
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-component catalysts with integrated MWW-type layers and mixed oxide domains for glucose-to-formic acid oxidation
Cristina
Esteban
,
Alexandra
Velty
 * and
Urbano
Díaz
*
* and
Urbano
Díaz
*
Instituto de Tecnología Química, Universitat Politècnica de València, Agencia Estatal Consejo Superior de Investigaciones Científicas, Valencia 46022, Spain. E-mail: avelty@itq.upv.es; udiaz@itq.upv.es
First published on 28th August 2025
Abstract
The development of efficient and multi-component catalysts with distinct active sites is essential to promote chemically and economically circular processes and to achieve sustainable development. In this work, various multi-component materials were synthesized combining the properties of MWW zeolitic layers and MgAl mixed oxides. Synthesis conditions were successfully developed and optimized to achieve the in situ formation of nanometric sub-domains of layered double hydroxides (LDHs) on the surfaces and within the microporous cavities and channels of MWW layers (methods I and II). Additionally, multi-component materials were prepared by mixing MWW lamellar precursors and preformed layered double hydroxides, using swelling and exfoliation techniques and combined with sonification and ball-milling techniques (method III). Calcination yielded materials effectively integrating zeolitic MWW layers with MgAl oxides, displaying distinct structural features depending on the synthesis method. The catalytic performance of these materials was evaluated in the oxidation of glucose to formic acid (FA) using H2O2 as the oxidant. Among the catalysts investigated, MWW@MgAl-in situ-40, which combined suitable textural properties and basicity, showed superior activity, achieving a 47.7% FA yield at 66.2% glucose conversion after 7.5 h at 363 K in dioxane.
1. Introduction
The exploration of multi-functional composite materials by combining different components in a well-designed structure is a prominent topic, as the combination can balance their drawbacks and improve their properties, leading to multi-functional materials with unique properties and applications. In fact, multi-component materials have received significant attention due to their ability to integrate complementary functionalities within a single framework, enabling synergistic interactions between components. This integration often leads to enhanced catalytic performance, adsorption properties, and energy storage applications. For instance, layered double hydroxides (LDHs) combined with porous supports such as zeolites or mesoporous silicas have demonstrated remarkable efficiency in catalytic oxidation, hydrogenation, and CO2 capture processes.1–4 Similarly, metal oxide composites incorporating active sites for redox and acid–base reactions have shown potential in biomass conversion and environmental remediation. The ability to precisely control the distribution and interaction of sub-domains within these materials is key to optimizing their multi-functional properties and tailoring them for specific applications. These advances highlight the importance of further research into the synthesis and characterization of multi-component systems.5–7These advanced materials for the sustainable and greener synthesis of chemicals and fuels by converting renewable raw materials play a crucial role in achieving sustainable development. Reducing the environmental footprint and addressing the limitations of fossil sources and the ever-increasing global demand for energy require the urgent implementation of carbon-neutral energy production and transport systems. In this context, the substitution of fossil fuels with biofuels, due to its uncertainty in CO2 balance and its competition with food production, is being reconsidered. Both legislation and industry are directing investments towards the production of renewable electricity, leveraging the existing electrical grid with its attractive features and moderate initial infrastructure investments. However, challenges arise in electrifying the transport sector due to the substantial requirements for on-board electrical energy storage, necessitating large batteries.8 In this respect, H2 emerges as a promising alternative for intermediate energy storage. H2 has a high volumetric capacity (33.3 kWh kg−1) and can be converted to energy in an internal combustion engine or fuel cells. Conventional H2 storage such as high-pressure compressed gas cylinders or cryogenic liquid suffer from excessive energy losses (compression, liquefaction and evaporation of H2) and low volumetric energy capacity. While physical adsorption of H2 on large surface area materials experience limitations in terms of temperature and pressure ranges providing lower gravimetric and volumetric energy densities. Formic acid (FA) emerges as an appealing alternative as a liquid hydrogen carrier. FA contains only 4.4 wt% H2, however due to its high density of 1.22 g cm−3 and volumetric capacity it reaches 53 gH2 L−1, which is equivalent to an energy density of 1.77 kWh L−1.9 Thus, FA represents the potential for a carbon-neutral H2 storage system, pending the development of efficient CO2 hydrogenation and selective FA dehydrogenation processes. Formic acid is produced mainly from carbon monoxide, either by heating it with sodium hydroxide to produce sodium formate, further acidified, or by the base-catalyzed reaction of CO and methanol to produce methyl formate, further hydrolyzed to acid. Formic acid is also one of the main by-products of acetic acid manufacture. Consequently, the biomass-based FA production from cellulosic biomass and sugars is an attractive route. Thus, two main chemocatalytic oxidation strategies have been explored to produce FA from carbohydrates using O2 or H2O2 as oxidant.10
Formic acid production from carbohydrate biomass occurs by oxidative cleavage of the C–C bond connecting an aldehyde group and a hydroxyl group. The terminal carbon is then oxidized to formic acid and the subsequent C5 intermediate will repeat this degradation until all C–C bonds have been cleaved (Scheme 1), and six formic acid molecules can be obtained. However, the selective cleavage of the C–C bonds is challenging, as several side reactions occur with CO2 being the main by-product of the decarboxylation of the acid intermediate.
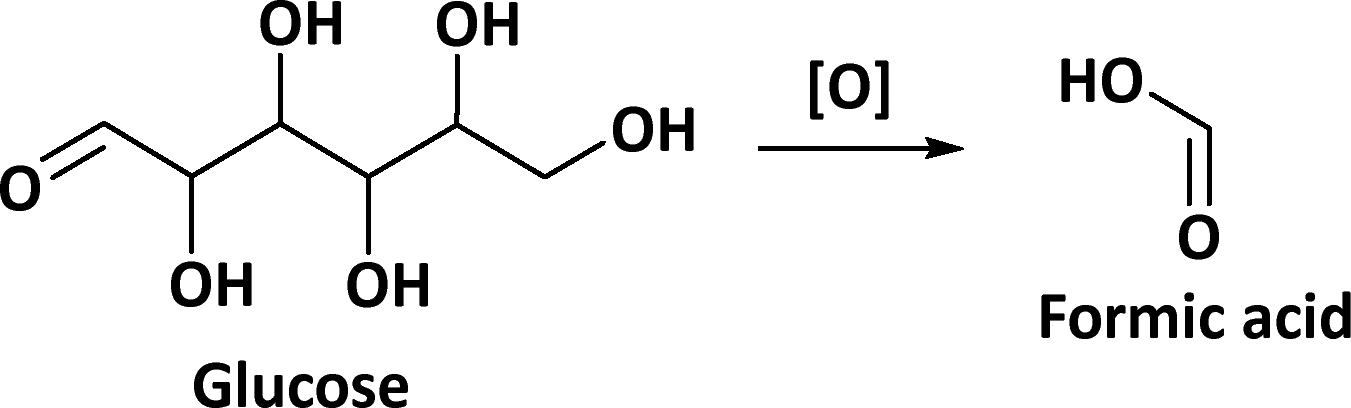 | ||
| Scheme 1 Oxidation of glucose to formic acid. | ||
The oxidation of carbohydrate to FA using molecular oxygen (O2) as oxidant have been performed in the presence of vanadium-based catalysts,11,12 such as NaVO3 and polyoxometalates (POM), with high activity for glucose oxidation due to their strong Brønsted acidity and high oxidation ability,13,14 but with important drawbacks such as long reaction times and high O2 pressures. Another approach employs hydrogen peroxide (H2O2) as oxidant for the oxidative decomposition of glucose to FA and catalyzed by homogeneous bases (Scheme 2).15,16 The degradation of an aldohexose begins with the addition of a hydroperoxide anion to the aldehyde carbon atom of the aldohexose acyclic form. This reaction produces an unstable adduct, which then decomposes to yield the next lower aldose (a pentose) and formic acid. The degradation process proceeds stepwise, ultimately breaking down the hexose into six moles of formic acid, unless interrupted by a glycosidic linkage lacking a hydroxyl group on the carbon adjacent to the peroxide, which prevents further decomposition.17,18 Ketose degradation can also take place in the same way, taking into account the lower electrophile character of ketone respect to aldehyde.
 | ||
| Scheme 2 Oxidation mechanism of aldose and ketose by hydroperoxide anion into formic acid. | ||
In the last decade, different studies using heterogeneous based catalysts have been reported using metallic oxide19 and mixed oxides derived from layered double hydroxides (LDH), because of the environmentally friendly nature of their recyclable nature attributed to simple separation and recovery, as well as its applicability to continuous reactor operations.18,19 The oxidation of monosaccharides was carried out using calcined Mg–Al hydrotalcite and H2O2, in ethanol, at 343 K, within for 5 h, reaching high 78% FA yield. The hydrotalcite-based catalyst was reusable twice. More recently, the efficient MgO–H2O2 catalytic performance was reported, achieving very good 78.6% FA yield with 90.5% glucose conversion at 323 K, in water. Moreover, the catalyst could be recycled five times without activity decline.19
Unlike conventional LDH–zeolite composites, which often rely on mechanical mixing or deposition methods with limited structural control, this work explores the potential of combining lamellar inorganic building blocks, MWW-type zeolitic layers and MgAl layered double hydroxides (LDHs), to construct well-integrated multi-component catalytic materials. The layered nature of both components facilitates the formation of interconnected architectures, where the degree of interaction and spatial distribution of active domains can be modulated by the selected synthesis strategy. By comparing in situ and ex situ approaches, we aim to establish how the synthetic route governs the structural integration and accessibility of catalytic sites, ultimately enabling a rational design of LDH–zeolite hybrid materials with tailored properties for base-catalyzed reactions involving biomass-derived substrates. In this line, advancing in the use of heterogeneous catalysts based on mixed oxides of LDH phases, we have recently reported the synthesis of different families of multi-component materials by combining MWW@type zeolitic layers with nanoscale MgAlCe oxide sub-domains using different synthetic approaches.20 After calcination, multi-component materials with integrated zeolitic MWW layers and MgAlCe oxides were obtained, revealing significant structural differences across the developed methods. Thus, in a first section of this study, we report the preparation of different multi-component materials combining the properties of individual MWW@layers and MgAl mixed oxides, following the same strategies. Developing optimized and variable synthesis conditions, it is possible to prepare in situ nanometric sub-domains of layered double hydroxides (LDHs) on the surface and inside the microporous half cavities and channels of MWW individual layers (methods I and II). Alternative approach (method III) uses swelling and intercalation of MWW layers with preformed LDH phases as intercalation units in the interlamellar space. Both ball milling and ultrasonication have been applied to enhance the intercalation and mixing of both precursors in order to enhance the interaction between MWW and LDH films and the effective combination of both phases at the nanometer scale. Ball milling is a high-energy solid-state grinding process that reduces particle size, modifies material properties and improves mixing. Ultrasonication is another effective homogenization technique that improves mixing and dispersion of materials by ultrasonic cavitation, breaking up particles and agglomerates. After calcination, materials integrating zeolitic layers and MgAl oxides were obtained, revealing significant structural differences between the different methods.
The obtained materials were characterized showing that contain accessible Lewis acidic and basic sites, showing their potential as multi-functional catalysts. In particular, method I would be based on an in situ deposition of Mg species in the presence of the MWW precursor, without the addition of external aluminium sources, so that any aluminium contributing to the formation of MgAl domains would be originated solely from partial extraction of framework Al. This would result in the generation of more Mg-rich domains, potentially with stronger basicity. Method II also would follow an in situ route, but with the co-precipitation of both Mg and Al salts within the MWW network, allowing for better control of the Mg/Al ratio and promoting the formation of homogeneously dispersed LDH-type phases. Finally, method III would involve the ex situ incorporation of preformed MgAl-LDHs through either ultrasound-assisted mixing or ball milling, enabling an assessment of how physical mixing strategies affect domain dispersion, porosity retention, and interfacial connectivity. These three approaches would be expected to impact the catalytic performance by modulating not only the basicity and strength of active sites, but also their accessibility and interaction with the zeolitic framework. Specifically, the homogeneous distribution of active sites within the porous zeolitic framework would facilitate synergetic interactions between active centres of the zeolite structure and metallic oxides located in the external layers or confined in the microporous channels present in each individual MWW layers. Moreover, the small particle size and high dispersion of MgAl oxides should improve both the accessibility to these active sites and their concentration.
Finally, in the second section of this paper the differences in the catalytic performance of the multi-component materials prepared following the three developed methods was explored for the oxidation of glucose to formic acid (FA) using H2O2. Therefore, the impact of the different structural features of the different multi-component materials over the FA yield and glucose conversion was discussed. A study of reaction conditions allowed producing FA with a moderate 47.7% yield at 66.2% glucose conversion, after 7.5 h, at 363 K, in dioxane.
2. Experimental section
2.1. Synthesis of MWW@Mg multi-component materials
All samples were prepared by a two-step procedure. The zeolite precursor was synthesized according to the method previously reported in the literature.20 In the second step, solution A was prepared by dispersing the zeolitic MWW precursor (Si/Al = 15) (0.5 g) in distilled water (300 mL) and then the desired amount of Mg(NO3)2 (1.1 g) was added. Next, aqueous solution B formed by NaOH (7.2 g), NaNO3 (1.9 g) and distilled water (200 mL) was added to solution A drop by drop to adjust and maintain the pH = 12, with constant stirring at room temperature. Finally, the obtained slurry was aged at 373 K for 24 h. Then, the precipitate was filtered, washed with distillate water and dried at 373 K overnight. These precursors were calcined at 853 K for 5 h in presence of air to obtain the final materials. Obtained solids were labelled as MWW@Mg.2.2. Synthesis MWW@MgAl-in situ multi-component materials
For obtaining MWW@MgAl-in situ materials, the synthesis of MWW precursors was carried out as previously described.20 An aqueous solution A (300 mL) consisting of Mg(NO3)2 (0.39–0.64 g), Al(NO3)3 (0.11–0.29 g) was prepared. Then, the pH was adjusted with solution B (7.2 g NaOH and 1.9 g NaNO3 in 200 mL distilled water), to pH = 12, with constant stirring at room temperature. After 10 min, the MWW zeolitic precursor (Si/Al = 15) (0.5 g) was added to the solution and maintain the stirring for 30 min. Finally, the obtained slurry was aged at 373 K for 24 h. Then, the precipitate was filtered, washed with distilled water and dried at 373 K overnight. The sample containing cerium is prepared in the same way by adding 0.06 g of Ce (NO3)3 to solution A. These precursors were calcined at 853 K for 5 h in presence of air to obtain the final materials. Obtained solids were labelled as MWW@MgAl-in situ-30, MWW@MgAl-in situ-40, MWW@MgAlCe-in situ-40 and MWW@MgAl-in situ-50, according to the LDH content in the samples. Characterization details of the sample containing cerium were included in (Fig. S1–S4).2.3. Synthesis MWW@LDH multi-component materials
In the synthesis of MWW@LDH type materials, the zeolitic MWW precursor (Si/Al = 15) and LDH (MgAl) are previously synthesized, according to the literature.20 For the swelling process, 1 g of MCM-22(P) was dispersed in 6 g of H2O milliQ and 20 g of the CTMAOH (25 wt%, 30% exchanged OH–/Br–) and 6 g of TPAOH (40 wt%, 30% exchanged OH–/Br–) were added. The obtaining slurry was heated at 353 K with constant stirring and reflux for 16 h. The solution was cooled at room temperature. The LDH (0.5 g) was added and continued stirring for 24 h, after the slurry being sonicated for 1 h. The solid was collected by centrifugation and dried at 373 K overnight. This precursor was calcined at 853 K for 5 h in the presence of air to obtain the final material, labelled as MWW@MgAl-US.In addition, 1 g of MWW precursor (Si/Al = 15) was milled in a ball mil with 0.5 g of LDH (MgAl) previously synthesized, according to the literature. The weight of the zirconium beads was 5 × grams of solid. Then, the samples were screened at room temperature and 20 rpm. The samples were calcined at 853 K in presence of air to obtain the final materials. Obtained solids were labelled as MWW@MgAl-BM.
2.4. General procedure for glucose oxidation
In a typical reaction procedure, glucose (0.96 mmol), the catalyst, the solvent (4.75 mL) and 30 wt% H2O2 (5–10 equivalent) were weighed into a 5 mL microwave vial reactor. Then, the reaction was carried out in a Biotage® Initiator + microwave apparatus at different temperatures ranging from 363 to 403 K. The reaction was monitored taking samples at different time of reaction. The catalyst was filtered off using a Milex syringe filter (0.20 μm). The samples were analyzed by high performance liquid chromatography (HPLC, Shimadzu Prominence-i LC-2030C 3D Plus) equipped with a RID-20A detector from Shimadzu Corporation, using an Aminex HPX-87H column (Bio-Rad Lab. Inc.). Aqueous H2SO4 (5 mM) at a flow rate of 0.6 mL min−1 was run through the column maintained at 333 K. Calibration curve were established for all reaction components using pure compounds (glucose, fructose, erythrose, arabinose, formic acid and glycolaldehyde dimer).Conversion, yield and selectivity were calculated following the equations shown as below:



For the recyclability test, the catalyst was filtered, washed with ethanol (20 mL) and acetone (20 mL) and calcined at 813 K for 4 h and reused for the next run.
2.5. Characterization techniques
Powder X-ray diffraction (XRD) patterns of the different samples were measured on a Philips X’PERT diffractometer equipped with a proportional detector and a secondary graphite monochromator. Data were collected stepwise over the angle range 2° < 2θ < 90° using Cu Kα (λ = 1.54178 Å) radiation.The chemical composition of the sample was measured on a Spectra AA 10 Plus, Varian, after the dissolution of the solids in HNO3/HCl/HF aqueous solution, heating in a microwave for 2 h.
Thermal gravimetric analysis and differential thermal analyses (TGA–DTA) were conducted in air streams with a Mettler Toledo TGA/SDTA 851E analyzer.
Nitrogen adsorption isotherms were measured in a Micromeritics ASAP 2010 instrument using approximately 200 mg of the adsorbent placed in a sample holder that was immersed in a liquid circulation thermo-static bath for precise temperature control. Before each measurement, the sample was treated overnight at 673 K under a vacuum. The Brunauer–Emmett–Teller (BET) method was followed for surface area and pore volume determinations. Surface area was calculated with the data in the range from 0.04 to 0.2. The total pore volume was obtained from the amount of N2 adsorbed at a relative pressure of 0.99. Finally, the external surface area and microporous volume were estimated using the t-plot method in the t range from 3.2 to 5.
Transmission electron microscopy (TEM) was performed in a JEOL model JEM1400Flash with a STEM detector. For the analysis, a small amount of the sample was dispersed in dichloromethane. Then, a drop was placed on a copper grid and dried under a lamp for 2 h. Field-emission scanning electron microscopy (FESEM) micrographs were recorded on a ZEISS Ultra 55 microscope and scanning electron microscopy (SEM) images were carried out on a Carl Zeiss Gemini 500 field emission scanning electron microscope.
The UV-Vis measurements were performed in a Cary 5 spectrometer equipped with a diffuse reflectance accessory.
Solid-state MAS-NMR spectra were recorded at room temperature under magic angle spinning (MAS) in AC-400 and AV-III HD-400 spectrometers. The single pulse 29Si and 27Al spectra were obtained at 79.5 MHz and 103.8 MHz, respectively, and with a 7 mm Bruker BL-7 probe. 29Si were referred to as tetramethylsilane, and 27Al were referred to as aluminum nitrate.
The nature of acid sites was studied using TPD of pre-adsorbed and desorbed NH3 on an Autochem II (Micromeritics). Approximately, 100 mg samples were pretreated at 473 K for 30 min under an O2 flow (10 mL min−1), then outgassed for 15 min under an He flow (100 mL min−1) and cooled to 373 K. Then, NH3 adsorption was performed at 373 K, using pulses of a known volume, until saturation of the solid. Finally, the sample was heated up to 1073 K (10 K min−1) in He flow (100 ml min−1). NH3 desorption was analyzed with a thermal conductivity detector and a mass spectroscopy (OnmiStar, Pfeiffer).
FTIR spectra were recorded with a Thermo Nicolet 8700 FTIR spectrophotometer and a conventional quartz infrared cell connected to a vacuum dosing system. The catalyst powder was pressed into self-supporting wafers (10 mg) and activated at 523 K in vacuum for 1 h, before the adsorption experiments. Adsorption of deuterated chloroform (4–90 mbar) was performed at room temperature with a calibrated volume (1.55 cm3), followed by time-controlled evacuation at the same temperature. In all cases, FTIR spectra of the unloaded catalyst sample were recorded as reference spectra. IR bands at 2004 and 1880 cm−1 on the reference sample were used for normalization of the spectra. Deconvolution of the IR spectra was done with the ORIGIN software program and Gaussian-type curves were considered.
3. Results and discussion
Three distinct synthesis strategies were employed to synthesize the multi-component materials, integrating MWW zeolitic layers with MgAl oxide sub-domains. These strategies were designed to optimize the structural and functional properties of the resulting composites while enabling flexibility in the incorporation and distribution of active sub-domains. Method (I) involved the in situ formation of MgAl layered double hydroxides (LDHs) directly on the external surfaces and within the microporous cavities of MWW layers. This approach utilized the intrinsic aluminum from the MWW framework as a source for the LDH formation, avoiding the introduction of external aluminum precursors. Method (II) extended this strategy by introducing an external source of aluminum, along with magnesium precursors, to facilitate the in situ formation of LDHs. This approach allowed the generation of well-dispersed nanometric LDH sub-domains that strongly interacted with the MWW layers, enhancing the structural integration between the two building components. Method (III) employed a combination of swelling and intercalation steps together with the use of advanced physical techniques such as ball-milling and ultrasonication to improve the interaction and dispersion of MWW layers with preformed LDH phases. The swelling process separated the individual MWW layers, creating space for LDH insertion. Subsequently, ultrasonication was applied to achieve intimate mixing and reduce particle sizes, promoting close contact between the lamellar components. Ball-milling further enhanced this process by breaking agglomerates and achieving a uniform dispersion of the precursors. This methodology enabled the preparation of composites with a highly interconnected structure and evenly distributed active sub-domains. These synthesis strategies collectively highlight the versatility of the methodologies in tailoring the structural features of the multi-component materials (Scheme 3). | ||
| Scheme 3 Synthesis methodologies applied to prepare lamellar multi-component materials based on MWW-type zeolitic layers and MgAl oxide sub-domains combined at the nanometric scale. | ||
3.1. Physico-chemical and textural properties of multi-component MWW@LDH-type materials
From XRD analysis of multi-component MWW@LDH-type materials synthesized via method (I) (Fig. 1), it was observed that the as-synthesized materials maintained the characteristic reflections of the laminar MWW precursor (MCM-22(P)), indicating that the synthesis conditions preserved the zeolitic framework. However, no clear diffraction peaks corresponding to MgAl layered double hydroxide (LDH) phases were observed, suggesting that under these synthesis conditions, the in situ formation of well-structured hydrotalcite domains was not favored. Instead, the presence of diffraction peaks attributed to Mg(OH)2 suggested the formation of magnesium hydroxide species rather than well-defined LDH phases. This result indicated that, apparently, aluminum from the zeolite framework did not contribute effectively to the direct formation of MgAl hydrotalcite-like structures in the as-synthesized state. After calcination, notable structural changes were observed. The disappearance of Mg(OH)2 reflections confirmed the thermal decomposition of hydroxide species, leading to the formation of mixed MgAl oxides. The emergence of new diffraction peaks associated with MgAlO2 suggested that the aluminum initially present in the tetrahedral framework of the MWW layers contributed to the formation of MgAl mixed oxides (MgAl MO) upon heating. This transformation would favor to the generation of catalytically oxide phases, which were expected to exhibit improved acid–base and redox properties. Despite these changes, the MWW-related reflections remain visible, confirming that the zeolitic framework was largely retained after calcination. However, slight modifications in peak intensity and broadening suggested subtle alterations in the interlayer structure, possibly due to the dispersion of oxide species within or on the surface of the MWW layers. | ||
| Fig. 1 XRD patterns of multi-component MWW@Mg materials obtained following synthesis method (I): (top) as-synthesized samples; (bottom) calcined samples. | ||
The chemical analysis of the MWW@Mg materials synthesized via method I provided valuable insights into the incorporation of Mg and Al like additional oxide phases integrating the multi-component structure (Table 1). The results indicated a significant incorporation of magnesium, suggesting that the synthesis conditions, particularly the alkaline environment, facilitate the formation of Mg-based oxide sub-domains, while the majority of aluminum remained within the zeolitic framework. The observed reduction in the Si/Al molar ratio compared to the theoretical value for the parent MWW zeolite indicated that partial desilication may have occurred during synthesis. This structural modification likely contributed to the enhanced integration of Mg species into the zeolitic layers, affecting both the textural and acidic properties of the material. This is consistent with the hypothesis that Mg is predominantly present in oxide sub-domains, while aluminum remains largely within the zeolite framework. These findings confirmed that method I promoted the incorporation of high amount of Mg into the MWW structure, with limited extraction of zeolitic tetrahedral aluminum contributing to the segregation of oxide phases.
| Sample | %Si | %Al | %Mg | %Ce | Si/Al | Al/(Mg + Al + Ce) | ||
|---|---|---|---|---|---|---|---|---|
| Theor. | Exp. | Theor.a | Exp.a | |||||
| a For these molar ratio values, aluminum from MWW zeolitic material is not considered (Si/Al molar ratio of the MWW precursor is 11, based on the chemical analysis results obtained of this material). | ||||||||
| MWW@Mg materials | ||||||||
| MWW@Mg | 19.9 | 2.2 | 18.2 | — | 15 | 8.7 | 0.11 | 0.02 |
| MWW@MgAl-in situ materials | ||||||||
| MWW@MgAl-in situ-30 | 16.0 | 4.2 | 10.1 | — | 10 | 3.7 | 0.20 | 0.20 |
| MWW@MgAl-in situ-40 | 15.2 | 5.4 | 11.0 | — | 7 | 2.7 | 0.20 | 0.25 |
| MWW@MgAl-in situ-50 | 14.7 | 5.7 | 12.9 | — | 7 | 2.5 | 0.20 | 0.20 |
| MWW@MgAlCe-in situ-40 | 17.3 | 5.5 | 11.6 | 3.3 | 8 | 3.0 | 0.20 | 0.23 |
| MWW@LDH materials | ||||||||
| MWW@MgAl-US | 13.8 | 5.1 | 10.2 | — | 15 | 2.6 | 0.23 | 0.27 |
| MWW@MgAl-BM | 26.1 | 3.6 | 6.8 | — | 15 | 7.0 | 0.23 | 0.20 |
The structural integrity of the MWW zeolitic framework after the incorporation of MgAl oxide sub-domains was evaluated through solid-state 29Si and 27Al NMR spectroscopy (Fig. 2). The 29Si spectra confirmed that the characteristic signals of MWW zeolites persisted, showing resonances associated with the Q3 and Q4 silicon environments. This indicates that the fundamental zeolitic structure remained intact throughout the synthesis process. However, a notable increase in the intensity and alteration of the Q2 and Q3 signals is observed in the multi-component MWW@Mg material, suggesting a greater presence of surface silanol groups and partially disrupted silicate environments. This shift would indicate that the formation of rich Mg oxide sub-domains could induce local modifications in the zeolitic framework, likely due to interactions between the oxide species and external silanol groups. The 27Al NMR spectra revealed the presence of both tetrahedral and octahedral aluminum species. The tetrahedral coordination corresponds to aluminum atoms integrated within the zeolite framework, while the octahedral contribution is attributed to extra-framework aluminum species, likely associated with MgAl oxyhydroxide domains formed during synthesis. Given that with this methodology the only aluminum source was the MWW structure itself, these results indicated that part of aluminum was mobilized from the zeolitic framework and contributed to the formation of new oxide domains.
 | ||
| Fig. 2 29Si BD MAS NMR (left) and 27Al MAS NMR (right) spectra. (a) As-synthesized multi-component MWW@Mg materials obtained through method (I). (b) As-synthesized multi-component MWW@MgAl-in situ materials obtained following synthesis method (II) with different LDH content. (c) As-synthesized multicomponent MWW@LDH materials obtained following synthesis method (III) using ultrasonic and ball milling techniques. | ||
The N2 adsorption isotherm of the MWW@Mg sample revealed significant differences in textural properties compared to the pristine MCM-22 zeolite, aligning more closely with the behavior observed for the LDH (MgAl) reference material (Fig. 3). This suggested that the formation of MgAl-based oxide sub-domains significantly altered the porosity and surface characteristics of the MWW framework. A notable reduction in microporous contribution was expected due to the deposition of Mg-based oxide phases within the zeolitic layers and interlayer spaces, which lead to partial blockage of micropores (Table 2). This effect is consistent with the trend observed in similar multi-component materials, where the incorporation of metal oxide sub-domains restricts access to the internal pore system of the zeolite.20 Given that this material was synthesized at a pH of approximately 12.5, the structural evolution observed in its textural properties was in agreement with previous findings, where a higher pH promoted the formation of more extensive oxide sub-domains, leading to a denser and more compact structure with reduced microporosity. These findings highlighted the structural transformation of the MWW@Mg samples, where the formation of MgAl oxide sub-domains modified the original zeolitic texture.
 | ||
| Fig. 3 N2 adsorption isotherms. (a) Multi-component MWW@Mg materials obtained through method (I). (b) Multi-component MWW@MgAl-in situ materials obtained through method (II) with different LDH contents. (c) Multi-component MWW@LDH materials obtained following synthesis method (III). | ||
| Sample | BET surface area (m2 g−1) | t-Plot micropore area (m2 g−1) | t-Plot micropore volume (cm3 g−1) | V total (cm3 g−1) |
|---|---|---|---|---|
| MCM-22 | 487 | 408 | 0.162 | 0.490 |
| LDH (MgAl) | 226 | 140 | 0.075 | 0.550 |
| MWW@Mg materials | ||||
| MWW@Mg | 269 | 181 | 0.088 | 0.413 |
| MWW@MgAl-in situ materials | ||||
| MWW@MgAl-in situ-30 | 391 | 254 | 0.105 | 0.480 |
| MWW@MgAl-in situ-40 | 378 | 192 | 0.076 | 0.519 |
| MWW@MgAl-in situ-50 | 388 | 226 | 0.095 | 0.643 |
| MWW@MgAlCe-in situ-40 | 347 | 156 | 0.066 | 0.501 |
| MWW@LDH materials | ||||
| MWW@MgAl-US | 293 | 264 | 0.108 | 0.318 |
| MWW@MgAl-BM | 429 | 372 | 0.156 | 0.424 |
In the case of multi-component materials prepared from method (II), XRD patterns of the as-synthesized MWW@MgAl-in situ materials revealed the characteristic reflections of the laminar MWW precursor (MCM-22(P)), confirming that the layered zeolitic framework remained intact following the synthesis procedure (Fig. 4). Unlike the materials obtained via method (I), these samples exhibited additional diffraction peaks corresponding to well-defined MgAl layered double hydroxides (LDHs). The presence of these signals, particularly in the low-angle region, indicated that under the synthesis conditions of method (II), the formation of LDH structures was probably achieved. This result contrasted with method (I), where no LDH phases were detected in the as-synthesized samples, and only Mg(OH)2 was observed. The difference suggests that the introduction of an external aluminum source in method (II) plays a crucial role in driving LDH formation, rather than relying solely on aluminum from the MWW framework. Moreover, the distinct diffraction signals suggested that these LDH domains were more homogeneously distributed within the composite, potentially intercalating into the interlayer space of the MWW structure rather than simply depositing on its external surface. Following calcination, the disappearance of LDH-related diffraction peaks confirmed the thermal decomposition of these phases into mixed oxides, primarily MgAlO2. As with the evolution observed in method (I), the formation of these oxide phases was accompanied by changes in peak intensities and slight broadening, indicating subtle modifications within the interlamellar regions of the MWW layers. In contrast, a key difference was that in method (II), the initial presence of LDH structures could lead to a more defined transformation pathway, likely resulting in a more uniform dispersion of the mixed oxides. On the other hand, although the MWW structure remains largely preserved, the peak broadening suggests that the thermal treatment may induce partial structural rearrangements, possibly due to strong interactions between the newly formed oxide domains and the zeolitic layers.
 | ||
| Fig. 4 XRD patterns of multi-component MWW@MgAl-in situ materials obtained following synthesis method (II): (top) as-synthesized samples; (bottom) calcined samples. | ||
The chemical analysis of the MWW@MgAl-in situ materials confirmed the successful incorporation of Mg and Al into the multi-component structures through the in situ formation of LDH sub-domains within the MWW framework (Table 1). The higher use of inorganic precursors during synthesis was reflected in the increasing levels of Mg and Al in the final materials, indicating the progressive development of MgAl LDH phases as the synthesis parameters were adjusted. These sub-domains would likely be distributed within the interlayer spaces and internal voids of the MWW structure, contributing to both the structural and chemical modification of the materials. Compared to the materials synthesized via method I, where the incorporation of Mg was prominent but the extraction of Al from the zeolitic framework was limited, the materials obtained via method II exhibited a more balanced integration of both Mg and Al. This suggests that the use of external aluminum sources in method II enhanced the formation of LDH phases, while in method I, the low aluminum availability restricted LDH development, favoring instead the formation of separate Mg oxide sub-domains. Moreover, the Si/Al molar ratios in the MWW@MgAl-in situ materials showed a more pronounced decrease compared to the parent MWW structure, indicating greater chemical modification due to the in situ formation of LDH sub-domains, associated with the presence of external aluminum sources during the synthesis process.
The 29Si NMR spectra confirmed that the structural integrity of the MWW framework was maintained after synthesis, as evidenced by the presence of characteristic Q-type resonances (Fig. 2). Similarly, to the materials obtained via method I, where appreciable alterations in the Q2 and Q3 signals were observed, the in situ formation of MgAl LDH sub-domains in method II lead to a slight increase in intensity and broadening of these signals, associated to the interaction between the zeolitic layers and the newly formed LDH phases, potentially through interactions with surface silanol groups. The 27Al NMR spectra provided further insight into these structural differences. In both methods, tetrahedral aluminum species associated with the zeolite framework remained present, confirming that the layered structure was largely preserved. However, while the materials obtained via method I primarily exhibit tetrahedral aluminum with only a limited presence of octahedral species, method II leads to a more significant contribution of octahedral aluminum (∼15% for method I front ∼50% for method II). This indicated that the introduction of an external aluminum source in method II facilitated the formation of LDH sub-domains, effectively integrating MgAl phases within the MWW structure. These results highlighted that while method I relied solely on the aluminum present in the MWW framework, limiting the extent of LDH formation, method II successfully generated well-dispersed MgAl LDH sub-domains, leading to a more homogeneous distribution of octahedral aluminum. This structural distinction may play a key role in influencing the catalytic properties of the materials, which will be explored in the following section. The N2 adsorption isotherms of the MWW@MgAl-in situ materials showed significant modifications in their textural properties compared to the pristine MCM-22 zeolite, reflecting the influence of the in situ formation of MgAl LDH phases and their transformation into mixed oxides upon calcination (Fig. 3). A notable decrease in specific surface area and micropore volume was observed in these materials, indicating that the formation of MgAl phases within the zeolitic structure resulted in a more compact network with reduced microporosity. This effect was particularly evident when comparing the micropore area and volume values, which were lower than those of MCM-22, suggesting that the newly formed oxide domains contributed to the partial blockage of microporous cavities and channels in the zeolitic framework (Table 2). Similar to what was observed for method I, the reduction in microporosity would likely be related with the deposition of MgAl phases within the zeolitic layers, limiting access to the internal pore network, and with the homogeneous distribution of oxide sub-domains, which formed a denser structure that restricted nitrogen adsorption in microporous regions. However, despite the loss of microporosity, an increase in total pore volume was observed, particularly in samples with a higher content of in situ formed LDH phases. This phenomenon could be attributed to the formation of smaller oxide crystallites upon calcination, leading to the generation of interparticle porosity, together with the presence of additional mesoporous, resulting from the arrangement of MgAl phases within the MWW layers.
For the multi-component materials prepared from method (III), XRD patterns of the as-synthesized materials confirmed the retention of the characteristic reflections of the laminar MWW precursor (MCM-22(P)), indicating that the swelling and intercalation processes did not compromise the integrity of the zeolitic structure (Fig. 5). Unlike method I, where LDH phases were not detected prior to calcination, and method II, where they were generated in situ, method III successfully incorporates preformed MgAl LDH phases into the interlamellar spaces between MWW layers. This was evidenced by the presence of diffraction peaks corresponding to LDH structures, confirming their integration within the composite. When comparing the two processing techniques employed in method III (ultrasonication and ball milling), no significant structural differences are observed in the as-synthesized state. In both cases, the LDH reflections remain well-defined, suggesting that the intercalation process was similarly effective in both treatments. After calcination, the disappearance of LDH-related peaks confirmed the thermal decomposition of these phases, leading to the formation of mixed MgAl oxides. As seen in methods I and II, diffraction peaks corresponding to MgAlO2 emerged, indicating the transformation of LDH sub-domains into catalytically active oxide species. A significant difference between the two processing techniques becomes evident after calcination because the sample prepared via ultrasonication (MWW@MgAl-US) exhibited broader and less intense diffraction bands compared to the ball-milled sample (MWW@MgAl-BM). This could suggest a greater degree of dispersion and/or a lower degree of crystallinity in the multi-component materials obtained by ultrasonication, which may influence the textural properties and catalytic performance of the final material.
 | ||
| Fig. 5 XRD patterns of multi-component MWW@LDH materials obtained following synthesis method (III): (top) as-synthesized samples; (bottom) calcined samples. | ||
The chemical analysis of the MWW@LDH materials synthesized via method III revealed significant modifications in the Si/Al molar ratios compared to the theoretical value of the MWW zeolite precursor (Table 1). While the starting MWW material had a Si/Al ratio of 15, the intercalated materials exhibit substantially lower Si/Al ratios, indicating a pronounced alteration of the zeolitic framework due to the intercalation of preformed LDH phases. The ultrasonicated samples (MWW@MgAl-US) showed a marked reduction in the Si/Al ratio, suggesting that the cavitation effects during ultrasonication promoted significant desilication together with the incorporation of MgAl species into the zeolitic framework. This pointed to a deeper interaction between the LDH phases and the MWW layers, leading to substantial chemical modification. In contrast, the ball-milled samples (MWW@MgAl-BM) exhibited a higher Si/Al ratio compared to the ultrasonicated material, although still significantly lower than the theoretical value. This could indicate that ball milling preserved the zeolitic framework more effectively, limiting the extent of desilication. The mechanical forces in ball milling likely enhanced the dispersion of LDH phases without deeply penetrating or chemically altering the MWW layers. In comparison to method I, where aluminum incorporation was limited and Mg oxide phases predominated, the materials synthesized via method III exhibited a more pronounced alteration of the zeolitic framework due to the intercalation of preformed LDH phases. Similarly, method III showed parallels with method II, where in situ LDH formation facilitated a controlled and homogeneous integration of Mg and Al into the final multi-component structures. Method III also induces significant framework modifications, particularly in the samples processed via ultrasonication, where the intercalation process led to a more aggressive chemical alteration of the solids.
The 29Si NMR spectra confirmed that the MWW zeolitic structure remained intact after the swelling and intercalation process, as evidenced by the characteristic Q3 and Q4 silicon resonances (Fig. 2). This observation aligns with the results obtained for the materials synthesized via methods I and II, where the integrity of the zeolitic framework was also preserved. However, in contrast, above all, to method I, where Q2 and Q3 signals were altered due to interactions with in situ formed LDH phases, the materials synthesized via method III did not exhibit a significant modification in these resonances. This suggested that while the LDH domains were effectively intercalated by physical mixing procedures, the interactions of the LDH domains with the surface zeolitic silanols were less strong compared to the LDH domains generated in situ. The 27Al NMR spectra revealed the coexistence of tetrahedral and octahedral aluminum environments, a trend observed across all synthesis methods. The tetrahedral aluminum species corresponded to framework aluminum in the MWW layers, confirming that the zeolitic structure is preserved. The octahedral aluminum contribution, in this case, was attributed to the preformed MgAl LDH phase incorporated into the MWW matrix. Compared to method I, where only a limited amount of octahedral aluminum was detected due to the absence of an external aluminum source, method III exhibited a more pronounced octahedral signal, similar to what was observed in method II. This confirms that introduction of preformed LDH phases ensured a stable integration of MgAl sub-domains within the composite. When comparing both techniques used in method III, ultrasonication versus ball milling, no significant differences in the NMR spectra are observed, indicating that both approaches lead to a similar aluminum coordination environment. Unlike method I and II, where in situ formation of LDH phases resulted in a more chemically interactive environment, the intercalated LDH phases in method III appeared to be structurally well-incorporated but with potentially weaker interactions with the zeolitic framework.
The N2 adsorption–desorption isotherms of the MWW@LDH materials synthesized via method III, through ultrasonication technique (MWW@MgAl-US), revealed a marked decrease in specific surface area and microporosity compared to pristine MCM-22, highlighting the impact of the intercalation of MgAl LDH phases within the interlayer space of the zeolitic framework (Fig. 3 and Table 2). This reduction in textural parameters was primarily attributed to the partial blockage of internal porosity, as the inserted LDH domains, in the interlamellar region, prevent access to the microporous channels of the MWW layers. These textural modifications confirmed that the swelling and intercalation steps introduced significant structural rearrangements, leading to a more compact network where the zeolitic and oxide subdomains coexist. A comparison between the both techniques in method III (ultrasonication vs ball milling process) revealed significant differences in textural properties. In fact, the ball-milled sample (MWW@MgAl-BM) exhibited a higher BET surface area (429 m2 g−1) and micropore volume (0.156 cm3 g−1) compared to the ultrasonicated sample (MWW@MgAl-US), which presented a lower surface area (293 m2 g−1) and micropore volume (0.108 cm3 g−1). This would suggest that ball milling was more effective in preserving microporosity, potentially by enhancing the dispersion of LDH phases and preventing excessive aggregation phenomena. The ultrasonicated sample, in contrast, showed a greater reduction in microporosity, indicating that the LDH domains might be more compactly arranged, further restricting access to zeolitic channels.
While all three synthesis approaches lead to modifications in the textural properties of the materials, the extent of these changes varies depending on the synthesis conditions and the nature of the oxide sub-domains formed. In method I (MWW@Mg samples), the MgAl oxide sub-domains were deposited on the external surface of the MWW layers, which could result in a marked reduction of microporosity while still retaining accessible textural properties. In method II (MWW@MgAl-in situ samples), the formation of MgAl LDH phases within the zeolitic framework introduces also significant structural modifications. However, as different LDH loadings were used, the resulting materials exhibited a range of textural properties, with some samples retaining features closer to the MCM-22 precursor, while others showed a more pronounced shift toward the behavior of LDH-derived oxides. In method III (MWW@LDH intercalated with MgAl-LDH phases), the insertion of preformed LDH phases within the interlayer space tended to produce materials with lower microporosity, as the oxide sub-domains physically occupied part of the available zeolitic channels. However, the extent of this effect varied depending on the intercalation technique used, ultrasonication or ball milling, which influenced the degree of porosity retention. Overall, these results suggest that the degree of porosity modification is not solely dependent on the synthesis method but also on the specific conditions used within each approach, such as LDH loading, oxide dispersion, and intercalation efficiency.
In addition, the thermal stability of the multi-component materials was evaluated by thermogravimetric analysis (TGA) and their corresponding derivative (DTG) curves (Fig. S5), evidencing that all samples exhibited good thermal stability with major decomposition steps occurring below 823 K, i.e. within the temperature range relevant to most catalytic applications.
3.2. Field emission scanning electron microscopy (FE-SEM) and high-resolution FE-SEM (HR-FE-SEM) analysis
Field emission scanning electron microscopy (FE-SEM) and high-resolution FE-SEM (HR-FE-SEM) were employed to capture microstructural features of the different materials following the three synthesis methods (Fig. 6). The obtained images illustrate the structural differences among the calcined multi-component materials, highlighting the effects of the synthesis approach on morphology and achieved organization. The MgAl MO material consists of round-shaped platelets, a characteristic feature of hydrothermally treated LDH phases. In contrast, MWW exhibits rose-like morphology, composed of irregularly packed individual layers. Images of MWW@Mg and MWW@MgAl-in situ-40 reveal clusters of disordered MWW sheets, also rose-shaped, with rounded platelets of mixed MgAl oxides probably deposited on their outer surface and in the inner pores. The MgAl platelets in these materials appear highly dispersed and smaller in size compared to those in MgAl MO. On the other hand, the MWW@MgAl-BM and MWW@MgAl-US samples show a homogenous mixture of irregularly packed MWW sheets and small clusters of MgAl MO platelets. In addition, the multi-component particle sizes of MWW@MgAl-BM and MWW@MgAl-US are smaller than those of the pristine MWW materials, confirming the decrease in particle size induced by ball-milling and ultrasonication techniques. HR-FE-SEM allowed also distinguishing the scalped and irregular edges of the MWW zeolitic sheets for MWW@MgAl-BM samples, providing evidence of the mechanical stress from the ball milling process (Fig. 6(f)). Accordingly, FE-SEM analysis suggests that method II may lead to the formation of multi-component materials where the MgAl-based sub-domains are more intimately associated with the zeolitic framework, as evidenced by the apparent deposition of oxide platelets around the MWW crystals, forming partially continuous coatings (Fig. 6(d)). This could be indicative of stronger interfacial interactions between both components. In contrast, the composites obtained through method III, based more on physical intercalation strategies, appear to show a more heterogeneous distribution of the MgAl domains, with the oxide and zeolitic phases being more discernible and exhibiting a less uniform structural integration. The images from the ultrasonicated and ball-milled samples suggest a physical mixture where the components are well dispersed but retain more individual morphological features. Although these interpretations are based on qualitative observations, they are supported by and consistent with the trends observed in the XRD and chemical analysis results. | ||
| Fig. 6 HR-FE-SEM and FE-SEM micrographs for the multi-component materials obtained using methods (I), (II) and (III): (a) MWW zeolitic precursor, (b) MgAl MO, (c) MWW@Mg, (d) MWW@MgAl-in situ-40, (e) MWW@MgAl-US and (f) MWW@MgAl-BM. | ||
3.3. Acidity and basicity
The acidity of the multi-component MWW@ materials synthesized via the three methods was evaluated by NH3 temperature-programmed desorption (NH3-TPD) to assess both the acidity and strength of the acid sites present in the materials (Fig. 7 and Fig. S6–S10). Based on desorption temperatures, acid sites can be classified as weak (423–523 K), medium (523–623 K) and strong (623–723 K).21 | ||
| Fig. 7 NH3 thermoprogrammed desorption (TPD) curves. (a) Multi-component MWW@Mg materials obtained through method (I). (b) Multi-component MWW@MgAl-in situ materials obtained through method (II) with different LDH content. (c) Multi-component MWW@LDH materials obtained through method (III). | ||
The desorption profile of the pristine MWW sample exhibited a desorption band around 492 K, likely associated with physisorbed NH3 interacting with silanol groups on the external surface of the MWW layers. A more intense and broad desorption peak centered at ∼653 K corresponding to medium to strong acid sites strength, likely linked to the tetrahedral aluminum within the zeolite framework, was deconvoluted into two bands centered at ∼666 K and ∼835 K corresponding to Brønsted and Lewis acidity, respectively (Fig. S6). For the MgAl MO sample, a broad desorption band centered at ∼633 K was observed, characteristic to medium-to-strong Lewis acid sites, arising from aluminum species in the MgAl mixed oxides.
The MWW@Mg material revealed three primary desorption regions ∼552 K, ∼657 K and ∼710 K. The low-temperature band attributed to physisorbed NH3 was suppressed, while the higher temperature desorption peaks were ascribed to the combination of Lewis acidity from the zeolite and the MgAl oxide phases. Notably, the total NH3 desorbed (114.2 cm3 g−1) was significantly higher than that of the pristine MCM-22 precursor (29.3 cm3 g−1), indicating the formation of additional acid sites due to incorporation of oxide phase (Table S1). The NH3-TPD profiles of the MWW@MgAl-in situ, MWW@MgAl-US and MWW@MgAl-BM materials all revealed four primary desorption regions. A low-temperature band at 446–451 K was attributed to physisorbed NH3 while the higher temperature desorption bands were consistent with acid sites derived from both the zeolite and the MgAl oxide phases. As it is supported below by IR study in the OH stretching region (4000–3400 cm−1), where practically all bands associated with Brønsted acidity disappeared in all the Mg-containing MWW@samples. Consequently, the NH3-TPD bands are mostly attributed to Lewis acidity.
In the MWW@MgAl-in situ series (Fig. 7(b)), the intensity of the desorption band in the range 493–643 K varied with LDH content. Samples with higher in situ MgAl LDH loading exhibited increased NH3 desorption, again confirming the introduction of additional acid sites through MgAl oxides incorporation (Table S1). Interestingly, despite the higher MgAl oxides content in the materials synthesized via method II, the total NH3 desorbed was lower than in method I (∼50–60 cm3 g−1). This unexpected result may be attributed to differences in acid site accessibility. In method I, MgAl oxides formed primarily on the outer surface of the MWW layers, likely leading to higher proportion of easily accessible acid sites. In contrast, method II resulted in more homogeneously dispersed MgAl subdomains within the interlamellar regions, leading to partial sites confinement and reduced accessibility. These findings suggest that method I, although producing fewer oxide domains, generates a greater proportion of accessible and stronger Lewis acid sites, as evidenced by the more intense NH3-desorption band at ∼710 K.
For materials synthesized via method III using ultrasonication and ball-milling-assisted physical mixing, NH3-TPD profiles were also similar, showing four distinct NH3 desorption regions (Fig. 7(c)). The MWW@MgAl-US material showed a total NH3 desorption of 67.7 cm3 g−1, higher than that the pristine MWW precursor, confirming additional Lewis acidity introduced through MgAl oxide incorporation. However, the total number of accessible acid sites was lower than that observed for materials prepared via method I. MWW@MgAl-BM material exhibited a desorption profile resembling a superposition of the individual MWW and MgAl MO components, suggesting limited interaction and primarily physical mixing. The high NH3 desorption (111.9 cm3 g−1) further supports this interpretation, indicating cumulative acidity from both phases (Table S1).
The basicity of the different multi-component MWW@samples was explored using IR spectroscopy with deuterated chloroform (CDCl3) absorption at room temperature (Fig. 8). Depending on the strength of the basic sites that interacts with CDCl3, different bands corresponding to C–D stretching vibrations appear in the range 2100–2300 cm−1. Studies on CDCl3 adsorption upon metallic oxides have identified two characteristic bands in the 2210–2220 cm−1 and 2245–2250 cm−1 ranges.22,23 The first band is associated with the interaction of CDCl3 with strongly basic surface oxygen anions (state of type C, Scheme S1), while the second band corresponds to the adsorption involving weakly basic OH groups (state of type B and B′, Scheme S1). In the absence of hydroxyl groups, the bands are assigned to type C and type D (Scheme S1). In general, IR spectra showed νCD frequency at ∼2193 and ∼2217 cm−1 which corresponded to strong basic sites and νCD frequency ∼2235 cm−1 corresponding to medium basic sites. Additionally, a band was registered at ∼2250 cm−1 attributed to the presence of weak basic sites associated to oxygen atoms of OH groups such as Al–OH.24 A study on the incorporation of silica to alumina also revealed a shift of the weak basic sites band at 2255–2258 cm−1, a range close to the basicity of silica gel OH groups.25,26
 | ||
| Fig. 8 FT-IR spectra of the different MWW@samples at maximum coverage of CDCl3 at 298 K and 90 mbar. | ||
Herein, the obtained results show that MMW sample exhibits an intensive band at ∼2256 cm−1 that corresponds to the presence of weak basic sites associated with alumina (extra-framework aluminum, EFAL) and aluminosilicate species. Studies on alumina basicity described that strong to medium basic sites arise from bridging oxygen atoms (Al–O–Al) (Fig. S11). MgAl-MO CDCl3 adsorption spectra showed a broad absorption band in the range 2260–2160 cm−1, which, upon deconvolution using a Gaussian fitting method, was decomposed into three peaks at 2182, 2220 and 2245 cm−1. These peaks correspond to strong, medium and weak basics sites,27 respectively (Fig. S12). Notably, MgAl MO sample exhibited the highest intensities for medium and strong basic sites (Fig. 8), indicating its high overall basicity. Similarly, MWW@MgAl-BM CDCl3 adsorption spectra displayed a broad absorption band in the range 2260–2160 cm−1, which was deconvoluted into three peaks at 2166, 2219 and 2258 cm−1, corresponding to strong, medium and weak basics sites, respectively (Fig. S13). MWW@MgAl-in situ (Fig. S14), MWW@MgAl-US (Fig. S15) and MWW@Mg CDCl3 (Fig. S16) adsorption spectra all showed an intense band at ∼2256 cm−1, which, after deconvolution, was resolved into two peaks at ∼2256 and ∼2223 cm−1, corresponding to weak and medium basics sites, respectively. In all multi-component MWW@samples, the weak basic site are most likely oxygen atoms in surface OH groups, a combination of MWW and MgAl MO species, while the medium strength basic sites are O2− ions associated to MgAl MO species.
A comparative analysis of the CDCl3 absorbance for weak, medium and strong basic sites and the combination of strong and medium basic sites, (S + M), for the different MWW@samples, is shown in the Fig. S17 and S18. The MgAl MO sample exhibited the highest basicity in term of (S + M) values. Among the prepared samples the (S + M) basicity follows the decreasing order: MWW@MgAl-US > MWW@Mg > MWW@MgAl-in situ > MWW@MgAl-BM. Particularty, the MWW@MgAl-BM sample, synthesized via under ball milling method, differs among the other MWW@samples in two aspects. First it presents strong basic sites, and second, its medium and strong basic sites band exhibited a shift of the νCD frequencies by 1–16 cm−1 to lower values in comparison to MgAl MO sample. This shift suggests an enhancement in basicity for this sample. When comparing the overall basicity of the MWW@samples with MgAl MO, it appears that MgAl MO not only possesses a greater total number of active sites (M + S), but also has high proportion of strong basic sites (34%). In contrast, MWW@samples lack strong active sites or, in the case of MWW@MgAl-BM, which contains the lowest number of active sites, 15% are strong.
On the other hand, the IR analysis of the OH shechting region (4000–3400 cm−1) confirmed the presence of silanol groups, which are associated with silicon coordination defects for the activated MWW@samples (Fig. S19). For MWW sample, the IR spectrum in this range showed the presence of silanols as possible defects and acid sites due to aluminum present in the zeolitic framework. Bands associated with presence of terminal silanols, geminal silanols, aluminum hydroxide (Al–OH) and the presence of Brønsted acid sites (Al–OH–Si) were registered at 3748, 3728, 3668 and 3620 cm−1, respectively.7 A distinct band at ∼3748 cm−1 was recorded for the MWW@MgAl-in situ and MWW@MgAl-BM samples, indicating the presence of silanol groups. However, in the case of MWW@MgAl-US and MWW@Mg this band was practically vanished, suggesting a significant reduction or elimination of these groups. Moreover, the bands associated with Brønsted acidity and aluminum hydroxide disappeared for all the synthesised MMW@samples. Additionally, a broad IR band was registered in the range 3740 and 3400 cm−1 for the MgAl MO sample, which can be attributed to isolated OH groups. These groups may exist in mono- and multicoordinated forms, depending on the thermal treatment applied. Similarly, a broad adsorption feature was observed in the range 3700 and 3500 cm−1 in the spectra of MWW@MgAl-in situ, MWW@MgAl-BM, WW@MgAl-US and MWW@Mg samples further supporting the presence of hydroxyl groups in these materials associated with the presence of Mg and Al oxides.
3.4. Study of catalytic performance of the multi-component materials for the catalyzed glucose oxidation to formic acid (FA) using H2O2 as oxidant
Two general chemocatalytic oxidation strategies have been followed to produce FA from carbohydrates. One, using molecular oxygen (O2) as oxidant, while another approach employs hydrogen peroxide (H2O2) for the oxidative decomposition of glucose to FA and catalyzed by homogeneous bases (Scheme 2).15,16 Different studies using heterogeneous basic catalysts have reported the use of MgO and MgAl mixed oxides derived from layered double hydroxides (LDH). LDHs have the general molecular formula [M1−x2+Mx3+(OH)2]x+ (Ax/nn−) mH2O, and structurally have brucite-like [Mg(OH)2] sheets where isomorphous replacement of Mg2+ by a trivalent cation like Al3+ induces an excess of positive charge in the layered network, balanced by interlayers anions, while the remaining space in the interlayer is occupied by water of crystallization. It has been widely accepted that LDHs pure phases are obtained when 0.20 < x < 0.33.28 The properties of LDHs are closely related to the location in the Periodic Table of the layer cations.29 Under controlled thermal decomposition, LDHs are converted to mixed oxides with high specific surface areas and strong Lewis basic sites.30 In the case of MgAl mixed oxides, Lewis active basic sites are associated with O2−–Mg2+ acid–base pairs. The basicity of these mixed oxides can be modified by varying the characteristics of the cation, for example by replacing Mg2+ with a more electropositive ion, such as Li+ or Ca2+, increasing the negative charge density on the oxygen and thus enhancing the basicity of the resulting mixed oxide. Li and Ca present similar electronegativity by Pauling's scale close to 1 while Mg present higher one around 1.3.31 Other groups of LDHs are obtained with the introduction of Li+ ([LiAl2(OH)6]+Ay−1/y·nH2O) and Ca2+, located in octahedral positions in the structure.32,33 After calcination, amorphous basic mixed oxides are obtained (Li(Al)Ox and Ca(Al)Ox). Therefore, the influence of mixed oxide basicity on glucose conversion and FA yield was explored by preparing mixed oxides of MgAl, AlLi and AlCa and investigating their catalytic performance. The catalytic results registered for glucose oxidation in ethanol for the three samples are displayed in the Fig. 9, indicating that the less basic MgAl mixed oxide exhibited the best catalytic performance reaching highest 47% FA yield and 83% glucose conversion, while Ca–Al and Li–Al mixed oxide catalysts exhibited lower and similar results accordingly to their electronegativity. The result can be due to strong adsorption of glucose and intermediates on the active sites and their subsequent blocking. Accordingly, the catalytic performance of Mg–Al containing multi-component materials herein prepared, following the different methods, was examined for the catalytic oxidation of glucose using H2O2 as oxidizing agent. The preparation of multi-functional composite materials by combining different components in a well-designed structure is a prominent topic, as the combination can balance their drawbacks and improve their properties, leading to materials with unique properties and applications. The characterization study showed that method II allowed the co-precipitation of MgAl LDH sub-domains on MWW zeolite sheets and promoted strong interactions between the LDH phases and the MWW structure, leading to modify the chemical environment of the zeolitic structure. | ||
Fig. 9 Catalytic oxidation of glucose to formic acid in the presence LDH based catalysts. Reaction conditions: glucose (0.96 mmol), catalyst (0.15 g), EtOH (4.75 mL), 20% H2O2 (5.76 mmol), 363 K, 5 h.  erythrose yield%, erythrose yield%,  glycolaldehyde dimer yield%, glycolaldehyde dimer yield%,  arabinose yield%, arabinose yield%,  fructose yield%, fructose yield%,  FA yield%, FA yield%,  glucose conversion%. glucose conversion%. | ||
On the other hand, the catalytic system, based on glucose oxidation, was deliberately chosen not only to probe the intrinsic basicity of the multi-component materials but also to evaluate how effectively the synthetic methods generate accessible active sites for bulky reactants like carbohydrates. In this type of reaction, both the strength of the basic sites and their accessibility, directly influenced by the porosity and morphology of the catalysts, play a crucial role in determining catalytic performance. Therefore, the oxidation of glucose to FA serves as a suitable test reaction to simultaneously assess the chemical nature of the active sites and the impact of structural parameters such as hierarchical porosity and domain dispersion, which are at the core of the synthetic strategies under comparison.
Likewise, the catalytic performance for glucose oxidation in ethanol was investigated using three catalysts, MWW@MgAl-in situ-40, MgAl MO (a mixed oxide derived from MgAl LDH over calcination), and MWW@MgAlCe-in situ-40, using the same loading of MgAl mixed oxide in each reaction. Glucose, erythrose, glycolaldehyde dimer, arabinose, fructose and FA were analyzed and quantified. Glucose and fructose retroaldol can be envisaged in the presence of basic mixed oxide (Scheme 4). However, neither glyceraldehyde nor dihydroxyacetone were detected in the reaction medium. Nevertheless, glucose retroaldol cannot be completely disregarded since erythrose and glycolaldehyde dimer were reaction products. These results suggest that the main mechanism of glucose degradation to FA follows the mechanism described in Scheme 2.
 | ||
| Scheme 4 Retroaldol of fructose and glucose in the presence of basic catalyst. | ||
The catalytic results shown in Fig. 10 indicate that MgAl MO provides the highest 65.5% glucose conversion and 33.5% FA yield. MWW@MgAlCe-in situ-40 sample exhibited the lower catalytic performance for the conversion of glucose to FA. 44% glucose conversion was obtained while fructose was the main reaction product (16.9%). The lowest FA yield is likely due to the rapid consumption of H2O2 ascribed to the oxygen chemisorption on the ceria surface. This result can be ascribed to the ceria high oxygen transport and storage abilities, which arises from the presence of oxygen vacancies that has been extensively described.34,35 In the presence of MWW@MgAl-in situ-40 catalyst, a product composition similar to that obtained in the presence of Mg/Al MO catalyst was obtained, reaching 20.9% FA yield and 51% glucose conversion. The effect of MgAl LDH content within the multi-component catalyst on glucose oxidation and FA yield was also explored, revealing an optimal content close to 40 wt% (Fig. S20). This suggests a balance between number of active sites and their accessibility, which may be influenced by the pore blockage within the catalyst structure.
 | ||
Fig. 10 Catalytic oxidation of glucose to formic acid in the presence LDH derived based catalysts, (a) Mg/Al MO, (b) MWW@MgAl-in situ-40 and (c) MWW@MgAlCe-in situ-40.  Erythrose yield%, Erythrose yield%,  glycolaldehyde dimer yield%, glycolaldehyde dimer yield%,  arabinose yield%, arabinose yield%,  fructose yield%, fructose yield%,  FA yield%, FA yield%,  glucose conversion%. Reaction conditions: glucose (0.96 mmol), catalyst (0.066 g mixed oxide phase), EtOH, (4.75 mL), 30% H2O2 (5.76 mmol), 363 K. glucose conversion%. Reaction conditions: glucose (0.96 mmol), catalyst (0.066 g mixed oxide phase), EtOH, (4.75 mL), 30% H2O2 (5.76 mmol), 363 K. | ||
In order to explore the catalytic behavior of MWW@MgAl-in situ-40 sample and improve its catalytic performance, a complete reaction engineering study was performed, varying reaction parameters such as reaction time, solvent, temperature, catalyst loading and H2O2 addition mode. First, a study of the effect of water miscible solvent (water, ethanol, diglyme, dioxane, isopropanol (IPA) and acetonitrile (ACN)) but with different polarity, boiling point and glucose solubility on the catalytic performance of MWW@MgAl-in situ-40 was carried out. The solvent polarity that is measured owing to dielectric constant influences the adsorption/desorption of the products and reactants. Protic solvents, such as water, solvate anions such as hydroperoxide anion (negatively charged solutes) or sugars strongly via hydrogen bonding. Among checked solvents, H2O, EtOH and IPA are polar protic solvents while diglyme and dioxane are polar aprotic solvents with small dipole moments and low dielectric constants, and ACN is a polar aprotic solvent with large dielectric constant (Table S2). The composition of reaction medium as well as glucose conversion are depicted in the Fig. 11, at end reaction time (7.5 h) and 363 K for all solvents. The catalytic results revealed that the highest catalytic performance was achieved in the presence of polar aprotic solvent and the best 34.5% FA yield was achieved in dioxane at 52.4% glucose conversion. These results may indicate that aprotic solvents are not involved in the reaction and/or solvation of reaction intermediate and, moreover, that they do not compete with the adsorption of glucose.
 | ||
Fig. 11 Catalytic results for the oxidation of glucose to FA in the presence MWW@MgAl-in situ-40 catalyst in different solvents. Reaction conditions: glucose (0.96 mmol), catalyst (0.066 g mixed oxide phase), solvent, (4.75 mL), 20% H2O2 (5.76 mmol), 363 K, 7.5 h.  erythrose yield%, erythrose yield%,  glycolaldehyde dimer yield%, glycolaldehyde dimer yield%,  arabinose yield%, arabinose yield%,  fructose yield%, fructose yield%,  FA yield%, FA yield%,  glucose conversion%. glucose conversion%. | ||
Further, the glucose conversion in the presence of MWW@MgAl-in situ-40 in dioxane at 363 K without H2O2 was explored. Over 7.5 hours, the monitorization of the composition of the reaction medium revealed that 24% glyceraldehyde yield and 37% of glycolaldehyde dimer were obtained via retro-aldol reaction at 88.5% glucose conversion, with only 1.2% FA yield. In contrast, in the absence of catalyst but with H2O2, 10.7% glucose conversion was registered with 4.9% FA yield. These findings confirm that glucose oxidation follows an oxidation mechanism involving the formation and addition of a hydroperoxide anion catalyzed by MWW@MgAl-in situ-40 sample with basic properties.
Then, the effect of temperature on the catalytic performance of the MWW@MgAl-in situ-40 catalyst in dioxane (Fig. S21a) and water (Fig. S21b) was studied. For both solvents, the best catalytic data were registered at lower temperature. This trend can be ascribed to the hydrogen peroxide decomposition with temperature increase, leading to lower availability of the oxidant concentration in the reaction medium. The activation energy for the catalytic oxidation of glucose to formic acid in both solvents was determined using the Arrhenius equation, k = Ae−Ea/RT (Fig. S22a and b). Similar values were calculated, 87 kJ mol−1 for dioxane and 85 kJ mol−1 for water. In contrast, a lower activation energy of 45.8 kJ mol−1 activation energy was recently reported for glucose oxidation over MgO.19 These results suggest that MWW@MgAl-in situ-40 exhibits lower catalytic activity than MgO, likely due to the lower basicity of their active sites.
From industrial point of view the use of H2O2 involves different consideration in addition to the consumption optimization such as safety. H2O2 is unstable with highly exothermic hazards and the initial decomposition temperature of H2O2 ranges from 320 to 354 K, with decomposition heat about 192–1079 J g−1.36 A study of H2O2/glucose ratio was conducted, considering that complete glucose oxidation to FA requires 5 moles of H2O2. The effect of addition mode (initial, fractionated and continuous) was also evaluated at 363 K and 383 K. As shown in Fig. 12, the catalytic results indicate that highest glucose conversion and formic acid yield were achieved when 6–8 equivalents of H2O2 were used. The mode of addition did not result in significant improvements, as fractionated or continuous addition of H2O2 showed no notable advantage over initial addition. At 363 K, maximum 34.5% FA yield and 65.9% selectivity at 52.4% glucose conversion were reached for initial addition of 6 equivalents of H2O2. A similar trend was observed at 383 K, where maximum 38.1% FA yield and 59.5% selectivity at 63.9% glucose conversion were obtained for initial addition of 8 equivalents of H2O2. These findings suggests that under the reaction conditions, the availability and concentration of H2O2 are adequate and are not the most important factor in improving the process efficiency. However, for safety reasons, slow or fractionated addition may be used.
 | ||
Fig. 12 Catalytic results for the oxidation of glucose in the presence MWW@MgAl-in situ-40 based catalyst at 363 and 383 K and different addition mode and ratio H2O2/glucose. Reaction conditions: glucose (0.96 mmol), catalyst (0.066 g mixed oxide phase), dioxane (4.75 mL).  Erythrose yield%, Erythrose yield%,  glycolaldehyde dimer yield%, glycolaldehyde dimer yield%,  arabinose yield%, arabinose yield%,  fructose yield%, fructose yield%,  FA yield%, FA yield%,  glucose conversion%, glucose conversion%,  FA selectivity%. FA selectivity%. | ||
Herein, we describe different approaches for the preparation of multi-component materials by combining MWW-type zeolitic layers with MgAl oxide sub-domains at nanometric scale. Methods I and II involve the co-precipitation of MgAl hydroxide using sources of Mg2+ and Al3+, in the presence of MWW zeolite. Method III utilizes swelling and intercalation of MWW with preformed LDH phases combined with sonification and ball-milling techniques. The first two methods resulted in MWW individual layers, incorporating MgAl MO on their surface and inner porous system with a strong interaction between the two phases, as shown by the characterization study. The third method resulted in a highly homogeneous and dispersed physical mixture of both phases, with high contact together with smaller particle size and lower basicity (Fig. S18). The influence of preparation method of MWW@LDH composites over their catalytic performance for the conversion of glucose to FA was explored in dioxane at 363 K. The catalytic results obtained in the presence of MWW@Mg obtained from method I, MWW@MgAl-in situ-40 obtained from method II, MWW@MgAl-US and MWW@MgAl-BM obtained from method III under ultrasonication and by ball-milling solid-state mixing, respectively, are depicted in the Fig. 13. The catalytic performance of the different catalysts, evaluated in terms of glucose conversion and FA yield, followed the trend MWW@MgAl-in situ-40 ∼ MWW@MgAl-US > MWW@Mg > MWW@MgAl-BM. However, IR analysis of CDCl3 adsorption revealed a different trend in basicity that decreased in the following order: MWW@MgAl-US > MWW@Mg > MWW@MgAl-in situ-40 > MWW@MgAl-BM. This ranking was established based on the CDCl3 IR absorbance for medium and strong basic sites (S + M) (Fig. S18). After calcination, MgAl MO exhibits Lewis active basic sites from O2−–Mg2+ acid–base pairs. Their basicity varies with the coordination of the O2− anions, the oxygen atoms at the corners exhibiting a higher basicity than those at the edges or faces due to their lower coordination. Because of the presence of nanoscopic MgAl MO sub-domains with smaller particle size than standard MgAl MO, derived from calcination of LDHs, in the calcined MWW@MgAl-in situ samples, an overall increase in basicity could be expected due to a higher number of low-coordination Lewis sites. Nevertheless, IR study of CDCl3 showed that MWW@MgAl-in situ sample exhibited lower number of active sites and lower strength basicity than MgAl MO. In contrast, the BET area decreased in the following order: MWW@MgAl-BM > MWW@MgAl-in situ-40 > MWW@MgAl-US > MWW@Mg. These results may suggest that not all active basic sites are accessible for glucose conversion, likely due to geometric constraints and differences in the adsorption and desorption properties of the catalytic surface.
 | ||
Fig. 13 Catalytic results for the oxidation of glucose in the presence MWW@LDH materials prepared following different synthesis method, at 363 K, in dioxane with 6 equivalents of H2O2. Reaction conditions: glucose (0.96 mmol), catalyst (0.066 g mixed oxide phase), 20% H2O2 (5.76 mmol), dioxane (4.75 mL).  Erythrose yield%, Erythrose yield%,  glycoaldehyde dimer yield%, glycoaldehyde dimer yield%,  arabinose yield%, arabinose yield%,  fructose yield%, fructose yield%,  FA yield%, FA yield%,  glucose conversion%. glucose conversion%. | ||
As depicted in the Fig. S19, the MWW@MgAl-in situ-40 sample and MWW@MgAl-BM exhibited a shoulder at 3748 cm−1, attributed to silanol groups, which enhance the hydrophilicity of the catalytic surface and could promote the adsorption of the polar reactant on the catalytic active sites. In contrast, this band is almost absent in the spectra of MWW@MgAl-US and MWW@Mg indicating lower surface hydrophilicity. These findings suggest that catalytic performance of the multi-component materials is influenced by a balance between overall basicity, active sites accessibility and surface hydrophilicity. Similar arguments can be applied to explain the low catalytic performance of MWW@Mg catalyst, achieving a low 20% FA yield. The MWW@MgAl-BM catalyst exhibited the lowest catalytic behavior, likely due to the loss of active centers caused by the intense ball-milling process, which led to a reduction in the number of basic sites. This effect was previously evidenced by the reduced Mg incorporation in this sample (Table 1).
These results highlight that method II produces a multi-component material with the most favorable catalytic properties offering a material with highly dispersed LDH/mixed oxide sub-domains in the porous structure and surface of the MWW zeolite with lower platelets size, which provides accessible and proper basic active sites for glucose oxidation to FA. Similarly, method III combined with US resulted in a multi-component material with notable catalytic properties, similar to those of MWW@MgAl-in situ-40 material.
To continue, the influence of the MWW@MgAl-in situ-40 catalyst loading on glucose conversion, FA yield and selectivity was investigated. As shown in Fig. 14, increasing the catalyst amount led to a progressive improvement in catalytic performance, reaching a glucose conversion of 66.2%, an FA yield of 47.7%, and an FA selectivity of 72.1%.
 | ||
Fig. 14 Catalytic results for the oxidation of glucose varying the MWW@MgAl-in situ-40 loading. Reaction conditions: glucose (0.96 mmol), catalyst (0.066 g mixed oxide phase), 20% H2O2 (5.76 mmol), dioxane (4.75 mL).  Erythrose yield%, Erythrose yield%,  glycoaldehyde dimer yield%, glycoaldehyde dimer yield%,  arabinose yield%, arabinose yield%,  fructose yield%, fructose yield%,  FA yield%, FA yield%,  glucose conversion%. glucose conversion%. | ||
Finally, a catalyst recycling study was performed using MWW@MgAl-in situ-40 catalyst at 383 K and 8 equivalents of H2O2. During the reaction, the Mg–Al mixed oxide sub-domains can at least partially recover the LDH layered structure due to the presence of water, a phenomenon well known as memory effect. As a result, the catalyst gradually loses its activity as a mixed oxide. To mitigate this deactivation, the spent catalyst underwent a regeneration process that included ethanol washing, drying overnight in an oven at 373 K and calcination at 813 K before reuse. The catalytic results, shown in the Fig. 15(a), demonstrated that the catalyst was reusable for two cycles without loss in FA yield. However, after the third use the glucose conversion and FA yield decreased drastically. This decline aligns with literature attributing the change in the performance of calcined LDHs to the leaching of Mg2+ (≤5%) under acidic conditions requiring a regeneration via calcination in air and rehydration in Na2CO3.37–39 A catalyst deactivation was also observed for the continuous-flow catalysis over MgO and calcined-rehydrated hydrotalcite. Therefore, it was determined that Mg2+ leaching induced the formation of an Al(OH)3 passive layer, serving as a barrier to prevent further leaching under mild acidic conditions.39 These phenomenon could explaining the decrease in the catalytic activity from the second to third cycle. The local metallic species leaching induced local changes in mixed metallic oxide structure and so in the basicity of the LDH based catalysts. Additionally, catalyst agglomeration may play a role in performance loss upon reuse, as increased crystallite size can reduce both the accessibility and total number of active sites. However, from XRD pattern of the used sample, no notable change could be observed (Fig. S23). To further investigate catalyst stability, the recycling study was performed at 363 K. After the third use, a pronounced decrease in the catalytic activity. To attempt regeneration, the spent catalyst was treated overnight at 363 K in aqueous solution of NaOH (pH ∼ 9), in order to remove-surface adsorbed species and restore the surface metallic hydroxides. Unfortunately, during the fourth use the activity could not be recovered and continued to decline (Fig. 15(b)).
 | ||
Fig. 15 Study of MWW@MgAl-in situ-40 recycling for the catalytic oxidation of glucose to formic acid catalyst at 383 K (a) and 363 K (b) and 8 equivalents of H2O2. Reaction conditions: glucose (0.96 mmol), catalyst (0.066 g mixed oxide phase), dioxane (4.75 mL).  Erythrose yield%, Erythrose yield%,  glycoaldehyde dimer yield%, glycoaldehyde dimer yield%,  arabinose yield%, arabinose yield%,  fructose yield%, fructose yield%,  FA yield%, FA yield%,  glucose conversion%. glucose conversion%. | ||
It is important to remark that these catalytic results, together with the observed product distribution, further support the mechanistic proposal outlined in Scheme 4. The formation of fructose, arabinose, erythrose, and glycolaldehyde dimer, as well as the progressive accumulation of FA, align with a cascade of base-catalyzed reactions involving isomerization, retro-aldol fragmentation, and oxidative cleavage steps. The quantification of these intermediates across all catalysts confirms the relevance of these pathways. Although a detailed kinetic analysis was beyond the scope of this work, the combined analysis of product profiles and material properties strongly suggests that the accessibility and basic strength of surface-active sites govern the efficiency of the initial C–C bond cleavage steps, which are generally considered rate-determining in similar oxidation processes.
A comparison of the catalytic performance of MWW@MgAl-in situ catalyst for the oxidation of glucose to FA with H2O2, relative to the published results, is summarized in Table S3. The data indicate that, in terms of yield and selectivity, the catalytic performance of the MWW@MgAl-in situ materials remains lower. This reduced catalytic activity can be attributed to its lower basicity strength, as evidenced by the IR study of CDCl3 adsorption. Indeed, a comparison of the overall basicity between the MWW@samples and MgAl MO, clearly demonstrated that MgAl MO not only possesses a greater total number of active sites (M + S), but also a higher proportion of strong basic sites (34%), whereas the MWW@samples lacked strong active sites. To enhance the basicity strength of the MWW@MgAl-in situ catalyst, further optimization of its chemical composition is currently underway. Future work will focus on modifying synthesis parameters and tuning the metallic composition to improve the catalyst structural and chemical properties and achieve higher catalytic performance.
As conclusion, considering the catalytic results obtained, it is possible to affirm that the trends observed in glucose oxidation to formic acid can be attributed to a combination of physicochemical parameters derived from the synthetic strategies employed. The presence of strong basic sites alone does not guarantee high activity, as the accessibility and distribution of these sites, largely influenced by the porosity and dispersion of the MgAl domains, play a decisive role in the catalytic outcome. Method II materials, particularly MWW@MgAl-in situ-40, exhibited the best compromise between site strength and accessibility, facilitated by the in situ formation of well-dispersed LDH sub-domains on and within the MWW framework. In contrast, methods I and III showed lower activity, which correlates with either a deficiency in accessible basic sites (method I) or a more heterogeneous distribution of active phases with limited interface contact (method III). These findings highlight that catalytic performance in this system arises from the synergy between basicity, hierarchical porosity, and the spatial integration of the functional domains within the hybrid architecture.
4. Conclusions
In this work, a series of multi-component catalysts combining MWW-type lamellar zeolites and MgAl-based hydrotalcite phases were successfully synthesized using three different approaches. Each method introduced distinct structural, compositional, and textural modifications into the final solids, influencing their Lewis acid–base characteristics and catalytic performance for the oxidation of glucose to formic acid.Method I, based on the in situ generation of MgAl oxide phases using the zeolite as the sole aluminum source, led to limited incorporation of Al into the mixed oxide domains. Although the MWW structure was largely preserved, the formation of Mg-rich oxide domains were detected. These catalysts showed moderate activity, likely due to the lower density and dispersion of active sites.
Method II enabled a more effective integration of Mg and Al through the in situ formation of hydrotalcite-like phases using external precursors. This approach promoted greater chemical interaction between the LDH domains and the zeolitic framework, as evidenced by a stronger alteration in the spectroscopic and textural properties of the resulting solids. Among all the synthesized materials, those obtained via this method demonstrated the most promising catalytic performance, associated to a better dispersion of active sites and favourable acid–base surface properties. Specifically, the sample MWW@MgAl-in situ-40 showed the highest performance, achieving a formic acid yield of 47.7% with 66.2% glucose conversion after 7.5 hours at 363 K in dioxane.
Method III, which employed swelling and intercalation of preformed MgAl-LDH phases followed by ultrasonic or ball-milling treatment, produced catalysts with lower interfacial integration but retained hierarchical porosity. Notably, the method of intercalation significantly impacted the dispersion and accessibility of the basic sites, which translated into differences in catalytic activity.
Definitively, the study confirms that the synthesis route plays a critical role in determining the textural, chemical, and acid–base properties of the final materials based on LDH sub-domains into zeolitic structures, showing that a careful selection of synthesis conditions enables the design of efficient and robust multi-component catalysts. The results provide a solid basis for further optimization of these materials for base-catalyzed reactions and other applications where tailored Lewis acid–base properties are essential. In particular, the selective transformation of glucose into formic acid, an attractive hydrogen carrier and renewable biobased molecule, highlights the potential of these materials to contribute to carbon-neutral energy strategies and sustainable chemical production from biomass-derived feedstocks.
Author contributions
C. E.: investigation, data curation, formal analysis, methodology, visualization, and writing – original draft; A. V.: conceptualization, data curation, validation, project administration, supervision, and writing – review & editing; U. D.: conceptualization, data curation, validation, funding acquisition, project administration, supervision, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI. Additional characterization and catalytic results of multicomponent materials are included in the SI. See DOI: https://doi.org/10.1039/d5ma00633cAcknowledgements
The authors are grateful for the financial support from the Spanish Ministry (projects PID2023-146114NB-C21 and CEX2021-001230-S, funded by MCIN/AEI/10.13039/501100011033). This study forms part of the Advanced Materials program and was supported by MCIN with funding from the European Union NextGeneration (PRTRC17.I1) and by Generalitat Valenciana (MFA/2022/003). C. E. has been partially supported by the Spanish Government through the FPI pre-doctoral scholarship number PRE2019-089547 under project SEV-2016-0683-19-3. The authors acknowledge the Microscopy Service of the Universitat Politècnica de València.References
- N. Kumar, S. Sauraj and N. C. Joshi, Solid Base Catalysts: Synthesis, Characterization, and Applications, 2024, pp. 279–315 Search PubMed.
- Y. Gao, Z. Zhang, J. Wu, X. Yi, A. Zheng, A. Umar, D. O’Hare and Q. Wang, J. Mater. Chem. A, 2013, 1, 12782–12790 RSC.
- E. Mandela, A. G. Margellou, A. Kotsaridou, G. E. Marnellos, M. Konsolakis and K. S. Triantafyllidis, Catalysts, 2025, 15, 323 CrossRef CAS.
- U. A. Mohanty, D. P. Sahoo, L. Paramanik and K. Parida, Sustainable Energy Fuels, 2023, 7, 1145–1186 RSC.
- U. Díaz, D. Brunel and A. Corma, Chem. Soc. Rev., 2013, 42, 4083–4097 RSC.
- R. Ye, J. Zhao, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, Nat. Catal., 2018, 1, 318–325 CrossRef.
- G. Kumar, E. Nikolla, S. Linic, J. W. Medlin and M. J. Janik, ACS Catal., 2018, 8, 3202–3208 CrossRef CAS.
- L. Zhang, C. Jia, F. Bai, W. Wang, S. An, K. Zhao, Z. Li, J. Li and H. Sun, Fuel, 2024, 355, 129455 CrossRef CAS.
- J. Eppinger and K. W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- M. Achour, D. Álvarez-Hernández, E. Ruiz-López, C. Megías-Sayago, F. Ammari, S. Ivanova and M. Á. Centeno, Tetrahedron Green Chem., 2023, 2, 100020 CrossRef.
- W. Wang, M. Niu, Y. Hou, W. Wu, Z. Liu, Q. Liu, S. Ren and K. N. Marsh, Green Chem., 2014, 16, 2614–2618 RSC.
- N. V. Gromov, T. B. Medvedeva, I. A. Lukoyanov, V. N. Panchenko, S. A. Prikhod’ko, V. N. Parmon and M. N. Timofeeva, Results Eng., 2023, 17, 100913 CrossRef CAS.
- J. Albert, D. Lüders, A. Bösmann, D. M. Guldi and P. Wasserscheid, Green Chem., 2013, 16, 226–237 RSC.
- J. Reichert and J. Albert, ACS Sustainable Chem. Eng., 2017, 5, 7383–7392 CrossRef CAS.
- H. S. Isbell and R. G. Naves, Carbohydr. Res., 1974, 36, C1–C4 CrossRef CAS.
- C. Wang, X. Chen, M. Qi, J. Wu, G. Gözaydin, N. Yan, H. Zhong and F. Jin, Green Chem., 2019, 21, 6089–6096 RSC.
- H. S. Isbell, H. L. Frush and E. W. Parks, Carbohydr. Res., 1976, 51, C5–C9 CrossRef CAS.
- R. Sato, H. Choudhary, S. Nishimura and K. Ebitani, Org. Process Res. Dev., 2015, 19, 449–453 CrossRef CAS.
- L. Wu, Y. Yang, J. Cheng, X. Shi, H. Zhong and F. Jin, ACS Sustain, Chem. Eng., 2022, 10, 15423–15436 CAS.
- C. Esteban, A. Velty and U. Díaz, Mater. Adv., 2024, 5, 9684–9698 RSC.
- M. Dixit, M. Mishra, P. A. Joshi and D. O. Shah, J. Ind. Eng. Chem., 2013, 19, 458–468 CrossRef CAS.
- Y. Ono and H. Hattori, Solid Base Catalysis, Springer, Berlin, Heidelberg, 2011, vol. 101 Search PubMed.
- E. A. Paukshtis, N. S. Kotsarenko and L. G. Karakchiev, React. Kinet. Catal. Lett., 1979, 12, 315–319 CrossRef CAS.
- E. A. Paukshtis and E. N. Yurchenko, Russ. Chem. Rev., 1983, 52, 242–258 CrossRef.
- G. Yang and L. Zhou, ACS Catal., 2018, 8, 6691–6698 CrossRef CAS.
- O. B. Belskaya, I. G. Danilova, M. O. Kazakov, R. M. Mironenko, A. V. Lavrenov and V. A. Likholobov, Infrared Spectroscopy – Materials Science, Engineering and Technology, InTech, 2012 Search PubMed.
- E. V. Korneeva, A. S. Ivanova, V. M. Bondareva, L. M. Plyasova and T. S. Glazneva, Kinet. Catal., 2015, 56, 605–616 CrossRef CAS.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- H. Yan, X. J. Zhao, Y. Q. Zhu, M. Wei, D. G. Evans and X. Duan, Struct. Bonding, 2019, 179, 89–120 CrossRef.
- A. Vaccari, Catal. Today, 1998, 41, 53–71 CrossRef CAS.
- A. L. Allred, J. Inorg. Nucl. Chem., 1961, 17, 215–221 CrossRef CAS.
- A. V. Besserguenev, A. M. Fogg, R. J. Francis, S. J. Price, D. O’Hare, V. P. Isupov and B. P. Tolochko, Chem. Mater., 1997, 9, 241–247 CrossRef CAS.
- M. Mora, M. I. López, C. Jiménez-Sanchidrián and J. R. Ruiz, Catal. Lett., 2010, 136, 192–198 CrossRef CAS.
- E. Aneggi, C. de Leitenburg, M. Boaro, P. Fornasiero and A. Trovarelli, Cerium Oxide (CeO2): Synthesis, Properties and Applications, Elsevier, 2020, pp. 45–108 Search PubMed.
- C. T. Campbell and C. H. F. Peden, Science, 2005, 309, 713–714 CrossRef CAS PubMed.
- K. Y. Chen, C. M. Lin, C. M. Shu and C. S. Kao, J. Therm. Anal. Calorim., 2006, 85, 87–89 CrossRef CAS.
- I. Delidovich and R. Palkovits, J. Catal., 2015, 327, 1–9 CrossRef CAS.
- I. K. M. Yu, A. Hanif, D. C. W. Tsang, J. Shang, Z. Su, H. Song, Y. S. Ok and C. S. Poon, Chem. Eng. J., 2020, 383, 122914 CrossRef CAS.
- M. Jobbágy and A. E. Regazzoni, Appl. Clay Sci., 2011, 51, 366–369 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |