
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and evaluation of acetylcholine-antitumor lipid hybrids led to identification of a potential anticancer agent disrupting the CDK4/6-Rb pathway in lung cancer†
Ahmed H. E.
Hassan
 abcd,
Eun Seo
Bae
e,
Youngdo
Jeong
f,
Chae Won
Ock
e,
Selwan M.
El-Sayed
ag,
Minji
Kim
f,
Mohamed F.
Radwan
h,
Tarek S.
Ibrahim
hi,
Jun-Young
Cho
f,
Boyoung Y.
Park
fj,
Jaehoon
Sim
bcd,
Sang Kook
Lee
*e and
Yong Sup
Lee
*bf
abcd,
Eun Seo
Bae
e,
Youngdo
Jeong
f,
Chae Won
Ock
e,
Selwan M.
El-Sayed
ag,
Minji
Kim
f,
Mohamed F.
Radwan
h,
Tarek S.
Ibrahim
hi,
Jun-Young
Cho
f,
Boyoung Y.
Park
fj,
Jaehoon
Sim
bcd,
Sang Kook
Lee
*e and
Yong Sup
Lee
*bf
aDepartment of Medicinal Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura, 35516, Egypt
bDepartment of Pharmacy, College of Pharmacy, Kyung Hee University, Seoul 02447, Republic of Korea. E-mail: kyslee@khu.ac.kr
cDepartment of Regulatory Science, Graduate School, Kyung Hee University, Seoul 02447, Republic of Korea
dInstitute of Regulatory Innovation through Science, Kyung Hee University, Seoul 02447, Republic of Korea
eNatural Products Research Institute, College of Pharmacy, Seoul National University, Seoul 08826, Republic of Korea. E-mail: sklee61@snu.ac.kr
fDepartment of Fundamental Pharmaceutical Sciences, Kyung Hee University, Seoul 02447, Republic of Korea
gDepartment of Medicinal Chemistry, Faculty of Pharmacy, Mansoura National University, Gamasa 7731168, Egypt
hDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, King Abdulaziz University, Jeddah 21589, Saudi Arabia
iDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Zagazig University, Zagazig 44519, Egypt
jDepartment of Biomedical and Pharmaceutical Sciences, Kyung Hee University, Seoul 02447, South Korea
First published on 7th March 2025
Abstract
Hybridization of acetylcholine with antitumor lipids (ATLs) was explored to achieve novel potential anticancer agents. The combination with a 2-stearoxyphenyl moiety substantially enhanced the anticancer activity of the acetylcholine hybrids. Compounds 6, 8, 9 and 10 exhibited pronounced anticancer activities higher than edelfosine and stPEPC and NSC43067. Compounds 6, 8, 9 and 10 also showed broad-spectrum anticancer activity against diverse cancer cells including lung, ovarian, renal, prostate, leukaemia, colon, CNS, melanoma, and breast cancer cells. Compounds 6 and 8 were potent compounds eliciting single digit low micromolar GI50 values. Compound 6 was the most potent against non-small cell lung cancer, ovarian cancer, renal cancer, and prostate cancer. Meanwhile, compound 8 was the most potent against leukaemia, colon cancer, CNS cancer, melanoma, and breast cancer. Exploration of the mechanism of action of compound 6 in A549 non-small cell lung cancer cells showed that it triggers cell cycle arrest in the G0/G1 phase via disruption of the CDK4/6-Rb pathway and induces apoptosis via the activation of caspases, upregulation of BAX and cleavage of PARP. Overall, the results present acetylcholine-ATL hybrids 6 and 8 as potential anticancer agents for possible further development.
1. Introduction
According to the WHO's world health statistics 2023, cancer is the second major fatal non-communicable disease (NCD) after cardiovascular diseases, accounting for about 9.3 million global deaths.1 Therefore, cancer treatment remains a significant focus for researchers as the global burden of cancer continues to increase.2–7 Despite the global efforts to develop anticancer therapies, the optimum anticancer therapeutic agent has not been achieved yet. Furthermore, the heterogeneity of cancer diseases and evolvement of resistance limit the efficacy of current available therapeutics agents. Hence, there is a crucial need for new therapeutic anticancer molecules to overcome such obstacles.2,8Natural compounds, including primary or secondary metabolites, could have indispensable functions within biological systems. Because of their influential impact on the biological system, natural products also play an important role in drug discovery and development.9–13 As per their definition, natural products are not limited to compounds produced by plants or microorganisms, but extend to those produced by any living organism including animals such as insects or mammals. Despite lipids having received relatively low attention, they are among the promising natural products for drug discovery and development.14–16 Although they are primarily recognized as structural building units, such as lipids in cell membranes or as a mean for energy storage in the body, lipids such as fatty acids, eicosanoids and phospholipids play essential roles in biological functions including cellular regulation and signalling.17,18 In fact, bioactive lipidic molecules can control and alter health and disease.
Cyclin-dependent kinases such as CDK4 and CDK6 are key players controlling the cell cycle of proliferating cells and, hence, are interesting targets for cancer therapy, including lung cancer and others. Developing therapeutics targeting CDKs can be achieved through developing inhibitor molecules that bind to and prevent the activity of CDKs. In this regard, several heterocyclic scaffolds have afforded CDK inhibitors, such as trilaciclib, palbociclib, and abemaciclib (Fig. 1).19 Cellular reduction or depletion of the target CDK protein levels through induction of protein degradation using PROTAC or molecular glue degraders, such as TMX-2138 or ML 1–71, respectively (Fig. 1), is an alternative strategy.20,21 In addition, some compounds inducing CDK protein aggregation, such as NSC43067 and NSC63002, were discovered as a novel class of compounds reducing the cellular levels of CDKs (Fig. 1).22
 | ||
| Fig. 1 Examples of literature-known compounds belonging to different categories of compounds targeting CDKs. | ||
Among the successful drug design techniques is the hybridization approach.23–30 Compounds designed by this strategy inherit some structural features from parent molecules.31 Hence, they might elicit bioactivity via combining more than one pathway.2 This might result in a beneficial higher efficacy and lower resistance evolvement rates. Several reports have reported successful stories for this approach for the development of therapeutics against various diseases. Successful examples of such developed hybrid drugs that reached markets include sunitinib, lapatinib, ladostigil, and trioxaquine (Fig. 2).32
 | ||
| Fig. 2 Examples of successful reports of the development of hybrid drugs. | ||
Among cancer diseases, lung cancer shows the highest incidence and mortality rates.33–35 This might be contributed by the fact that more than 85% of lung cancer cases are non-small lung cancers, which are less responsive to therapy and furthermore insidious and mostly diagnosed in advanced stages. In combination with the fact that resistance to currently used anticancer agents could evolve, these factors render lung cancer one of the most difficult cancers to treat with poor prognosis and low survival rates. Together, these factors raise urgent needs to discover and develop new anticancer agents against lung cancer and other cancers. In lieu of these needs, we endeavour in the current work to develop new anticancer agents inspired by natural products and following a molecular hybridization approach. Herein, we report our interesting results.
2. Results and discussion
2.1. Design rational
Acetylcholine and its congener butyrylcholine (1 and 2 respectively, Fig. 3) are natural compounds that are biosynthesized within the body and are well recognized as neurotransmitters. However, they have also other functions including their engagement in oncogenic signalling pathways.36 It was reported that acetylcholine functions as an autocrine growth factor for lung cancer cell proliferation and acetylcholine signalling modulation impacts the resistance and hinders relapse of EGFR-mutant lung cancer.37–39 In addition, down-stream targets of acetylcholine are involved in proliferation of various other cancer diseases.40,41 Due to the reported connection between acetylcholine and cancer, several compounds modulating acetylcholine and butyrylcholine signalling elicit anticancer activity against various cancer diseases.42–46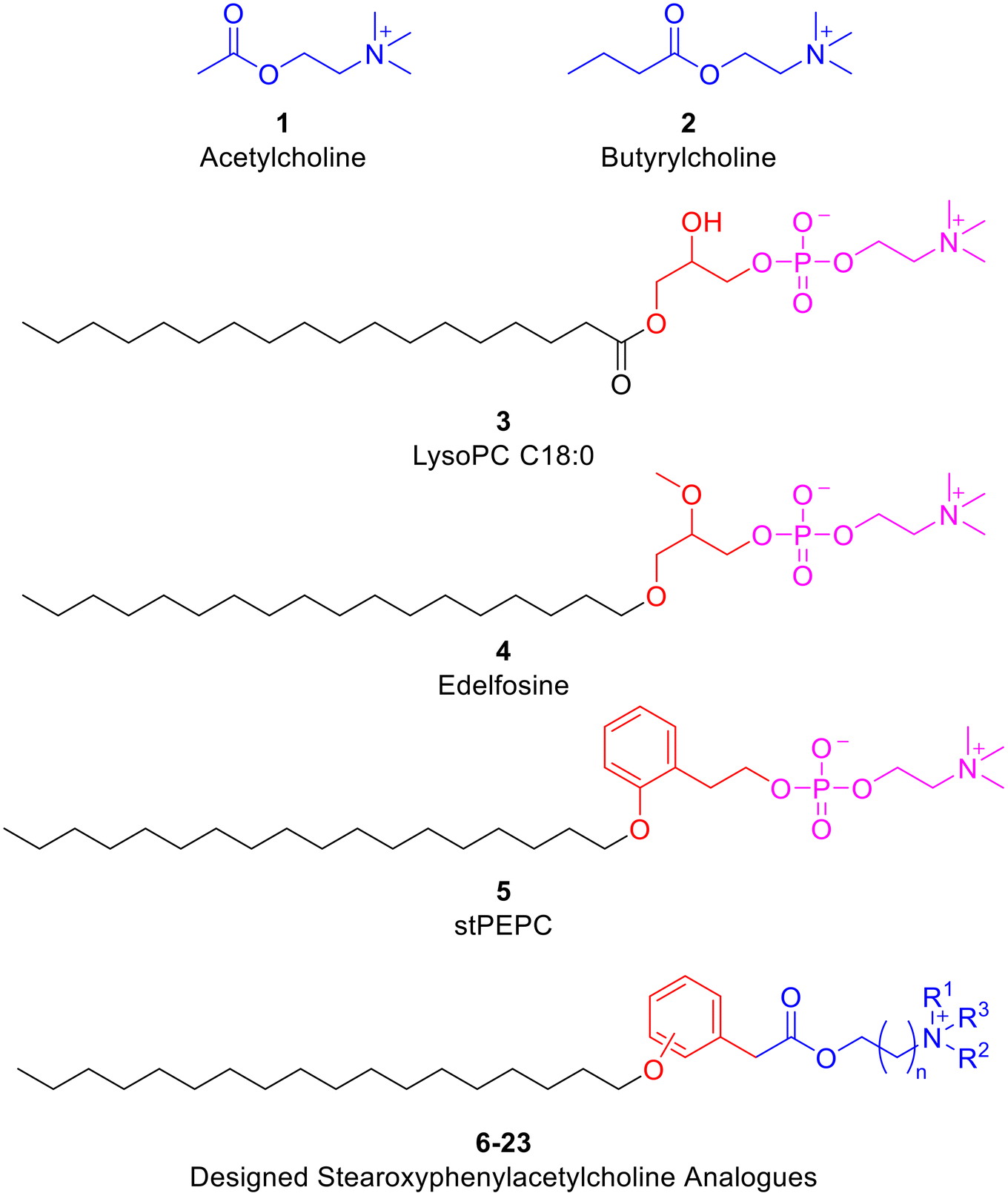 | ||
| Fig. 3 Previously known bioactive compounds and the design rational of the targeted derivatives of phenylacetylcholine analogues. | ||
Lysophosphatidylcholines (LysoPCs), such as LysoPC C18:0 (3; Fig. 3), are natural compounds biosynthesized through partial hydrolysis of the glycerophospholipids phosphatidylcholines. Investigations of the biological activities of LysoPCs have unveiled their roles in tumour invasion, metastasis and prognosis.47 In fact, anticancer effects of LysoPCs against several cancers including lung and breast cancers have been reported.48,49 However, LysoPCs are metabolically labile. Consequently, edelfosine (4; Fig. 3), the metabolically more stable analogue of the natural LysoPC C18:0, was developed as a potential anticancer agent.50–52 Employing edelfosine as a starting point, stPEPC (5; Fig. 3) was recently developed as an anticancer agent via replacing the central glycerol moiety by a phenethyl moiety.14,53 In the search for more promising anticancer agents, a molecular hybridization approach was implemented herein to design the targeted derivatives of phenylacetylcholine analogues (6; Fig. 3). Thus, the designed derivatives of phenylacetylcholine analogues (6–23) incorporate an acetylcholine or acetylcholine-like moiety instead of the methylphosphocholine fragment of LysoPC (3) and edelfosine (4) or ethylphosphocholine fragment of stPEPC (5). Meanwhile, replacement of the central moiety of LysoPC (3) or edelfosine (4) by an aromatic phenyl ring-containing moiety was maintained in analogy to stPEPC (5). While the central glycerol moiety is conformationally flexible, the phenyl ring in the designed derivatives of phenylacetylcholine analogues (6–23) can serve as a conformational lock and thus o-, m-, or p-positional scanning of the C18 alkoxy substituent at the phenyl ring might be beneficial. Therefore, restricting the flexibility near the head and such positional scanning were applied in the designed molecules to evaluate their effect regarding biological activity. Furthermore, variation of the quaternary trimethylammonium moiety within the designed derivatives of phenylacetylcholine analogues (6–23) by the more steric triethylammonium or cyclic moieties was evaluated. Finally, replacing the two-carbon chain choline moiety by its homologous homocholine moiety that has a longer three-carbon chain was explored.
2.2. Chemical synthesis
Straightforward and concise synthesis of the targeted compounds was achieved in five linear steps employing commercially available hydroxyphenylacetic acid derivatives 24–26 (Scheme 1). The initial step involved esterification to access methyl ester derivatives 27–29. Thus, methanolic solutions of starting materials 24–26 were treated with acetyl chloride to generate in situ hydrochloric acid that promoted an acid-catalysed esterification reaction. The conversion into methyl esters was elucidated by the appearance of a methyl group NMR peak as a singlet within the 3.77–3.72 ppm range in the isolated products 27–29. The second step involved O-alkylation of the phenolic hydroxyl group to access the corresponding stearoxy derivatives 30–32. This was performed using stearyl tosylate in a nucleophilic substitution reaction in the presence of a catalytic amount of potassium iodide. The conversion into stearoxy derivatives was elucidated by the appearance of NMR peaks in the 3.96–3.93, 1.78–1.73, 1.47–1.24, and 0.89–0.87 ppm ranges corresponding to a stearoxy moiety. The third step was hydrolysis into acid derivatives 33–35 in preparation for choline-like moiety installation in the following steps. Next, installation of the choline-like moieties was started by esterification with appropriate alkanolamines to access ester derivatives 36–53. This was performed via in situ generation of Vilsmeier reagent using stoichiometric oxalyl chloride and catalytic DMF to convert the acid derivatives 33–35 into acid chlorides, which were reacted with the appropriate alkanolamine to afford derivatives 36–53. The conversion into aminoalkyl esters was elucidated by the appearance of NMR peaks corresponding to the aminoalkyl moieties. For example, compound 36 showed two triplets at 4.22 and 2.58 ppm, each equivalent to methylene protons, in addition to a singlet at 2.27 ppm equivalent to six protons of the two methyl groups. Finally, the aminoalkyl esters 36–53 were converted into the targeted choline analogue derivatives 6–23via quaternization of the tertiary amino nitrogen through stirring their solutions with the appropriate alkyl iodide. The conversion was confirmed by the increase in the chemical shift of protons on the carbons attached to the quaternary nitrogen atom and NMR peak of the introduced alkyl moiety. For example, the chemical shift of the NMR peaks of the methyl and methylene protons of compound 6 increased to 3.04 and 3.62 ppm, respectively, in comparison with 2.27 and 2.58 ppm for the precursor compound 36. The analytical and spectroscopical data of the synthesized target compounds were in agreement with the expected values, which elucidated their structures.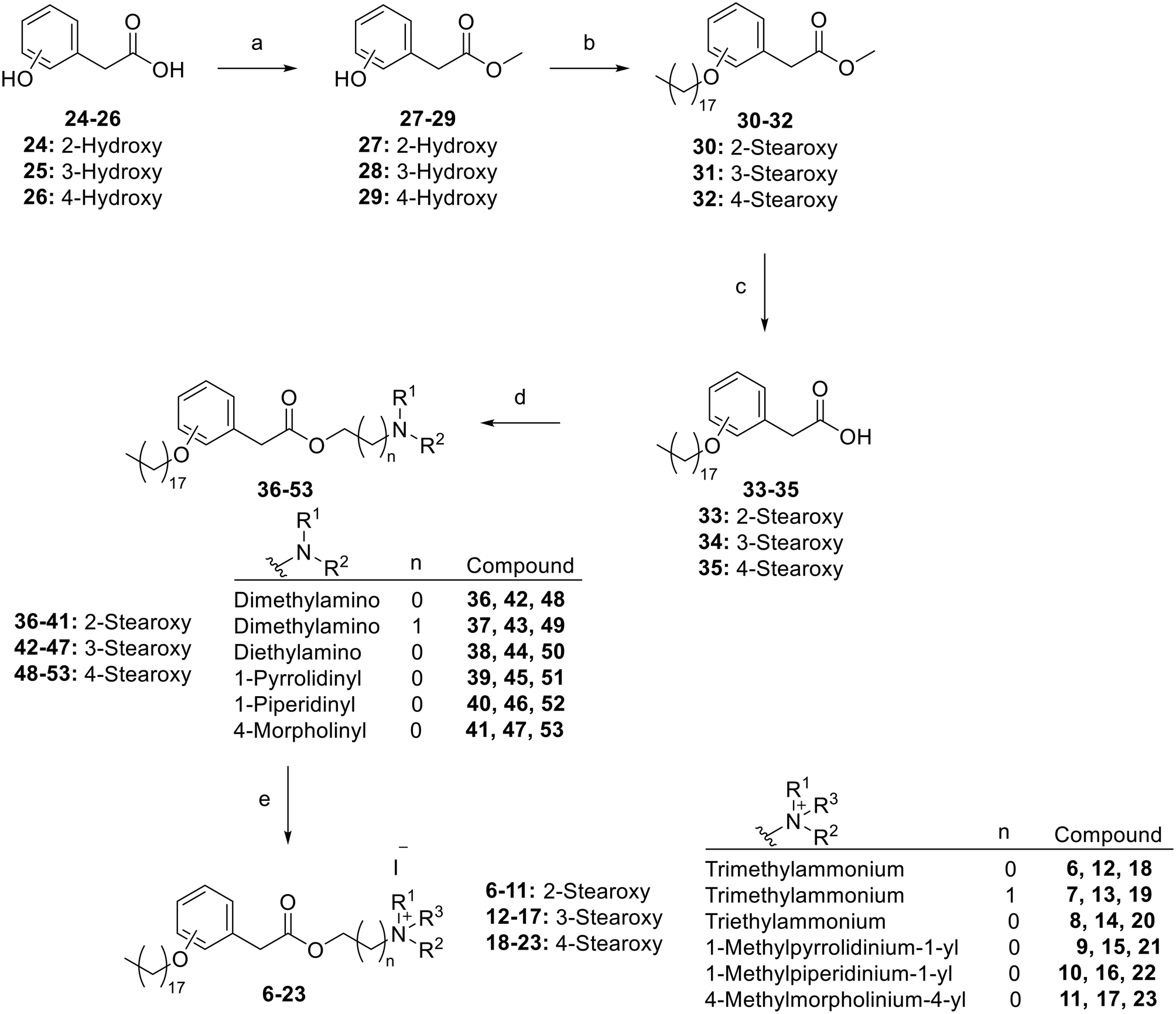 | ||
| Scheme 1 Synthesis of the targeted derivatives of phenylacetylcholine analogues: a) acetyl chloride, MeOH, 65 °C, 2 h; b) stearyl tosylate, K2CO3, cat. KI, DMF, 60 °C, 20 h; c) NaOH, THF, MeOH, 80 °C, 3 h; (d) 1) oxalyl chloride, cat. DMF, anhydrous CH2Cl2, 25 °C, 3 h, 2) alkanolamine, CH2Cl2, 25 °C, 12 h; (e) alkyl iodide, acetonitrile, ethyl acetate, 25 °C, 24 h. | ||
2.3. Biological evaluations
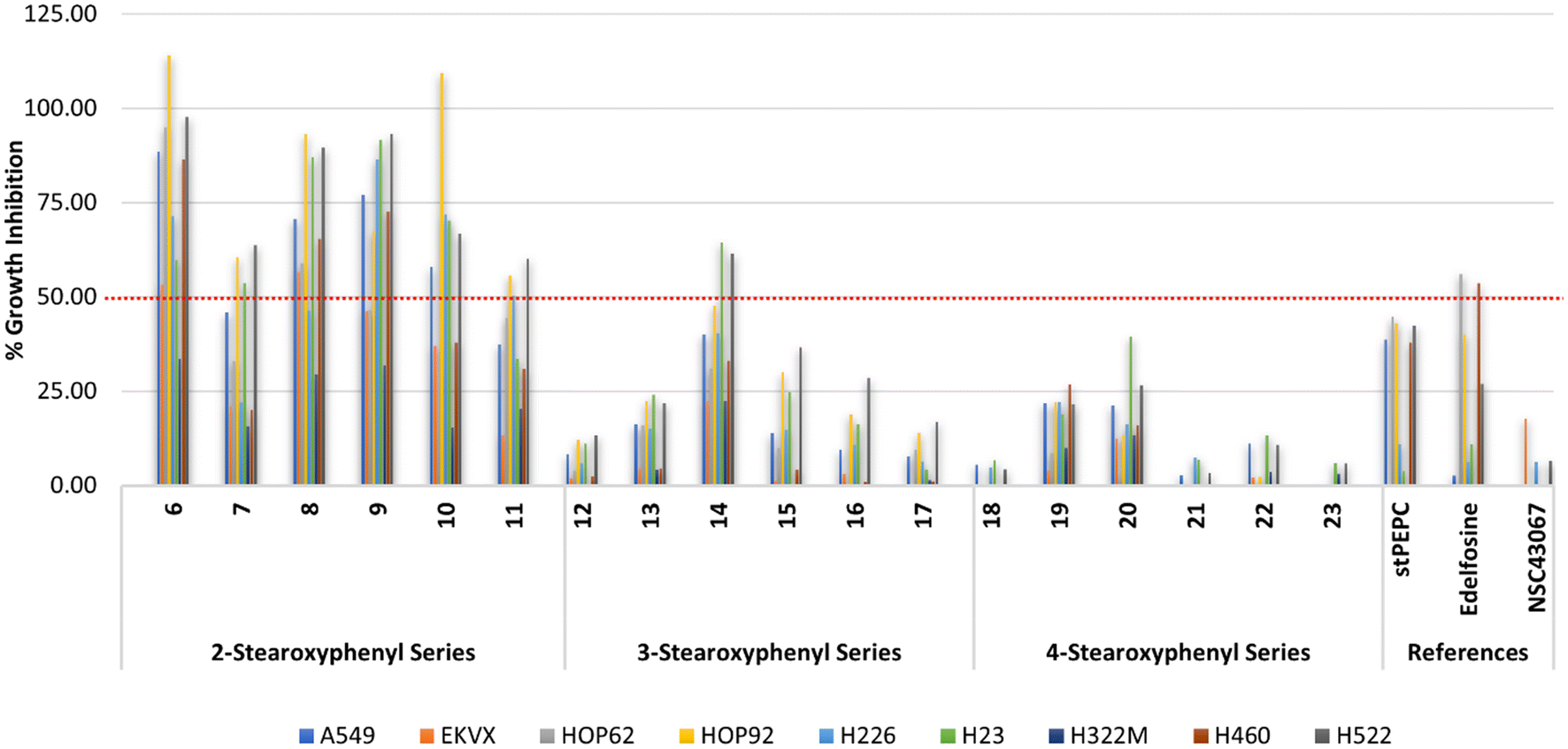 | ||
| Fig. 4 % Growth inhibition of diverse non-small cell lung cancer cell lines triggered by 10 μM concentrations of the prepared compounds 6–23, stPEPC and edelfosine. | ||
As shown in Fig. 4, the reference compound NSC43067 exhibited low activities at 10 μM concentration against only three cell lines (H460, EKVX, and H522; Table S1†). Meanwhile, both edelfosine and stPEPC at 10 μM concentration, in general, showed modest activity lower than 50% inhibition of NSCLC cell lines. Thus, the average percent inhibition values over the employed 6 adenocarcinoma cell lines, the metastatic adenocarcinoma, and the large cell and squamous carcinomas were 28.03 and 27.69% for edelfosine and stPEPC, respectively. The metastatic adenocarcinoma, the squamous carcinoma and EKVX adenocarcinoma cell lines were the least responsive to both edelfosine and stPEPC (Table S1†). Conversely, the 2-stearoxyphenyl-based series of compounds 6–11 were more active at the tested 10 μM concentration, as shown in Table S1† and Fig. 4. In particular, compound 6 with a calculated value of 77.74% possessed the highest average activity over the tested nine NSCLC cells. It showed its highest activity against NSCLC large cell carcinoma and four out of the tested six NSCLC adenocarcinomas (88.49–114.01% growth inhibition against H460, A549, HOP62, HOP92, and H522 cell lines; Table S1†). In addition, it elicited good activity against NSCLC squamous carcinoma and average activity against two NSCLC adenocarcinomas (Table S1†). However, it elicited low activity against NSCLC metastatic adenocarcinoma (33.49% growth inhibition; Table S1†). Meanwhile, its homologous compound 7 showed a relatively lower activity than compound 6 but a higher activity than both edelfosine and stPEPC (Table S1†). Thus, compound 7 showed an average activity above 50% against the three NSCLC adenocarcinomas (HOP92, H23 and H522), which is higher than both edelfosine and stPEPC. Nevertheless, the activity of compound 7 was very low against the tested NSCLC metastatic adenocarcinoma, large cell carcinoma, and squamous carcinoma cell lines. Potential anticancer activity was still observed for analogous compounds 8, 9, and 10 (Fig. 4). As displayed in Table S1,† compound 8 elicited excellent to average activity against all tested six NSCLC adenocarcinoma cell lines, while the activities of compounds 9 and 10 ranged from excellent to average against four out of the tested six NSCLC adenocarcinoma cell lines (Table S1†). Moreover, compound 8 showed a good activity against NSCLC large cell carcinoma (H460), while compound 10 possessed a good activity against NSCLC squamous carcinoma (H226) and compound 9 was potentially active against both types (Table S1†). Nevertheless, the activities of compounds 8, 9, and 10 against metastatic NSCLC adenocarcinoma (H322) were low in a similar pattern to compound 6 and their average growth inhibition values over all tested nine NSCLC cell lines were more than 55% (Table S1†). An exception of the good activity of analogous compounds 6, 8, 9, and 10 was compound 11, which possessed an average growth inhibition activity over all tested NSCLC cells lower than 40%. It showed growth inhibition of more than 50% against only three NSCLC cells out of the tested NSCLC cells.
Analysis of the results of the 3-stearoxyphenyl and 4-stearoxyphenyl-based series of compounds 12–17 and 18–23, respectively, revealed that these two series in general have low activity at the tested concentration (Fig. 4). Among them, only compound 14 showed better activity than the references edelfosine and stPEPC at the tested 10 μM concentration. However, compound 14 was less active than the corresponding 4-stearoxyphenyl-based compound 8, showing growth inhibition above 50% against only two out of the tested nine NSCLC cells (Table S1†). As Fig. 4 illustrates, the 4-stearoxyphenyl-based series of compounds 18–23 were in general even less active than the 3-stearoxyphenyl-based series of compounds 12–17. Together, these results show that the 2-stearoxyphenyl-based compounds 6–11 are the most potentially active against diverse NSCLC cells, including NSCLC adenocarcinoma, large cell carcinoma and squamous carcinoma, but not metastatic lung adenocarcinoma. In addition, four compounds (6, 8, 9, and 10) out of the 2-stearoxyphenyl-based compounds were found to possess potential activity and might be worth further investigation.
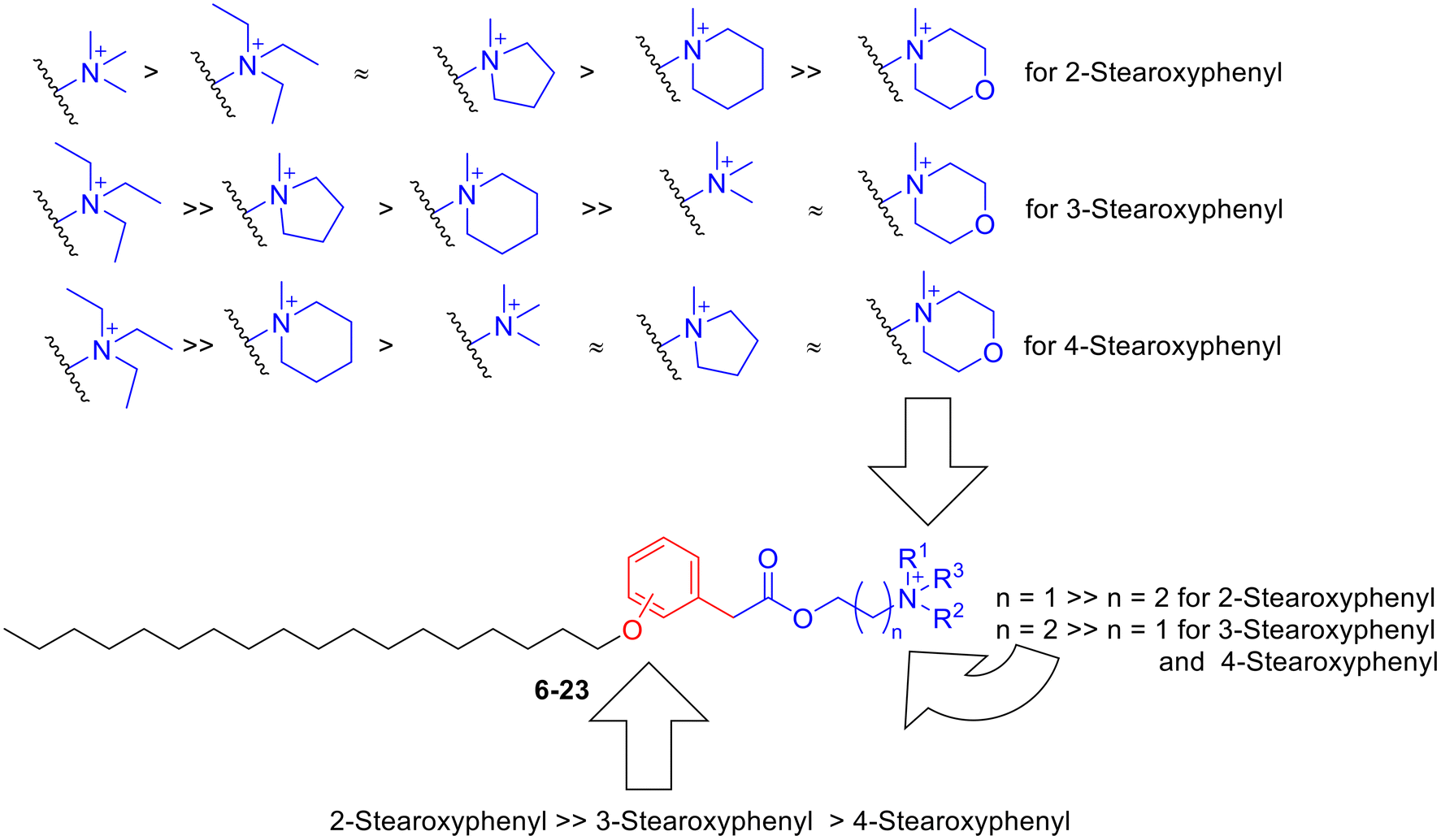 | ||
| Fig. 5 The relation between structure and anticancer activity of compounds 6–23 against human lung cancer. | ||
 | ||
| Fig. 6 Evaluation results of the spectrum of anticancer activity of compounds 6, 8, 9 and 10 at 10 μM concentration against nine panels of human cancer cells belonging to diverse cancer diseases. | ||
 | ||
| Fig. 7 Results of GI50 evaluation for the anticancer activities of compounds 6, 8, 9, 10 and edelfosine against several cancer cells representing diverse cancer diseases: (A) potencies against non-small cell lung cancers; (B) potencies against leukemic cancers; (C) potencies against colon cancers; (D) potencies against CNS cancers; (E) potencies against ovarian cancers; (F) potencies against melanomas; (G) potencies against renal cancers; (H) potencies against prostate and breast cancers. | ||
Analysis of the potencies against non-small lung cancer showed that the reference edelfosine possessed GI50 values within the range of 7.05–46.77 μM and a calculated average GI50 of 25.20 μM; all these values are above the cut-off value of 3 μM for the displayed results in Fig. 7A. Conversely, compound 6 possessed much more superior potencies against all employed cell lines with low GI50 values within the range of 1.25–2.17 μM and a calculated average GI50 of 1.62 μM. This represents an average 15.5-fold increase compared to the potency of edelfosine. With a calculated average GI50 of 1.80 μM and a GI50 range of 1.10–4.03 μM, compound 8 possessed excellent potency close to that of compound 6. Although compounds 9 and 10 elicited much more superior potencies than the reference edelfosine (a calculated average GI50 of 2.67 and 2.35 versus 25.20 μM and range of 1.22–8.22 and 1.32–5.78 versus 7.05–46.77 μM for compounds 9, 10 and edelfosine, respectively), they were notably less potent than compounds 6 and 8. Only compound 6 maintained a potent GI50 value less than the applied cut-off value against all employed lung cancer cell lines, including the metastatic adenocarcinoma cell line H322M (Fig. 7A). Together, these results suggest consideration of compound 6 as a potential agent against NSCLC for further investigations.
Analysis of the results against four leukemic cancer cells showed that edelfosine possessed GI50 values within the range of 3.94–44.26 μM and a calculated average GI50 of 16.82 μM. In comparison, all of compounds 6, 8, 9 and 10 elicited potencies much superior to edelfosine, showing GI50 values less than the applied cut-off value of 3 μM. The most potent was compound 8, eliciting an average GI50 value of 1.29 μM and a range of 0.93–1.50 μM over the used four leukemic cancer cells. This represents an average 13-fold increase compared to the potency of edelfosine. The second most potent was compound 6, which showed GI50 values comparable to those of compound 8. Compounds 9 and 10 maintained good potencies with GI50 less than the applied cut-off value, but were slightly less potent than compounds 8 and 6 as antileukemic agents (Fig. 7B). Based on these results, compounds 8 and 6 might be candidates for further development of antileukemic agents.
The results of potency evaluation against colon cancer cell lines showed than edelfosine elicited GI50 within the cut-off value against only two cell lines out of the used seven cell lines (2.38 and 2.65 μM against HCC2998 and COLO205, respectively; Fig. 7C). Over the used seven cell lines, edelfosine showed a calculated average GI50 of 7.80 μM and a range of 2.38–14.72 μM. Conversely, compounds 6, 8, 9 and 10 were more potent. Among them, only compound 6 maintained GI50 values within the applied cut-off value (range of 1.59–2.96 μM) and showed an average GI50 value of 1.91 μM. This represents an average 4-fold increase compared to the potency of edelfosine. Although compound 8 showed GI50 outside the applied cut-off window against the colon adenocarcinoma HCT15 cell line, it was potent against all other tested colon cancer cell lines showing a calculated average GI50 of 1.88 μM and a range of 1.04–4.39 μM. Similarly, compounds 9 and 10 showed GI50 outside the applied cut-off window against only the colon adenocarcinoma HCT15 cell line, but were potent against all other tested colon cancer cell lines. Compounds 9 and 10 were of almost equal potencies to each other (average GI50 values of 2.17 and 2.16 μM for compounds 9 and 10, respectively) but, in general, were slightly less potent than compounds 6 and 8. Collectively, these results present compound 6 as the most promising compound for further development of agents against colon cancer.
Investigation of the potencies against six CNS cancer cell lines showed that edelfosine possessed GI50 values above the applied cut-off against all tested cell lines with a calculated average GI50 of 21.40 μM and a range of 8.75–58.48 μM. Conversely, all determined GI50 values for compounds 6, 8, 9 and 10 were within the applied cut-off value (Fig. 7D). Compound 8 was the most potent among the investigated compounds, possessing a calculated average GI50 value of 1.35 μM and a range of 0.97–1.62 μM. This represents a more than 15-fold increase compared to the potency of edelfosine. The potencies of the other three compounds 6, 9 and 10 were, in general, close to each other and to compound 8 (average GI50 of 1.59, 1.82, and 1.92 μM with ranges 1.23–2.03, 1.26–2.83, and 1.39–2.93 μM for compounds 6, 9 and 10, respectively). Interestingly, compounds 6, 9 and 10 were less potent against CNS cancer cell line SF295 relative to other CNS cancer cell lines, which is an opposite trend to the determined GI50 values of edelfosine where the second best GI50 for edelfosine against CNS cancers was against SF295 cells. This might be a reflection of differences between edelfosine and these compounds in their mechanisms of action. Together, these results might nominate compound 8 as the best candidate among the studied compounds as the most promising compound for further development of agents against CNS cancer.
Regarding ovarian cancer, edelfosine possessed GI50 outside the applied cut-off, with a GI50 range of 6.67–42.27 μM and an average of 25.15 μM. Edelfosine showed its highest potency against the ovarian IGROV1 cell line and least potency against the doxorubicin-resistant ovarian ADRRES cell line. Compounds 6 and 8 possessed similar activity patterns, but with higher potencies (Fig. 7E). The most potent was compound 6 possessing a calculated average GI50 of 2.64 μM with a range of 0.65–9.84 μM. This represents a 9.5-fold increase compared to the potency of edelfosine. Close to compound 6 was compound 8 as the second most potent, eliciting a calculated average GI50 of 2.67 μM with a range of 0.92–7.58 μM. Compounds 9 and 10 were significantly less potent, showing calculated average GI50 values of 6.30 and 6.28 μM with a range of 1.52–18.90 and 1.54–18.20 μM, respectively. Furthermore, their activity trend was different from that of edelfosine, as they were least potent against the ovarian cancer SKOV3 cell line, rather than the doxorubicin-resistant ovarian ADRRES cells. These differences suggest variations in the mode of action of these compounds. Collectively, these results present compounds 6 and 8 as the most potent among the tested compounds for further development of agents against ovarian cancer.
Analysis of the potencies against melanoma showed that edelfosine possessed high GI50 values against all nine employed melanoma cell lines, which are outside the applied cut-off value. Edelfosine possessed a calculated average GI50 of 38.69 μM with a range of 15.38–98.40 μM. Compounds 6, 8, 9 and 10 were notably much more potent than edelfosine with all determined GI50 values less than the applied cut-off value (Fig. 7F). Compound 8 was identified as the most potent with a calculated average GI50 of 1.33 μM and range of 0.88–1.90 μM. This represents an average 29-fold increase compared to the potency of edelfosine. Compounds 6, 9 and 10 were also potent but with lower potencies than compound 8, showing calculated average GI50 values of 1.73, 2.07, and 2.07 μM and ranges of 1.57–2.34, 1.63–2.82, and 1.70–2.79 μM for compounds 6, 9 and 10, respectively. As illustrated in Fig. 7F, these results present compound 8 as a promising candidate for the development of antimelanoma agents.
Assessment of potencies against eight renal cancer cells showed that edelfosine had a calculated average GI50 value of 25.02 μM and range of 12.11–72.78 μM. All these values were higher than the applied cut-off value. In contrast, compound 6 showed much superior potencies that were within the cut-off value against all employed renal cancer cell lines (Fig. 7G). Compound 6 possessed a calculated average GI50 value of 1.35 μM with a GI50 range of 0.49–1.75 μM. This represented an average 18.5-fold increase compared to the potency of edelfosine. Compounds 8, 9 and 10 were less potent relative to compound 6 showing almost an average half its potency. Furthermore, the GI50 values of compounds 8, 9 and 10 against two renal cancer cell lines, UO31 and ACHN, were more than the applied cut-off value, which reflected their relatively lower potencies. Hence, among the tested compounds, compound 6 might be the best candidate for further development of agents against renal cancer.
The potency evaluation results against two prostate cancer cell lines and five breast cancer cell lines showed that edelfosine has high calculated average GI50 values of more than 53.60 and 67.20 μM against prostate and breast cancers, respectively. In comparison, compounds 6, 8, 9 and 10 were much more potent than edelfosine (Fig. 7H). Generally, compound 8 was the most potent against breast cancer, while compound 6 was the most potent against prostate cancer. Compound 8 possessed a calculated average GI50 value of 1.43 μM against breast cancer, which is around a 47-fold increase compared to the potency of edelfosine. Meanwhile, compound 6 possessed a calculated average GI50 value of 1.60 μM against prostate cancer, which is a 33.5-fold increase compared to the potency of edelfosine. Despite being relatively less potent against prostate and breast cancers, the potencies of compounds 9 and 10 were still comparable to the most potent compounds (compound 6 against prostate cancer and compound 8 against breast cancer). From these results, compound 6 might be nominated for further development of potential agents against prostate cancer, while compound 8 might be nominated for further development of potential agents against breast cancer.
2.3.6.1 Compound 6 triggers G0/G1 cell cycle arrest in A549 lung cancer cells. Cells undergo different phases during the proliferation cycle, and each phase of this cycle is distinguished by having varying quantities of DNA. Quantifying the DNA content per cell using the fluorescent stain propidium iodide enables classification of a population of cells according to their phase of the cell cycle. As a single copy of DNA exists when cells are in the G0/G1 phase, a one-fold fluorescence should be measured for cells in this phase. Because two-DNA copies exist when cells are in the G2/M phase, two-fold fluorescence should be detected for these cells. Meanwhile, DNA is being synthesized by cells in the S phase and, hence, the detected fluorescence is more than one-fold but less than two fold. Using flow cytometric analysis techniques, the cell cycle distributions of A549 lung cancer cells treated with different concentrations of compound 6 were determined and compared (Fig. 8). The results revealed that the counts of cells in G0/G1 phase increased dose-dependently, while the counts of cells in the S and G2/M phases decreased in a dose-dependent pattern. This outcome showed that compound 6 inhibits the proliferation of A549 lung cancer cells via arresting the cells in the G0/G1 phase and preventing their progression into the next S phase.
 | ||
| Fig. 8 Changes in cell cycle distributions of A549 cells upon treatment with compound 6. (A) Flow cytometric analysis of the cell cycle distributions of A549 cells treated with different concentrations of compound 6 after propidium iodide staining. (B) Graphs showing the quantified changes in cell cycle distributions of A549 cells upon treatment with different concentrations of compound 6. | ||
2.3.6.2 Compound 6 induces apoptotic death in A549 lung cancer cells. According to some literature reports, G0/G1 cell cycle arrest can trigger a subsequent apoptosis, which is a programmed death of cells.54 Meanwhile, some other reports claimed that it can confer resistance against apoptosis.55 Accordingly, whether compound 6 might trigger apoptosis or not in A549 lung cancer cells was investigated. As shown in Fig. 9, fluorescence-activated cell sorting (FACS) flow cytometry post staining with annexin-V and propidium iodide was used to sort A549 cells after treatment with different concentrations of compound 6. The results showed an increase in the portion of cells that entered the early apoptosis state, and a lower portion entered late apoptosis, while no significant necrosis was detected. The increase in apoptosis was proportional to the employed concentration of compound 6. It might be concluded that compound 6 triggers G0/G1 cell cycle arrest and induces apoptosis in A549 lung cancer cells.
 | ||
| Fig. 9 Apoptosis induction in A549 cells treated with compound 6. (A) FACS flow cytometric analysis of cell populations treated with different concentrations of compound 6 after annexin-V and propidium iodide staining. (B) The percentages of each cell population (live cell, early apoptosis, late apoptosis, and necrosis) were quantified and presented as a bar graph. | ||
2.3.6.3 G0/G1 cell cycle arrest triggered by compound 6 in A549 lung cancer cells is mediated by disruption of the CDK4/6-Rb pathway. To understand the mechanism by which compound 6 triggers G0/G1 cell cycle arrest in A549 lung cancer cells, western blotting was performed to investigate proteins involved in cycle regulation that might be responsible for arresting the cells in G0/G1 phase. Retinoblastoma (Rb) protein is a tumour suppressor protein that controls the transition from G1 to S phase in the cell cycle.56 Phosphorylation of Rb releases the transcription factor E2F from the Rb-E2F complex, prompting the cell to leave G1 phase and enter S phase.57 Inhibition of Rb phosphorylation would prevent the cell from entering S phase and eventually arrests the cell in the G0/G1 phase. As shown in Fig. 10A, the formation of phospho-Rb (p-Rb) was inhibited dose-dependently. Rb protein is monophosphorylated by cyclin D-CDK4/6, which is further hyperphosphorylated by CDK2. Interestingly, it was reported that the combined loss of CDK2 and CDK4 results in hypophosphorylation of Rb protein.58 Accordingly, western blotting was performed for CDK2, CDK4, CDK6 and cyclin D1 to investigate their involvement. The results showed dose-dependent inhibition of all these regulator proteins, suggesting that the triggered G0/G1 cell cycle arrest by compound 6 is mediated through disruption of the CDK4/6-Rb pathway. Western blotting was also addressed for other CDK isoforms that showed no dose-dependent inhibition of CDK1, CDK7 and CDK8 upon treatment by compound 6 (Fig. S1†). It might be concluded that despite inhibition of CDK4/6 levels mediating the cytotoxic activity of compound 6, other CDK isoforms including CDK1, CDK7 and CDK8 do not contribute to its cytotoxic activity.
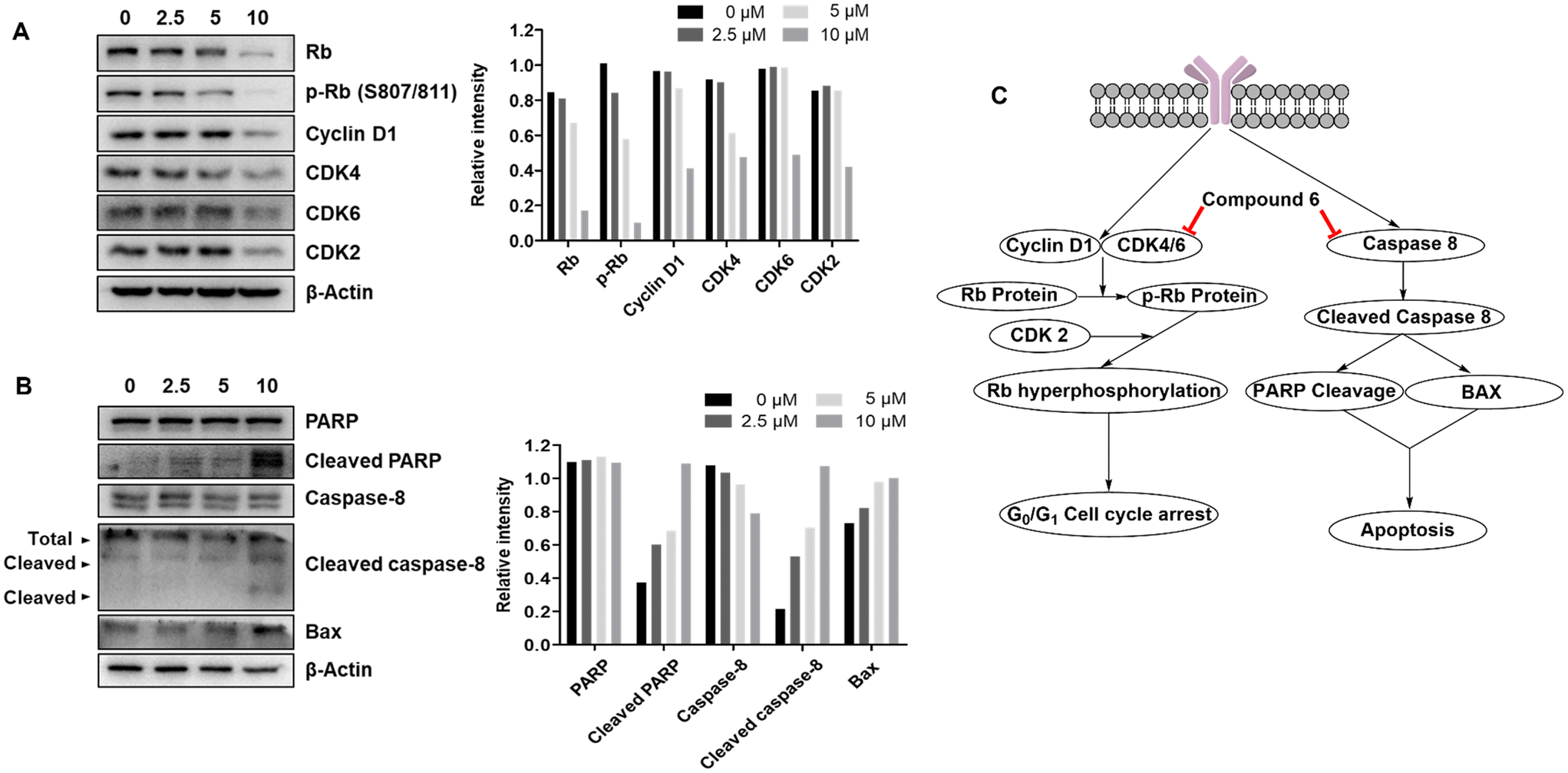 | ||
| Fig. 10 Western blot analysis of regulatory proteins levels in A549 cells upon treatment with different concentrations of compound 6. (A) Western blotting and bar representation of quantified regulatory proteins involved in the G0/G1 phase of the cell cycle. (B) Western blotting and bar representation of quantified regulatory proteins involved in induction of apoptosis. (C) Schematic representation of pathways contributing to G0/G1 cell cycle arrest and induction of apoptosis in A549 lung cancer cells. | ||
2.3.6.4 Apoptosis triggered by compound 6 in A549 lung cancer cells is mediated via activating the caspase pathway. As the results showed, apoptosis is induced subsequent to G0/G1 cell cycle arrest in A549 lung cancer cells by compound 6. Apoptosis is a programmed cell death that results in DNA fragmentation and cell death.10,24,33 PARP is a key regulator implicated in DNA repair.59 Cleavage of PARP results in loss of its function to repair DNA and subsequent DNA fragmentation and apoptosis. Western blotting of PARP and cleaved PARP confirmed that compound 6 induced dose-dependent PARP cleavage (Fig. 10B). PARP cleavage is a pathway downstream of caspases that includes several members. Among them, caspase 8 is a key member that is involved after cleavage results in activation of other caspases either directly or through upregulation of BAX protein. The latter is a protein that induces opening of mitochondrial voltage-dependent anion channels, loss of mitochondrial potential and further activation of the caspase pathway and eventually PARP cleavage. As shown in Fig. 10, western blots confirmed dose-dependent upregulation of BAX and cleavage of caspase 8 upon treatment with compound 6. These results suggested that compound 6 induced apoptosis in A549 lung cancer cells through activation of the caspase pathway.
3. Conclusion
Three series of hybrids of acetylcholine derivatives with edelfosine and stPEPC were designed and synthesized. Evaluation of their anticancer activity against a panel of non-small cell lung cancer cell lines showed that compounds belonging to the 2-stearoxyphenyl series were more active than the corresponding compounds from the 3-stearoxyphenyl or 4-stearoxyphenyl series. Four compounds (6, 8, 9 and 10) were identified as potential inhibitors of non-small cell lung cancer. Further analysis of the spectrum of anticancer activity of compounds 6, 8, 9 and 10 against cells representing diverse cancer diseases disclosed their broad-spectrum anticancer activities. Evaluation of their potencies revealed the low micromolar potencies of compounds 6 and 8. Compound 6 was the most potent against non-small cell lung cancer, ovarian cancer, renal cancer, and prostate cancer. Meanwhile, compound 8 was the most potent against leukemia, colon cancer, CNS cancer, melanoma, and breast cancer. Further evaluation of the mechanism of action of compound 6 in A549 non-small cell lung cancer cells showed that it arrested cells in the G0/G1 phase of the cell cycle and induced apoptosis of cells. The triggered cell cycle arrest was mediated via disruption of the CDK4/6-Rb pathway, which controls cells' transition from G1 to S phase, while the induced apoptosis was mediated through activation of the caspase pathway and subsequent upregulation of BAX and cleavage of PARP. Collectively, these results showed that acetylcholine hybrids incorporating a 2-stearoxyphenyl moiety possessed a substantially enhanced anticancer activity and presented acetylcholine hybrids 6 and 8 as potential anticancer agents for possible further development.4. Experimental section
4.1. Chemistry
Synthesis of the compounds was conducted as detailed in the ESI.†4.2. Biological evaluation
Biological evaluations were conducted as detailed in the ESI.†Data availability
All data is included in the manuscript and/or the ESI.†Author contributions
Conceptualization: A. H. E. H., J. S., S. K. L., and Y. S. L.; data curation: S. K. L. and Y. S. L.; formal analysis: A. H. E. H., E. S. B., Y. J., S. M. E.-S., M. K., M. F. R., and T. S. I.; funding acquisition: J. S., S. K. L., and Y. S. L.; investigation: A. H. E. H., E. S. B., Y. J., C. W. O., M. K., M. F. R., T. S. I., J.-Y. C., and B. Y. P.; methodology: A. H. E. H., J. S., S. K. L., and Y. S. L.; project administration: Y. S. L.; resources: J. S., S. K. L., and Y. S. L.; validation: S. M. E.-S., M. K., and B. Y. P.; visualization: A. H. E. H., E. S. B., Y. J., S. M. E.-S.; all authors contributed to drafting and writing.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2021R1C1C1010044). National Cancer Institute, Bethesda, Maryland, USA is acknowledged for performing the NCI-60 human tumour cell line screen. The A549 cancer cell line was obtained from the American Type Culture Collection (Manassas, VA, USA). The MRC-5 cell line was provided by the Korean Cell Line Bank (Seoul, Korea).References
- WHO, World Health Statistics 2023. A visual summary, https://www.who.int/data/stories/world-health-statistics-2023-a-visual-summary/, (accessed April, 2024, 2024).
- M. Szumilak, A. Wiktorowska-Owczarek and A. Stanczak, Molecules, 2021, 26, 2601 CAS.
- D. E. Thurston and I. Pysz, Chemistry and pharmacology of anticancer drugs, CRC press, 2nd edn, 2021 Search PubMed.
- W. A. Zaki, S. M. El-Sayed, M. Alswah, A. El-Morsy, A. H. Bayoumi, A. S. Mayhoub, W. H. Moustafa, A. A. Awaji, E. J. Roh, A. H. E. Hassan and K. Mahmoud, Pharmaceuticals, 2023, 16, 1593 CAS.
- A. H. E. Hassan, C. Y. Wang, C. J. Lee, H. R. Jeon, Y. Choi, S. Moon, C. H. Lee, Y. J. Kim, S. B. Cho, K. Mahmoud, S. M. El-Sayed, S. K. Lee and Y. S. Lee, Pharmaceuticals, 2023, 16, 1597 CAS.
- A. H. E. Hassan, C. Y. Wang, H. J. Lee, S. J. Jung, Y. J. Kim, S. B. Cho, C. H. Lee, G. Ham, T. Oh, S. K. Lee and Y. S. Lee, Eur. J. Med. Chem., 2023, 256, 115421 CAS.
- A. K. Farag, A. H. E. Hassan, B. S. Ahn, K. D. Park and E. J. Roh, J. Enzyme Inhib. Med. Chem., 2020, 35, 311–324 CAS.
- R. Nussinov, C.-J. Tsai and H. Jang, Drug Resistance Updates, 2021, 59, 100796 CAS.
- A. H. E. Hassan, H. J. Kim, S. J. Jung, S.-Y. Jang, S. M. El-Sayed, K.-T. Lee and Y. S. Lee, Eur. J. Med. Chem., 2023, 258, 115566 CAS.
- J.-H. Won, K.-S. Chung, E.-Y. Park, J.-H. Lee, J.-H. Choi, L. A. Tapondjou, H.-J. Park, M. Nomura, A. H. E. Hassan and K.-T. Lee, Molecules, 2018, 23, 3306 Search PubMed.
- A. H. E. Hassan, K.-T. Lee and Y. S. Lee, Eur. J. Med. Chem., 2020, 187, 111965 CrossRef CAS PubMed.
- A. H. E. Hassan, H. J. Kim, M. S. Gee, J.-H. Park, H. R. Jeon, C. J. Lee, Y. Choi, S. Moon, D. Lee, J. K. Lee, K. D. Park and Y. S. Lee, J. Enzyme Inhib. Med. Chem., 2022, 37, 768–780 CrossRef CAS PubMed.
- A. Belal, M. S. Elballal, A. A. Al-Karmalawy, A. H. E. Hassan, E. J. Roh, M. M. Ghoneim, M. A. M. Ali, A. J. Obaidullah, J. M. Alotaibi, S. Shaaban and M. A. Elanany, Front. Chem., 2024, 12, 1425485 CrossRef CAS PubMed.
- A. H. E. Hassan, Y. I. Oh, C. H. Lee, Y. J. Kim, S. B. Cho, M. M. Alam, S.-E. Park, K.-S. Chung, K.-T. Lee and Y. S. Lee, J. Enzyme Inhib. Med. Chem., 2023, 38, 2217695 Search PubMed.
- A. H. E. Hassan, H. R. Park, Y. M. Yoon, H. I. Kim, S. Y. Yoo, K. W. Lee and Y. S. Lee, Bioorg. Chem., 2019, 84, 444–455 CAS.
- M. M. Alam, A. H. E. Hassan, Y. H. Kwon, H. J. Lee, N. Y. Kim, K. H. Min, S.-Y. Lee, D.-H. Kim and Y. S. Lee, Arch. Pharmacal Res., 2018, 41, 35–45 CAS.
- M. K. Pandey, Int. J. Mol. Sci., 2023, 24, 13223 Search PubMed.
- J. Park, J. Choi, D.-D. Kim, S. Lee, B. Lee, Y. Lee, S. Kim, S. Kwon, M. Noh, M.-O. Lee, Q.-V. Le and Y.-K. Oh, Biomol. Ther., 2021, 29, 465–482 CAS.
- S. Lv, J. Yang, J. Lin, X. Huang, H. Zhao, C. Zhao and L. Yang, Eur. Respir. Rev., 2024, 33, 230145 Search PubMed.
- M. Teng, J. Jiang, Z. He, N. P. Kwiatkowski, K. A. Donovan, C. E. Mills, C. Victor, J. M. Hatcher, E. S. Fischer, P. K. Sorger, T. Zhang and N. S. Gray, Angew. Chem., Int. Ed., 2020, 59, 13865–13870 CAS.
- M. Lim, T. D. Cong, L. M. Orr, E. S. Toriki, A. C. Kile, J. W. Papatzimas, E. Lee, Y. Lin and D. K. Nomura, ACS Cent. Sci., 2024, 10, 1318–1331 CAS.
- P. Corsino, N. Horenstein, D. Ostrov, T. Rowe, M. Law, A. Barrett, G. Aslanidi, W. D. Cress and B. Law, J. Biol. Chem., 2009, 284, 29945–29955 CAS.
- A. H. E. Hassan, C. Y. Wang, T. Oh, G. Ham, S. K. Lee and Y. S. Lee, Bioorg. Chem., 2024, 145, 107178 CAS.
- J. Y. Hong, K.-S. Chung, J.-S. Shin, J.-H. Lee, H.-S. Gil, H.-H. Lee, E. Choi, J.-H. Choi, A. H. E. Hassan, Y. S. Lee and K.-T. Lee, Cancers, 2019, 11, 1927 CAS.
- A. K. Farag, A. H. E. Hassan, K.-S. Chung, J.-H. Lee, H.-S. Gil, K.-T. Lee and E. J. Roh, Bioorg. Chem., 2020, 103, 104121 CAS.
- M. M. Alam, A. H. E. Hassan, K. W. Lee, M. C. Cho, J. S. Yang, J. Song, K. H. Min, J. Hong, D.-H. Kim and Y. S. Lee, Bioorg. Chem., 2019, 84, 51–62 CrossRef CAS PubMed.
- A. H. E. Hassan, E. Choi, Y. M. Yoon, K. W. Lee, S. Y. Yoo, M. C. Cho, J. S. Yang, H. I. Kim, J. Y. Hong, J.-S. Shin, K.-S. Chung, J.-H. Lee, K.-T. Lee and Y. S. Lee, Eur. J. Med. Chem., 2019, 161, 559–580 CrossRef CAS PubMed.
- A. H. E. Hassan, K. Mahmoud, T.-N. Phan, M. A. Shaldam, C. H. Lee, Y. J. Kim, S. B. Cho, W. A. Bayoumi, S. M. El-Sayed, Y. Choi, S. Moon, J. H. No and Y. S. Lee, Eur. J. Med. Chem., 2023, 250, 115211 CrossRef CAS PubMed.
- A. H. E. Hassan, W. A. Bayoumi, S. M. El-Sayed, T.-N. Phan, T. Oh, G. Ham, K. Mahmoud, J. H. No and Y. S. Lee, Pharmaceuticals, 2023, 16, 1594 CrossRef CAS PubMed.
- A. H. E. Hassan, W. A. Bayoumi, S. M. El-Sayed, T.-N. Phan, Y. J. Kim, C. H. Lee, S. B. Cho, T. Oh, G. Ham, K. Mahmoud, J. H. No and Y. S. Lee, J. Enzyme Inhib. Med. Chem., 2023, 38, 2229071 CrossRef PubMed.
- A. K. Singh, A. Kumar, H. Singh, P. Sonawane, H. Paliwal, S. Thareja, P. Pathak, M. Grishina, M. Jaremko, A.-H. Emwas, J. P. Yadav, A. Verma, H. Khalilullah and P. Kumar, Pharmaceuticals, 2022, 15, 1071 CrossRef CAS PubMed.
- S. Choudhary, P. K. Singh, H. Verma, H. Singh and O. Silakari, Eur. J. Med. Chem., 2018, 151, 62–97 CrossRef CAS PubMed.
- H.-S. Gil, J.-H. Lee, A. K. Farag, A. H. E. Hassan, K.-S. Chung, J.-H. Choi, E.-J. Roh and K.-T. Lee, Cancers, 2021, 13, 5849 CrossRef CAS PubMed.
- J.-H. Lee, H.-H. Lee, K. D. Ryu, M. Kim, D. Ko, K.-S. Chung, A. H. E. Hassan, S. H. Lee, J. Y. Lee and K.-T. Lee, J. Clin. Med., 2020, 9, 704 CrossRef CAS PubMed.
- K. C. Thandra, A. Barsouk, K. Saginala, J. S. Aluru and A. Barsouk, Contemp. Oncol., 2021, 25, 45–52 CAS.
- H. Xu, Z. Shen, J. Xiao, Y. Yang, W. Huang, Z. Zhou, J. Shen, Y. Zhu, X.-Y. Liu and L. Chu, BMC Cancer, 2014, 14, 668 CrossRef PubMed.
- J. R. Friedman, S. D. Richbart, J. C. Merritt, K. C. Brown, N. A. Nolan, A. T. Akers, J. K. Lau, Z. R. Robateau, S. L. Miles and P. Dasgupta, Pharmacol. Ther., 2019, 194, 222–254 CrossRef CAS.
- M. Nie, N. Chen, H. Pang, T. Jiang, W. Jiang, P. Tian, L. Yao, Y. Chen, R. J. DeBerardinis, W. Li, Q. Yu, C. Zhou and Z. Hu, J. Clin. Invest., 2022, 132, e160152 CrossRef CAS PubMed.
- H.-J. Xi, R.-P. Wu, J.-J. Liu, L.-J. Zhang and Z.-S. Li, Thorac. Cancer, 2015, 6, 390–398 CrossRef CAS PubMed.
- A. L. Aronowitz, S. R. Ali, M. D. E. Glaun and M. Amit, Adv. Biol., 2022, 6, 2200053 CrossRef CAS PubMed.
- H. Yu, H. Xia, Q. Tang, H. Xu, G. Wei, Y. Chen, X. Dai, Q. Gong and F. Bi, Sci. Rep., 2017, 7, 40802 CAS.
- I. C. Vaaland Holmgard, A. González-Bakker, E. Poeta, A. Puerta, M. X. Fernandes, B. Monti, J. G. Fernández-Bolaños, J. M. Padrón, Ó. López and E. Lindbäck, Org. Biomol. Chem., 2024, 22, 3425–3438 CAS.
- M. Hudáčová, S. Hamuľaková, E. Konkoľová, R. Jendželovský, J. Vargová, J. Ševc, P. Fedoročko, O. Soukup, J. Janočková, V. Ihnatova, T. Kučera, P. Bzonek, N. Novakova, D. Jun, L. Junova, J. Korábečný, K. Kuča and M. Kožurková, Int. J. Mol. Sci., 2021, 22, 3830 Search PubMed.
- U. Karmacharya, P. Chaudhary, D. Lim, S. Dahal, B. P. Awasthi, H. D. Park, J.-A. Kim and B.-S. Jeong, Bioorg. Chem., 2021, 110, 104805 CrossRef CAS PubMed.
- Z. Sun, M. Zhangsun, S. Dong, Y. Liu, J. Qian, D. Zhangsun and S. Luo, Mar. Drugs, 2020, 18, 61 CrossRef CAS PubMed.
- A. Saeed, P. A. Mahesar, S. Zaib, M. S. Khan, A. Matin, M. Shahid and J. Iqbal, Eur. J. Med. Chem., 2014, 78, 43–53 CAS.
- P. Liu, W. Zhu, C. Chen, B. Yan, L. Zhu, X. Chen and C. Peng, Life Sci., 2020, 247, 117443 CrossRef CAS.
- L. Zhang, X. Liu, Y. Liu, F. Yan, Y. Zeng, Y. Song, H. Fang, D. Song and X. Wang, Clin. Transl. Med., 2023, 13, e1180 CrossRef CAS PubMed.
- D. Mohamad Ali, K. Hogeveen, R.-M. Orhant, T. L. G. de Kerangal, F. Ergan, L. Ulmann and G. Pencreac'h, Nutrients, 2023, 15, 2137 CrossRef CAS PubMed.
- N. P. Tambellini, V. Zaremberg, S. Krishnaiah, R. J. Turner and A. M. Weljie, J. Proteome Res., 2017, 16, 3741–3752 CrossRef CAS PubMed.
- E. L. H. Dakir, C. Gajate and F. Mollinedo, Biomed. Pharmacother., 2023, 167, 115436 CrossRef CAS.
- F. T. Sarah, P. R. Cecilia, J. S. C. Cícero, N. P. Thais, A. D. A. Ricardo, I. M. Lisley, D. J. Salomão, N. R. Rodrigo, S. F. Emer, A. M. B. José and K. F. Adilson, Anti-Cancer Agents Med. Chem., 2018, 18, 865–874 CrossRef PubMed.
- S.-E. Park, K.-S. Chung, S.-W. Heo, S.-Y. Kim, J.-H. Lee, A. H. E. Hassan, Y. S. Lee, J. Y. Lee and K.-T. Lee, Life Sci., 2023, 334, 122227 CrossRef CAS PubMed.
- H. Liu, Z. Li, S. Huo, Q. Wei and L. Ge, Mol. Clin. Oncol., 2020, 12, 9–14 CAS.
- M. Kluska, A. W. Piastowska-Ciesielska and P. Tokarz, Curr. Issues Mol. Biol., 2023, 45, 6325–6338 CrossRef CAS PubMed.
- A. Sun, L. Bagella, S. Tutton, G. Romano and A. Giordano, J. Cell. Biochem., 2007, 102, 1400–1404 CrossRef CAS PubMed.
- N. Sobhani, A. D'Angelo, M. Pittacolo, G. Roviello, A. Miccoli, S. P. Corona, O. Bernocchi, D. Generali and T. Otto, Cell, 2019, 8, 321 CrossRef CAS PubMed.
- C. Berthet, K. D. Klarmann, M. B. Hilton, H. C. Suh, J. R. Keller, H. Kiyokawa and P. Kaldis, Dev. Cell, 2006, 10, 563–573 CrossRef CAS PubMed.
- A. Ray Chaudhuri and A. Nussenzweig, Nat. Rev. Mol. Cell Biol., 2017, 18, 610–621 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md01007h |
| This journal is © The Royal Society of Chemistry 2025 |