
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanostructured MnS-based thin films deposited from propylamine solutions of elemental sulfur and manganese
Jiri
Jancalek
 *,
Michal
Kurka
,
Jhonatan
Rodriguez-Pereira
,
Stanislav
Slang
and
Milos
Krbal
*,
Michal
Kurka
,
Jhonatan
Rodriguez-Pereira
,
Stanislav
Slang
and
Milos
Krbal
Center of Materials and Nanotechnologies, Faculty of Chemical Technology, University of Pardubice, Studentska 95, Pardubice, 532 10, Czech Republic. E-mail: jiri.jancalek@upce.cz
First published on 19th September 2025
Abstract
The exploration of novel methods for depositing MnS thin films is crucial due to their huge potential for optoelectronic, energy storage, and other advanced technological applications. This study proposes a novel solution-processing approach for the Mn–S-based nanostructured thin film fabrication using dissolved elemental Mn and S in propylamine-based solvents. The Mn/S ratio in the solution dictates the resulting film morphology, chemical composition, and molecular structure. Utilizing a 1/1 Mn/S ratio in propylamine and propylamine–methanol mixture solutions yielded nanoporous network structures formed mainly from Mn2+ sulfides and hydroxides. Conversely, using 1/5 Mn/S ratio solutions under the same solvent conditions resulted in ∼85 nm spherical nanoparticle films made from Mn2+/Mn4+ sulfides and hydroxides, polysulfides, and organic residues. Utilizing a propylamine–acetonitrile mixture, regardless of the Mn/S ratio, led to films formed from spherical/oval nanoparticles (∼126–136 nm). All nanoparticle-based films annealed at 300 °C exhibited photocatalytic activity, as evidenced by the methylene blue degradation under UV light illumination. Sulfur-rich films demonstrated the highest photocatalytic efficiency, indicating a promising route for tailored Mn–S photocatalysts.
Introduction
Research on manganese sulfide-based materials as p-type wide bandgap (Eg ∼ 2.8–3.2 eV (ref. 1)) diluted magnetic semiconductors is currently receiving a lot of scientific attention as they are promising candidates for photocatalysis applications.2,3 The specific application depends on the crystal modification and the material form. MnS exists in three crystalline polymorphs: stable cubic rock salt-like α-MnS modification, metastable cubic zinc blende-like structure β-MnS, and metastable hexagonal wurtzite structure γ-MnS.4 While the metastable polymorphs can exhibit distinct electronic properties, the thermodynamic stability of the α-MnS phase offers a crucial advantage for developing robust and durable photocatalysts suitable for long-term applications. A key scientific challenge, therefore, lies in developing synthesis routes that allow for precise control over the morphology of the stable α-MnS phase-based materials.5–7 The development of high surface area structures, such as the nanoporous networks examined in this study, is imperative for maximizing the number of catalytically active sites and enhancing light-harvesting efficiency, which are crucial for superior photocatalytic performance.Manganese sulfide thin films have already been synthesized and grown using various techniques, including both vapor phase deposition techniques, such as vacuum thermal evaporation,8,9 sputtering,10–12 or chemical vapor deposition,13–15 and solution-based techniques employing primarily chemical bath deposition16–18 or SILAR.19–21 Although vapor phase deposition provides high quality thin films with high reproducibility, research on Mn–S-based thin film deposition has primarily focused on the solution-based deposition approach in recent years. In this way, the Mn/S stoichiometry can be excellently controlled along with large variations in the morphology and crystal structure depending on the solution composition and deposition or post-deposition conditions.22–24 In general, the solution approach to Mn–S and other chalcogenide materials uses precursors based on Mn2+ salts (e.g. Mn(Ac),19,24,25 MnSO4,17,26,27 MnCl228–31 or MnNO320,32) and S2− precursors, either again in the form of salts (e.g. Na2S19,26 and Na2S2O317) or as organic compounds (thiourea,31,33–36 thioacetamide,37 and 1-dodecanethiol38) and elemental sulfur39,40 in an aqueous or alcoholic solvent. The use of aqueous media often leads to the unintentional incorporation of oxygen, resulting in the formation of oxides/hydroxides within the film. Furthermore, the resulting morphologies are generally limited to particulate films, which coalesce into granular or cauliflower-like structures. Achieving highly ordered, non-particulate architectures through these methods remains a significant challenge.
In this work, we demonstrate the preparation of Mn–S-based solution-processed nanostructured films from high-purity elements using an anhydrous environment. We confirmed the assumptions of J.W. Thompson et al.,41 describing the possibility of dissolving a pure metal at room temperature in the environment of alkylammonium polysulfide salts, formed as a dissolution product of the elemental sulfur in aliphatic amines.42,43 The most significant advantage of our approach, however, lies in the unprecedented morphological control it affords. While previous studies using this chemistry focused on powder synthesis,44,45 we have adapted it for the direct deposition of thin films, demonstrating the formation of a unique, self-assembled nanoporous network structure, a morphology not reported in the aforementioned salt-based studies. We prepared compositionally and morphologically distinct nanostructured films, ranging from nanoporous networks to spherical nanoparticles, by systematically varying the initial Mn/S ratio, concentration, and solvent composition. We provide detailed protocols on how the morphology, composition, and molecular structure of such prepared films can be controlled. Finally, we discuss the role of the above-described parameters in the photocatalytic activity of the deposited nanostructured MnS-based films.
Experimental
Chemicals
Manganese (4N) was purchased from HiChem Spol. Native surface oxides were dissolved in diluted nitric acid (p.a., 68%, Lach:Ner, diluted by demineralized water to ∼15% HNO3). The purified manganese was rinsed with demineralized water and isopropyl alcohol. To further reduce the oxygen content, the acid-treated manganese was placed into a quartz ampoule, which was subsequently evacuated (10−3 Pa) and annealed at a temperature of 600 °C for 1 hour. All other operations with the purified manganese were carried out in an inert atmosphere of a nitrogen-filled glovebox LabMaster Pro_MB200 (MBraun). Despite the applied procedure, it was not possible to completely remove all residual oxygen from the material (see Table S1). Sulfur (99.999%, AlfaAesar), n-propylamine (99.5%, Sigma Aldrich), methanol (p.a., 99.5%, Lach:Ner), acetonitrile (for HPLC, ≥99.9%, Honeywell) and Methylene blue (certificated by the Biological Stain Commission, Sigma Aldrich) were used as received.Mn–S solutions and thin film preparation
The purified manganese was crushed, and its corresponding amount was weighted into vials together with elemental sulfur. The Mn/S molar ratio was varied from 1/1 up to 1/5 while the final molar manganese concentration in solution was 0.3 M, 0.5 M, and 1 M. The powders were co-dissolved in three different solvents: pure n-propylamine (PA), a mixture of 90 vol% n-propylamine with 10 vol% methanol (PA-MeOH) and a mixture of 90 vol% n-propylamine with 10 vol% of acetonitrile (PA–AN). The selection of cosolvents was based on criteria including higher polarity of MeOH and AN (dielectric constants ε: 5.1 for PA,46 32.7 for MeOH47 and 35.1 for AN48), the capacity of MeOH to form hydrogen bonds, and the aprotic nature of AN and its capacity to form coordination bonds. To prevent unwanted oxidation or solution hydrolysis, the entire dissolution process was carried out inside a nitrogen-filled glovebox for 7 days. Before the deposition, the unreacted manganese portion, along with agglomerates of dissolution products (Fig. S2A), were filtered through a syringe filter with a 0.22 mm pore size. FTIR and EDS analysis of the filtered agglomerates confirmed that they are composed of organic salts, manganese sulfide19 and manganese oxide, concretely Mn3O4,49 and the results are presented in Fig. S2B and Table S2C.Thin films were deposited from the filtered solution by dynamic spin-coating (spin-coater Laurel WS-650Mz23NPPB) at 2000 rpm for 80 s in a nitrogen atmosphere onto the soda-lime, fused silica and silicon substrates. Immediately after deposition, thin films were annealed at 60 °C for 1 h to evaporate the chemically non-bonded solvent. Subsequently, thin films were annealed at 150 °C for 1 h in the nitrogen atmosphere of the glovebox to decompose and remove the main content of the chemically bound solvent in the dissolution products. The annealing at 300 °C was performed in a five-times flushed nitrogen (4.6N) tube furnace to crystallize the prepared thin films.
Scanning electron microscopy (SEM) and energy dispersive spectroscopy (EDS)
The scanning electron microscopy (SEM) scans and elemental composition of the prepared thin films were obtained using a scanning electron microscope LYRA 3 (Tescan) equipped with an EDS analyzer AZtec X-Max![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 (Oxford Instruments). The samples annealed at 60 and 150 °C were deposited on gold-coated (20 nm) conductive soda-lime substrates, while the samples annealed at 300 °C were deposited on fused silica substrates and subsequently overcoated with a 20 nm carbon layer. To obtain compositional information by EDS analysis, thin films were scraped from the substrate onto conductive carbon tape. The particle size distribution was determined from the obtained SEM images in Fiji software.
20 (Oxford Instruments). The samples annealed at 60 and 150 °C were deposited on gold-coated (20 nm) conductive soda-lime substrates, while the samples annealed at 300 °C were deposited on fused silica substrates and subsequently overcoated with a 20 nm carbon layer. To obtain compositional information by EDS analysis, thin films were scraped from the substrate onto conductive carbon tape. The particle size distribution was determined from the obtained SEM images in Fiji software.
Atomic force microscopy (AFM)
Thin film morphology was also studied by atomic force microscopy (AFM) in semi-contact mode using an NTEGRA (NT-MDT) microscope equipped with NSG10 tips (AppNano). The structure's heights were evaluated from obtained scans (2.5 × 2.5 μm) using the corresponding z-axis histograms.X-ray diffraction (XRD)
The crystalline nature of synthesized Mn–S-based materials was probed by X-ray diffraction (XRD) using a Cu Kα (λ = 1.5406 Å) X-ray source and a diffractometer (Empyrean Malvern Panalytical) operated in the glancing angle geometry under an incidence angle of 0.5°.Raman spectroscopy
The Raman spectra of pure solvents, MnS solutions, sulfur solutions (1 M and 5 M concentrations), and elemental sulfur as well as spin-coated films were obtained using a MultiRam (Bruker) FT-Raman spectrometer utilizing a 1064 nm Nd:YAG laser excitation beam (2 cm−1 resolution, averaging of 200 scans). Raman spectra were normalized by the most intensive band in the region of 100–500 cm−1. Due to the high luminescence levels, the data from the film annealed at 150 and 300 °C could not be provided.Fourier transform infrared (FTIR) spectroscopy
The ATR-FTIR spectra of the studied samples were recorded in the 4500–50 cm−1 region (resolution 4 cm−1, 100 scans) using a Vertex 70v FTIR spectrometer (Bruker) equipped with a single-bounce diamond ATR crystal. The samples were measured in powder form by scraping the film from the substrate with a razor blade. The resulting spectra were subsequently normalized to the intensity of the most dominant band within each individual spectrum.X-ray photoelectron spectroscopy (XPS)
The surface chemical composition of MnS thin films was evaluated by X-ray photoelectron spectroscopy at room temperature (XPS, ESCA 2SR, Scienta Omicron) using a monochromatic Al Kα (1486.7 eV) X-ray source. The X-ray source was operated at 200 W. A charge neutralizer (CN 10) was used to control the surface charge, operated at 5 mA and 1 eV. The binding energy scale was referenced to adventitious carbon (284.8 eV). CasaXPS software (Casa software Ltd) was used to analyze the data and the quantitative analysis was performed using the elemental sensitivity factors provided by the manufacturer.Photocatalytic efficiency
The methylene blue (MB) photodegradation was performed to determine photocatalytic efficiency of the prepared thin films annealed at 300 °C. In the first step, the samples were immersed in the MB solution in demineralized water (conductivity ∼0.1 μS cm−1) with an initial concentration of 1 × 10−5 mol L−1 for 30 min to achieve absorption–desorption equilibrium. In the following step, the MB solution with the sample was illuminated by an LED array (365 nm, 10 W), and the photodegradation of MB solution was examined in situ as a change of absorbance A at a wavelength of 664 nm. For in situ analysis, a fiber spectrometer AvaSpec-3648 (AVANTES) and a halogen lamp SL1 (Stellarnet), were used. When a low concentration of organic compound is applied, photocatalysis can be considered a first-order reaction.50 Therefore, MB photocatalytic degradation with an initial concentration 1 × 10−5 mol L−1 can be described as linear dependence expressed by the following equation:50where C0 is the MB solution concentration at the initial time, C is the MB solution concentration at time t and k is the rate constant. According to the Lambert–Beer law, the concentration can be expressed as

where A is the absorbance of the solution, l is the optical path and ε is the molar extinction coefficient. In the case where the molar extinction coefficient and optical path are constant, it is possible to substitute the A/A0 ratio for the C/C0 ratio and otherwise. For this reason, photocatalysis efficiency is presented here as the dependence of ln(A/A0) on time.

Results and discussion
The morphology of the resulting films annealed at 300 °C depends upon the employed Mn/S ratio, as well as the MeOH or AN presence in the source amine solution (Fig. 1). In the case of pure PA solvent and a stoichiometric Mn/S ratio (Mn/S = 1/1), the formation of a porous network was discovered with pore sizes in the tens of nanometers. It was further found that the MeOH addition in the PA solvent partially disintegrates this porous network, thereby increasing the pore size. In contrast, adjusting the Mn/S ratio to favor sulfur overstoichiometry results in spherical nanoparticles with a diameter of 85 ± 9 nm from pure PA or 84 ± 9 nm from the PA–MeOH solvent. | ||
| Fig. 1 Top-view SEM images of Mn–S-based films deposited from solutions of various ratios of Mn/S (1/1 and 1/5) and various solvents (pure PA, the mixture of 90% PA with 10% MeOH, and the mixture of 90% PA with 10% AN) after annealing at 300 °C. | ||
On the other hand, the films deposited from PA–AN solutions consist of larger nanoparticles with an equivalent diameter of 135 ± 11 nm from 1/1 Mn/S ratio PA–AN solution or 126 ± 20 nm from 1/5 Mn/S ratio PA–AN solution. The divergent morphologies of the PA–AN films are likely connected to the formation of Mn2+ acetonitrile complexes.51
We assume that the nanoporous network formation is probably thermodynamically disadvantageous and is associated with the incomplete reaction in the source solution. Our hypothesis is supported by the dissolution time dependence of the morphology transition for the thin films with sulfur overstoichiometry in the Mn/S ratio = 1/2.5, as presented in the SI (see Fig. S3). This phenomenon can also be observed directly through the gradual shift of the polysulfide bands in the Raman spectrum (see Fig. S4–S9). As the dissolution time increases, a shift in the polysulfide bands becomes evident, particularly towards S32− (bands at 444 and 184 cm−1 (ref. 52)) and S42− (234 and 503 cm−1 (ref. 53 and 54)) polysulfides. Subsequent to the reaction with Mn, a shift in band positions are observed after approximately 72 hours, which can be attributed to the formation of MnSx complexes. Furthermore, Raman spectral analysis indicates that the presence of methanol accelerates the decomposition of polysulfides, particularly at S32−, which is likely attributable to an increased ionic character resulting from the higher polarity of methanol. The decomposition of polysulfides is then reflected in the observed changes in the morphology, i.e. disintegration and enlargement of the rings of the porous networks.
In a similar way, we can control the nanostructured nature of Mn–S-based thin films by the concentration of the prepared solution, which is documented by a series of SEM images taken on samples with an intermediate Mn/S ratio = 1/2.5 deposited from pure PA (see Fig. S10). Shallow porous network structures are formed at a concentration of c = 0.3 M. Further increasing the concentration to 0.5 M results in the formation of spherical nanoparticles, while the highest concentration of 1 M transforms the film into a semi-continuous structure with pronounced roughness and non-spherical nanoparticles.
The topography of porous network structures obtained from pure PA and PA–MeOH solutions at a ratio of Mn/S = 1/1 was studied by AFM (Fig. 2). We found that the network porosity size significantly depends on the used solution. Specifically, the pure PA solution generates meshes with smaller openings and higher density in comparison with those obtained from the PA–MeOH mixture. On the other hand, the height of the mesh wall has the opposite trend with values of 39.0 ± 13.3 nm and 59.1 ± 17.1 nm from pure PA and PA–MeOH solution, respectively.
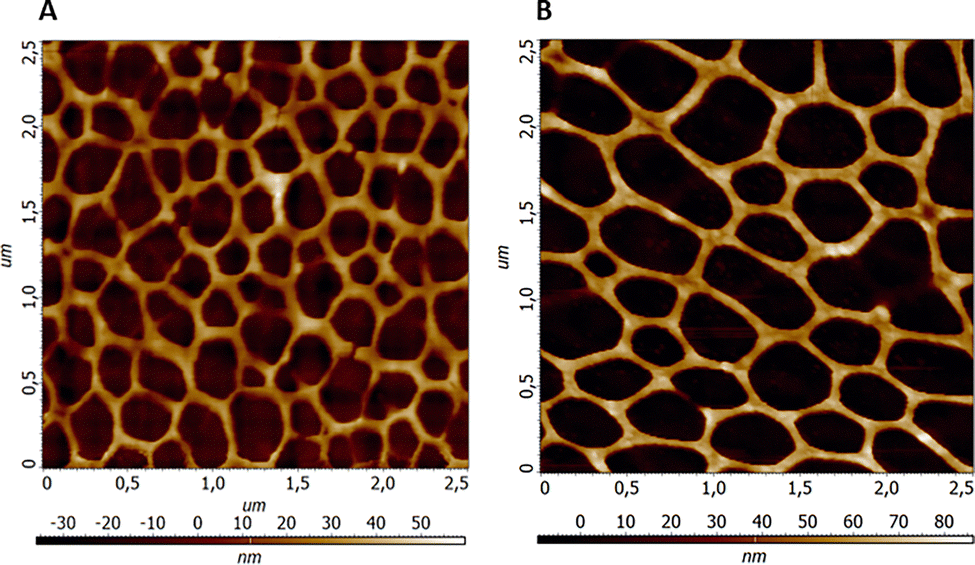 | ||
| Fig. 2 The representative AFM scans of the Mn–S-based porous network of films annealed at 300 °C deposited from pure PA (A) and PA–MeOH (B) solutions (Mn/S = 1/1; 1 M). | ||
A nanoporous network is formed by the self-assembly of the material during the post-deposition stabilization process at 60 °C (Fig. 3). Annealing at higher temperatures (150 and 300 °C) induces the evaporation of a part of the chemically bound solvent in polysulfide ammonium salts, which has a significant effect on increased porosity. On top of that, cracking of the nanoporous network occurs simultaneously in samples deposited from PA–MeOH solution, probably due to the leakage of residual organic vapors from the volume of the structures.
 | ||
| Fig. 3 Top-view SEM images of Mn–S-based thin films deposited from PA, PA–MeOH and PA–AN solutions of the 1/1 Mn/S ratio depending on annealing temperature. | ||
In contrast, the annealed Mn–S-based thin films deposited from the PA–AN solutions and sulfur-rich PA and PA–MeOH solutions result in a structure consisting of spherical nanoparticles (Fig. 3 and 4). The SEM image of sulfur-rich Mn–S deposited from PA–MeOH solution and stabilized at 60 °C revealed that part of the Mn–S in the film's volume is converted into irregular and differently sized nanoparticles. However, annealing at higher temperatures seems to be necessary for the spherical nanoparticle formation. It is evident from Fig. 4 that the sizes of Mn–S-based nanoparticles prepared from different solutions are not the same. At first glance, the nanoparticles synthesized from the PA–AN solutions are significantly larger compared to those synthesized from sulfur-rich PA and PA–MeOH solutions. Particle size distributions for films annealed at 300 °C are shown in Fig. S11.
 | ||
| Fig. 4 Top-view SEM images of Mn–S-based thin films deposited from PA, PA–MeOH and PA–AN solution of the 1/5 Mn/S ratio depending on annealing temperature. | ||
Regardless of the Mn/S ratio or the employed solvent, the annealed films at 300 °C crystalize in the cubic α-MnS phase (JCPDS 03-065-0891)55 with the preferential growth along (200) and (220) reflection planes at 2θ = 34.4 and 49.3°, respectively (see Fig. S12). However, the development of the crystalline phase depends on the Mn/S ratio reflecting the initial nanostructure. Fig. 5 (Left) shows that the α-MnS phase was observed in nanoporous networks (Mn/S = 1/1) only after annealing at 300 °C. Below this temperature, XRD patterns are featureless, indicating that the long-range order is missing. On the other hand, the XRD pattern of Mn–S-based films prepared from the PA solution with sulfur excess (Mn/S = 1/5) and stabilized at 60 °C exhibits a narrow and intense characteristic Bragg reflection along (002) peak at 28.5°, which could be indexed to the γ-MnS phase (JCPDS card No. 00-003-1065)12 (see Fig. 5 Right). The thermodynamically unstable γ-MnS is transformed into stable α-MnS at 150 °C, which is in good agreement with previous studies.4,56,57 The intensities of the Bragg reflections corresponding to α-MnS become more pronounced when the annealing temperature is further increased to 300 °C. The average crystallite size (D) was calculated using the Scherrer equation (the dimensionless shape factor K was selected as 0.94, based on the cubic lattice of α-MnS and the nearly spherical shape of the nanoparticles observed at SEM). Microstrain and dislocation density were calculated according to equations described in ref. 58 and 59. Calculated parameters are present in Table S12b. The determined crystallite sizes clearly indicate that the prepared nanostructures are formed by agglomerates of the formed small crystallites (D = 4 − 7 nm).
 | ||
| Fig. 5 XRD patterns of Mn–S-based thin films deposited from PA Mn/S = 1/1 solution (left) and PA Mn/S = 1/5 solution (right) annealed at 60, 150 and 300 °C. | ||
The elemental composition of the thin films was studied using EDS analysis (Table 1). The results of the analysis show not only the expected presence of manganese and sulfur but also the oxygen together with carbon and nitrogen as traces of solvent residuals. Moreover, the oxygen, nitrogen, and carbon signals were present even after annealing at 300 °C. The oxygen signal found in deposited thin films from PA or PA–AN (i.e. MeOH-free) solvents indicates that the primary source of oxygen is manganese oxide, which could not be fully removed through purification of the initial manganese metal (see Table S1). A secondary potential source of oxygen is surface oxidation during transport to the SEM chamber through the ambient air.
| Solvent | Mn/S ratio (solution) | Annealing temperature (°C) | Atomic concentration (at%) | Mn/S ratio (from EDS) | ||||
|---|---|---|---|---|---|---|---|---|
| Mn | S | O | N | C | ||||
| PA | 1/1 | 60 | 12.6 | 17.7 | 21.0 | 6.5 | 42.2 | 0.71 |
| 150 | 17.0 | 16.2 | 25.8 | 4.2 | 36.8 | 1.05 | ||
| 300 | 25.8 | 9.6 | 24.2 | 0.9 | 39.5 | 2.69 | ||
| 1/5 | 60 | 2.1 | 23.8 | 6.8 | 6.9 | 60.5 | 0.09 | |
| 150 | 6.8 | 14.2 | 5.8 | 10.2 | 63.1 | 0.48 | ||
| 300 | 12.4 | 22.1 | 5.7 | 8.5 | 51.3 | 0.56 | ||
| PA–MeOH | 1/1 | 60 | 12.6 | 15.4 | 17.5 | 7.0 | 47.5 | 0.82 |
| 150 | 15.6 | 17.7 | 20.4 | 4.8 | 41.6 | 0.88 | ||
| 300 | 18.9 | 13 | 22.5 | 0.6 | 45.0 | 1.45 | ||
| 1/5 | 60 | 2.4 | 17 | 7.5 | 8.6 | 64.6 | 0.14 | |
| 150 | 7.4 | 13.8 | 5.5 | 10.1 | 63.1 | 0.54 | ||
| 300 | 11.0 | 18.7 | 5.5 | 9.2 | 55.6 | 0.59 | ||
| PA–AN | 1/1 | 60 | 10.5 | 17.5 | 17.4 | 6.9 | 47.6 | 0.60 |
| 150 | 16.5 | 17.0 | 26.1 | 4.5 | 35.9 | 0.97 | ||
| 300 | 12.5 | 10.2 | 22.1 | 1.2 | 54.0 | 1.23 | ||
| 1/5 | 60 | 2.5 | 13.5 | 7.9 | 11.3 | 64.8 | 0.19 | |
| 150 | 5.1 | 13.0 | 5.8 | 9.9 | 66.2 | 0.39 | ||
| 300 | 8.2 | 16.6 | 7.0 | 9.1 | 59.2 | 0.49 | ||
As demonstrated in Table 1, the chemical composition of thin films depends on the Mn/S ratio in the source solution. Thin films deposited from solutions with a 1/1 Mn/S ratio exhibit a sulfur overstoichiometry after postdeposition stabilization at 60 °C, which, depending on the annealing temperature (150 or 300 °C), shifts into Mn overstoichiometry. In the case of thin films prepared from solutions with sulfur overstoichiometry (Mn/S = 1/5), a high sulfur overstoichiometry remains even after annealing at 300 °C.
The thin films with nanoporous networks exhibit a rapid decrease in the nitrogen content with increasing annealing temperature. This decrease is closely connected to the release of sulfur and could be attributed to the decomposition and evaporation of alkylammonium polysulfide-based residues in the form of decomposition products such as H2S, NH3, etc.60 On the other hand, the oxygen and carbon contents in thin films remain nearly unchanged.
The results of the EDS analysis provide information regarding the entire volume of the prepared thin films. For the eventual use of such prepared nanostructures in catalytic applications, the composition and the chemical state of the top surface itself are crucial information. Significant insight into this issue was gained by XPS measurements of the annealed films at 300 °C (see Table 2 and Fig. 6, and Fig. S13–S18).
| Solvent | Mn/S ratio (solution) | Atomic concentration (at%) | Mn/S ratio (from XPS) | |||||
|---|---|---|---|---|---|---|---|---|
| Mn | S | O | N | C | Si | |||
| PA | 1/1 | 11.8 | 14.0 | 26.9 | 3.4 | 24.0 | 19.9 | 0.84 |
| 1/5 | 4.8 | 13.0 | 10.2 | 11.0 | 53.6 | 7.3 | 0.37 | |
| PA–MeOH | 1/1 | 7.2 | 8.8 | 36.0 | 2.5 | 18.5 | 27.0 | 0.82 |
| 1/5 | 5.2 | 12.7 | 11.0 | 10.9 | 53.1 | 7.1 | 0.41 | |
| PA–AN | 1/1 | 12.4 | 13.4 | 22.8 | 3.7 | 31.1 | 16.6 | 0.93 |
| 1/5 | 6.3 | 15.5 | 5.1 | 12.6 | 58.1 | 2.4 | 0.41 | |
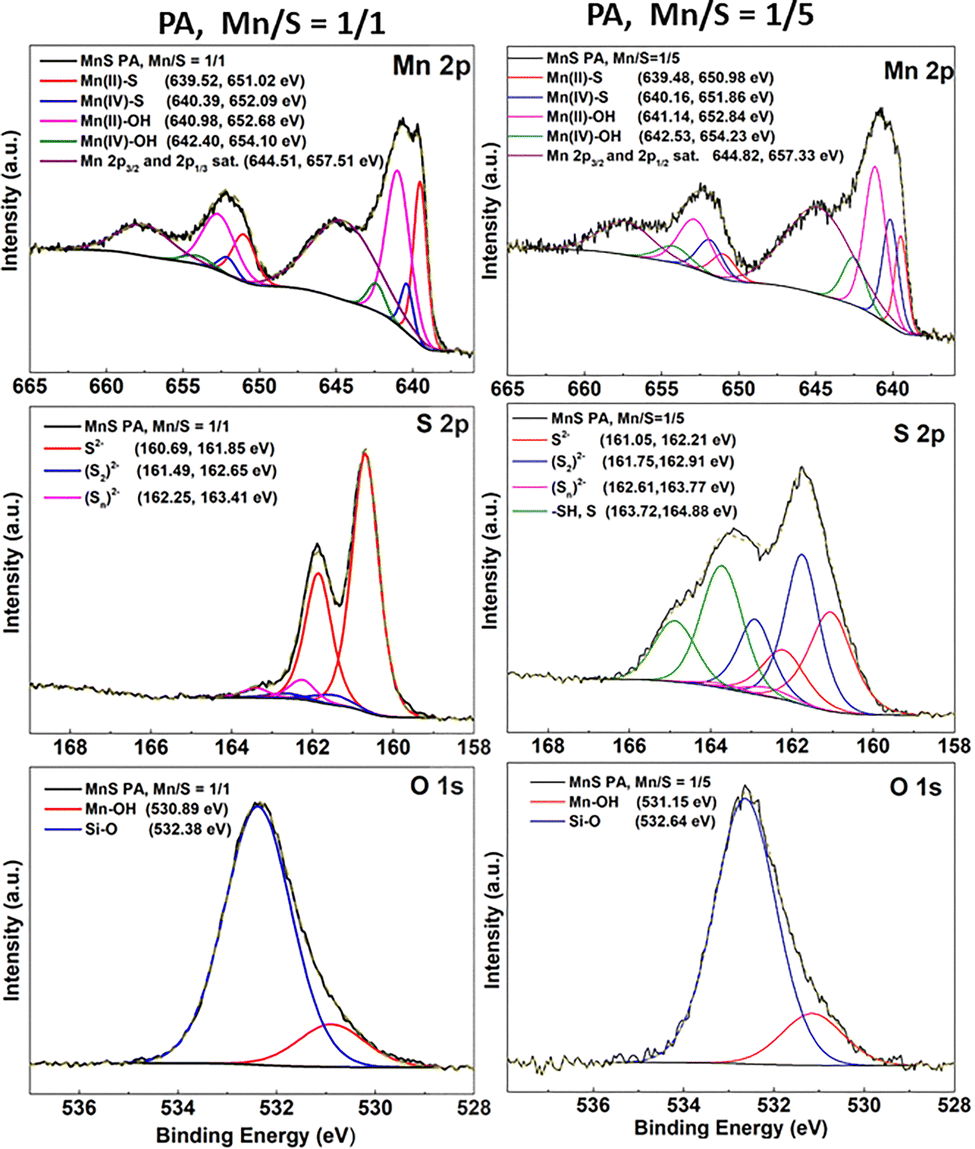 | ||
| Fig. 6 Mn 2p, S 2p and O 1s peak fitted high-resolution XPS spectra of Mn–S-based films deposited from PA Mn/S = 1/1 and Mn/S = 1/5 solutions annealed at 300 °C. | ||
The XPS results revealed that the surface of thin films with porous networks (see Fig. 6 and Fig. S13 and S14) is mainly formed from a mixture of manganese sulfides and hydroxides with a dominant Mn2+ oxidation form as well as a less common Mn4+ oxidation form. A minor excess of sulfur is present in the form of S22− and Sn2− polysulfides, while its majority is bonded in sulfide S2− form. Due to the porous film's structure, most of the measured oxygen signal originates from Si–O bonds from the silicon substrate. However, oxygen is also bound to Mn in the form of hydroxides and to carbon originating from alkylammonium salt residues. Nitrogen and carbon are present in the thin film as alkylammonium salt residues. However, it is important to note that neither carbon nor nitrogen is directly chemically bound to sulfur or manganese.
The surface of the 1/5 Mn/S ratio nanoparticle-based films from PA (Fig. 6 and Fig. S16) and PA–MeOH (Fig. S17) solutions is again formed from the mixture of Mn2+ and Mn4+ sulfides and hydroxides. However, the content of sulfur and residual dissolution products is significantly higher. The significant excess of sulfur, as also evidenced by EDS analysis, is reflected in the thin films by the dominant presence of sulfur in the form of S22− and Sn2− polysulfides. Moreover, the distinctive thiol –SH component was identified as well, contributing to the number of identified sulfur species. It should be noted that no –SH bonds were observed in the case of porous network nanostructures. Similar to the EDS analysis, the content of oxygen in nanoparticle-based thin films is significantly lower in comparison with the network structures, which can be attributed to the passivation of the film surface by polysulfides. The presence of polysulfides probably hinders the reaction of oxygen with manganese sulfur species. Nevertheless, the overall residual content of alkylammonium salts, represented by both carbon and nitrogen contents, is significantly higher in comparison with the porous network structures, probably due to the formation of an impermeable layer of polysulfides on the surface.
Despite their different morphology, XPS spectra of thin films from both PA–AN Mn/S = 1/1 and Mn/S = 1/5 solutions (Fig. S15 and S18) do not differ significantly from those of their analogues from PA or PA–MeOH.
The Raman and FTIR spectra, shown in Fig. 7 and Fig. 8, were obtained to provide complementary structural information to surface XPS analysis. Due to the high luminescence in Raman spectroscopy, the annealed film at 150 °C does not provide any structural information about the sample. Due to the very low thickness and good adhesion to the substrate, the thin films annealed at 300 °C could not be prepared in sufficient quantity in powder form for Raman and FTIR analyses, and therefore, they are not presented here.
 | ||
| Fig. 7 Raman spectra of Mn–S-based solution as well as used solvents and sulfur (left); Raman spectra of sulfur powder and Mn–S-based films annealed at 60 °C (right). | ||
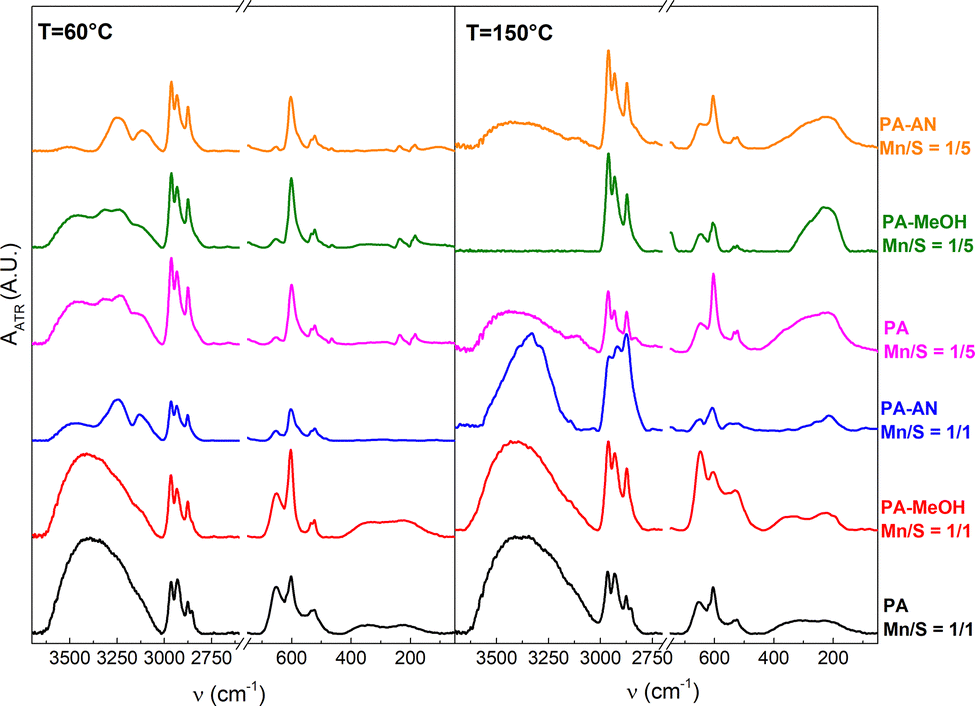 | ||
| Fig. 8 FTIR spectra of deposited Mn–S-based thin films annealed at 60 °C (left) and 150 °C (right). | ||
The Raman spectra of the solutions (Fig. 7, left) clearly indicate the presence of polysulfides (S32− and S42−) regardless of the Mn/S ratio. Sulfur-rich solutions (Mn/S = 1/5), compared to stoichiometric solutions (Mn/S = 1/1), show polysulfide bands more clearly and show more pronounced shifts of the bands located at 130 and 184 cm−1 to higher values and of the band located at 444 cm−1 to lower values. This indicates the probable binding of manganese to form complexes based on [MnS3]. An increase in the intensity of the bands at 234 cm−1 (S42− (ref. 54)) and 266 cm−1 (S62− (ref. 61)), and a significant decrease in the band at 402 cm−1 (S82− (ref. 54 and 61)) compared to sulfur solutions indicate the continued shortening of polysulfide chains. However, after the film deposition, unification occurs again, and the resulting layers (Fig. 8, right) show identical rhombohedral S8 bands (84, 153, 184, 220, 248, 437 and 474 cm−1)54,61 in the Raman spectra, which correspond to the source powdered sulfur. Other bands are not visible in the layers' spectra, due to the presence of weak luminescence masking the signal, as well as the S8 sulfur bands' superior cross-section compared to any other species present.
The FTIR results of all other samples clearly confirm the presence of both Mn–S bonds, represented by bands at 512 and 599 cm−1,19,62,63 while the bands at 650 cm−1 show the presence of Mn–OH vibrations.19,62 The typical bands of manganese oxides represented by MnO (270 and 550 cm−1 (ref. 5 and 64)), Mn3O4 (446, 499 and 501 cm−1 (ref. 49)), and MnO2 or MnOOH (446, 488, 592 and 645 cm−1 (ref. 65 and 66)) were not detected by FTIR analysis in all studied films. We, therefore, assume that all oxygen atoms are associated with Mn2+ and Mn4+ hydroxides, which corroborates the XPS analysis.
FTIR analysis also reveals that the porous network films (PA Mn/S = 1/1 and PA–MeOH Mn/S = 1/1) exhibit significantly more intense bands of –OH bonds (i.e., the broad band around 3400 cm−1 (ref. 67) and the Mn–OH bands) regardless of annealing temperature. In contrast, the nanoparticle-based films exhibited more intense bands indicative of C–S, C–N, and NH4+ vibrations,68–71 attributable to a higher content of alkylammonium polysulfide salt residues. The presence of polysulfide bonds in thin films with sulfur overstoichiometry is supported by bands at 181, 235 and 464 cm−1.59,72 Their disappearance, along with the decrease in the intensity of C–N and NH4+ bands after annealing at 150 °C, is associated with the decomposition of alkylammonium polysulfide salts. These observations are in excellent agreement with the EDS analysis results of the oxygen, sulfur and nitrogen contents.
To verify the photocatalytic efficiency of the prepared films annealed at 300 °C, the photocatalytic degradation of methylene blue (MB) was explored (see Fig. 9 and Fig. S19). As demonstrated in Fig. 9, the photofading of the MB under the UV light (365 nm) exposure, along with the low-intense halogen probe beam, is negligible, which validates the applicability of the in situ experiment. Measured kinetic dependences of the absorbance decrease over time clearly show that Mn–S-based nanoparticle films exhibit photocatalytic activity. In contrast, porous networks either do not show photocatalytic activity or it is significantly lower than that of nanoparticle films. We assume that the increased surface area of the nanoparticle films, compared to the porous networks, is the contributing factor to the higher activity. However, the sulfur-rich films' surface composition plays a crucial role in their enhanced photocatalytic activity. The presence of mixed-valence Mn2+/Mn4+ states and various sulfur species, particularly polysulfides, likely provides efficient pathways for photogenerated charge separation73–75 and facilitates the redox reactions involved in methylene blue (MB) degradation. However, the influence of other elements contained on the surface of nanoparticles cannot be ignored, as evidenced by constantly lower kinetic rates for thin films deposited from PA–AN solutions. We assume that the photocatalytic efficiency of PA–AN is significantly reduced because the photoactive surface can be blocked by carbon-based organic residues. Our hypothesis arises from a higher carbon content in both the surface and volume of Mn–S-based thin films from PA–AN solutions in comparison to pure PA and PA–MeOH inferred from the XPS and EDS analysis results. The kinetic rate values for nanoparticle thin films are listed in Table 3.
 | ||
| Fig. 9 First order kinetics plot of ln(A/A0) as a function of irradiation time for photo-assisted degradation of methylene blue under UV light diode exposure catalyzed using Mn–S-based films annealed at 300 °C. | ||
| Sample | K × 10−4 [min−1] |
|---|---|
| PA–AN, Mn/S = 1/1 | 4.84 ± 5 × 10−2 |
| PA, Mn/S = 1/5 | 15.6 ± 7 × 10−2 |
| PA–MeOH, Mn/S = 1/5 | 15.4 ± 4 × 10−2 |
| PA–AN, Mn/S = 1/5 | 12.4 ± 3 × 10−2 |
Conclusions
In the present study, a novel approach for the solution processing of Mn–S-based thin films has been developed, which employs elemental Mn and S dissolved in propylamine-based solvents. The solutions with various Mn/S ratios exert a substantial influence on the morphology, chemical composition, and molecular structure of the prepared thin films. The deposition of solutions based on pure propylamine or propylamine–methanol mixtures with Mn/S = 1/1 produces thin films with a nanoporous network structure. This nanoporous network is primarily composed of manganese sulfides and hydroxides with a Mn2+ oxidation state. Conversely, deposition employing solutions derived from propylamine and a propylamine–methanol mixture with Mn/S = 1/5 produces spherical nanoparticles with a diameter of approx. 85 nm. These nanoparticles are formed by a combination of manganese sulfides and hydroxides (both in Mn2+ and Mn4+ oxidation states), polysulfides, and residues of dissolution products. In contrast, the deposition of a solution formed by dissolving elemental manganese and sulfur in a propylamine–acetonitrile mixture, regardless of the Mn/S ratio, leads to the formation of larger spherical and oval nanoparticles with equivalent diameters of 136 ± 11 nm for Mn/S = 1/1 and 126 ± 20 nm for Mn/S = 1/5 with chemical compositions close to their PA and PA–MeOH analogues, respectively. All nanoparticle films annealed at 300 °C showed photocatalytic activity, as evidenced by the photodegradation of methylene blue during UV exposure.Author contributions
J. J. designed the experiment with the contribution of M. K., prepared solutions and Mn–S based films, conducted Raman and FTIR measurement and interpretation, and wrote the first draft of the manuscript; M. K. performed AFM and photocatalytic measurements and interpretations and evaluation of nanoparticles size distributions, and edited the manuscript; J. R. P. performed XPS measurements and data interpretations and edited the manuscript; S. S. performed SEM/EDS measurements and edited the manuscript; M. K. performed XRD measurements, participated in experiment design, edited the manuscript, and supervised the experiment. The manuscript was written with contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article, including raw spectroscopic data (EDS, FTIR, XRD, and UV-vis), AFM scan images, SEM images, and XPS raw data and fits are available at Zenodo repository at https://doi.org/10.5281/zenodo.15373035.The data supporting this article have also been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ma00519a.
Acknowledgements
All authors appreciate the financial support from grant LM2023037 from the Ministry of Education, Youth and Sports of the Czech Republic.References
- R. Woods-Robinson, Y. Han, H. Zhang, T. Ablekim, I. Khan, K. A. Persson and A. Zakutayev, Chem. Rev., 2020, 120, 4007–4055 CrossRef CAS.
- S. Swathi, R. Yuvakkumar, P. Senthil Kumar, G. Ravi and D. Velauthapillai, Fuel, 2021, 303, 121293 CrossRef CAS.
- H. Li, L. Zhang and Y. Cao, Appl. Surf. Sci., 2021, 541, 148519 CrossRef CAS.
- G. A. Tigwere, M. D. Khan, L. D. Nyamen, A. A. Aboud, T. Moyo, S. T. Dlamini, P. T. Ndifon and N. Revaprasadu, Mater. Sci. Semicond. Process., 2022, 139, 106330 CrossRef CAS.
- M. Mylarappa, V. V. Lakshmi, K. R. V. Mahesh, H. P. Nagaswarupa and N. Raghavendra, IOP Conf. Ser.: Mater. Sci. Eng., 2016, 149, 012178 Search PubMed.
- Y. Yu, Y. Tao and S. Yan, Surf. Interfaces, 2024, 55, 105395 CrossRef CAS.
- M. Dan, Q. Zhang, S. Yu, A. Prakash, Y. Lin and Y. Zhou, Appl. Catal., B, 2017, 217, 530–539 CrossRef CAS.
- A. Hannachi, A. Segura and H. Maghraoui-Meherzi, Mater. Chem. Phys., 2016, 181, 326–332 CrossRef CAS.
- H. M. Ali, E. K. Shokr, A. A. Ismail, M. S. Kamel, H. A. Mohamed and M. F. Hasaneen, Opt. Mater., 2024, 148, 114821 CrossRef CAS.
- S. A. Mayén-Hernández, S. Jiménez-Sandoval, R. Castanedo-Pérez, G. Torres-Delgado, B. S. Chao and O. Jiménez-Sandoval, J. Cryst. Growth, 2003, 256, 12–19 CrossRef.
- C. Gümüş, C. Ulutaş and Y. Ufuktepe, Opt. Mater., 2007, 29, 1183–1187 CrossRef.
- P. Tiwari, J. Jaiswal and R. Chandra, J. Appl. Phys., 2019, 126, 213108 CrossRef.
- J. Wang, J. Tan, L. He, Z. Li, S. Li, Y. Zhang, H. Nong, Q. Wu, Q. Yu, X. Zou, H. M. Cheng and B. Liu, Adv. Powder Mater., 2024, 3, 100164 CrossRef.
- S. C. Riha, A. A. Koegel, X. Meng, I. S. Kim, Y. Cao, M. J. Pellin, J. W. Elam and A. B. F. Martinson, ACS Appl. Mater. Interfaces, 2016, 8, 2774–2780 CrossRef CAS.
- F. Srouji, M. Afzaal, J. Waters and P. O’Brien, Chem. Vap. Deposition, 2005, 11, 91–94 CrossRef CAS.
- S. H. Chaki, S. M. Chauhan, J. P. Tailor and M. P. Deshpande, J. Mater. Res. Technol., 2017, 6, 123–128 CrossRef CAS.
- R. B. Pujari, A. C. Lokhande, A. A. Yadav, J. H. Kim and C. D. Lokhande, Mater. Des., 2016, 108, 510–517 CrossRef CAS.
- M. Imran, A. M. Afzal, M. W. Iqbal, H. H. Hegazy, M. Z. Iqbal, S. Mumtaz and R. Qureshi, Mater. Sci. Semicond. Process., 2023, 158, 107366 CrossRef CAS.
- A. Admuthe, S. S. Kumbhar, S. K. Chougule, G. N. Padasare and M. M. Tonape, Macromol. Symp., 2020, 392, 2000186 CrossRef CAS.
- J. Luo, J. Sun, P. C. Guo, Z. S. Yang, Y. X. Wang and Q. F. Zhang, Mater. Lett., 2018, 215, 176–178 CrossRef CAS.
- S. An, Q. Gao, X. Zhang, X. Li, L. Duan and W. Lü, J. Alloys Compd., 2019, 799, 351–359 CrossRef CAS.
- J. Hu, P. Tao, W. Fan, R. Yang, H. Feng, B. Guo, H. Li, L. Xu and S. Li, Inorg. Chem. Front., 2024, 11, 4528–4551 RSC.
- A. Hannachi and H. Maghraoui-Meherzi, J. Solid State Chem., 2017, 247, 120–130 CrossRef CAS.
- Z. K. Heiba, M. B. Mohamed, A. Badawi and N. M. Farag, Chem. Phys. Lett., 2021, 779, 138877 CrossRef CAS.
- C. Ulutas, O. Erken, M. Gunes and C. Gumus, Phys. B, 2020, 588, 412175 CrossRef CAS.
- R. B. Pujari, A. C. Lokhande, A. R. Shelke, S. B. Kale, D. W. Lee and C. D. Lokhande, Solid State Sci., 2021, 111, 106449 CrossRef CAS.
- H. Zhang, B. Liu, Z. Lu, J. Hu, J. Xie, A. Hao and Y. Cao, Small, 2023, 19, 2207214 CrossRef CAS.
- W. Hussain, H. Malik, R. A. Hussain, H. Hussain, I. R. Green, S. Marwat, A. Bahadur, S. Iqbal, M. U. Farooq, H. Li and A. Badshah, J. Electron. Mater., 2019, 48, 2278–2288 CrossRef CAS.
- H. Moreno-García, J. O. Sigala-Valdez, M. del, R. Martínez-Blanco, I. Cruz Reyes, S. M. Durón-Torres, I. L. Escalante-García and A. Del Rio-De Santiago, Heliyon, 2024, 10, e26703 CrossRef.
- X. Yu, C. Li-Yun, H. Jian-Feng, L. Jia, F. Jie and Y. Chun-Yan, J. Alloys Compd., 2013, 549, 1–5 CrossRef.
- Y. Xi, X. Zeng, J. Chen, L. Ma, Z. Zhan, C. Chen, Y. Yuan, L. Liao, Z. Peng, L. Zheng, Y. Huang and L. Xu, Adv. Powder Technol., 2023, 34, 103838 CrossRef CAS.
- V. Vignesh, K. Subramani, M. S. Oh, M. Sathish and R. Navamathavan, Mater. Chem. Phys., 2019, 230, 249–257 CrossRef CAS.
- C. Zhang, Z. Lin, C. Huang, B. Zheng, Y. Li, J. Wang, M. Deng, S. Tang and Y. Du, ACS Appl. Energy Mater., 2019, 2, 6599–6607 CrossRef CAS.
- T. Dhandayuthapani, M. Girish, R. Sivakumar, C. Sanjeeviraja and R. Gopalakrishnan, Appl. Surf. Sci., 2015, 353, 449–458 CrossRef CAS.
- A. Goktas and İ. H. Mutlu, J. Electron. Mater., 2016, 45, 5709–5720 CrossRef CAS.
- A. Goktas, A. Tumbul, Z. Aba, A. Kilic and F. Aslan, Opt. Mater., 2020, 107, 110073 CrossRef CAS.
- A. Hannachi, S. Hammami, N. Raouafi and H. Maghraoui-Meherzi, J. Alloys Compd., 2016, 663, 507–515 CrossRef CAS.
- V. Mantella, S. B. Varandili, J. R. Pankhurst and R. Buonsanti, Chem. Mater., 2020, 32, 9780–9786 CrossRef.
- A. Mahajan, M. Jha and S. Ghosh, New J. Chem., 2024, 48, 4144–4154 RSC.
- F. Jamal, A. Rafique, S. Moeen, J. Haider, W. Nabgan, A. Haider, M. Imran, G. Nazir, M. Alhassan, M. Ikram, Q. Khan, G. Ali, M. Khan, W. Ahmad and M. Maqbool, ACS Appl. Nano Mater., 2023, 6, 7077–7106 CrossRef.
- J. W. Thomson, K. Nagashima, P. M. Macdonald and G. A. Ozin, J. Am. Chem. Soc., 2011, 133, 5036–5041 CrossRef PubMed.
- R. E. Davis and H. F. Nakshbendi, J. Am. Chem. Soc., 1962, 84, 2085–2090 CrossRef.
- V. Mauritz and R. W. Crisp, J. Mater. Chem. C, 2024, 12, 11319–11334 RSC.
- V. Dusastre, B. Omar, I. P. Parkin and G. A. Shaw, J. Chem. Soc., Dalton Trans., 1997, 3505–3508 RSC.
- S. Graham and P. Parkin Ivan, Main Group Met. Chem., 2001, 24, 195–204 Search PubMed.
- W. T. Cronenwett and L. W. Hoogendoorn, J. Chem. Eng. Data, 1972, 17, 298–300 CrossRef.
- P. S. Albright and L. J. Gosting, J. Am. Chem. Soc., 1946, 68, 1061–1063 CrossRef PubMed.
- C. V. V. Ramana, A. B. V. K. Kumar, M. A. Kumar and M. K. Moodley, J. Chem., 2013, 2013, 687106 CrossRef.
- S. Isber, E. Majdalani, M. Tabbal, T. Christidis, K. Zahraman and B. Nsouli, Thin Solid Films, 2009, 517, 1592–1595 CrossRef CAS.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS.
- H. Konieczna, D. Lundberg and I. Persson, Polyhedron, 2021, 195, 114961 CrossRef CAS.
- M. Hagen, P. Schiffels, M. Hammer, S. Dörfler, J. Tübke, M. J. Hoffmann, H. Althues and S. Kaskel, J. Electrochem. Soc., 2013, 160, A1205 CrossRef CAS.
- H.-L. Wu, L. A. Huff and A. A. Gewirth, ACS Appl. Mater. Interfaces, 2015, 7, 1709–1719 CrossRef CAS.
- F. P. Daly and C. W. Brown, J. Phys. Chem., 1975, 79, 350–354 CrossRef CAS.
- A. M. Alanazi, P. D. McNaughter, F. Alam, I. J. Vitorica-yrezabal, G. F. S. Whitehead, F. Tuna, P. O’Brien, D. Collison and D. J. Lewis, ACS Omega, 2021, 6, 27716–27725 CrossRef CAS PubMed.
- Y. Yuan, X. Yan, Y. Wang, Y. Xiong, C. Tian, Z. Lin and L. Chai, Surf. Interfaces, 2021, 23, 101015 CrossRef CAS.
- V. Natarajan, H. Singh, I. Arora, A. Sathya and P. K. Sharma, Surf. Interfaces, 2025, 68, 106705 CrossRef CAS.
- A. Göktaş and İ. H. Mutlu, J. Solgel Sci. Technol., 2014, 69, 120–129 CrossRef.
- A. Goktas, Phys. E, 2020, 117, 113828 CrossRef CAS.
- X. Zhang, W. Yang, W. Niu, P. Adams, S. Siol, Z. Wang and S. D. Tilley, ChemSusChem, 2021, 14, 3967–3974 CrossRef CAS.
- B. A. Trofimov, L. M. Sinegovskaya and N. K. Gusarova, J. Sulfur Chem., 2009, 30, 518–554 CrossRef CAS.
- Z. K. Heiba, M. B. Mohamed, N. M. Farag and S. I. Ahmed, Cryst. Res. Technol., 2021, 56, 2000201 CrossRef CAS.
- Z. K. Heiba, M. B. Mohamed, N. M. Farag and A. Badawi, J. Mater. Sci.: Mater. Electron., 2022, 33, 10388–10398 CrossRef CAS.
- H. L. Lu, G. Scarel, X. L. Li and M. Fanciulli, J. Cryst. Growth, 2008, 310, 5464–5468 CrossRef CAS.
- R. Yang, Z. Wang, L. Dai and L. Chen, Mater. Chem. Phys., 2005, 93, 149–153 CrossRef CAS.
- M. I. Said and B. Harbrecht, J. Nanopart. Res., 2019, 21, 68 CrossRef.
- M. Shamim and K. Dana, J. Mol. Struct., 2016, 1125, 27–35 CrossRef CAS.
- S. D. Lin, A. C. Gluhoi and B. E. Nieuwenhuys, Catal. Today, 2004, 90, 3–14 CrossRef CAS.
- H. Su, Z. Li, Q. Huo, J. Guan and Q. Kan, RSC Adv., 2014, 4, 9990–9996 RSC.
- S. D. Lin, A. C. Gluhoi and B. E. Nieuwenhuys, Catal. Today, 2004, 90, 3–14 CrossRef CAS.
- C. M. Leewis, W. M. M. Kessels, M. C. M. van de Sanden and J. W. Niemantsverdriet, Appl. Surf. Sci., 2006, 253, 572–580 CrossRef CAS.
- A. Rafie, R. Pereira, A. A. Shamsabadi and V. Kalra, J. Phys. Chem. C, 2022, 126, 12327–12338 CrossRef CAS.
- Y. Feng, H. Liu, Q. Lu, Y. Liu, J. Li, X. He, X. Liu and D. Mikhailova, J. Power Sources, 2022, 520, 230885 CrossRef CAS.
- H.-Y. Kai, K.-L. Wong and P. A. Tanner, Next Mater., 2025, 8, 100610 CrossRef.
- J. Gao, Z. Zhuang, X. Zhou, H. Xu, X. Xu and W. Li, Chem. Eng. J., 2024, 485, 149520 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |