
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnticancer potential of copper(I) complexes based on isopropyl ester derivatives of bis(pyrazol-1-yl)acetate ligands†
Maura
Pellei
 a,
Carlo
Santini
*a,
Miriam
Caviglia
a,
Jo'
Del Gobbo
a,
Chiara
Battocchio
b,
Carlo
Meneghini
b,
Simone
Amatori
b,
Chiara
Donati
c,
Eleonora
Zampieri
c,
Valentina
Gandin
*c and
Cristina
Marzano
c
a,
Carlo
Santini
*a,
Miriam
Caviglia
a,
Jo'
Del Gobbo
a,
Chiara
Battocchio
b,
Carlo
Meneghini
b,
Simone
Amatori
b,
Chiara
Donati
c,
Eleonora
Zampieri
c,
Valentina
Gandin
*c and
Cristina
Marzano
c
aSchool of Science and Technology, Chemistry Division, University of Camerino, Via Madonna delle Carceri (ChIP), 62032 Camerino, Italy. E-mail: carlo.santini@unicam.it
bDepartment of Science, Roma Tre University, Via della Vasca Navale 79, 00146 Roma, Italy
cDepartment of Pharmaceutical and Pharmacological Sciences, University of Padova, Via Marzolo 5, 35131 Padova, Italy. E-mail: valentina.gandin@unipd.it
First published on 13th November 2024
Abstract
In this paper, the isopropyl ester derivatives LOiPr and L2OiPr of bis(pyrazol-1-yl)acetic acid and bis(3,5-dimethyl-pyrazol-1-yl)acetic acid were used as chelators for the preparation of new Cu(I) phosphane complexes 1–4. They were synthesized by the reaction of [Cu(CH3CN)4]PF6 and triphenylphosphine or 1,3,5-triaza-7-phosphaadamantane with LOiPr and L2OiPr ligands, in acetonitrile or acetonitrile/methanol solution. The authenticity of the compounds was confirmed by CHN analysis, 1H-, 13C- and 31P-NMR, FT-IR spectroscopy, and electrospray ionization mass spectrometry (ESI-MS). Furthermore, the electronic and molecular structures of the selected Cu(I) coordination compound 3 were investigated by synchrotron radiation-induced X-ray photoelectron spectroscopy (SR-XPS), and the local structure around the copper ion site was studied combining X-ray absorption fine structure (XAFS) spectroscopy techniques and DFT modelling. Triphenylphosphine as a coligand confers to [Cu(LOiPr)(PPh3)]PF6 (1) and [Cu(L2OiPr)(PPh3)]PF6 (3) a significant antitumor activity in 3D spheroidal models of human colon cancer cells. Investigations focused on the mechanism of action evidenced protein disulfide-isomerase (PDI) as an innovative molecular target for this class of phosphane copper(I) complexes. By hampering PDI activity, copper(I) complexes were able to cause an imbalance in cancer cell redox homeostasis thus leading to cancer cell death – a non-apoptotic programmed cell death.
1. Introduction
Metal-based chemotherapy is a firmly established anticancer approach and copper-based complexes have demonstrated intriguing anti-tumor and anti-metastatic properties across various solid tumors.1,2 They operate through distinct mechanisms, significantly divergent from platinum-based drugs.3,4Copper is a trace element in the human body,5 playing a role in a broad array of biochemical processes.6,7 Intracellular copper levels are tightly regulated through active homeostatic mechanisms to prevent the harmful accumulation of free copper within cells.7,8 Disrupted copper homeostasis has been associated with cancer development.9,10 Copper chelators have been developed for suppressing cancer proliferation and metastasis by decreasing the intracellular copper concentration.11 Conversely, copper-ionophores, which transport extracellular copper ions into cells, have emerged as a distinct strategy in anticancer research and exceeded copper chelators in clinical studies.12 Even if recent research showed that copper is crucial for tumor angiogenesis and tends to accumulate in tumor tissues,13–16 the mechanisms of copper-induced cytotoxicity remain elusive.17–19 On the other hand, in recent years, the discovery of cuproptosis,20 a novel form of regulated cell death dependent on copper,21,22 has garnered attention, with many researchers investigating the pivotal link between cuproptosis and cancer.23 Based on these findings, the use of copper complexes has been highlighted for their therapeutic potential in treating various cancers,24,25 as researchers aim to develop more potent, clinically effective, and less toxic metal-based antiproliferative drugs with distinct anti-tumor properties.6,15,24,26–29 Indeed, copper complexes represent promising alternatives to platinum-based compounds, offering potential solutions to challenges such as inherent or acquired resistance and dose-limiting toxicity.30–32
Our investigations into anticancer Cu(I) and Cu(II) complexes, employing various classes of ligands with differing lipophilicity, have demonstrated the enhancement of bioavailability and cellular uptake of these complexes, leading to the discovery of novel modes of action.15,29,33–39 Recently, we reported on copper complexes exhibiting significant cytotoxic effects against various human tumor cell lines. These complexes were obtained from ligands prepared by conjugating the terminal COOH group of the bifunctional species bis(pyrazol-1-yl)acetic acid (LH) and bis(3,5-dimethyl-pyrazol-1-yl)acetic acid (L2H) with nitroimidazole, glucosamine, the non-competitive NMDA receptor antagonist and the antineoplastic drug lonidamine, demonstrating cytotoxic activity across a range of human tumor cell lines.40–45
To enhance understanding of the coordination chemistry and biological implications of copper coordination compounds in cancer, this study aimed to investigate the potential of small ester derivatives of bis(pyrazolyl)acetates species to form Cu(I) complexes with potential applications in anticancer therapy. Bis(pyrazol-1-yl)acetic acid (HC(pz)2COOH) and bis(3,5-dimethyl-pyrazol-1-yl)acetic acid (HC(pzMe2)2COOH) were converted into the isopropyl ester derivatives LOiPr and L2OiPr,46 respectively and they were used for the preparation of Cu(I) phosphane complexes 1–4 (Fig. 1). In particular, the lipophilic triphenylphosphine (PPh3) and the hydrophilic 1,3,5-triaza-7-phosphaadamantane (PTA), able to stabilize copper in +1 oxidation state, were selected as coligands in order to confer a different hydrophilic–lipophilic balance to the corresponding complexes.
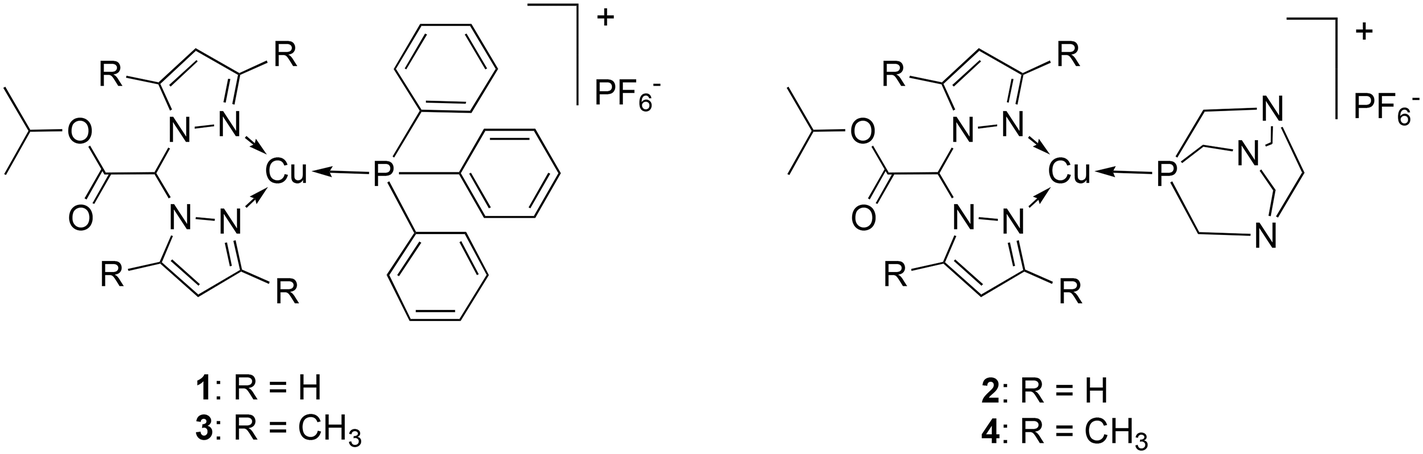 | ||
| Fig. 1 Chemical structures of complexes 1–4. | ||
The new Cu(I) complexes 1–4 and the corresponding uncoordinated ligands (LOiPr and L2OiPr) were investigated for their cytotoxic activity on a panel of human cancer cell lines, derived from different solid tumors, by means of both 2D and 3D cell viability studies. Mechanistic studies were performed to analyse the main molecular and cellular determinants accounting for their antitumor potential. Moreover, microscopy analysis allowed the assessment of the ability of selected Cu(I) complexes to provoke cancer cell death by means of an ER-stress driven cancer cell death alternative to apoptosis, such as paraptosis.
2. Results and discussion
2.1. Synthesis and characterization
Ligands LOiPr and L2OiPr were synthesized using the precursors bis(pyrazol-1-yl)acetic acid (LH) and bis(3,5-dimethyl-pyrazol-1-yl)acetic acid (L2H), respectively, and iPrOH as starting materials, according to the procedure in the literature.46 Ligands LOiPr and L2OiPr are soluble in n-hexane, CHCl3, EtOAc, and CH3CN. L2OiPr is also soluble in CH3OH, diethyl ether, THF and acetone.The triphenylphosphine Cu(I) complexes [Cu(LOiPr)(PPh3)]PF6 (1) and [Cu(L2OiPr)(PPh3)]PF6 (3) were prepared from the reaction of PPh3, [Cu(CH3CN)4]PF6, and the ligands LOiPr and L2OiPr respectively, following two steps in acetonitrile or acetonitrile/methanol solution. Analogously, the 1,3,5-triaza-7-phosphaadamantane Cu(I) complexes [Cu(LOiPr)(PTA)]PF6 (2) and [Cu(L2OiPr)(PTA)]PF6 (4) were prepared from the reaction of PTA, [Cu(CH3CN)4]PF6 and the related ligands, in acetonitrile/methanol solution. The same compounds have been obtained using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio between the phosphanes and the metal acceptors.
:1 stoichiometric ratio between the phosphanes and the metal acceptors.
The IR spectra obtained on solid samples of the Cu(I) complexes showed all the expected bands for the chelating ligand and the phosphane coligand: weak absorption bands due to the C–H stretching were observed in the range 2883–3242 cm−1, while the absorption bands due to the asymmetric stretching of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups were at 1749–1752 cm−1, in the typical range of the ester groups and they did not significantly vary with respect to the free ligands LOiPr and L2OiPr (1747 and 1748 cm−1, respectively).47–50 In a lower frequency region, the complexes showed a broad strong band at 832–837 cm−1 due to the stretching vibrations of the PF6− anion. The 1H-NMR spectra of Cu(I) complexes 1–3, recorded in CD3CN solution at room temperature, showed a single set of resonance frequencies for the pyrazole rings, indicating that the pyrazole protons were equivalent, with a slight shift due to the coordination to the metal center. The triphenylphosphine and 1,3,5-triaza-7-phosphaadamantane showed a characteristic series of peaks at δ 7.37–7.57 and 4.08–4.60 ppm, respectively, with an integration which confirmed the 1:1 stoichiometric ratio between the ligand and the phosphane coligand. The 1H-NMR spectrum of 4, recorded in DMSO solution, showed broad resonance frequencies for the pyrazole rings, together with the characteristic series of peaks of PTA at δ 4.08–4.62 ppm, with an integration which confirmed the 1:1 stoichiometric ratio ligand:PTA. In the 13C{1H}-NMR spectra of complexes 1–4, recorded in CD3CN, the CO signals are in the range 163.5–164.2 ppm, and the related peaks are at 164.5 and 164.3 ppm in the spectra of the free ligands LOiPr and L2OiPr. The room temperature 31P{1H}-NMR spectra of Cu(I) complexes 1–4, recorded in CD3CN solution, gave singlets downfield shifted with respect to the value of the free phosphanes PPh3 and PTA (δ = −4.85 and −102.07 ppm, respectively). In particular, the spectra of [Cu(LOiPr)(PPh3)]PF6 (1) and [Cu(L2OiPr)(PPh3)]PF6 (3) exhibited broad singlets centered at δ −1.16 and −0.48 ppm, respectively, while the spectra of [Cu(LOiPr)(PTA)]PF6 (2) and [Cu(L2OiPr)(PTA)]PF6 (4) gave broad singlets centered at δ −92.10 and −96.10 ppm, respectively. The signals of complexes 1–4 are in the same range of chemical shifts reported for analogous copper(I) monophosphane species.34,48 In all the spectra, the characteristic septets centered between −144.60 and −144.63 ppm were due to the PF6− counterion. The low temperature 31P-NMR spectra of complexes 1–4 confirm the presence of molecular species in the undissociated form.
O groups were at 1749–1752 cm−1, in the typical range of the ester groups and they did not significantly vary with respect to the free ligands LOiPr and L2OiPr (1747 and 1748 cm−1, respectively).47–50 In a lower frequency region, the complexes showed a broad strong band at 832–837 cm−1 due to the stretching vibrations of the PF6− anion. The 1H-NMR spectra of Cu(I) complexes 1–3, recorded in CD3CN solution at room temperature, showed a single set of resonance frequencies for the pyrazole rings, indicating that the pyrazole protons were equivalent, with a slight shift due to the coordination to the metal center. The triphenylphosphine and 1,3,5-triaza-7-phosphaadamantane showed a characteristic series of peaks at δ 7.37–7.57 and 4.08–4.60 ppm, respectively, with an integration which confirmed the 1:1 stoichiometric ratio between the ligand and the phosphane coligand. The 1H-NMR spectrum of 4, recorded in DMSO solution, showed broad resonance frequencies for the pyrazole rings, together with the characteristic series of peaks of PTA at δ 4.08–4.62 ppm, with an integration which confirmed the 1:1 stoichiometric ratio ligand:PTA. In the 13C{1H}-NMR spectra of complexes 1–4, recorded in CD3CN, the CO signals are in the range 163.5–164.2 ppm, and the related peaks are at 164.5 and 164.3 ppm in the spectra of the free ligands LOiPr and L2OiPr. The room temperature 31P{1H}-NMR spectra of Cu(I) complexes 1–4, recorded in CD3CN solution, gave singlets downfield shifted with respect to the value of the free phosphanes PPh3 and PTA (δ = −4.85 and −102.07 ppm, respectively). In particular, the spectra of [Cu(LOiPr)(PPh3)]PF6 (1) and [Cu(L2OiPr)(PPh3)]PF6 (3) exhibited broad singlets centered at δ −1.16 and −0.48 ppm, respectively, while the spectra of [Cu(LOiPr)(PTA)]PF6 (2) and [Cu(L2OiPr)(PTA)]PF6 (4) gave broad singlets centered at δ −92.10 and −96.10 ppm, respectively. The signals of complexes 1–4 are in the same range of chemical shifts reported for analogous copper(I) monophosphane species.34,48 In all the spectra, the characteristic septets centered between −144.60 and −144.63 ppm were due to the PF6− counterion. The low temperature 31P-NMR spectra of complexes 1–4 confirm the presence of molecular species in the undissociated form.
The ESI-MS study was performed by dissolving complexes 1–4 in CH3CN, recording the spectra in ion-positive and ion-negative mode. In the ESI-MS(+) spectra of complexes 1 and 2 the major peaks at m/z 559 and 454, due to the [Cu(LOiPr)(PPh3)]+ and [Cu(LOiPr)(PTA)]+ species, respectively, confirm the presence and the stability of the complexes. In addition, fragments at m/z 366, 587 and 261 are relative to the [Cu(PPh3)(CH3CN)]+, [Cu(PPh3)2]+ and [Cu(PTA)(CH3CN)]+ species, respectively, with the coordinative bond between copper and phosphanes. Analogously in the ESI-MS(+) spectra of complexes 3 and 4 peaks at m/z 615 and 510, due to the [Cu(L2OiPr)(PPh3)]+ and [Cu(L2OiPr)(PTA)]+ species, respectively, confirm the presence and the stability of the complexes with the L2OiPr ligand, too. In addition, fragments due to the dissociation of the phosphanes ([Cu(L2OiPr)(CH3CN)]+ and [Cu(L2OiPr)]+) or the scorpionate ligand ([Cu(PPh3)(CH3CN)]+, [Cu(PPh3)2]+ and [Cu(PTA)(CH3CN)]+) are present. In the negative-ion spectra of 1–4, [PF6]− was observed as the major peak at m/z 145.
2.2. Molecular and electronic structures of [Cu(L2OiPr)(PPh3)]PF6 (3): XPS investigation
The molecular structure, electronic structure, and local geometry around the Cu(I) ion were assessed by means of XPS and XAS spectroscopy for complex 3, considered representative for all synthesized Cu(I) coordination compounds.X-ray photoelectron spectroscopy (XPS) allowed both electronic and molecular structures of 3 to be probed. XPS spectra were collected at C1s, N1s, O1s, Cl2p, Cu2p, P2p and F1s core levels; the detailed data analysis results (binding energy (BE), full width half maximum (FWHM), and assignments), leading information about the molecular structure of the coordination compound and the stability of the ligand molecular structure upon coordination to copper, are summarized in Table S1 in the ESI.†
The C1s signal can be resolved by curve fitting analysis into several components corresponding to the different C atoms in the proposed molecular structure; as expected, the C1s signal, reported in Fig. 2a, has four components: aromatic and aliphatic C–C carbons (BE = 284.7 eV), superimposed to the C–P signal (component C1); C–N carbons of the pyrazole-like rings (BE = 286.0 eV, namely C2), C–O (BE = 287.6 eV, component C3), and COOR (BE = 288.6 eV, C4).51
 | ||
| Fig. 2 XPS spectra collected on complex 3 at C1s (a), N1s (b), P2p (c) and Cu2p (d) core levels. | ||
The coordination compound stability and molecular structure consistency with the hypothesised one is further confirmed by the N1s spectrum (Fig. 2b), showing a single component for the N atoms coordinating Cu at about 400 eV BE, as expected for the symmetrized pyrrole N in coordination compounds.40,48,50,52 What is more, the N/Cu atomic ratio is in extremely good agreement with the value calculated for the proposed theoretical structure (N/Cu exp. = 4.4, th. 4). P2p (Fig. 2c) and F1s signals also support the proposed chemical structure: a P2p3/2 component at 132.2 eV BE is attributed to P atoms of PPh3, and its stability and reproducibility confirm that no oxidation of phosphane takes place. The higher BE P2p signal (P2p3/2 BE = 136 eV) is due to PF6− ions; coherently, the F1s signal is observed around 686 eV BE.51 Finally, the Cu2p spectrum is reported in Fig. 2d. As expected, the very intense component at lower BE values is indicative of Cu(I) ions (Cu2p3/2 BE = 932.8 eV); however, a signal of small intensity is also observed at higher BE values (Cu2p3/2 BE = 935.3 eV), and attributed to about 10% of Cu(II) ions and compatible with Cu(OH)2, probably arising from sample degradation due to the preparation procedure for XPS analysis.53,54
2.3. Local structure around Cu ions: XAFS studies
The analysis of Cu K edge X-ray absorption spectra measured on complex 3 provides further details about the oxidation state, coordination chemistry and local atomic structure of the copper site. The near edge features (XANES) of the absorption spectrum provide information about the absorber valence state and local coordination geometry.55 The Cu K-edge XANES spectrum (Fig. 3a) of complex 3 exhibits distinct features, consistent with the Cu(I) valence state.56 In accordance with the reference, the Cu(I) site of complex 3 is characterized by a peak in the pre-edge around 8983 eV, assigned to the 1s–4s or 1s–4p transitions, whose position and intensity are directly correlated to the coordination number of Cu(I);56 in our case (peak height of approximately 0.6 at 8983 eV) they indicate a coordination number of 3, consistent with the structural model (see Fig. 4) that shows a trigonal planar configuration. Further details can be derived from the analysis of the extended (EXAFS) region of the spectrum. | ||
| Fig. 3 a) Normalized XANES spectrum of Cu K edge of 3 (blue), Cu2O (red) and Cu foil (black) as reference compounds. b) k2-Weighted experimental (black) and fit (red) curves for 3. c) Moduli of Fourier transforms (FT) of k2χexp(k) of experimental (blue) and best fit (black) curves; the FT curves have not been corrected for the phase shift effect, resulting in a main peak shift of approximately −0.5 Å with respect to the actual distance. | ||
 | ||
| Fig. 4 Optimized DFT model describing the local atomic structure around the Cu absorber for complex 3. | ||
To quantitatively analyse the Cu EXAFS data of 3, the k2-weighted experimental spectrum k2χexp was fitted to the theoretical curve k2χth in the 3–17 Å−1k range, the theoretical signals defined as a sum of partial contributions χth = 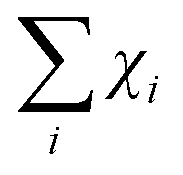 which represent the relevant single (SS) and multiple (MS) scattering photoelectron paths from the neighbouring atoms around the absorber. According to the relevant data, scattering paths were obtained from the DFT atomic cluster models;48 the χi were calculated according to the standard EXAFS formula,57,58 using the theoretical photoelectron amplitude and phase functions calculated using the FEFF8.4 program.59
which represent the relevant single (SS) and multiple (MS) scattering photoelectron paths from the neighbouring atoms around the absorber. According to the relevant data, scattering paths were obtained from the DFT atomic cluster models;48 the χi were calculated according to the standard EXAFS formula,57,58 using the theoretical photoelectron amplitude and phase functions calculated using the FEFF8.4 program.59
From the DFT model the main single and multiple contributions for 3 were identified and grouped together in shells having similar path lengths and types of neighbours. The preliminary trial and error procedure allowed the suitable set of χi to be defined; the coordination numbers were constrained to the model structure, while coordination distances (R) and mean square relative displacement (MSRD, σ2) factors were refined (Table 1). This approach allowed for a precise description of the Cu coordination shells in the complex till around 5 Å (Fig. 2b and c). More in detail, the EXAFS fitting required 5 SS and 1 MS contribution (see Table 1).
| Complex 3 | ||||
|---|---|---|---|---|
| Shell | N c | R (Å) | σ 2 [Å2] × 10−2 | Path |
| 1 | 2 | 2.03(1) | 0.39(3) | Cu–N1 |
| 2 | 1 | 2.16(2) | 0.88(5) | Cu–P |
| 3 | 3 | 3.02(2) | 1.3(2) | Cu–N2/C2 |
| 4 | 4 | 3.44(2) | 1.7(2) | Cu–C3/C4 |
| 5 | 5 | 4.00(3) | 0.8(3) | Cu–C5 |
| 6 | 12 | 4.27(3) | 3.5(5) | Cu–N1/N2–C5 |
2.4. Stability studies
The stability of the new complexes in 0.5% DMSO/saline solution was also evaluated by using 1H-NMR. Changes observed in the 1H-NMR spectra of the complexes over 72 h were insignificant or only minimal, thus indicating that these complexes are stable under physiological conditions (Fig. S35 in the ESI†).2.5. Biological studies
The Cu(I) complexes and the corresponding uncoordinated ligands were evaluated for their cytotoxic activity in an inhouse human cancer cell line panel including various cancer cell lines representative of different solid tumors. The cytotoxicity of PPh3 and PTA ligands has already been published.34 In particular, the inhouse cancer cell panel included examples of human ovarian (2008), cervical (A431), breast (MCF-7), colon (LoVo), pancreatic (PSN-1), thyroid (BCPAP), and small cell lung (U-1285) cancer cells as well as human colon cells endowed with resistance to oxaliplatin (LoVo OXP). The cytotoxicity parameters, expressed in terms of IC50 values obtained after 72 h of exposure to the MTT assay, are reported in Table 2. For comparison, the cytotoxicity of the reference metal-based chemotherapeutic drug cisplatin was assessed under the same experimental conditions.| IC50 (μM) ± S.D. | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 2008 | A431 | MCF-7 | U-1285 | PSN-1 | B-CPAP | LoVo | LoVo OXP (R.F.) | HEK-293 (S.I.) | |
| [Cu(LOiPr)(PPh3)]PF6 (1) | 9.3 ± 2.3 | 12.1 ± 1.1 | 6.7 ± 0.9 | 6.8 ± 0.8 | 17.3 ± 0.6 | 11.4 ± 0.1 | 6.6 ± 0.3 | 8.8 ± 1.3 (1.3) | 10.6 ± 0.26 (1.1) |
| [Cu(LOiPr)(PTA)]PF6 (2) | 24.5 ± 2.2 | 32.3 ± 1.1 | 20.7 ± 1.4 | 21.3 ± 2.3 | 30.1 ± 1.1 | 46.5 ± 2.2 | 11.0 ± 0.3 | 21.8 ± 3.4 (1.9) | >50 (>1.9) |
| [Cu(L2OiPr)(PPh3)]PF6 (3) | 9.7 ± 0.6 | 9.2 ± 0.6 | 7.2 ± 0.9 | 7.3 ± 1.1 | 21.9 ± 1.9 | 8.8 ± 0.9 | 6.5 ± 0.5 | 10.5 ± 0.3 (1.6) | 7.2 ± 0.29 (0.7) |
| [Cu(L2OiPr)(PTA)]PF6 (4) | 6.4 ± 1.9 | 11.3 ± 1.8 | 14.0 ± 1.7 | 11.9 ± 1.9 | 18.2 ± 0.8 | 14.3 ± 0.9 | 7.8 ± 1.5 | 14.9 ± 1.0 (1.9) | >50 (>4.0) |
| LOiPr | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | — |
| L2OiPr | >50 | >50 | >50 | >50 | >50 | >50 | >50 | >50 | — |
| Cisplatin | 2.2 ± 1.2 | 2.1 ± 0.6 | 13.9 ± 1.3 | 2.1 ± 0.8 | 12.1 ± 2.8 | 18.5 ± 4.1 | 7.6 ± 1.5 | — | 21.6 ± 3.5 (2.6) |
| Oxaliplatin | — | — | — | — | — | — | 1.1 ± 0.6 | 14.3 ± 2.1 (13) | — |
Both LOiPr and L2OiPr ligands proved to be completely ineffective against all tested cancer cell lines, whereas all tested Cu(I) complexes elicited a cytotoxic effect, although at different extents. On average, the PPh3 derivatives were more effective than complexes bearing the PTA moiety.
Both the triphenylphosphine derivatives 1 and 3 showed a similar pattern of response across the different human cancer cell lines, with average IC50 values of 10.0 and 10.1 μM, respectively. Noteworthily, against MCF-7 breast and B-CPAP thyroid cancer cells, both expressing estrogen receptors, 1 and 3 were about 2-fold more active than the reference metallodrug cisplatin. Conversely, in the case of PTA bearing complexes, the L2OiPr derivative 4 was 2-fold more effective than complex 2 bearing the LOiPr ligand (average IC50 values of 26.0 and 12.3 μM for 2 and 4 respectively); however, they were both less potent than cisplatin.
Interestingly, all the Cu(I) complexes showed a similar cytotoxicity profile against human colon cancer cells sensitive (LoVo) and resistant (LoVo OXP) to oxaliplatin, the platinum drug used as first-line chemotherapy in colorectal cancer. Actually, the resistant factors (R.F.; defined as IC50 resistant/parent line) calculated for all the Cu(I) complexes and reported in Table 2 were significantly lower than that calculated for oxaliplatin, thus attesting their ability to overcome oxaliplatin resistance. Several mechanisms underlying oxaliplatin resistance have been reported in previous studies in which some genes were found to be associated with oxaliplatin resistance and some were found to predict the treatment response and prognoses of colorectal cancer (CRC) patients.60–62 However, due to the molecular heterogeneity of CRC, the mechanisms underlying oxaliplatin resistance remain to be elucidated. Based on our previous experience on phosphane copper(I) complexes, the results obtained on the LoVo/LoVo OXP cell pair clearly propose for 1–4 a distinct mechanism of action from conventional Pt(II) drugs.
The antiproliferative activity of the new Cu(I) phosphane complexes was also evaluated on non-tumor human HEK-293 embryonic kidney cells (Table 2) with the aim to preliminarily assess their cell selectivity. As highlighted by the selectivity index values (SI = the quotient of the average IC50 toward normal cells divided by the average IC50 for the malignant cells), only [Cu(L2OiPr)(PTA)]PF6 (4) proved to be much more effective in affecting cancer cells with respect to non-cancer cells, whereas for all the other Cu(I) complexes no preferential cytotoxicity against cancer cells could be detected.
The in vitro antitumor activity of the newly developed Cu(I) derivatives was also assayed in a 3D cell culture model of LoVo colon cancer cells. Even if the 2D cell cultures are the most employed assays for in vitro drug screening due to the low cost, simplicity and reliability, 2D methods are unable to properly reproduce the characteristics of in vivo solid tumors. In contrast, 3D cell cultures, being more efficient in closely mimicking the heterogeneity and complexity of the tumor microenvironment, are more predictive for in vivo outcomes.63 LoVo spheroids were treated with the investigated compounds for 72 h, and cell viability was assessed by means of the acid phosphatase (APH) assay (Table 3).
| IC50 (μM) ± S.D. | |
|---|---|
| LoVo | |
| [Cu(LOiPr)(PPh3)]PF6 (1) | 20.4 ± 2.2 |
| [Cu(LOiPr)(PTA)]PF6 (2) | 156.2 ± 11.4 |
| [Cu(L2OiPr)(PPh3)]PF6 (3) | 16.6 ± 1.8 |
| [Cu(L2OiPr)(PTA)]PF6 (4) | 144.2 ± 7.8 |
| Cisplatin | 60.6 ± 6.3 |
The IC50 values obtained in this set of experiments were sensibly higher than those calculated in 2D monolayer cell cultures, due to the greater resistance to drug treatment generally showed by cells in 3D cultures after forming the spheroids. Nevertheless, the results clearly confirm that PPh3 complexes 1 and 3 were much more effective than PTA derivatives 2 and 4, and moreover, they were more potent than the reference drug cisplatin, thus confirming that human colorectal cancer cells are highly sensitive to copper complexes.15 In particular, compound 3 bearing the L2OiPr ligand was the most effective compound, with an IC50 value about 3.7 times lower than that calculated for cisplatin.
During the past three decades, several different molecular targets have been proposed for copper complexes. Among them, PDI has recently emerged as a potential target for several copper(I) and copper(II) complexes.64 On this basis, we also evaluated the ability of the newly synthesized complexes to act as PDI inhibitors. PDI enzyme was treated with 25 or 50 μM of tested complexes, and the ability to hamper its activity was assessed by a biochemical colorimetric method (Proteostat kit). As shown in Fig. 5A, copper derivatives 1–4 were able to inhibit PDI in a dose dependent manner. At the highest tested dose, the PPh3 derivatives 1 and 3 were able to reduce PDI catalytic activity by about 90%, whereas the PTA complexes 2 and 4 by about 80%. It is worth noting that all the complexes were much more effective than bacitracin, a well-known PDI inhibitor. The IC50 value calculated for complex 4 is 13.7 μM, as shown in Fig. 5A (right).
 | ||
| Fig. 5 A) Cell-free inhibition of PDI. PDI inhibition induced by 1–4 was measured by a Proteostat PDI assay kit. The PDI inhibitor bacitracin (0.5 mM) was used as a positive control. Error bars indicate S.D. On the right, the dose–response curve for PDI inhibition for compound 4 with indication of the IC50 value. B) Sulfhydryl content in LoVo cancer cells incubated for 36 h with 5 μM of tested compounds. The sulfhydryl group amount was determined by the DTNB assay. Error bars indicate S.D. *p < 0.05 compared with the control. C) ROS production. LoVo cells were preincubated in PBS/10 mM glucose medium for 20 min at 37 °C in the presence of 10 μM CM–H2DCFDA and then treated with the tested compounds at 25 μM. Antimycin (3 μM), a well-known inhibitor of mitochondrial complex III in the respiratory chain, was used as a positive control. | ||
As one of the key cellular functions of PDI is to catalyse the reduction of disulfide bonds and the oxidation of thiols, we thought it interesting to evaluate the levels of reduced thiols in LoVo colorectal cells after treatment with complexes 1–4 (Fig. 5B). Cells treated for 36 h with 5 μM of the tested complexes showed a significant increase in total sulfhydryl content, which even reached 200% in LoVo cancer cells treated with 4 (Fig. 5B). Accordingly, cells treated with copper complexes 1–4 showed no increase in ROS production/content (Fig. 6c). All together these results support the hypothesis that the newly developed copper(I) derivatives can effectively target PDI in colon cancer cells causing an imbalance in cellular redox homeostasis by shifting it towards a more “reduced state”.
 | ||
| Fig. 6 Hoechst staining of LoVo cells: a) control cells; b) cells incubated for 48 h with IC50 of cisplatin; c) cells incubated for 48 h with IC50 of 3; d) cells incubated for 48 h with IC50 of 4. | ||
We have recently reported that some copper complexes able to hamper PDI and to induce a cellular redox shift kill cancer cells by an alternative non-apoptotic cell death.64 With the aim to assess the cancer cell death mode induced by the most representative complexes 3 and 4, the Hoechst 33258 staining assay was performed to observe the morphological changes in treated cancer cells. LoVo cells were incubated with the IC50 of tested complexes for 48 h. Morphological analysis revealed no classical hallmarks of apoptosis, such as cell shrinkage, chromatin condensation or apoptotic bodies, suggesting a process of non-apoptotic cell death. Conversely, the classical morphological features of apoptosis induction (e.g. brightly stained nuclei, chromatin condensation and fragmentation) were evident on colorectal cells treated with the reference metallo-drug cisplatin (Fig. 6).
3. Materials and methods
3.1. Chemistry
The chemical shifts (δ) are reported in ppm, and the coupling constants (J) are reported in hertz (Hz). Abbreviations used in the analyses of the NMR spectra are as follows: br = broad, d = doublet, dd = doublet of doublets, m = multiplet, s = singlet, sbr = singlet broad, t = triplet, q = quartet and sept = septet. Electrospray ionization mass spectrometry (ESI-MS) spectra were recorded in positive- (ESI-MS(+)) or negative-ion (ESI-MS(−)) mode on a Waters Micromass ZQ Spectrometer, equipped with a single quadrupole (Waters Corporation, Milford, MA, USA), using an acetonitrile mobile phase. The compounds were added to reagent grade acetonitrile to give approximately 0.1 mM solutions. These solutions were injected (1 μL) into the spectrometer fitted with an autosampler. The pump delivered the solutions to the mass spectrometer source at a flow rate of 200 μL min−1, and nitrogen was employed both as a drying and nebulizing gas. Capillary voltage was typically 2500 V. The temperature of the source was 100 °C, while the temperature of the desolvation was 400 °C. In the analyses of the ESI-MS spectra, the confirmation of major peaks was supported by the comparison of the observed and predicted isotope distribution patterns, with the latter calculated using the IsoPro 3.1 computer software (T-Tech Inc., Norcross, GA, USA).
The precursors HC(pz)2COOH (LH)65 and HC(3,5-Me2pz)2COOH (L2H)66 were prepared by the literature method. The ligands [HC(pz)2COOiPr] (LOiPr) and [HC(3,5-Me2pz)2COOiPr] (L2OiPr) were prepared according to our procedures detailed in the literature46 and were fully characterized (FT-IR, 1H- and 13C-NMR: Fig. S1–S6†).
O); 1523w, 1481w (CC/CN); 1455w, 1436m, 1403m, 1377w, 1360w, 1289m, 1227m, 1205w, 1181w, 1148w, 1097s, 1058w, 1028w, 986w, 958w, 921w, 903w; 832vs (P–F); 798m, 745s. 1H-NMR (CD3CN, 293 K, Fig. S8†): δ 1.19 (d, 6H, CH(CH3)2), 5.05 (sept, 1H, CH(CH3)2), 6.39 (t, 2H, 4-CH), 7.26 (s, 1H, CHCO), 7.37–7.57 (m, 15H, CHar), 7.58 (d, 2H, 3- or 5-CH), 7.87 (d, 2H, 3- or 5-CH). 1H-NMR (CDCl3, 293 K, Fig. S9†): δ 0.94 (d, 6H, CH(CH3)2), 4.45 (sbr, 1H, CH(CH3)2), 6.29 (t, 2H, 4-CH), 7.13–7.29 (m, 10H, CHar), 7.33 (d, 2H, 3- or 5-CH), 7.43 (m, 5H, CHar), 7.67(s, 1H, CHCO), 8.30 (d, 2H, 3- or 5-CH). 13C{1H}-NMR (CD3CN, 293 K, Fig. S10†): 20.7 (s, CH3), 71.5 (s, CH(CH3)2), 73.5 (s, CHCO), 106.9 (s, 4-CH), 128.9 (d, J(C–P) = 9.1 Hz, PPh3), 130.4 (s, 3- or 5-CH), 132.2 (sbr, PPh3), 132.5 (d, J(C–P) = 32.2 Hz, PPh3), 133.4 (d, J(C–P) = 15.0 Hz, PPh3), 141.7 (s, 3- or 5-CH), 163.7 (s, CO). 31P{1H}-NMR (CD3CN, 293 K, Fig. S11†): δ −1.16 (br), −144.63 (sept, J(P–F) = 706 Hz, PF6). 31P{1H}-NMR (CD3CN, 243 K, Fig. S12†): δ −1.38 (sbr), −144.78 (sept, J(P–F) = 706 Hz, PF6). ESI-MS(+) (major positive ions, CH3CN, Fig. S13†), m/z (%): 145 (25) [Cu(CH3CN)2]+, 338 (35) [Cu(LOiPr)(CH3CN)]+, 366 (70) [Cu(PPh3)(CH3CN)]+, 559 (100) [Cu(LOiPr)(PPh3)]+, 587 (20) [Cu(PPh3)2]+. ESI-MS(−) (major negative ions, CH3CN), m/z (%): 145 (100) [PF6]−. Elemental analysis (%) calculated for C29H29CuF6N4O2P2: N 7.95, C 49.40, H 4.15; found: N 8.48, C 49.76, H 3.92.
O); 1653w, 1519w (CC/CN); 1452m, 1403m, 1390m, 1377m, 1292s, 1242m, 1227m, 1182w, 1147w, 1099s, 1045m, 1015m, 971s, 949s, 918w, 904w, 875m; 833vs (P–F); 797s, 753s. 1H-NMR (CD3CN, 293 K, Fig. S15†): δ 1.22 (d, 6H, CH(CH3)2), 4.08 (s, 6H, NCH2P), 4.51–4.60 (AB q, 6H, NCH2N), 5.12 (sept, 1H, CH(CH3)2), 6.39 (t, 2H, 4-CH), 7.24 (s, 1H, CHCO), 7.60 (d, 2H, 3- or 5-CH), 7.85 (d, 2H, 3- or 5-CH). 1H-NMR (CD3OD, 293 K, Fig. S16†): δ 1.20 (d, 6H, CH(CH3)2), 4.15 (s, 6H, NCH2P), 4.60–4.69 (AB q, 6H, NCH2N), 5.10 (m, 1H, CH(CH3)2), 6.45 (sbr, 2H, 4-CH), 7.51 (s, 1H, CHCO), 7.68 (sbr, 2H, 3- or 5-CH), 7.96 (d, 2H, 3- or 5-CH). 13C{1H}-NMR (CD3CN, 293 K, Fig. S17†): 20.7 (s, CH3), 52.1 (sbr, NCH2P), 71.2 (s, CH(CH3)2), 72.4 (sbr, NCH2N), 74.4 (s, CHCO), 106.8 (s, 4-CH), 130.4 (s, 3- or 5-CH), 140.5 (s, 3- or 5-CH), 164.2 (s, CO). 31P{1H}-NMR (CD3CN, 293 K, Fig. S18†): δ −92.10 (s), −144.60 (sept, J(P–F) = 706 Hz, PF6). 31P{1H}-NMR (CD3CN, 243 K, Fig. S19†): δ −95.77 (sbr), −144.77 (sept, J(P–F) = 706 Hz, PF6). ESI-MS(+) (major positive ions, CH3CN, Fig. S20†), m/z (%): 145 (100) [Cu(CH3CN)2]+, 158 (70) [PTA + H]+, 261 (45) [Cu(PTA)(CH3CN)]+, 297 (25) [Cu(LOiPr)]+, 338 (90) [Cu(LOiPr)(CH3CN)]+, 454 (100) [Cu(LOiPr)(PTA)]+. ESI-MS(−) (major negative ions, CH3CN), m/z (%): 145 (100) [PF6]−. Elemental analysis (%) calculated for C17H26CuF6N7O2P2: N 16.34, C 34.04, H 4.37; found: N 17.08, C 35.29, H 4.66.
O); 1673vw, 1561w (CC/CN); 1480w, 1463w, 1437w, 1422w, 1397w, 1377w, 1313w, 1299w, 1269w, 1241w, 1182vw, 1100m, 1043w, 998vw, 961w, 906vw, 880w; 837vs (P–F); 799w, 776w, 749w. 1H-NMR (CD3CN, 293 K, Fig. S22†): δ 0.93 (sbr, 6H, CH(CH3)2), 2.15 (s, 6H, CH3), 2.47 (s, 6H, CH3), 4.38 (sbr, 1H, CH(CH3)2), 6.19 (sbr, 2H, 4-CH), 6.87 (s, 1H, CHCO), 7.44–7.51 (m, 15H, CHar). 1H-NMR (CDCl3, 293 K, Fig. S23†): δ 1.18 (sbr, 6H, CH(CH3)2), 1.95 (s, 6H, CH3), 2.57 (s, 6H, CH3), 4.96 (sbr, 1H, CH(CH3)2), 6.07 (sbr, 2H, 4-CH), 6.98 (s, 1H, CHCO), 7.44–7.51 (m, 15H, CHar). 13C{1H}-NMR (CD3CN, 293 K, Fig. S24†): 10.4 (s, 3- or 5-CH3), 13.5 (s, 3- or 5-CH3), 20.7 (s, CH(CH3)2), 66.2 (s, CH(CH3)2), 72.2 (s, CHCO), 106.7 (s, 4-CH), 128.8 (d, J(C–P) = 9.5 Hz, PPh3), 130.3 (d, J(C–P) = 1.8 Hz, PPh3), 133.0 (d, J(C–P) = 33.2 Hz, PPh3), 133.4 (d, J(C–P) = 15.0 Hz, PPh3), 143.8 (s, 3- or 5-CH), 151.5 (s, 3- or 5-CH), 163.6 (s, CO). 31P{1H}-NMR (CD3CN, 293 K, Fig. S25†): δ −0.48 (br), −144.61 (sept, J(P–F) = 706 Hz, PF6). 31P{1H}-NMR (CD3CN, 243 K, Fig. S26†): δ −1.59 (sbr), −144.79 (sept, J(P–F) = 706 Hz, PF6). ESI-MS(+) (major positive ions, CH3CN, Fig. S27†), m/z (%): 145 (35) [Cu(CH3CN)2]+, 366 (100) [Cu(PPh3)(CH3CN)]+, 394 (40) [Cu(L2OiPr)(CH3CN)]+, 587 (20) [Cu(PPh3)2]+, 615 (75) [Cu(L2OiPr)(PPh3)]+. ESI-MS(−) (major negative ions, CH3CN), m/z (%): 145 (100) [PF6]−. Elemental analysis (%) calculated for C33H37CuF6N4O2P2: N 7.36, C 52.07, H 4.90; found: N 7.22, C 53.87, H 4.88.
O); 1565m (CC/CN); 1464sh, 1451m, 1418m, 1391w, 1376w, 1349w, 1315m, 1293w, 1272m, 1241s, 1186w, 1168w, 1148w, 1103s, 1037w, 1014m, 968s, 947m, 895w, 884w, 876w; 833vs (P–F); 741m, 716m. 1H-NMR (DMSO-d6, 293 K, Fig. S29†): δ 1.18 (s, 6H, CH(CH3)2), 2.18 (br, 6H, CH3), 2.33 (br, 6H, CH3), 4.08 (s, 6H, NCH2P), 4.43–4.62 (AB q, 6H, NCH2N), 5.00 (sbr, 1H, CH(CH3)2), 6.04 (sbr, 2H, 4-CH), 7.17 (s, 1H, CHCO). 1H-NMR (CD3CN, 293 K, Fig. S30†): δ 1.17 (d, 6H, CH(CH3)2), 2.29 (s, 6H, CH3), 2.42 (s, 6H, CH3), 4.10 (s, 6H, NCH2P), 4.49–4.57 (AB q, 6H, NCH2N), 5.01 (m, 1H, CH(CH3)2), 6.11 (sbr, 2H, 4-CH), 6.76 (s, 1H, CHCO). 13C{1H}-NMR (CD3CN, 293 K, Fig. S31†): 10.3 (s, CH3), 13.2 (s, CH3), 20.8 (s, CH(CH3)2), 50.6 (d, J(C–P) = 8.2 Hz, NCH2P), 66.1 (s, CH(CH3)2), 71.5 (s, CHCO), 72.7 (d, J(C–P) = 5.9 Hz, NCH2N), 106.4 (s, 4-CH), 143.7 (s, 3- or 5-CH), 151.3 (s, 3- or 5-CH), 163.5 (s, CO). 31P{1H}-NMR (CD3CN, 293 K, Fig. S32†): δ −96.10 (sbr), −144.62 (sept, J(P–F) = 707 Hz, PF6). 31P{1H}-NMR (CD3CN, 263 K, Fig. S33†): δ 96.27 (sbr), −144.79 (sept, J(P–F) = 707 Hz, PF6). ESI-MS(+) (major positive ions, CH3CN, Fig. S34†), m/z (%): 158 (20) [PTA + H]+, 261 (10) [Cu(PTA)(CH3CN)]+, 291 (50) [L2OiPr + H]+, 313 (70) [L2OiPr + Na]+, 353 (5) [Cu(L2OiPr)]+, 510 (100) [Cu(L2OiPr)(PTA)]+. ESI-MS(−) (major negative ions, CH3CN), m/z (%): 145 (100) [PF6]−. Elemental analysis (%) calculated for C21H34CuF6N7O2P2: N 14.95, C 38.45, H 5.22; found: N 15.20, C 39.65, H 5.65.
3.2. Spectroscopic methods
520 eV range. Two gas filled ionization chambers were used to measure incident (I0) and transmitted (I1) intensities. A reference spectrum was obtained simultaneously for each sample by placing a pure Cu foil downstream of the X-ray beam after I1, followed by a third ionization chamber (I2). The XAFS signal was calculated as  , with the reference signal obtained as
, with the reference signal obtained as  . The experimental data αexp were treated following standard procedures for background subtraction α′ = αexp − αpre, edge jump normalization and bare atom subtraction αb to extract the EXAFS structural signal
. The experimental data αexp were treated following standard procedures for background subtraction α′ = αexp − αpre, edge jump normalization and bare atom subtraction αb to extract the EXAFS structural signal 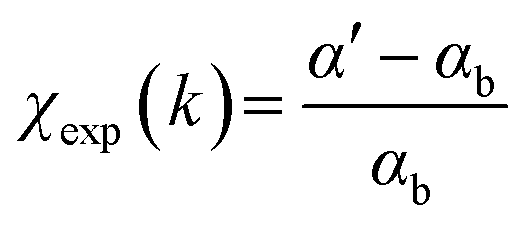 .71 For each spectrum the wavelength of the photoelectron
.71 For each spectrum the wavelength of the photoelectron  was calculated by selecting the photoelectron energy origin E0 at the first inflection point of the normalized absorption coefficient.
was calculated by selecting the photoelectron energy origin E0 at the first inflection point of the normalized absorption coefficient.
3.3. Experiments with cultured human cancer cells
Cu(I) complexes and the corresponding uncoordinated ligands were dissolved in DMSO just before the experiment, and a calculated amount of drug solution was added to the cell growth medium to a final solvent concentration of 0.5%, which had no detectable effects on cell viability. Cisplatin was dissolved in 0.9% sodium chloride solution. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), cisplatin and oxaliplatin were obtained from Sigma Chemical Co, St. Louis, MO, USA.The LoVo OXP cells were derived, using a standard protocol, by growing LoVo cells in increasing concentrations of OXP and following 17 months of selection of resistant clones, as previously described.74
Cell lines were maintained in the logarithmic phase at 37 °C in a 5% carbon dioxide atmosphere using RPMI-1640 medium (EuroClone, Milan, Italy) containing 10% fetal calf serum (EuroClone, Milan, Italy), antibiotics (50 units per mL penicillin and 50 μg mL−1 streptomycin) and 2 mM L-glutamine.
4. Conclusions
In the present study, the isopropyl ester derivatives of bis(pyrazol-1-yl)- and bis(3,5-dimethyl-pyrazol-1-yl)-acetic acid (LOiPr and L2OiPr) were used as chelators for the preparation of the new Cu(I) phosphane complexes 1–4. The hydrophilic 1,3,5-triaza-7-phosphaadamantane and the lipophilic triphenylphosphine were used for the synthesis of Cu(I) complexes, to stabilize Cu in +1 oxidation state and to modulate the hydrophilic–lipophilic balance of the related complexes. All the compounds were fully characterized in the solid state and in solution. As for the solid-state structural investigation, a multi-technique approach allowed the molecular stability of the ligands to be ascertained upon interaction with the copper ions, as well as determining the coordination geometry and copper ion oxidation state. More in detail, the molecular structure, electronic structure, and local geometry around the Cu(I) ion were probed by means of XPS and XAS spectroscopy for the selected Cu(I) coordination compound [Cu(L2OiPr)(PPh3)]PF6 (3). Specifically, XAS analysis suggested a trigonal planar geometry of the copper center, as hypothesized by the molecular structure, and XANES and XPS spectra confirmed the oxidation state (+1) for the metal ion.All the investigated phosphane Cu(I) complexes showed significant cytotoxic effects against a panel of human cancer cell lines. As in the case of the parent compounds based on the bis(pyrazol-1-yl)acetic acid (HC(pz)2COOH) and bis(3,5-dimethyl-pyrazol-1-yl)acetic acid (HC(pzMe2)2COOH),48 complexes bearing the more lipophilic PPh3 phosphane ligand were more effective than the corresponding PTA complexes. In addition, the presence of the 3,5-dimethyl-pyrazole ligand contributed to the enhancement of the cytotoxic potential of Cu(I) species, even if to a lesser extent relative to the parent compounds. Remarkably, [Cu(L2OiPr)(PPh3)]PF6 (3) showed a noticeable antitumor activity in 3D spheroidal models of human colon cancer cells, being about 2-fold more active than the reference metallodrug cisplatin. The cytotoxicity profile on the LoVo/LoVo OXP human colon cancer cell pair for complexes 1–4 suggested a distinct mechanism of action from conventional Pt(II) drugs. Actually, cellular mechanistic studies led to the identification of PDI as one of the crucial cellular targets of this class of phosphane copper(I) complexes. By hampering PDI activity, copper(I) complexes were able to cause an imbalance in cancer cell redox homeostasis thus leading to cancer cell death – a non-apoptotic programmed cell death. Hence, these results, confirming phosphane copper(I) complexes as PDI-targeting species, open a new perspective scenario for further research aimed to optimize the ability of copper compounds to modulate PDI activity in cancer cells.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by Unione Europea – NextGenerationEU (MUR-Fondo Promozione e Sviluppo – D.M. 737/2021, INVIRCuM, University of Camerino, FAR 2022 PNR, and NGEU PNRR, D.M. n. 351/2022 M4C1 I4.1) and by the University of Padova (PRID BIRD225980). Authors from Roma Tre – Dept. of Sciences gratefully acknowledge the Grant of Excellence Departments 2023-27 MIUR (ARTICOLO 1, COMMI 314–337 LEGGE 232/2016). The authors from Roma Tre University also acknowledge the CERIC-ERIC Consortium for the access to experimental facilities and financial support (experiment #20192011, XAFS Beamline).References
- C. S. Allardyce and P. J. Dyson, Dalton Trans., 2016, 45, 3201–3209 CAS.
- S. Medici, M. Peana, V. M. Nurchi, J. I. Lachowicz, G. Crisponi and M. A. Zoroddu, Coord. Chem. Rev., 2015, 284, 329–350 CAS.
- C. Molinaro, A. Martoriati, L. Pelinski and K. Cailliau, Cancers, 2020, 12, 2863 CAS.
- M. Zaki, F. Arjmand and S. Tabassum, Inorg. Chim. Acta, 2016, 444, 1–22 CAS.
- R. A. Festa and D. J. Thiele, Curr. Biol., 2011, 21, R877–R883 CAS.
- A. Erxleben, Coord. Chem. Rev., 2018, 360, 92–121 CAS.
- B. E. Kim, T. Nevitt and D. J. Thiele, Nat. Chem. Biol., 2008, 4, 176–185 CAS.
- E. J. Ge, A. I. Bush, A. Casini, P. A. Cobine, J. R. Cross, G. M. DeNicola, Q. P. Dou, K. J. Franz, V. M. Gohil, S. Gupta, S. G. Kaler, S. Lutsenko, V. Mittal, M. J. Petris, R. Polishchuk, M. Ralle, M. L. Schilsky, N. K. Tonks, L. T. Vahdat, L. Van Aelst, D. Xi, P. Yuan, D. C. Brady and C. J. Chang, Nat. Rev. Cancer, 2022, 22, 102–113 CAS.
- A. Gupte and R. J. Mumper, Cancer Treat. Rev., 2009, 35, 32–46 CAS.
- X. Wang, M. Zhou, Y. Liu and Z. Si, Cancer Lett., 2023, 561, 216157 CrossRef CAS PubMed.
- A. Steinbrueck, A. C. Sedgwick, J. T. Brewster, K.-C. Yan, Y. Shang, D. M. Knoll, G. I. Vargas-Zúñiga, X.-P. He, H. Tian and J. L. Sessler, Chem. Soc. Rev., 2020, 49, 3726–3747 RSC.
- V. Oliveri, Coord. Chem. Rev., 2020, 422, 213474 CrossRef CAS.
- P. Lelievre, L. Sancey, J. L. Coll, A. Deniaud and B. Busser, Cancers, 2020, 12, 3594 CrossRef CAS PubMed.
- M. Wehbe, A. W. Y. Leung, M. J. Abrams, C. Orvig and M. B. Bally, Dalton Trans., 2017, 46, 10758–10773 RSC.
- V. Gandin, C. Ceresa, G. Esposito, S. Indraccolo, M. Porchia, F. Tisato, C. Santini, M. Pellei and C. Marzano, Sci. Rep., 2017, 7, 13936 CrossRef PubMed.
- D. Denoyer, S. Masaldan, S. La Fontaine and M. A. Cater, Metallomics, 2015, 7, 1459–1476 CrossRef CAS PubMed.
- P. Tsvetkov, A. Detappe, K. Cai, H. R. Keys, Z. Brune, W. W. Ying, P. Thiru, M. Reidy, G. Kugener, J. Rossen, M. Kocak, N. Kory, A. Tsherniak, S. Santagata, L. Whitesell, I. M. Ghobrial, J. L. Markley, S. Lindquist and T. R. Golub, Nat. Chem. Biol., 2019, 15, 681–689 CAS.
- S. Tardito, I. Bassanetti, C. Bignardi, L. Elviri, M. Tegoni, C. Mucchino, O. Bussolati, R. Franchi-Gazzola and L. Marchiò, J. Am. Chem. Soc., 2011, 133, 6235–6242 CrossRef CAS PubMed.
- S. J. O'Day, A. M. M. Eggermont, V. Chiarion-Sileni, R. Kefford, J. J. Grob, L. Mortier, C. Robert, J. Schachter, A. Testori, J. Mackiewicz, P. Friedlander, C. Garbe, S. Ugurel, F. Collichio, W. Guo, J. Lufkin, S. Bahcall, V. Vukovic and A. Hauschild, J. Clin. Oncol., 2013, 31, 1211–1218 Search PubMed.
- P. Tsvetkov, S. Coy, B. Petrova, M. Dreishpoon, A. Verma, M. Abdusamad, J. Rossen, L. Joesch-Cohen, R. Humeidi, R. D. Spangler, J. K. Eaton, E. Frenkel, M. Kocak, S. M. Corsello, S. Lutsenko, N. Kanarek, S. Santagata and T. R. Golub, Science, 2022, 375, 1254–1261 CrossRef CAS.
- Y. Q. Wang, L. Zhang and F. F. Zhou, Cell. Mol. Immunol., 2022, 19, 867–868 CrossRef CAS.
- D. L. Tang, X. Chen and G. Kroemer, Cell Res., 2022, 32, 417–418 CrossRef PubMed.
- J. M. Xie, Y. N. Yang, Y. B. Gao and J. He, Mol. Cancer, 2023, 22, 46 CrossRef CAS PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- F. Tisato, C. Marzano, M. Porchia, M. Pellei and C. Santini, Med. Res. Rev., 2010, 30, 708–749 CrossRef CAS PubMed.
- O. Krasnovskaya, A. Naumov, D. Guk, P. Gorelkin, A. Erofeev, E. Beloglazkina and A. Majouga, Int. J. Mol. Sci., 2020, 21, 3965 CrossRef CAS PubMed.
- N. K. Singh, A. A. Kumbhar, Y. R. Pokharel and P. N. Yadav, J. Inorg. Biochem., 2020, 210, 111134 CrossRef CAS PubMed.
- A. Kellett, Z. Molphy, V. McKee and C. Slator, in Metal-based Anticancer Agents, ed. A. Casini, A. Vessières and S. M. Meier-Menches, The Royal Society of Chemistry, 2019, pp. 91–119 Search PubMed.
- M. Marinelli, C. Santini and M. Pellei, Curr. Top. Med. Chem., 2016, 16, 2995–3017 Search PubMed.
- S. Spreckelmeyer, C. Orvig and A. Casini, Molecules, 2014, 19, 15584–15610 Search PubMed.
- X. Y. Man, S. H. Li, G. Xu, W. J. Li, M. H. Zhu, Z. L. Zhang, H. Liang and F. Yang, J. Med. Chem., 2024, 67, 5744–5757 Search PubMed.
- M. Jiang, Z. L. Zhang, W. J. Li, X. Y. Man, H. B. Sun, H. Liang and F. Yang, J. Med. Chem., 2022, 65, 9447–9458 CrossRef CAS PubMed.
- F. Tisato, C. Marzano, V. Peruzzo, M. Tegoni, M. Giorgetti, M. Damjanovic, A. Trapananti, A. Bagno, C. Santini, M. Pellei, M. Porchia and V. Gandin, J. Inorg. Biochem., 2016, 165, 80–91 CAS.
- V. Gandin, F. Tisato, A. Dolmella, M. Pellei, C. Santini, M. Giorgetti, C. Marzano and M. Porchia, J. Med. Chem., 2014, 57, 4745–4760 CAS.
- M. Pellei, G. G. Lobbia, G. Papini and C. Santini, Mini-Rev. Org. Chem., 2010, 7, 173–203 CAS.
- C. Marzano, M. Pellei, D. Colavito, S. Alidori, G. G. Lobbia, V. Gandin, F. Tisato and C. Santini, J. Med. Chem., 2006, 49, 7317–7324 CrossRef CAS.
- C. Santini, M. Pellei, G. Gioia Lobbia, D. Fedeli and G. Falcioni, J. Inorg. Biochem., 2003, 94, 348–354 CrossRef CAS.
- C. Santini, M. Pellei, G. G. Lobbia, A. Cingolani, R. Spagna and M. Camalli, Inorg. Chem. Commun., 2002, 5, 430–433 CrossRef CAS.
- A. Karapetyan, C. Santini, M. Pellei, V. Grigoryan, A. Dallakyan and G. Khachatryan, Arch. Euromedica, 2022, 12, 5 Search PubMed.
- F. Del Bello, M. Pellei, L. Bagnarelli, C. Santini, G. Giorgioni, A. Piergentili, W. Quaglia, C. Battocchio, G. Iucci, I. Schiesaro, C. Meneghini, I. Venditti, N. Ramanan, M. De Franco, P. Sgarbossa, C. Marzano and V. Gandin, Inorg. Chem., 2022, 61, 4919–4937 CrossRef CAS PubMed.
- M. Pellei, L. Bagnarelli, L. Luciani, F. Del Bello, G. Giorgioni, A. Piergentili, W. Quaglia, M. De Franco, V. Gandin, C. Marzano and C. Santini, Int. J. Mol. Sci., 2020, 21, 2616 CrossRef CAS PubMed.
- M. B. Morelli, C. Amantini, G. Santoni, M. Pellei, C. Santini, C. Cimarelli, E. Marcantoni, M. Petrini, F. Del Bello, G. Giorgioni, A. Piergentili and W. Quaglia, New J. Chem., 2018, 42, 11878–11887 RSC.
- M. Pellei, V. Gandin, C. Cimarelli, W. Quaglia, N. Mosca, L. Bagnarelli, C. Marzano and C. Santini, J. Inorg. Biochem., 2018, 187, 33–40 CrossRef CAS PubMed.
- M. Giorgetti, S. Tonelli, A. Zanelli, G. Aquilanti, M. Pellei and C. Santini, Polyhedron, 2012, 48, 174–180 CrossRef CAS.
- M. Pellei, G. Papini, A. Trasatti, M. Giorgetti, D. Tonelli, M. Minicucci, C. Marzano, V. Gandin, G. Aquilanti, A. Dolmella and C. Santini, Dalton Trans., 2011, 40, 9877–9888 RSC.
- M. Pellei, L. Bagnarelli, S. Gabrielli, G. Lupidi, C. Cimarelli, F. Stella, A. Dolmella and C. Santini, Inorg. Chim. Acta, 2023, 544, 121234 CrossRef CAS.
- M. Pellei, C. Santini, L. Bagnarelli, M. Caviglia, P. Sgarbossa, M. De Franco, M. Zancato, C. Marzano and V. Gandin, Int. J. Mol. Sci., 2023, 24, 4091 CAS.
- M. Pellei, C. Santini, L. Bagnarelli, C. Battocchio, G. Iucci, I. Venditti, C. Meneghini, S. Amatori, P. Sgarbossa, C. Marzano, M. De Franco and V. Gandin, Int. J. Mol. Sci., 2022, 23, 9397 Search PubMed.
- L. Bagnarelli, A. Dolmella, C. Santini, R. Vallesi, R. Giacomantonio, S. Gabrielli and M. Pellei, Molecules, 2021, 26, 6271 CAS.
- S. Gabrielli, M. Pellei, I. Venditti, I. Fratoddi, C. Battocchio, G. Iucci, I. Schiesaro, C. Meneghini, A. Palmieri, E. Marcantoni, L. Bagnarelli, R. Vallesi and C. Santini, Dalton Trans., 2020, 49, 15622–15632 CAS.
- NIST X-ray Photoelectron Spectroscopy Database, Version 5.0 (National Institute of Standards and Technology), (https://srdata.nist.gov/xps/) Search PubMed.
- J. C. Klein, C. P. Li, D. M. Hercules and J. F. Black, Appl. Spectrosc., 1984, 38, 729–734 CAS.
- F. A. Akgul, G. Akgul, N. Yildirim, H. E. Unalan and R. Turan, Mater. Chem. Phys., 2014, 147, 987–995 CAS.
- M. E. Aguirre, R. X. Zhou, A. J. Eugene, M. I. Guzman and M. A. Grela, Appl. Catal., B, 2017, 217, 485–493 CAS.
- M. Benfatto and C. Meneghini, in Synchrotron Radiation: Basics, Methods and Applications, ed. S. Mobilio, F. Boscherini and C. Meneghini, Springer Berlin Heidelberg, Berlin, Heidelberg, 2015, pp. 213–240 Search PubMed.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CAS.
- D. E. Sayers, E. A. Stern and F. W. Lytle, Phys. Rev. Lett., 1971, 27, 1204–1207 CrossRef CAS.
- G. Bunker, Introduction to XAFS: A Practical Guide to X-ray Absorption Fine Structure Spectroscopy, Cambridge University Press, Cambridge, 2010 Search PubMed.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- Q. Lin, L. Luo and H. Wang, Front. Oncol., 2021, 11, 644956 CrossRef CAS PubMed.
- Y. Yin, S. Li, X. Liang, K. Li, M. Xie and B. Hu, Pharmaceuticals, 2022, 15, 1139 CrossRef CAS PubMed.
- W. J. Li, T. Li, Y. Pan, S. H. Li, G. Xu, Z. L. Zhang, H. Liang and F. Yang, J. Med. Chem., 2024, 67, 3843–3859 CrossRef CAS.
- L. A. Kunz-Schughart, J. P. Freyer, F. Hofstaedter and R. Ebner, J. Biomol. Screening, 2004, 9, 273–285 CrossRef CAS.
- F. Miglioli, M. De Franco, J. Bartoli, M. Scaccaglia, G. Pelosi, C. Marzano, D. Rogolino, V. Gandin and M. Carcelli, Eur. J. Med. Chem., 2024, 276, 116697 CrossRef CAS.
- N. Burzlaff, I. Hegelmann and B. Weibert, J. Organomet. Chem., 2001, 626, 16–23 CrossRef CAS.
- A. Beck, B. Weibert and N. Burzlaff, Eur. J. Inorg. Chem., 2001, 521–527 CrossRef CAS.
- V. Secchi, S. Franchi, M. Dettin, A. Zamuner, K. Beranova, A. Vladescu, C. Battocchio, V. Graziani, L. Tortora and G. Iucci, Nanomaterials, 2020, 10, 1151 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, Eden Prairie, 1996 Search PubMed.
- P. Swift, D. Shuttleworth and M. P. Seah, Practical surface analysis: by Auger and x-ray photoelectron spectroscopy, ed. D. Briggs and M. P. Seah, Wiley, Chichester, 1983, ch. 5 and appendix 3 Search PubMed.
- A. Di Cicco, G. Aquilanti, M. Minicucci, E. Principi, N. Novello, A. Cognigni and L. Olivi, J. Phys.: Conf. Ser., 2009, 190, 012043 CrossRef.
- C. Meneghini, F. Bardelli and S. Mobilio, Nucl. Instrum. Methods Phys. Res., Sect. B, 2012, 285, 153–157 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- N. Margiotta, C. Marzano, V. Gandin, D. Osella, M. Ravera, E. Gabano, J. A. Platts, E. Petruzzella, J. D. Hoeschele and G. Natile, J. Med. Chem., 2012, 55, 7182–7192 CrossRef CAS.
- M. P. Rigobello, V. Gandin, A. Folda, A.-K. Rundlöf, A. P. Fernandes, A. Bindoli, C. Marzano and M. Björnstedt, Free Radical Biol. Med., 2009, 47, 710–721 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4md00610k |
| This journal is © The Royal Society of Chemistry 2025 |