
Miniaturized click chemistry and direct screening facilitate the discovery of triazole piperazine SARS-CoV-2 Mpro inhibitors with improved metabolic stability†
Shenghua
Gao‡
 b,
Letian
Song‡
a,
Bing
Ye
a,
Mianling
Yang
a,
Junyi
Li
a,
Manyu
Gu
a,
Ann E.
Tollefson
c,
Karoly
Toth
*c,
Peng
Zhan
*a and
Xinyong
Liu
*a
b,
Letian
Song‡
a,
Bing
Ye
a,
Mianling
Yang
a,
Junyi
Li
a,
Manyu
Gu
a,
Ann E.
Tollefson
c,
Karoly
Toth
*c,
Peng
Zhan
*a and
Xinyong
Liu
*a
aDepartment of Medicinal Chemistry, Key Laboratory of Chemical Biology, Ministry of Education, School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Ji'nan, 250012, China. E-mail: zhanpeng1982@sdu.edu.cn; xinyongl@sdu.edu.cn; Tel: +86 531 88382005 Tel: +86 531 88380270
bShenzhen Research Institute of Shandong University, A301 Virtual University Park in South District of Shenzhen, Guangdong, PR China
cDepartment of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, 63103, St. Louis, Missouri, USA. E-mail: karoly.toth@health.slu.edu
First published on 18th October 2024
Abstract
The continuous mutational nature of SARS-CoV-2 and its inter-species' similarities emphasize the urgent need to design and develop more direct-acting antiviral agents against highly infectious variants. Herein, we report on the efficient discovery of potent non-covalent non-peptide-derived Mpro inhibitors using miniaturized click chemistry and direct screening. Based on the privileged piperazine scaffold, 68 triazole-containing derivatives were assembled and screened. Notably, representative compound C1N46 (IC50 = 1.87 μM, EC50 = 6.99 μM, CC50 > 100 μM) displayed potent inhibition activity against Mpro and showed promising anti-SARS-CoV-2 properties in vitro. Additionally, C1N46 exhibited improved liver microsome stability compared to lead compound GC-14. Docking studies predicted a multi-site binding mode of the triazole-based compounds. In conclusion, our studies validate the efficacy and feasibility of click chemistry in rapidly discovering antiviral agents.
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is still a serious threat to human life and health, affecting socio-economic development, and the corona virus disease 2019 (COVID-19) infection has not yet ended. The SARS-CoV-2 virus exhibits rapid mutation rates and possesses significant immune evasion capabilities, enabling it to overcome the protective barriers established by vaccines or prior infections in the human body, thereby leading to multiple reinfections.1 It is highly likely that SARS-CoV-2 will persist alongside humans for an extended duration akin to influenza viruses.2 Despite the current approval of several drugs, variant strains rapidly develop resistance against existing therapy. The continuous mutational nature of SARS-CoV-2 and its inter-species similarities emphasize the urgent necessity for designing and developing more direct-acting antiviral agents to complement vaccines against highly infectious variants.3,4The SARS-CoV-2 main protease plays a crucial role in the replication cycle of the virus and is widely conserved among different species of coronaviruses. The sequence homology of Mpro between SARS-CoV-2 and SARS-CoV is 96%.5 Moreover, the highly conserved sequence and structure of Mpro make it an optimal target for the development of anti-SARS-CoV-2 drugs.
To date, a plethora of Mpro inhibitors have been reported and gradually advanced into clinical trials or the market. The majority of these inhibitors are covalent peptide-like compounds, including nirmatrelvir, simnotrelvir, leritrelvir, and FB2001 (Fig. 1),6–9 which exhibit similar structures and significant homogeneity. Conversely, there is a scarcity of reports on non-peptide inhibitors with only ensitrelvir being currently approved in Japan, while WU-04 is undergoing phase III clinical trials (Fig. 1).10 Given the escalating utilization of these drugs coupled with the high mutation rate of SARS-CoV-2, drug resistance has become an increasingly pressing concern. Recently, nirmatrelvir-resistant mutant strains (E166V/T21I and E166V/L50F double mutants) have emerged in clinical settings.11,12 Consequently, there is an urgent imperative to discover novel anti-SARS-CoV-2 drugs possessing unique scaffolds that can also serve as reserves and provide technical support for potential future coronavirus outbreaks.
 | ||
| Fig. 1 SARS-CoV-2 Mpro inhibitors currently approved or in clinical studies. | ||
Our previous study has led to the discovery of GC-14 (IC50 = 0.4 μM, EC50 = 1.1 μM),13 a promising non-peptide non-covalent main protease inhibitor with a novel piperazine skeleton. However, its anti-SARS-CoV-2 activity and pharmacokinetics require improvement for clinical application. The co-crystal of GC-14 and Mpro (PDB ID: 8ACL) provides a foundation for further structural modifications; through meticulous analysis of the co-crystal structure (Fig. 2), it is evident that GC-14 does not fully occupy the hydrophobic S4/S5 cavity and lacks interaction with crucial amino acids Gln189, Thr190 and Gln192. Consequently, our objective is to investigate the unexplored chemical space within the S4/S5 cavity utilizing GC-14 as a lead compound, with an aim of establishing additional hydrogen bonds with key amino acids to enhance the anti-SARS-CoV-2 activity.
 | ||
| Fig. 2 (A) Binding mode of GC-14 in the SARS-CoV-2 Mpro active center, highlighting the unoccupied S4/P5 site; (B) design concept of triazole-containing Mpro inhibitors; (C) workflow of click-chemistry assisted lead discovery. | ||
On the other hand, we conducted an analysis on the potential metabolic sites of GC-14. The results revealed that the thiophene-2-methyl amide moiety is susceptible to metabolism, whereas the 1,2,3-triazole structure derived from copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) exhibits enhanced metabolic stability as a bioisostere of amide. This structural feature plays a crucial role in facilitating the discovery of bioactive compounds.14,15 It is worth noting that the compound library generated from this methodology can be directly utilized for high-throughput screening without requiring purification. In other words, it enables direct screening based on microplate technology which offers advantages such as rapidity, traceability, sensitivity, and cost-effectiveness.16 Therefore, we aim to efficiently explore the uncharted chemical space of the S4/S5 cavity using click chemistry and the direct screening technique, thereby establishing additional interactions and enhancing target affinity while improving pharmacokinetic properties. By employing the CuAAC reaction between alkynes and azides, we constructed large-scale combinatorial libraries without the need for laborious synthesis and purification. Active candidates identified through screening were subsequently synthesized at a milligram scale, followed by determination of concentration-dependent enzyme inhibitory activities and cell culture-based antiviral activities. The specific design concept is illustrated in Fig. 2A and B.
2. Experimental section
2.1 General information and materials
All chemical reagents and reaction solvents were purchased from commercial suppliers. All melting points were determined on a micro melting point apparatus (RY-1G, Tianjin TianGuang Optical Instruments). 1H NMR and 13C NMR spectra were recorded in DMSO-d6 on a Bruker AV-600 spectrometer, with tetramethyl silane (TMS) as the internal standard. Coupling constants were given in hertz, and chemical shifts were reported in δ values (ppm) from TMS; signals were abbreviated as s (singlet), d (doublet), t (triplet), q (quarter), and m (multiplet). The mass spectrometry (MS) spectra were acquired on an LTQ Orbitrap XL (Thermo Fisher). All reactions were routinely monitored by thin layer chromatography (TLC) on silica gel GF254. Flash column chromatography was performed on columns packed with silica gel (200–300 mesh), purchased from Biotage. The purity of representative final compounds was tested on an Agilent 1260 prim HPLC system. HPLC conditions: Agilent ZORBAX, SB-C18 column (250 mm × 4.6 mm × 5 μm). methanol/water with 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20; flow rate 1.0 mL min−1; temperature, 30 °C; injection volume, 10 μL. The purity of all tested compounds are >95%.
:20; flow rate 1.0 mL min−1; temperature, 30 °C; injection volume, 10 μL. The purity of all tested compounds are >95%.
2.2 Synthesis
General synthesis procedure for intermediates 9–13. A 250 mL round-bottom flask was charged with compound 7 (7.8 g, 41.1 mmol), compound 8 (4.4 g, 20.5 mmol) and 80 mL dichloromethane. Anhydrous cupric acetate (3.7 g, 20.7 mmol) and pyridine (3.2 g, 41.0 mmol) were then added to the mixture simultaneously. The mixture was stirred under oxygen atmosphere and at room temperature for 12 h, then washed with water (4 × 60 mL) to remove copper salt and pyridine. The resulting organic phase was separated, dried over anhydrous Na2SO4, then evaporated in vacuo to produce a dark-green oil as the residue. Purification through flash chromatography (EtOAc
:hexane = 1:15) gave compound 9 (4.8 g) as a colorless thick oil. ESI-MS: m/z 389.2 [M + H]+. C17H22Cl2N2O4 (388.1) (Scheme 1).
 | ||
| Scheme 1 The general route of synthesizing targeted compounds. Reagents and conditions: (i) Cu(OAc)2, O2, pyridine, dichloromethane (DCM), r.t.; (ii) Dess–Martin periodinane, DCM, 0 °C–r.t.; (iii) dimethyl (1-diazo-2-oxopropyl)phosphonate, K2CO3, MeOH, r.t.; (iv) 2 M HCl/dioxane, DCM, r.t.; (v) nicotinic acid, HATU, DIPEA, DCM, r.t.; (vi) R–N3, CuSO4·5H2O, sodium ascorbate, 55 °C, THF/H2O = 1:1 (v/v). | ||
A solution of compound 9 (4.8 g, 13.3 mmol) in dichloromethane (120 mL) was cooled in an ice bath under stirring. Dess–Martin periodinane (11.3 g, 26.6 mmol, 2.0 eq.) was added in three portions over 10 min. Then, the mixture was stirred at room temperature for 2 h. After the reaction was monitored by TLC, saturated Na2S2O3 and NaHCO3 solution were added simultaneously to quench the reaction. The organic phase was separated, washed with saturated NaCl solution, dried over anhydrous Na2SO4, and then concentrated under reduced pressure to yield the crude product of 10 (4.28 g) for the following procedures without purification. ESI-MS: m/z 359.1 [M + H]+. C17H22Cl2N2O4 (358.1).
Compound 10 (4.0 g, 11.2 mmol) was dissolved in 80 mL methanol with stirring and under an ice bath. Potassium carbonate (3.1 g, 22.4 mmol) and dimethyl(1-diazo-2-oxopropyl)phosphonate (3.2 g, 16.8 mmol) were added to the solution successively. The reaction was warmed to room temperature and stirred for 6 hours. Subsequently, methanol was removed under vacuum. The residual solid was then combined with DCM (100 mL) and water (100 mL). The organic phase was collected, dried over anhydrous Na2SO4, and evaporated in vacuo. The crude product was purified through flash column chromatography to afford 11 (2.89 g, 73% yield). ESI-MS: m/z 355.1 [M + H]+. C17H22Cl2N2O4 (354.1).
Compound 11 (2.8 g, 7.9 mmol) was dissolved in 70 mL dichloromethane under stirring. A solution of HCl in dioxane (2M, 23.7 mL) was added dropwise into the solution. The reaction was stirred for 6 h at room temperature, then evaporated in vacuo to remove the volatile component. The residual solid was re-suspended in EtOAc/hexane. The precipitant was collected by filtration and dried to give compound 12 in the form of hydrochloride (2.46 g, 96% yield.). ESI-MS: m/z 254.1 [M + H]+. C17H22Cl2N2O4 (254.0).
Nicotinic acid (0.85 g, 6.7 mmol) and HATU (3.50 g, 9.2 mmol) were mixed in DCM (45 mL) under an ice bath and vigorous stirring. After 30 minutes, diisopropylethylamine (3.15 g, 24.4 mmol) and compound 12 (2.0 g, 6.1 mmol) were added. The reaction was warmed to ambient temperature and allowed to react for 12 hours, then quenched with water. The organic phase was separated, washed with saturated NaCl solution, dried over anhydrous Na2SO4, and concentrated in vacuo. Purification by flash column chromatography gave the key intermediate 13 (1.71 g, 78%). ESI-MS: m/z 359.1 [M + H]+. C17H22Cl2N2O4 (359.0).
Compound 13 (0.1 g, 0.28 mmol) and azide fragments (0.28 mmol) were added to a mixture solution of tetrahydrofuran/water (1:1). To this, CuSO4·5H2O (0.028 mmol, 0.1 eq.) and sodium ascorbate (0.14 mmol) were added. The reaction was stirred under 55 °C heating for 24 hours. Upon competition, the reaction was extracted with EtOAc (3 × 10 mL). The organic phase was collected, dried over anhydrous Na2SO4 and then removed under reduced pressure. Crude products are purified through flash column chromatography to yield the target compounds C1N31, C1N43, C1N46, and C1N47.
(S)-(3-(1-(Benzo[d][1,3]dioxol-5-yl)-1H-1,2,3-triazol-4-yl)-4-(3,4-dichlorophenyl) piperazin-1-yl)(pyridin-3-yl)methanone (C1N31). Yield: 72.3%. Melting point 90–91 °C. White solid. 1H NMR (600 MHz, DMSO) δ 8.75–8.44 (m, 2H, pyridine-H), 8.34 (d, J = 59.3 Hz, 1H, pyridine-H), 7.53 (s, 1H, triazole-H), 7.46 (s, 1H), 7.38 (d, J = 9.0 Hz, 1H), 7.31 (d, J = 7.3 Hz, 1H), 7.18 (s, 1H), 7.09 (d, J = 8.4 Hz, 1H), 6.95 (d, J = 19.7 Hz, 1H), 6.14 (s, 2H, O-CH2-O), 5.38 (d, J = 133.1 Hz, 1H, piperazine-CH), 4.54 (d, J = 136.8 Hz, 1H, piperazine-CH2), 3.91 (d, J = 10.0 Hz, 1H, piperazine-CH2), 3.74 (s, 1H, piperazine-CH2), 3.54 (d, J = 32.3 Hz, 2H, piperazine-CH2), 3.26 (s, 1H, piperazine-CH2). 13C NMR (150 MHz, DMSO) δ 167.98, 151.04, 149.55, 148.64 (C × 2), 147.89, 135.29, 131.98, 131.34 (C × 2), 130.97, 123.82, 120.16, 115.56, 114.30, 109.09 (C × 2), 102.62 (C × 2), 102.40, 51.14, 46.01, 41.82, 40.52. ESI-MS: m/z 523.33 [M + H]+. C25H20Cl2N6O3 (522.1). HPLC purity: 99.41%.
(S)-(4-(3,4-Dichlorophenyl)-3-(1-mesityl-1H-1,2,3-triazol-4-yl)piperazin-1-yl) (pyridin-3-yl)methanone (C1N43). Yield: 75%. Melting point 88–89 °C. White solid. 1H NMR (600 MHz, DMSO) δ 8.68 (s, 2H, pyridine-H), 8.49 (d, J = 111.9 Hz, 1H, pyridine-H), 8.09 (d, J = 66.5 Hz, 1H, triazole-H), 7.84 (d, J = 38.8 Hz, 1H), 7.55–7.49 (m, 1H), 7.38 (d, J = 8.1 Hz, 1H), 7.21–7.08 (m, 1H), 7.05 (s, 2H), 7.01 (d, J = 8.0 Hz, 1H), 5.37 (d, J = 136.4 Hz, 1H, piperazine-CH), 4.65–4.36 (m, 1H, piperazine-CH2), 3.96 (d, J = 12.2 Hz, 1H, piperazine-CH2), 3.67–3.58 (m, 2H, piperazine-CH2), 3.50 (d, J = 9.6 Hz, 1H, piperazine-CH2), 3.01 (d, J = 11.7 Hz, 1H, piperazine-CH2), 2.30 (s, 3H, CH3), 1.74 (s, 6H, 2CH3). 13C NMR (150 MHz, DMSO) δ 167.99, 151.09, 149.76, 148.05, 139.93, 135.24, 134.84, 133.70, 132.01, 131.84, 131.08, 130.82 (C × 2), 129.26 (C × 2), 124.06, 122.06, 120.46, 118.40, 117.17, 79.60, 51.98, 48.62, 40.52, 21.08, 16.99. ESI-MS: m/z 521.7 [M + H]+. C27H26Cl2N6O (520.1). HPLC purity: 99.80%.
(S)-(4-(3,4-Dichlorophenyl)-3-(1-(4-(hydroxymethyl)phenyl)-1H-1,2,3-triazol-4-yl) piperazin-1-yl)(pyridin-3-yl)methanone (C1N46). Yield: 76%. Melting point 163–164 °C. White solid. 1H NMR (600 MHz, DMSO) δ 8.65 (s, 1H, pyridine-H), 8.59 (s, 1H, pyridine-H), 8.39 (d, J = 107.6 Hz, 1H, pyridine-H), 7.86 (s, 1H, triazole-H), 7.84–7.75 (m, 2H), 7.45 (dd, J = 78.4, 8.9 Hz, 4H), 7.19 (s, 1H), 6.96 (d, J = 20.4 Hz, 1H), 5.56–5.23 (m, 2H, piperazine-CH and OH), 4.57 (d, J = 5.4 Hz, 2H, CH2OH), 4.45 (s, 1H), 3.93 (d, J = 8.0 Hz, 1H), 3.75 (s, 1H), 3.55 (d, J = 43.2 Hz, 2H), 3.33 (s, 1H). 13C NMR (150 MHz, DMSO) δ 167.95, 150.89, 149.54, 147.90, 143.75, 135.52, 134.86, 133.76, 131.99, 131.11, 130.98, 130.62, 128.03 (C × 3), 123.82, 122.15, 120.30, 118.52, 116.51, 62.65, 51.74, 51.18, 42.54, 40.52. ESI-MS: m/z 509.41 [M + H]+. C25H22Cl2N6O2 (508.1). HPLC purity: 99.88%.
(S)-(4-(3,4-Dichlorophenyl)-3-(1-(4-methoxyphenyl)-1H-1,2,3-triazol-4-yl)piperazin-1-yl)(pyridin-3-yl)methanone (C1N47). Yield: 73%. Melting point 80–81 °C. White solid. 1H NMR (600 MHz, DMSO) δ 8.64 (d, J = 55.6 Hz, 2H, pyridine-H), 8.34 (d, J = 54.9 Hz, 1H, triazole-H), 7.81 (d, J = 77.6 Hz, 2H), 7.53 (d, J = 5.7 Hz, 1H), 7.43 (dd, J = 63.9, 9.0 Hz, 2H), 7.21 (d, J = 36.2 Hz, 1H), 7.15–7.09 (m, 2H), 7.02–6.92 (m, 1H), 5.39 (d, J = 133.6 Hz, 1H, piperazine-CH), 4.82–4.38 (m, 2H, pyridine-H), 3.91 (d, J = 10.5 Hz, 1H, pyridine-H), 3.82 (s, 3H, CH3), 3.79–3.69 (m, 1H, pyridine-H), 3.64–3.53 (m, 2H, pyridine-H). 13C NMR (150 MHz, DMSO) δ 168.15, 159.74, 151.07, 149.77, 147.85, 143.75, 135.03, 133.82, 132.01, 131.08, 130.97, 130.34, 128.12, 124.05, 122.24, 118.39, 117.18, 116.51, 115.32 (C × 2), 79.60, 56.03, 51.21, 48.63, 40.52. ESI-MS: m/z 509.52 [M + H]+. C25H22Cl2N6O2 (508.1). HPLC purity: 99.9%.
 | ||
| Fig. 3 Structures of the in-house azide library (N1–N69). | ||
| Reagents | Order of addition | Concentration & solvent | Volume | Final concentration |
|---|---|---|---|---|
| Azides | 1 | 35 mM, DMSO | 20 μL | 7 mM |
| Alkyne | 2 | 25 mM, DMSO | 20 μL | 5 mM |
| TBTA | 3 | 10 mM, DMSO | 10 μL | 1 mM |
| CuSO4·5H2O | 4 | 4 mM, water | 25 μL | 1 mM |
| Sodium ascorbate | 5 | 20 mM, water | 25 μL | 5 mM |
2.3 SARS-CoV-2 Mpro inhibition screening method
A fluorescence resonance energy transfer (FRET) method was applied to measure Mpro inhibition of the tested compounds. Black 96-well plates, SARS-CoV-2 main protease solution (1 mg mL−1, 500 mM Tris, 150 mM NaCl, 1 mM EDTA, 50% glycerol) and fluorescent substrate MCA-AVLQSGFR-Lys(Dnp)-Lys-NH2 (20 mM in DMSO) were purchased from Beyotime Biotechnology (Shanghai, China). Mpro was diluted to 1.5 μM using assay buffer (50 mM Tris–HCl, 150 mM NaCl, 20% glycerol, pH = 7.3) and preserved at −20 °C. The fluorescent substrate was diluted to 500 μM in assay buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, pH = 7.3). The compound solutions from the established library were diluted to desired concentration with assay buffer. Each well of a black 96-well plate was then filled with 50 μL assay buffer, followed by adding 20 μL of diluted enzyme solution and 20 μL inhibitor solution. Control wells (no inhibitor) and blank wells (no enzyme) were measured in parallel experiments.The 96-well plate was then incubated at 37 °C in a shaking incubator for 10 minutes. Following the incubation, the substrate solution (10 μL per well) was added per well to initiate the reaction. The fluorescence signal was measured every 10 s for a duration of 10 min, with a SpectraMax iD5 multimode plate reader (molecular devices) with an excitation wavelength of 320 nM and an emission wavelength of 405 nM. Data from the first 60 seconds were linear-fitted to calculate slope values (V0 for the control well, Vb for the blank well, Vi for the test well). % Inhibition (i%) in each well can thus calculated using the equation below:
| i% = 1 − (Vi − Vb)/(V0 − Vb) × 100% |
IC50 value determination: for each compound, the inhibition rate against SARS-CoV2 Mpro was measured at 5 different concentrations (0.1 μM, 0.5 μM, 1 μM, 5 μM, 10 μM), and three independent experiments were performed. All experimental data were analyzed using GraphPad Prism 8.0 to calculate IC50 values.
2.4 In vitro antiviral activity evaluation method
Vero E6 cells (CRL-1856™, from African green monkey kidney) were supplied by American Type Culture Collection (ATCC®). Cells were plated at 1.7 × 104 cells per well into 96-well plates 24 hours prior to infection. Test compounds were prepared to a 10 mM stock solution in DMSO. Nirmatrelvir (Bidepharm) and GC-14 (from in-house library) were used as the positive controls and blank wells with no compounds served as the negative controls. The test compounds were diluted to 80 micromolar concentration in Dulbecco's MEM/5% fetal bovine serum, followed by a 1:3 dilution on a dilution plate (each compound was done in triplicate), to form a series of gradients. After removing the medium from the cells, 50 μL of the compound dilutions were added to each well of the cell plate. The plates were then transferred to the BSL-3 facility for infection with SARS-CoV-2. The virus was diluted to approximately 100 focus forming units (infectious particles) per 50 μL (the volume of diluted virus added per well). Following virus addition, the plates were incubated at 37 °C, 5% CO2 for 1 hour. After the incubation, the medium containing methyl cellulose was added to each well (100 μL). The total final volume in each well was 200 μL. The final compound concentration range was 20 μM to 0.0274 μM. After the plates were incubated for 25–26 hours, the methyl cellulose overlay was removed and plates were washed twice with PBS. Subsequently, a solution of 5% paraformaldehyde in PBS was added to each well and the entire plates were submerged in 5% formalin for 15 minutes. Afterward, the plates were submerged in PBS for another 15 minutes before fresh PBS was added to each well and the plates were transferred out of the BSL-3 laboratory. Plates carrying immobilized Vero E6 cells were rinsed with focus forming assay buffer, and incubated with a guinea pig anti-SARS-CoV-2 antibody generated against virions. The secondary antibody was goat anti-guinea pig IgG (horseradish peroxidase conjugate). The substrate was TrueBlue and the infected cells per foci appeared as individual or clusters of blue cells. After staining, plates were read on an EliSpot reader. The relative surface area that was stained was used to calculate the inhibition activity of a certain concentration gradient of the drug, and generate the EC50 curves. EC50 values were generated by curve fitting using Graphpad Prism 8.0.
2.5 In vitro cellular toxicity evaluation method
Vero E6 cells were plated on 96-well plates (Greiner) at 1.7 × 104 cells per well in DMEM (Dulbecco’ s Modified Eagle Medium) high glucose with 10% FBS (fetal bovine serum). Cells were incubated for 24 h in 37 °C and 5% CO2 at saturating humidity. Cells were treated with the compound in DMEM high glucose with 5% FBS at a final concentration of 1% DMSO for 26 h. Cellular viability was measured using the CellTiter 96 aqueous non-radioactive cell proliferation assay (Promega). A solution containing 2 mg mL−1 3-(4,5-dimethylthiazol-2-yl)-5- (3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS), and 0.043 mg mL−1 phenazine methosulfate (PMS) was added to the cells. Cells incubated with the mixture for 4 h at 37 °C and absorbance was measured at 490 nm. Background absorbance values from media-only wells were subtracted and data were converted to percent cell viability, as defined by the DMSO vehicle-treated wells. Three or more replicate assays were done to calculate the average values. The 50% cytotoxic concentration (CC50) values were determined with GraphPad Prism 8.0 using the four parameter variable slope algorithm with the bottom set to zero.2.6 Human liver microsome stability experiment
A master solution was prepared using a 100 mM phosphate buffer, which contained 0.5 mg mL−1 liver microsome and 1 μM test compounds (final concentration). Diclofenac was set as the positive control. The mixture was pre-warmed at 37 °C for 5 minutes before starting the reaction with NADPH regeneration solution. The test compound sample without NADPH was used as a negative control. The incubation solution was incubated in water batch at 37 °C. At time points of 0, 5, 15, 30, 60 minutes, the reaction was stopped by adding cold MeOH:acetonitrile = 1:1 with IS (50 ng mL−1 labetalol, 50 ng mL−1 tolbutamide) in a volume of 200 μL. The samples were centrifuged at 4000 rpm for 10 minutes. A 100 μL aliquot of the resulting supernatant was mixed with 100 μL of H2O and subsequently utilized for LC-MS/MS analysis. The slope value, denoted as k, was obtained through linear regression analysis of the natural logarithm of the remaining percentage of the parent drug against incubation time curve. The in vitro half-life (in vitro t1/2) was determined from the slope value:| in vitro t1/2 = −0.693/k |
Conversion of the in vitro t1/2 (min) into the in vitro intrinsic clearance (in vitro CLint, in μL min−1 mg−1 protein) was done using the following equation (mean of triplicate determinations). The microsomal protein concentration is 0.5 mg mL−1, while the scaling factor of rat liver microsomes is 1800.
| CLint (mL min−1 kg−1) = (0.693/T1/2) × (microsomal protein concentration)−1 × Scaling Factors |
2.7 Molecular docking studies
The protein structures for computational simulation were downloaded from the Protein Database Bank (PDB). PDB ID: 8ACL was selected as the docking receptor. All the calculation processes were supported by the corresponding modules of Schrodinger 2021-4 suite (https://www.schrodinger.com) and were performed on the DELL Precision T5500 workstation. Firstly, the compounds were optimized with the Ligprep model with default parameters, and only the (S)- chiral isomer was generated for each compound. The ionic state under the physiological conditions of the ligand (pH = 7.3) was added; the OPLS4 force field was selected to optimize and obtain the ligand molecules required for screening. The preparation of the protein was completed by the protein preparation wizard module. A series of processes including hydrogenation, charging, elimination of conflicting amino acid residues and energy minimization of the protein crystal structure were carried out with default parameters, and then the GC-14 molecule of the composite crystal structure is extracted to obtain the receptor protein. The binding position of GC-14 was used to locate the receptor grid for ligands. Finally, the Glide module was used to dock the optimized ligands with the receptor protein with extra precision (XP). According to the established docking model, the Schrodinger 2021-4 Glide XP module was used for molecular docking of the compounds. The docking poses were visualized by Pymol (Schrödinger, LLC. DeLano Scientific, San Francisco, CA, USA, https://pymol.org).3. Results and discussion
3.1 Design of 1,2,3-triazole-containing piperazine derivatives targeting Mpro
In our previous work, a comprehensive strategy including multi-site binding and privileged group assembly served as the inspiration for the discovery of tri-substituted piperazine analogs.13 Through multiple rounds of modification, GC-14 was identified with significant antiviral activity towards SARS-CoV-2, while demonstrating low cellular toxicity. However, there is still a disparity of antiviral activity between GC-14 and nirmatrelvir. Current piperazine derivatives need further improvement to develop oral bioavailable clinical candidates. The selection of GC-14 as the starting point of this study is driven by the objective of enhancing the antiviral activity and optimizing the pharmacokinetic profile.The co-crystal structure studies of the GC-14/Mpro complex revealed that the sidearm group partially occupied the S4 site within the active pocket of Mpro. Additionally, the carboxyl amide formed additional interaction with Glu166. However, the thiophenyl group in GC-14 failed to fully penetrate the S4 cavity, leaving the far-end S4/P5 site unoccupied.17 Meanwhile, the thiophene is considered as a susceptible site for metabolism.13 Therefore, these findings suggest that introducing better groups onto the piperazine ring could potentially improve both activity and metabolic stability of GC-14. Here, we incorporated 1,2,3-triazole to newly designed molecules as an amide group mimic,18 which could potentially enhance drug-like properties and improve active site occupancy. By employing a highly efficient and convenient CuAAC reaction, we successfully constructed a focused library of piperazine derivatives containing triazole moieties. Guided by Mpro inhibition screening results, four compounds exhibiting potent inhibitory activity against SARS-CoV-2 Mpro were identified and evaluated through subsequent in vitro assays, liver microsome stability experiment and molecular modeling simulation.
3.2 Enzyme inhibition screening of targeted compounds
The newly synthesized compounds in the crude library were evaluated for Mpro inhibition using a FRET-based assay, as previously described. The results of primarily screening under 5 μM compound concentration are presented in Fig. 4. Overall, the newly synthesized compounds exhibited varying extent of Mpro inhibition activities, with several compounds showing comparable inhibition to that of GC-14 at the same concentration. Compounds featuring an aromatic ring with a hydrogen bond acceptor on the triazole group displayed the highest activity among those with newly introduced S4 groups, exemplified by four top candidates, C1N47, C1N46, C1N31, and C1N43. The rigid aromatic ring might have guaranteed the contact between the H-bond acceptor (typically oxygen) and hydrophilic P5 site. In contrast, substituted benzyls are less likely to enhance compound activity. Specific acetyl amide derivatives like C1N23 and C1N35 also lead to modest potency. Meanwhile, those bulkier groups (C1N20, C1N24, C1N40) are doubtlessly disfavored, due to the limited tolerability of the S4 pocket. A structure–activity relationship analysis of crude library screening consisted with our design concept, and hinted for further modification of the triazole analogs. Blank controls consisting of alkyne fragment 7 and a mixture solution of TBTA/CuSO4/sodium ascorbate were tested and did not show significant enzyme inhibition. | ||
| Fig. 4 Primary screening results of the crude compound library. | ||
For further biological evaluation, C1N47, C1N46, C1N31, and C1N43 were synthesized and purified at the milligram scale. Lead compound GC-14 and the approved drug nirmatrelvir were employed as positive references. As indicated in Table 2, these compounds exhibited low-micromolar activities against Mpro, but were not as potent as GC-14.
| Compound | IC50a (μM) | Compound | IC50 (μM) |
|---|---|---|---|
| a Enzyme (SARS-CoV-2 Mpro) inhibitory activity determined by a standard fluorescence resonance energy transfer (FRET) assay. IC50 values are means ± SD of at least three independent experiments performed in triplicate. b GC-14 and nirmatrelvir were used as positive controls (n = 3 biological replicates). For structure of nirmatrelvir see Fig. 1. | |||
| Nirmatrelvirb | 0.074 ± 0.006 | GC-14 | 0.39 ± 0.03 |
| C1N31 | 2.23 ± 0.48 | C1N43 | 1.85 ± 0.14 |
| C1N46 | 1.87 ± 0.07 | C1N47 | 2.09 ± 0.27 |
3.3 Antiviral activity and cellular toxicity evaluation of target compounds
Next, the anti-SARS-CoV-2 activity and toxicity of the four hit compounds were measured by plaque assays in Vero E6 cells. As listed in Table 3, all the compounds displayed potent antiviral activity, with EC50 values in the micromolar range. C1N46, C1N47 and C1N31 showed similar activities, while C1N43 was less active, probably due to decreased solubility brought by the 2,4,6-trimethylphenyl group.| Compound | Structure | EC50a (μM) | CC50b (μM) |
|---|---|---|---|
| a Antiviral activity determined in Vero E6 cells using an Enzyme Linked Immunospot Assay (ELISPOT). b Cytotoxicity values (CC50) were measured in Vero E6 cells using the MTS method. | |||
| C1N31 |

|
6.32 | >100 μM |
| C1N43 |

|
12.3 | |
| C1N46 |

|
6.99 | |
| C1N47 |

|
7.82 | |
| GC-14 |

|
1.78 | |
| Nirmatrelvir |

|
0.49 | |
3.4 Liver microsome stability of C1N46 and GC-14
To investigate the potential enhancement of compound metabolic stability and druggability through the incorporation of a 1,2,3-triazole moiety, C1N46 and GC-14 were evaluated for their stability in rat liver microsomes with diclofenac as a control. C1N46 is still detectable in the system at up to 60 minutes, exhibiting a longer half-life than GC-14 (Table 4). Comparatively, GC-14 was completely degraded within approximately 15 minutes. These experimental findings indicated that the newly designed compounds incorporating the 1,2,3-triazole moiety may possess improved druggability for further developments.| Cpd. | Percent remaining (%)(mean, n = 2) | T 1/2 (min) | CLint (mL min−1 kg−1) | ||||
|---|---|---|---|---|---|---|---|
| 0 min | 5 min | 15 min | 30 min | 60 min | |||
| C1N46 | 100 | 70.7 | 36.5 | 13.3 | 5.1 | 10.3 | 242 |
| GC-14 | 100 | 22.7 | 1.56 | 0.00 | 0.00 | 2.51 | 994 |
| Diclofenac | 100 | 75.5 | 48.6 | 25.7 | 7.93 | 16.6 | 150 |
3.5 Molecular modeling to predict the binding mode of representative compounds
To elucidate the preferable binding mode of these active Mpro inhibitors, molecular docking was conducted based on the GC-14-Mpro complex (PDB ID: 8ACL). The resulting predicted docking poses are depicted in Fig. 5A–D. Consistent with GC-14, the novel compounds conserved the multi-site binding mode in S1, S2 and S4 subsites. The piperazine scaffold in the target compounds aligns well with that of GC-14, and crucial interactions within the S1/S2 cavities, including hydrogen bonds with His163/Gln142 and stacking with His41, remain undisturbed by the triazole moiety. The nitrogen atom in the triazole ring maintained the key hydrogen bond with Glu166 in all the compounds, unlike GC-14. Additionally, due to its rigid linear conformation (Fig. 6A and B) the newly introduced triazole group allowed for complete occupancy of the S4 cavity by the aromatic group from azide fragments. Furthermore, hydrogen bond acceptors, such as the methoxy group in C1N47 and the hydroxyl group in C1N46, formed hydrogen bonds with Gln189, thereby enhancing their binding affinity.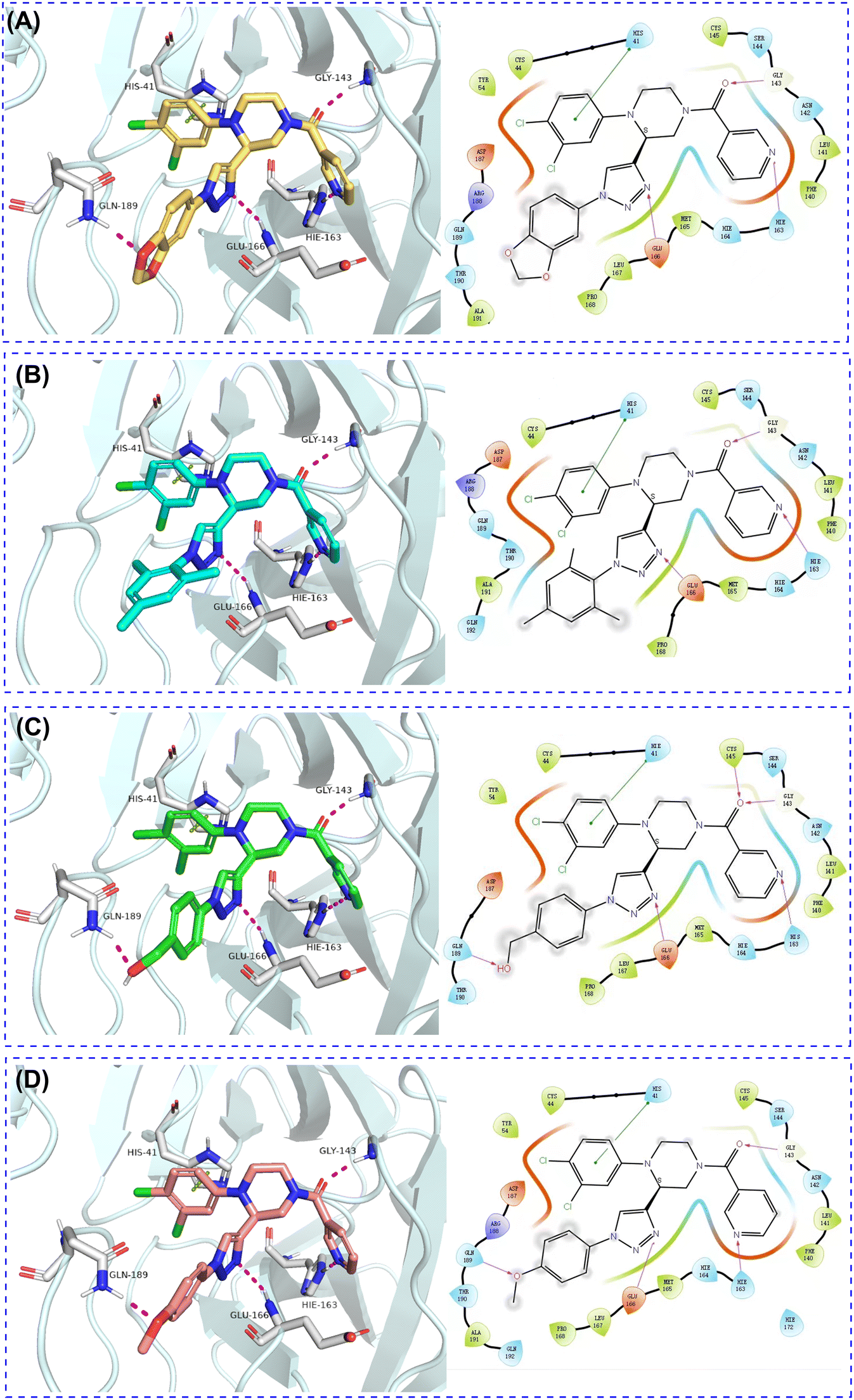 | ||
| Fig. 5 (A–D) Docking pose of C1N31, C1N43, C1N46, C1N47 with SARS-CoV-2 Mpro, respectively. PDB ID: 8ACL. | ||
 | ||
| Fig. 6 (A and B) Overlay of C1N31 (yellow), C1N43 (blue), C1N46 (green), C1N47 (pink) and GC-14 (white) in the SARS-CoV-2 Mpro active center, highlighting their space occupations at S1, S2 and S4 subsites. PDB ID: 8ACL. | ||
4. Conclusion
The increasing demand for effective anti-coronavirus agents necessitates the development of more efficient tools for compound derivation, library construction, and rapid screening. In this study, we selected SARS-CoV-2 Mpro as the antiviral target, and introduced a 1,2,3-triazole group onto the validated piperazine scaffold. A focused library containing desired compounds was efficiently constructed using miniaturized click-chemistry via the CuAAC reaction, and directly screened to identify representative compounds. As a result, four new compounds exhibited good activity in both enzymatic inhibition screening and antiviral assays, without cellular toxicity. Among them, C1N46 represented by a triazole-based derivative showed significantly higher liver microsome stability compared to GC-14. Additionally, docking simulation suggested that the target compounds extended space occupation in the Mpro active center, and revealed the contribution of 1,2,3-triazole in inhibitor binding. Furthermore, these findings suggest that triazole can be further applied to modify other coronavirus Mpro inhibitors19 to find broad-spectrum antiviral agents.Compared to the traditional synthesis workflow in medicinal chemistry, our study successfully synthesized the desired compounds with minimal time, space, and raw material costs. This demonstrates the effectiveness of lead compound discovery enhanced by miniaturized synthesis and direct screening. In future studies, this paradigm can be applied for inhibitor discovery against a variety of antiviral targets, or used to assemble multifunctional antiviral molecules including PROTAC degraders.20 Additionally, emerging techniques, including novel diazotizing reagents,21 protein-templated in situ screening22 and droplet microarray,23 will contribute to click chemistry-based lead discovery by increasing library throughput and accuracy.
Data availability
Miniaturized click chemistry and direct screening facilitate the discovery of triazole piperazine SARS-CoV-2 Mpro inhibitors with improved metabolic stability. All data supporting the findings of this study are available within the paper and its ESI.†Author contributions
Peng Zhan and Xinyong Liu conceived the project and designed the experiments. Shenghua Gao and Letian Song synthesized the compounds and performed enzymatic experiments. Letian Song carried out computational experiments. Ann Tollefson and Karoly Toth completed the antiviral activity evaluation and cytotoxicity studies in Vero E6 cells. Mianling Yang and Bing Ye assisted in confirming the structure of the compounds. Shenghua Gao prepared the manuscript with input from all authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge financial support from Guangdong Basic and Applied Basic Research Foundation (No. 2021A1515110740), Youth Fund of National Natural Science Foundation Project (No. 22307067), the Key Research and Development Program, Ministry of Science and Technology of the People's Republic of China (Grant No. 2023YFC2606500), General Program of National Natural Science Foundation of China (No. 82373727), Shandong Laboratory Program (SYS202205) and Major Basic Research Project of Shandong Provincial Natural Science Foundation (No. ZR2021ZD17).References
- P. Qu, K. Xu, J. N. Faraone, N. Goodarzi, Y. M. Zheng, C. Carlin, J. S. Bednash, J. C. Horowitz, R. K. Mallampalli, L. J. Saif, E. M. Oltz, D. Jones, R. J. Gumina and S. L. Liu, Immune evasion, infectivity, and fusogenicity of SARS-CoV-2 BA.2.86 and FLip variants, Cell, 2024, 187, 585–595 CrossRef CAS PubMed.
- Y. Chen, Q. Liu, L. Zhou, Y. Zhou, H. Yan and K. Lan, Emerging SARS-CoV-2 variants: Why, how, and what's next?, Cell Insight, 2022, 1, 100029 CrossRef PubMed.
- X. Ji, X. Meng, X. Zhu, Q. He and Y. Cui, Research and development of Chinese anti-COVID-19 drugs, Acta Pharm. Sin. B, 2022, 12, 4271–4286 CrossRef CAS.
- A. M. Carabelli, T. P. Peacock, L. G. Thorne, W. T. Harvey, J. Hughes, COVID-19 Genomics UK Consortium, S. J. Peacock, W. S. Barclay, T. I. de Silva, G. J. Towers and D. L. Robertson, SARS-CoV-2 variant biology: immune escape, transmission and fitness, Nat. Rev. Microbiol., 2023, 21, 162–177 CAS.
- S. Gao, T. Huang, L. Song, S. Xu, Y. Cheng, S. Cherukupalli, D. Kang, T. Zhao, L. Sun, J. Zhang, P. Zhan and X. Liu, Medicinal chemistry strategies towards the development of effective SARS-CoV-2 inhibitors, Acta Pharm. Sin. B, 2022, 12, 581–599 CrossRef CAS.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. E. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Wei, Q. Yang and Y. Zhu, An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19, Science, 2021, 374, 1586–1593 CrossRef CAS.
- X. Jiang, H. Su, W. Shang, F. Zhou, Y. Zhang, W. Zhao, Q. Zhang, H. Xie, L. Jiang, T. Nie, F. Yang, M. Xiong, X. Huang, M. Li, P. Chen, S. Peng, G. Xiao, H. Jiang, R. Tang, L. Zhang, J. Shen and Y. Xu, Structure-based development and preclinical evaluation of the SARS-CoV-2 3C-like protease inhibitor simnotrelvir, Nat. Commun., 2023, 14, 6463 CrossRef CAS PubMed.
- X. Chen, X. Huang, Q. Ma, P. Kuzmič, B. Zhou, S. Zhang, J. Chen, J. Xu, B. Liu, H. Jiang, W. Zhang, C. Yang, S. Wu, J. Huang, H. Li, C. Long, X. Zhao, H. Xu, Y. Sheng, Y. Guo, C. Niu, L. Xue, Y. Xu, J. Liu, T. Zhang, J. Spencer, Z. Zhu, W. Deng, X. Chen, S. H. Chen, N. Zhong, X. Xiong and Z. Yang, Preclinical evaluation of the SARS-CoV-2 Mpro inhibitor RAY1216 shows improved pharmacokinetics compared with nirmatrelvir, Nat. Microbiol., 2024, 9, 1075–1088 CrossRef CAS PubMed.
- W. Shang, W. Dai, C. Yao, L. Xu, X. Tao, H. Su, J. Li, X. Xie, Y. Xu, M. Hu, D. Xie, H. Jiang, L. Zhang and H. Liu, In vitro and in vivo evaluation of the main protease inhibitor FB2001 against SARS-CoV-2, Antiviral Res., 2022, 208, 105450 CrossRef CAS PubMed.
- N. Hou, L. Shuai, L. Zhang, X. Xie, K. Tang, Y. Zhu, Y. Yu, W. Zhang, Q. Tan, G. Zhong, Z. Wen, C. Wang, X. He, H. Huo, H. Gao, Y. Xu, J. Xue, C. Peng, J. Zou, C. Schindewolf, V. Menachery, W. Su, Y. Yuan, Z. Shen, R. Zhang, S. Yuan, H. Yu, P. Y. Shi, Z. Bu, J. Huang and Q. Hu, Development of Highly Potent Noncovalent Inhibitors of SARS-CoV-2 3CLpro, ACS Cent. Sci., 2023, 9, 217–227 CAS.
- Y. Duan, H. Zhou, X. Liu, S. Iketani, M. Lin, X. Zhang, Q. Bian, H. Wang, H. Sun, S. J. Hong, B. Culbertson, H. Mohri, M. I. Luck, Y. Zhu, X. Liu, Y. Lu, X. Yang, K. Yang, Y. Sabo, A. Chavez, S. P. Goff, Z. Rao, D. D. Ho and H. Yang, Nature, 2023, 622, 376–382 CrossRef CAS PubMed.
- S. Iketani, H. Mohri, B. Culbertson, S. J. Hong, Y. Duan, M. I. Luck, M. K. Annavajhala, Y. Guo, Z. Sheng, A. C. Uhlemann, S. P. Goff, Y. Sabo, H. Yang, A. Chavez and D. D. Ho, Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir, Nature, 2023, 613, 558–564 CrossRef CAS PubMed.
- S. Gao, K. Sylvester, L. Song, T. Claff, L. Jing, M. Woodson, R. H. Weiße, Y. Cheng, L. Schäkel, M. Petry, M. Gütschow, A. C. Schiedel, N. Sträter, D. Kang, S. Xu, K. Toth, J. Tavis, A. E. Tollefson, C. E. Müller, X. Liu and P. Zhan, Discovery and Crystallographic Studies of Trisubstituted Piperazine Derivatives as Non-Covalent SARS-CoV-2 Main Protease Inhibitors with High Target Specificity and Low Toxicity, J. Med. Chem., 2022, 65, 13343–13364 CrossRef CAS PubMed.
- X. Wang, B. Huang, X. Liu and P. Zhan, Discovery of bioactive molecules from CuAAC click-chemistry-based combinatorial libraries, Drug Discovery Today, 2016, 21, 118–132 CrossRef CAS PubMed.
- Y. Itoh, P. Zhan, T. Tojo, P. Jaikhan, Y. Ota, M. Suzuki, Y. Li, Z. Hui, Y. Moriyama, Y. Takada, Y. Yamashita, M. Oba, S. Uchida, M. Masuda, S. Ito, Y. Sowa, T. Sakai and T. Suzuki, Discovery of Selective Histone Deacetylase 1 and 2 Inhibitors: Screening of a Focused Library Constructed by Click Chemistry, Kinetic Binding Analysis, and Biological Evaluation, J. Med. Chem., 2023, 66, 15171–15188 CrossRef CAS PubMed.
- M. Whiting, J. Muldoon, Y. C. Lin, S. M. Silverman, W. Lindstrom, A. J. Olson, H. C. Kolb, M. G. Finn, K. B. Sharpless, J. H. Elder and V. V. Fokin, Inhibitors of HIV-1 protease by using in situ click chemistry, Angew. Chem., Int. Ed., 2006, 45, 1435–1439 CrossRef CAS.
- Y. Zhao, Y. Zhu, X. Liu, Z. Jin, Y. Duan, Q. Zhang, C. Wu, L. Feng, X. Du, J. Zhao, M. Shao, B. Zhang, X. Yang, L. Wu, X. Ji, L. W. Guddat, K. Yang, Z. Rao and H. Yang, Structural basis for replicase polyprotein cleavage and substrate specificity of main protease from SARS-CoV-2, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2117142119 CrossRef CAS.
- Q. Guan, S. Xing, L. Wang, J. Zhu, C. Guo, C. Xu, Q. Zhao, Y. Wu, Y. Chen and H. Sun, Triazoles in Medicinal Chemistry: Physicochemical Properties, Bioisosterism, and Application, J. Med. Chem., 2024, 67, 7788–7824 CrossRef CAS PubMed.
- Z. Zeng, C. Liao and L. Yu, Molecules for COVID-19 treatment, Chin. Chem. Lett., 2024, 35, 109349 Search PubMed.
- Y. Zhou, S. Xu, N. López-Carrobles, D. Ding, X. Liu, L. Menéndez-Arias and P. Zhan, Recent advances in the molecular design and applications of proteolysis targeting chimera-based multi-specific antiviral modality, Acta Mater. Med., 2023, 2, 285–298 Search PubMed.
- G. Meng, T. Guo, T. Ma, J. Zhang, Y. Shen, K. B. Sharpless and J. Dong, Modular click chemistry libraries for functional screens using a diazotizing reagent, Nature, 2019, 574, 86–89 CrossRef CAS PubMed.
- R. Wamser, X. Zhang, B. Kuropka, C. Arkona and J. Rademann, Protein-Templated Ugi Reactions versus In Situ Ligation Screening: Two Roads to the Identification of SARS-CoV-2 Main Protease Inhibitors, Chemistry, 2024, 30, e202303940 CrossRef CAS.
- W. Lei, K. Demir, J. Overhage, M. Grunze, T. Schwartz and P. A. Levkin, Droplet-Microarray: Miniaturized Platform for High-Throughput Screening of Antimicrobial Compounds, Adv. Biosyst., 2020, 4, e2000073 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional spectral data of the target compounds (PDF). See DOI: https://doi.org/10.1039/d4md00555d |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2025 |