guanidine series (compound 2–Cu(II)) and the amine series (compound 6–Cu(II)) was analyzed by EPR spectroscopy (Fig. 7). | ||
| Fig. 7 The spectra shown above are as follows: (A) solid state 2–Cu(II) at 300 K (gperpendicular = 2.11; gparallel = 2.05, Aparallel(63,65Cu) = 69.0 G); (B) solid state 6–Cu(II) at 300 K (gperpendicular = 2.04; gparallel = 2.12, Aparallel(63,65Cu) = 65.0 G). | ||
Data availability
Data for this article, including experimental details, purity determination of the lead compound, and NMR and HRMS spectra, are available in the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by research grants from the Israel Science Foundation founded by the Israel Academy of Sciences and Humanities (grant no. 670/20 for T. B.) and by The Israel Innovation Authority (KAMIN Program, grant no. 75044 for T. B.). We acknowledge Dr. Boris Tumansky for his assistance with the acquisition and interpretation of EPR spectra, and Dr. Guy Kamnesky for running HPLC analysis. This work was supported in part by a Technion postdoctoral fellowship for T. P. V. B. acknowledges financial support from the Ministry of Immigration Absorption and the Ministry of Science and Technology, Israel (Kamea Program).References
- S. Reardon, Nature, 2014 DOI:10.1038/nature.2014.15135.
- J. H. Kwon and W. G. Powderly, Science, 2021, 373, 471 CrossRef CAS PubMed.
- J. Murugaiyan, P. Anand Kumar, G. S. Rao, K. Iskandar, S. Hawser, J. P. Hays, Y. Mohsen, S. Adukkadukkam, W. A. Awuah, R. A. M. Jose, N. Sylvia, E. P. Nansubuga, B. Tilocca, P. Roncada, N. Roson-Calero, J. Moreno-Morales, R. Amin, B. Krishna Kumar, A. Kumar, A. R. Toufik, T. N. Zaw, O. O. Akinwotu, M. P. Satyaseela and M. B. M. van Dongen, Antibiotics, 2022, 11, 200 CrossRef CAS PubMed.
- J. Ye and X. Chen, Antibiotics, 2023, 12, 67 CAS.
- M. Terreni, M. Taccani and M. Pregnolato, Molecules, 2021, 26, 2671 CAS.
- H. L. Stennett, C. R. Back and P. R. Race, Antibiotics, 2022, 11, 1237 CAS.
- Z. Yu and J. A. Cowan, Chem. – Eur. J., 2017, 23, 14113–14127 CrossRef CAS PubMed.
- M. N. Goldmeier, S. Katz, F. Glaser, V. Belakhov, A. Khononov and T. Baasov, ACS Infect. Dis., 2021, 7, 608–623 CAS.
- J. C. Joyner, W. F. Hodnick, A. S. Cowan, D. Tamuly, R. Boyd and J. A. Cowan, Chem. Commun., 2013, 49, 2118–2120 CAS.
- J. L. Alexander, Z. Thompson, Z. Yu and J. A. Cowan, ACS Chem. Biol., 2019, 14, 449–458 CAS.
- M. D. J. Libardo, C. De La Fuente-Nuñez, K. Anand, G. Krishnamoorthy, P. Kaiser, S. C. Pringle, C. Dietz, S. Pierce, M. B. Smith, A. Barczak, S. H. E. Kaufmann, A. Singh and A. M. Angeles-Boza, ACS Infect. Dis., 2018, 4, 1623–1634 CrossRef CAS PubMed.
- M. D. J. Libardo, A. A. Bahar, B. Ma, R. Fu, L. E. McCormick, J. Zhao, S. A. McCallum, R. Nussinov, D. Ren, A. M. Angeles-Boza and M. L. Cotten, FEBS J., 2017, 284, 3662–3683 CrossRef CAS PubMed.
- C. M. Agbale, J. K. Sarfo, I. K. Galyuon, S. A. Juliano, G. G. O. Silva, D. F. Buccini, M. H. Cardoso, M. D. T. Torres, A. M. Angeles-Boza, C. De La Fuente-Nunez and O. L. Franco, Biochemistry, 2019, 58, 3802–3812 CAS.
- A. Zhao, J. Sun and Y. Liu, Front. Cell. Infect. Microbiol., 2023, 13, 1137947 CrossRef CAS PubMed.
- B. Smolkin, A. Khononov, T. Pieńko, M. Shavit, V. Belakhov, J. Trylska and T. Baasov, ChemBioChem, 2019, 20, 247–259 CrossRef CAS PubMed.
- M. N. Goldmeier, A. Khononov, V. Belakhov, T. Pieńko, N. Orbach, Y. Gilad Barzilay and T. Baasov, J. Med. Chem., 2022, 65, 14049–14065 CrossRef CAS PubMed.
- J. A. Collins and N. Osheroff, ACS Infect. Dis., 2024, 10, 1097–1115 CrossRef CAS PubMed.
- A. A. Oviatt, E. G. Gibson, J. Huang, K. Mattern, K. C. Neuman, P. F. Chan and N. Osheroff, ACS Infect. Dis., 2024, 10, 1137–1151 CrossRef CAS PubMed.
- L. Tjioe, T. Joshi, C. M. Forsyth, B. Moubaraki, K. S. Murray, J. Brugger, B. Graham and L. Spiccia, Inorg. Chem., 2012, 51, 939–953 CrossRef CAS PubMed.
- S. Mahboobi, W. Wagner and T. Burgemeister, Arch. Pharm., 1995, 328, 371–376 CrossRef CAS.
- M. I. Page and W. P. Jencks, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1678–1683 CrossRef CAS PubMed.
- J. C. Joyner, J. Reichfield and J. A. Cowan, J. Am. Chem. Soc., 2011, 133, 15613–15626 CAS.
- W. Xu, X. Yang, L. Yang, Z. L. Jia, L. Wei, F. Liu and G. Y. Lu, New J. Chem., 2010, 34, 2654–2661 RSC.
- M. Fang, L. Wei, Z. Lin and G. Y. Lu, Chin. J. Chem., 2014, 32, 142–150 CAS.
- V. Pokrovskaya, V. Belakhov, M. Hainrichson, S. Yaron and T. Baasov, J. Med. Chem., 2009, 52, 2243–2254 CAS.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CAS.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 CAS.
- S. Grimme, F. Bohle, A. Hansen, P. Pracht, S. Spicher and M. Stahn, J. Phys. Chem. A, 2021, 125, 4039–4054 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- T. Gerlach, S. Müller, A. G. de Castilla and I. Smirnova, Fluid Phase Equilib., 2022, 560, 113472 CAS.
- C. A. Fitch, G. Platzer, M. Okon, B. E. Garcia-Moreno and L. P. McIntosh, Protein Sci., 2015, 24, 752–761 CrossRef CAS PubMed.
- A. Marlin, A. Koller, E. Madarasi, M. Cordier, D. Esteban-Gómez, C. Platas-Iglesias, G. Tircsó, E. Boros, V. Patinec and R. Tripier, Inorg. Chem., 2023, 62, 20634–20645 CrossRef CAS PubMed.
- L. Pazderová, V. Kubíček, J. Kotek and P. Hermann, Z. Anorg. Allg. Chem., 2021, 647, 1261–1268 CrossRef.
- D. A. Case, H. M. Aktulga, K. Belfon, D. S. Cerutti, G. A. Cisneros, V. W. D. Cruzeiro, N. Forouzesh, T. J. Giese, A. W. Götz, H. Gohlke, S. Izadi, K. Kasavajhala, M. C. Kaymak, E. King, T. Kurtzman, T. S. Lee, P. Li, J. Liu, T. Luchko, R. Luo, M. Manathunga, M. R. Machado, H. M. Nguyen, K. A. O'Hearn, A. V. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, A. Risheh, S. Schott-Verdugo, A. Shajan, J. Swails, J. Wang, H. Wei, X. Wu, Y. Wu, S. Zhang, S. Zhao, Q. Zhu, T. E. Cheatham, D. R. Roe, A. Roitberg, C. Simmerling, D. M. York, M. C. Nagan and K. M. Merz, J. Chem. Inf. Model., 2023, 63, 6183–6191 CrossRef CAS PubMed.
- C. M. Dupureur, Curr. Opin. Chem. Biol., 2008, 12, 250–255 CrossRef CAS PubMed.
- W. J. Allen, T. E. Balius, S. Mukherjee, S. R. Brozell, D. T. Moustakas, P. T. Lang, D. A. Case, I. D. Kuntz and R. C. Rizzo, J. Comput. Chem., 2015, 36, 1132–1156 CrossRef CAS PubMed.
- A. Wohlkonig, P. F. Chan, A. P. Fosberry, P. Homes, J. Huang, M. Kranz, V. R. Leydon, T. J. Miles, N. D. Pearson, R. L. Perera, A. J. Shillings, M. N. Gwynn and B. D. Bax, Nat. Struct. Mol. Biol., 2010, 17, 1152–1153 CrossRef CAS PubMed.
- D. R. Roe, C. Bergonzo and T. E. Cheatham, J. Phys. Chem. B, 2014, 118, 3543–3552 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 Search PubMed.
- S. Izadi, R. Anandakrishnan and A. V. Onufriev, J. Phys. Chem. Lett., 2014, 5, 3863–3871 Search PubMed.
- A. Sengupta, Z. Li, L. F. Song, P. Li and K. M. Merz, J. Chem. Inf. Model., 2021, 61, 869–880 CAS.
- M. Zgarbová, J. Šponer and P. Jurečka, J. Chem. Theory Comput., 2021, 17, 6292–6301 CrossRef PubMed.
- S. Izadi and A. V. Onufriev, J. Chem. Phys., 2016, 145, 074501 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CAS.
- R. M. Betz and R. C. Walker, J. Comput. Chem., 2015, 36, 79–87 CAS.
- Y. Tao, T. J. Giese, Ş. Ekesan, J. Zeng, B. Aradi, B. Hourahine, H. M. Aktulga, A. W. Götz, K. M. Merz Jr. and D. M. York, J. Chem. Phys., 2024, 160, 224104 CAS.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- B. Hourahine, B. Aradi, V. Blum, F. Bonafé, A. Buccheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Dumitric, A. Dominguez, S. Ehlert, M. Elstner, T. Van Der Heide, J. Hermann, S. Irle, J. J. Kranz, C. Köhler, T. Kowalczyk, T. Kubař, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, A. J. Page, A. Pecchia, G. Penazzi, M. P. Persson, J. Řezáč, C. G. Sánchez, M. Sternberg, M. Stöhr, F. Stuckenberg, A. Tkatchenko, V. W. Z. Yu and T. Frauenheim, J. Chem. Phys., 2020, 152, 124101 CAS.
- D. M. Perreault, L. A. Cabell and E. V. Anslyn, Bioorg. Med. Chem., 1997, 5, 1209–1220 CAS.
- Y. A. Kondratenko, A. A. Nikonorova, A. A. Zolotarev, V. L. Ugolkov and T. A. Kochina, J. Mol. Struct., 2020, 1207, 127813 CrossRef CAS.
- S. Ullrich, Z. Nazir, A. Büsing, U. Scheffer, D. Wirth, J. W. Bats, G. Dürner and M. W. Göbel, ChemBioChem, 2011, 12, 1223–1229 CrossRef CAS PubMed.
- R. Salvio, S. Volpi, R. Cacciapaglia, F. Sansone, L. Mandolini and A. Casnati, J. Org. Chem., 2016, 81, 9012–9019 CrossRef CAS PubMed.
- D. Wirth-Hamdoune, S. Ullrich, U. Scheffer, T. Radanovic, G. Dürner and M. W. Göbel, ChemBioChem, 2016, 17, 506–514 CrossRef CAS PubMed.
- S. Alkhader, A. Ezra, J. Kasparkova, V. Brabec and E. Yavin, Bioconjugate Chem., 2010, 21, 1425–1431 CrossRef CAS PubMed.
- M. R. Redinbo, L. Stewart, P. Kuhn, J. J. Champoux and W. G. J. Hol, Science, 1998, 279, 1504–1513 CrossRef CAS PubMed.
- S. H. Wan, F. Liang, X. Q. Xiong, L. Yang, X. J. Wu, P. Wang, X. Zhou and C. T. Wu, Bioorg. Med. Chem. Lett., 2006, 16, 2804–2806 CrossRef CAS PubMed.
- J. Li, J. Zhang, Q. S. Lu, Y. Yue, Y. Huang, D. W. Zhang, H. H. Lin, S. Y. Chen and X. Q. Yu, Eur. J. Med. Chem., 2009, 44, 5090–5093 CrossRef CAS PubMed.
- M. Q. Wang, J. L. Liu, J. Y. Wang, D. W. Zhang, J. Zhang, F. Streckenbach, Z. Tang, H. H. Lin, Y. Liu, Y. F. Zhao and X. Q. Yu, Chem. Commun., 2011, 47, 11059–11061 Search PubMed.
- X. Sheng, X. M. Lu, J. J. Zhang, Y. T. Chen, G. Y. Lu, Y. Shao, F. Liu and Q. Xu, J. Org. Chem., 2007, 72, 1799–1802 CrossRef CAS PubMed.
- M. W. Göbel, C. D. Roussev and U. Scheffer, Helv. Chim. Acta, 2014, 97, 215–227 CrossRef.
- R. Hettich and H.-J. Schneider, J. Am. Chem. Soc., 1997, 119, 5638–5647 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- CLSI, Performance Standards for Antimicrobial Susceptibility Testing. 29th Ed. CLSI supplement M100, Clinical and Laboratory Standards Institute, Wayne PA, 2019 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization of compounds, additional biological assay results, theoretical methods and additional computational results. See DOI: https://doi.org/10.1039/d4md00984c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |