
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Composite hydrogels of phenylalanine dipeptides with trivalent metal cations†
Melissa
Kemesies‡
,
Vanessa
Jerschabek‡
,
Chife
Ekene Fidelis
,
Jonas
Volmer
,
Anna Franziska
Roth
,
Christian
Schwieger
 ,
Annette
Meister
,
Haleh
Hashemi Haeri
and
Dariush
Hinderberger
*
,
Annette
Meister
,
Haleh
Hashemi Haeri
and
Dariush
Hinderberger
*
Institute of Chemistry, Physical Chemistry – Complex Self-Organizing Systems, Martin Luther University Halle-Wittenberg, Von-Danckelmann-Platz 4, 06120 Halle (Saale), Germany. E-mail: dariush.hinderberger@chemie.uni-halle.de
First published on 8th July 2025
Abstract
We studied multicomponent hydrogels from a short model peptide, Fmoc-protected diphenylalanine (Fmoc–FF), in the presence of trivalent cations of aluminium (Al) and iron (Fe). Additionally, we investigated the implicit effects of small molecules on the self-assembly of Fmoc–FF by buffering the model system with three commonly used buffers: HEPES, TRIS, and sodium phosphate. The formation and stability of the resulting hydrogels were analyzed through TEM imaging and rheological characterization, and changes in the secondary structure of Fmoc–FF due to addition of metal cations were monitored using ATR-IR spectroscopy. Our results suggest that complexation occurs between the metal cations and the amide groups of the peptide. Furthermore, a spin-probing electron paramagnetic resonance (EPR) strategy, employing persistent nitroxyl radicals TEMPO, TEMPO-benzoate, and Fmoc-TOAC reveals differences in the solvation shell of probes and peptides induced by the presence of metal cations. TEM images revealed different fibrilation mechanisms in the presence of the two cations; while Al-cations lead to formation of water droplets and liquid–liquid phase separation (LLPS), ferric cations form helical superstructures. These differences were then correlated with the toughness of the hydrogels and the distinct types of interactions within them allowing to draw conclusions on the internal nanostructure of these composite materials.
Introduction
Since the pioneering work by Gazit and co-workers, diphenylalanine (FF) and its derivatives have been extensively used as minimalistic peptide building blocks capable of self-assembling into larger functional structures.1–8 Self-assembly of hydrophobic dipeptides (like Phe–Phe) is also environment-dependent and has, e.g., been discussed for the case of microporous materials by Görbitz.9 The self-assembly process of FF dipeptides is primarily driven by hydrogen bonding yet in its N-protected form (9-fluorenylmethoxycarbonyl) Fmoc–FF, the self assembly is driven mostly by π–π interactions, referring to the attractive interactions between the aromatic fluorenyl rings. Imaging techniques such as transmission and scanning electron microscopy (TEM and SEM, respectively), and atomic force microscopy (AFM) have revealed that FF dipeptides self-assemble into rigid nanotubular structures, with diameters ranging from 500 to 2000 nm and lengths up to 10 μm. These structures can be altered through co-assembly with other components or fragments in the gel. Another imaging method (PAINT) presented by Fuents et al.10 shows the possibility of imaging hydrogels in their native state. It appears to be obvious that peptide-based hydrogels possess attractive properties for biomedical applications such as biocompatibility, high water content, and injectability.11–13 It has been found that partial folding or misfolding of peptides and proteins lead to the formation of insoluble amyloids.These amyloids display structural transitions from random coil or α-helix conformations to adopt β-sheet structures. Such transitions can be triggered by various physical and chemical stimuli, including changes in temperature, pH, electrostatic interactions, and chemical additives such as metal ions. Furthermore, even the presence of small molecules, such as buffers, can significantly influence the self-assembly or disassembly of peptides and should be taken into account. However, transitions like peptide self-assembly and aggregation are not always associated with harmful processes, as is often assumed. Hauser et al.14 demonstrated that aggregation and amyloid-like structures can play essential roles in certain biological contexts, such as peptide hormone storage or microbial biofilm formation. This highlights the importance of expanding our understanding of the peptide self-assembly process.
In this work, we focus on metal-containing peptide hydrogels (MCH) and on questions related to gelation in different buffer systems. On a nanoscopic scale, MCHs benefit from a combination of covalent interactions (such as polymer connectivity and metal coordination) and on complex non-covalent interaction patterns (including electrostatic forces, hydrogen bonding, and van der Waals interactions). This combination gives rise to a novel class of hydrogels with unique and tunable properties.15–17 From a biological perspective, metals can act as stimuli, inducing structural changes in biological scaffolds, such as the aforementioned transformation of α-helices into β-sheetsleading to amyloid formation.18,19 In nature, metal ions often serve as catalytic centers in enzymes, so that MCH could be considered simplified enzyme models or catalytic nanoreactors.20–23 Even small amounts of metal cations not only influence the physicochemical properties of MCH materials but also significantly enhance the toughness of the hydrogels. This is particularly valuable, as purely organic hydrogels are often mechanically weak.
According to the LD50 values,24 metals such as Fe2+, Fe3+, Al3+, Eu3+, Ca2+, Cu2+, Zn2+, and Mg2+ are considered safe for incorporation into bio-hybrid materials.25–28 When cations have the same charge, those with a larger ionic radius typically exhibit a stronger cross-linking effect inducing a tougher polymeric or bio-based gel structure. While numerous studies have described a wide range of applications involving monovalent and divalent cations (such as Ca2+, Cs+, Na+, Mg2+) in combination with FF or Fmoc–FF hydrogels,29–33 relatively limited research is available on the role of trivalent cations in hydrogels. Most existing studies have focused on aluminium and ferric ions.34–37
Aluminium is commonly used to enhance the self-healing and toughness of polyacrylic acid (PAA) gels, where it serves as an ionic cross-linker during the polymerization process.35 Similarly, there are reports of PAA gels being physically and/or covalently cross-linked with ferric ions.38 However, studies that investigate the role of these cations as additional cross-linkers remain limited. As an example, Strachota and co-workers explored the crosslinking effects of Al3+, Fe3+, and La3+ on gels prepared from LAPONITE® clay.39 In a physiological context, iron homeostasis is crucial for the daily functioning of living cells.40,41 Aluminium, on the other hand, has been implicated in some forms of dementia, including Alzheimer's disease. However, it remains unclear whether Al3+ contributes to the formation of abnormal neurofibrillary tangles in the brain or merely forms complexes with them.42
The effects of ferric cations (Fe3+) have been mostly investigated for hydrogels based on poly (acrylic acid) (pAA) or polysaccharides (like chitosan or alginates). Based on the different complexation behaviour, ferric cations are stronger cross linking agents when compared to ferrous cations. While Fe2+ prefers binding to neutral ligands due to its soft cationic nature, Fe3+ strongly binds to carboxylate groups and drives the transition from soluble to gel sate.37,43–46
The self-assembly of the short model peptide Fmoc–FF (9-fluorenylmethoxycarbonyl-FF) in the presence of buffers has been the focus of several attempts. Reports of the self-assembly of Fmoc–FF, its derivatives, or similar structures in buffers such as borax (sodium tetraborate) under relatively basic conditions (pH 8.5–9.5), PBS (phosphate-buffered saline) or MES (2-(N-morpholino)ethanesulfonic acid) buffer for BPmoc-FF hydrogels (p-borono-phenylmethoxycarbonyl) can be found. These reports mainly examine the effects of charge, salt-induced changes, or replacing the Fmoc-group during the self-assembly process.47–52
In this study, we first investigated the self-assembly and gelation of the reference Fmoc–FF dipeptide in different commonly used biological buffers, focusing on gelation pH.53,54 The formation of Fmoc–FF hydrogels in buffered solutions that contain metal–cations is currently under investigation and will be discussed in a subsequent report. Here, we explore the multicomponent Fmoc–FF dipeptides prepared in water with trivalent metal ions, Fe3+ and Al3+, using TEM imaging, rheological characterization, ATR-IR, and EPR spectroscopy with a spin-probing approach. In spin-probing EPR spectroscopy, persistent nitroxyl-based radicals with different noncovalent interaction patterns (amphiphilic, hydrophilic, aromatic, or amino-acid-based) are admixed during the gelation process and report on local interactions and structures. We have established this approach for protein and polymer gels and for self-assembling, thermoresponsive elastin-like polypeptides and polymers derived from those.55–59 Complementary to the nanoscale characterization, the mechanical aspects of self-assembly in and gelation of buffered Fmoc–FF are examined thoroughly using rheological characterization techniques.
Materials and methods
Lyophilized Fmoc–FF-OH (hereafter referred to as Fmoc–FF) was obtained from Bachem, and Fmoc-TOAC was purchased from IRIS Biotech. TEMPO and TEMPO-benzoate were purchased from Sigma Aldrich. Aluminium chloride and iron chloride were provided by Fluka Analytical and Carl Roth. HEPES (N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid) buffer was purchased from Sigma-Aldrich. TRIS hydrochloride (tris(hydroxymethyl)aminomethane) and sodium phosphate (NaH2PO4) and sodium chloride buffers were provided from Carl Roth. All samples were prepared using the solvent exchange method.54,60 A batch of 100 mg of Fmoc–FF was dissolved in 1 mL of dimethyl sulfoxide (DMSO) to create a stock solution. Examining the critical gel concentration (CGC), we obtained gelation concentration of ∼0.5 wt% (5 mg mL−1) in accordance to the CGC values reported in the literature (see ESI,† Fig. S1).Therefore, Fmoc–FF samples with a gelator concentration of 5.0 mg mL−1 were prepared by adding 50 μL of the stock solution to 950 μL of Milli-Q ultrapure water, followed by vortexing for one minute. For the metal-containing gels, Fmoc–FF was dissolved in a metal salt solution at the appropriate concentration to achieve a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.001 Fmoc–FF:M3+ composition. Gel formation started and proceded notably within less than 5 minutes at room temperature and were clear and transparent. Formation of the gel samples was first examined visually using the inverted vial test (see Fig. 1). The gel formation kinetics are discussed in more detail in the Results and discussion section. The gel samples for TEM imaging were prepared similarly, although at a lower concentration of 2 mg mL−1.
:0.001 Fmoc–FF:M3+ composition. Gel formation started and proceded notably within less than 5 minutes at room temperature and were clear and transparent. Formation of the gel samples was first examined visually using the inverted vial test (see Fig. 1). The gel formation kinetics are discussed in more detail in the Results and discussion section. The gel samples for TEM imaging were prepared similarly, although at a lower concentration of 2 mg mL−1.
 | ||
| Fig. 1 Left: Chemical structures of the used materials, (A) Fmoc protected FF dipeptide, (B)–(D) used spin probes: Fmoc-TOAC, TEMPO and TEMPO-benzoate, (E)–(G) buffer molecules: HEPES, TRIS and PBS. Right: Examples of prepared hydrogels (inversion test). Samples are numbered according to Table 1. | ||
To study the spin-probed hydrogels with EPR spectroscopy, in addition to the common spin probes TEMPO (2,2,6,6-tetramethylpiperidinyl-N-oxyl) and TEMPO-benzoate (4-hydroxy-2,2,6,6-tetramethylpiperidinyl-N-oxyl-benzoate), we also employed the non-natural amino acid TOAC (2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4-carboxylic acid). The latter was used in its amino-protected form, Fmoc-TOAC (2,2,6,6-Tetramethylpiperidine-N-oxyl-4-(9-fluorenylmethyloxycarbonyl-amino)-4-carboxylic acid). Stock solutions of TEMPO (in water), Fmoc-TOAC, and TEMPO-benzoate (hereafter referred to as TB, both in DMSO) were prepared at a concentration of 0.1 M and then diluted with Milli-Q ultrapure water to achieve a final concentration of 200 μM in the mixture. This dilution is necessary to avoid the collision-induced broadening effect of EPR spectra due to high concentrations, known as Heisenberg spin exchange broadening. The chemical structures of the spin probes used are shown in Fig. 1. A list of the prepared samples is provided in Table 1.
| Nr. | Sample | Gelation pH |
|---|---|---|
| 1 | Fmoc–FF | 5 |
| 2 | Fmoc–FF-Al | 6–7 |
| 3 | Fmoc–FF-Fe | 7–8 |
| 4 | TEMPO | |
| 5 | Fmoc–FF-TEMPO | 5 |
| 6 | Fmoc–FF-TEMPO-Fe | 5–6 |
| 7 | Fmoc–FF-TEMPO-Al | 6–7 |
| 8 | TEMPO-benzoate (TB) | |
| 9 | Fmoc–FF-TB | 5 |
| 10 | Fmoc–FF-TB-Al | 7–8 |
| 11 | Fmoc–FF-TB-Fe | 7–8 |
| 12 | Fmoc-TOAC | |
| 13 | Fmoc–FF–Fmoc-TOAC | 4 |
| 14 | Fmoc–FF–Fmoc-TOAC-Al | 6–7 |
| 15 | Fmoc–FF–Fmoc-TOAC-Fe | 6–7 |
Rheometry
Rheological characterization of the hydrogels was conducted using an Anton Paar Physica MCR 301 rheometer (Anton Paar, Graz, Austria) equipped with a 25 mm cone-plate (CP25-2/TG-SN9120) geometry. The CP system was selected since the samples are not highly viscoous and technically the shear rate is independent from measuring system and therefore a consistent shear rate over sample is ensured. The samples were carefully placed on the surface of the lower plate, and the upper plate was lowered to achieve a gap distance of 0.051 mm. Amplitude sweeps were performed to determine the linear regime of viscoelastic (LVR) behavior. No variation in storage and loss moduli (G′ and G′′) could be observed up to strain of 1% (see Fig. S2, ESI†). Therefore, measuring parameters were adjusted so the LVR criteria is fulfilled. Frequency sweeps were performed to examine the dynamic moduli of all samples with frequencies increasing from 0.1 to 100 (1 s−1) in 31 steps. All prepared samples exhibited steady and stable behavior across the frequency range. Time sweeps were measured as well to obtain kinetics of gel formation, with the angular frequency set to 0.1 rad s−1 (see Fig. S3, ESI†). Finally, the viscosity profiles of the hydrogel samples were checked to obtain a qualitative notion of the hydrogel microstructure (see Fig. S4, ESI†). The samples were incubated for 24 hours at room temperature before rheological measurements were conducted.ATR-IR spectroscopy
IR spectra were measured using a Bio-ATR II unit in a Vertex 70 IR spectrometer (BRUKER Optics, Ettlingen, Germany) equipped with a K10 thermostat (Thermo Fisher Scientific, Schwerte, Germany). 30 μL of the samples were placed on the zinc selenide/silica hybrid crystal of the ATR-IR device, and the sample cell was sealed. The peptide samples were measured with the empty ATR crystal as a reference. All experiments were conducted at a constant temperature of 20 °C, maintained by a circulation water bath. 256 scans were accumulated with a spectral resolution of 4 cm−1 and Fourier transformed with a zero-filling factor of 2. In all spectra the atmospheric water vapor was compensated and spectra of pure water recorded at the same conditions were subtracted. Afterwards, the ATR IR spectra were transformed into transmission spectra for representation and normalized in the spectral region of 900–1100 cm−1 to a vector norm of 1 to eliminate the influence of concentration differences between the samples. Spectra recording and data analysis were performed using the OPUS software (Bruker Optics, Ettlingen, Germany).Negative-stain transmission electron microscopy (EM)
EM grids were prepared by loading 10 μL hydrogel (2 mg mL−1) onto glow-discharged copper TEM grids with continuous 10–12 nm carbon film coating (300 mesh size; Quantifoil Micro Tools, Großlöbichau, Germany). Excess liquid was blotted off with a strip of filter paper after 45 s followed by two washing steps with water and staining with 10 μL 2% (w/v) aqueous uranyl acetate solution. Specimens were dried and examined in an EM 900 transmission electron microscope (Carl Zeiss Microscopy, Oberkochen, Germany), and micrographs were recorded with an SM-1k-120 slow-scan charge-coupled device (slow-scan CCD) camera (TRS, Moorenweis, Germany).EPR spectroscopy
Room temperature continuous wave EPR measurements (CW-EPR) at X-band frequency (9.4 GHz) were performed on a Magnettech MiniScope MS400 benchtop spectrometer (Magnettech GmbH, Berlin, Germany, now Bruker Biospin). Spectra were recorded with a microwave power of 3 mW, a modulation frequency of 100 kHz, a modulation amplitude of 0.1 mT, and 4096 data points.Q-band CW-EPR (33.9 GHz) measurements at 298 K were conducted on a Bruker EMX-plusQ spectrometer, using an ER5106QT resonator. A microwave power of 1–3 mW was applied for sample and background measurements. Unless noted otherwise, a modulation amplitude of 0.1–0.3 mT was used during measurements, with a modulation frequency of 100 kHz. EPR simulations were carried out using the MATLAB-based Easyspin program package.61,62
Results and discussion
1. TEM imaging of self-assembly of Fmoc–FF in different buffers and MCHs
TEM images of Fmoc–FF in different buffers are shown in Fig. 2(A)–(D). We observed that in water, Fmoc–FF self-assembles into fibers (see A1, white arrows) and helical fibers (see A1, white arrowheads) with a pitch of approximately 100 nm (A2). Additionally, both thin and thick stiff ribbons are observed (see A1, A3, black arrows). In TRIS (B1–B3), PBS (C1–C3), and HEPES (D1–D3), stiff ribbons of varying thicknesses are the exclusively found aggregates. Higher magnification images (see B3, C3, D3, black arrows) reveal that these ribbons are composed of multiple layers forming terraced structures of varying height. Helical fibers are absent in all buffer-containing hydrogels. This suggests that the absence of ions is a prerequisite for helical fiber formation, where individual fibers may assemble into helical superstructures, likely driven by van der Waals (London dispersion) interactions and π–π stacking. In contrast, the presence of buffer ions appears to screen the charges of Fmoc–FF, favouring the formation of tightly packed, parallel ribbon structures instead.8,54 Notably, helical superstructures of Fmoc–FF have not been previously reported in the literature. Therefore, further studies will focus on elucidating the molecular arrangement and identifying the driving forces behind helical fiber formation. | ||
| Fig. 2 TEM images of 2 mg mL−1 Fmoc–FF gels in water (A1–3), TRIS (B1–3), PBS (C1–3), and HEPES (D1–3) at different magnifications. White arrows point to thin fibers and white arrow heads indicate helical fibers with a pitch (p) of around 100 nm. Black arrows point to ribbons with terraces. Corresponding invert test tube of each buffered gel is shown on the right side. | ||
TEM images of MCHs are presented in Fig. 3(A) and (B). In water, Fmoc–FF-Fe self-assembles into fibers (see (A1–A3), white arrows) and helical fibers (see A1, A3, white arrowheads). These helical fibers consist of multiple individual fibers (see A3, white arrows) bundled together to form a helical superstructure (see A3, white arrowhead). Additionally, short and narrow stiff ribbons are observed (see A1, A2, black arrows).
 | ||
| Fig. 3 TEM images of 2 mg mL−1 Fmoc–FF-Fe hydrogels (A1–3) and Fmoc–FF-Al hydrogels (B1–3) at different magnifications. White arrows point to thin fibers and white arrow heads indicate helical fibers with a pitch (p) of around 100 nm. Black arrow heads point to LLPS droplets. | ||
In contrast, Fmoc–FF-Al primarily forms droplets in water (see B1, B2, black arrowheads), indicative of liquid–liquid phase-separated (LLPS) regions.30,63 A limited number of individual thin fibers and thicker helical fibers with a pitch of approximately 100 nm are also present (see B3). These findings support different self-assembly mechanisms in the presence of Fe3+ and Al3+ cations.
2. Mechanical properties of MCHs
The mechanical properties of the prepared hydrogels were examined using Rheometry. Due to the dynamic nature of metal–coordinated bonds, these properties depend on the applied deformation rate.29,64,65 Stress sweeps on the pure hydrogel in water as a reference (Fmoc–FF), the hydrogels in buffered solutions, as well as metal-containing samples, were performed to find the linear viscoelastic region, LVR (see Fig. 4 and Fig. S1, ESI†). The linear part of LVR region was also checked for TEMPO spin probed metal containing samples, as well (Fig. S1, ESI†). When the strain (γ) was increased to 10%, we observed a significant loss in viscoelastic properties. Therefore, all the rheological characterizations (frequency sweep, time sweep and viscosity) were measured with a strain of 1%. Viscosity profiles for all samples revealed shear thinning behavior (Fig. 1 and Fig. S2, ESI†). | ||
| Fig. 4 Mechanical properties of Fmoc FF-hydrogels prepared in water and different buffers. | ||
The final pH of the hydrogel was approximately 7.0. Gel preparation in TRIS buffer did not show immediate negative effects on gelation, either, and the gel appeared to be self-supporting when the vial was inverted. However, it was sensitive to external stress (e.g., gently shaking the vial), causing the gel to collapse into a liquid. The final pH of this hydrogel was 4.5. In contrast, the gelation process in HEPES buffer was insufficient, resulting in a thick, viscous solution of white color (final pH ∼6.8). Similar to the TRIS-buffered case, this sample also exhibited sensitivity to stress. For all examined hydrogels, the storage modulus (G′) dominated over the loss modulus (G′′), which is characteristic of elastic, gel-like materials (see Fig. 4). When monitoring absolute stability, the reference gel prepared in water exhibited both viscous and elastic properties, with a moderate stability of ∼764 Pa (see Table 2).
| Medium | pH | Total stability/Pa | tan(δ) |
|---|---|---|---|
| Water | 4.96 | 764 | 0.18 |
| HEPES | 6.87 | 383 | 0.36 |
| PBS | 7.01 | 348 | 0.32 |
| TRIS | 4.53 | 3174 | 0.15 |
In this context, total stability values exceeding 1000 Pa are considered desirable, as they are at least on the level of the reference gel. The hydrogel prepared in TRIS buffer remarkably exhibited the strongest elastic properties, with higher values for both G′ and G′′. However, as mentioned earlier, the TRIS-buffered hydrogel was sensitive to external stress and, therefore, was not considered long-term stable against mechanical stimuli. In comparison, both the sodium phosphate buffer and HEPES buffer exhibited adverse effects on the elastic properties of the hydrogel. In both cases, the values of G′ and G′′ were low and relatively close to each other, indicating higher viscous and lower elastic properties compared to the reference gel prepared in water.
Our results reveal that the choice of buffer as the gelation medium significantly affects hydrogel formation, with the specific type of buffer playing a crucial role. Both TRIS and HEPES samples were sensitive to external stress, with the HEPES sample requiring a longer time to form a gel.
In contrast, the sample prepared in sodium phosphate buffer exhibited neither of these effects, making it the most favorable option based on visual observations alone. Rheological analysis of the hydrogels revealed that TRIS buffer had a positive effect on static network stability, whereas sodium phosphate and HEPES buffers had a negative influence. A possible explanation for these observations is the formation of microgels. Microgels are known to have a high water content, similar to hydrogels, but they also possess a larger surface area. There are few studies about the relationship between gelation behavior and surface area. In a recent study, e.g., the relation between gelation of PEO polymers on silica nanoparticles has been discussed.66 The authors found that gelation behavior depends on the available surface area and the amount of gelator per unit area. In our case, the formation of microgels increases the surface area, but at the same time, it hinders networking and cross-linking between gelator sites. As a result, the formed gel is more easily destabilized, particularly when external stress is applied. Given these findings, it is also worth considering whether the molecular structure of the buffer—such as the zwitterionic form of HEPES—could disrupt the self-assembly process of the hydrogel. In conclusion, better gelation does not necessarily correlate with higher stability. Stability is influenced not only by the structure of the gel but also by its water/solution content. Taking into account the working pH range of these buffers and the pH at which gelation occurs, sodium phosphate emerges as the best option for controlling pH while minimizing structural effects. Further research on buffered, metal-containing peptide gels is currently performed in our lab and will be in the focus of a future publications. The gelation kinetics of buffered gels were monitored using time sweep measurements (Fig. S3, ESI†). For all samples except for the HEPES-buffered one, the storage modulus (G′) and the loss modulus (G′′) exhibited similar behavior up to 50 seconds. After this point, G′ showed a sharp increase and began to diverge from G′′, indicating that the gel transitioned to being primarily governed by elastic properties. This process continued until around 300 seconds, after which both G′ and G′′ reached a plateau. Clearly, the gelation onset point occurs within the first minute.
The viscosity profiles of the buffered gels demonstrated a decrease in viscosity with increasing shear rate, indicating shear-thinning behavior. This suggests that the microstructure of the gels facilitates flow. As shown by TEM imaging (Fig. 2), the gels are predominantly composed of large, flat ribbons, which tend to align with the direction of flow, giving a nanoscale indication for their shear-thinning characteristics.
We discuss the various properties of these gels in the following sections. To assess the effect of steric hindrance, we also used TB and Fmoc-TOAC spin probes. The Fmoc–FF-TB gel exhibited the highest mechanical strength, being an order of magnitude stiffer than the Fmoc–FF-TOAC gel. This highlights the significant role of steric hindrance in the gel formation process. One may conclude that by using TB instead of Fmoc-TOAC, there was no disruption and even a strengthening of the π–π interactions. Possibly, the amphiphilic TEMPO structure within TB may have separated the water molecules from the aromatic crosslinking points by hydrogen bonding, which may even have reinforced the gel-promoting interactions. We also found that addition of spin probes does neithert affect gelation kinetics nor viscosity. For example, the TEMPO containing Fmoc–FF gel, displayed the same kinetics and viscosity as the Fmoc–FF itself (see Fig. S3, ESI†).
The damping factor tan(δ) = G′′/G′ serves as a measure of elasticity or viscosity independent of the respective measuring setup. We found that all prepared gels exhibited elastic behavior, as indicated by tan(δ) < 1.
 | ||
| Fig. 5 Mechanical properties of Fmoc FF metal containing hydrogels. Amplitude sweeps of (A) Aluminium and Iron containing hydrogels. (B) Time evolution of elastic and viscous moduli for Fmoc–FF and metal containing hydrogels. (C) Viscosity profile of Fmoc–FF and its metal containing hydrogels. | ||
 | ||
| Fig. 6 Frequency-dependent rheological characteristics (storage modulus) of spin probed Fmoc–FF-hydrogels in combinations with Al3+ and the three spin probes used in this study. In figure legend, Fmoc is abbreviated as fm, for the sake of legend placement. | ||
Supplementing Fmoc–FF-Al gels with the Fmoc-TOAC spin label did not improve the toughness of the gel with and without Al3+. In fact, the metal-free Fmoc-TOAC-containing gels were slightly softer than the Fmoc–FF-Al gel. This could be due to the presence of additional bulky protecting Fmoc groups. While these groups strengthen π–π interactions, they also introduce steric hindrance for the cation crosslinking due to their large size. These two opposing effects appear to cancel each other out, resulting in a gel with properties similar to the Fmoc–FF-Al gel itself. Al3+-containing Fmoc–FF-TB gels exhibited reduced mechanical properties compared to the Fmoc–FF-TB gel (with G′ ∼ 11400 vs. 4260 Pa). This again suggests that negative interference with the strong gel network either by TB or Al3+ or both together introducing interactions that counteract gelation.
The most thermodynamically stable complex of Al3+ in chloride solutions, across a wide range of concentrations (0.2–3 M) and temperatures (25–125 °C), is the hexaaquaaluminium(III) ion, [Al(H2O)6]3+, as identified by Raman spectroscopic measurements.67 Theoretical calculations, including DFT and first-principles simulations, in agreement with X-ray data, have shown that hydrogen bonding (H-bond) interactions dominate over other forces in the first hydration shell, arranging water molecules in a trigonal coordination.68,69 Spectroscopic findings69 also estimated a high density of water molecules in the Al3+ cation's hydration shell, about 71% higher than in bulk water. These results suggest that the synergy of electrostatics, π–π interactions and H-bonding during gelation of Fmoc–FF-Al leads to a tougher gel compared to pure Fmoc–FF gels.
Examination of spin probe behavior reveals a somewhat different interaction between Fmoc–FF and metal cations. Aluminium-containing gels may be strengthened by an additional physical linker, in this case, TEMPO. H-Bonding interactions between TEMPO and the water shell surrounding the Al3+ cation are likely further enhanced, thereby boosting the gel's viscoelastic properties. For the other two spin probes, the presence of bulky groups destabilizes the H-bond interaction networks to a lesser extent in Fmoc–FF-Al gels containing TB compared to those with Fmoc-TOAC. As a result, the gels with Fmoc-TOAC are softer.
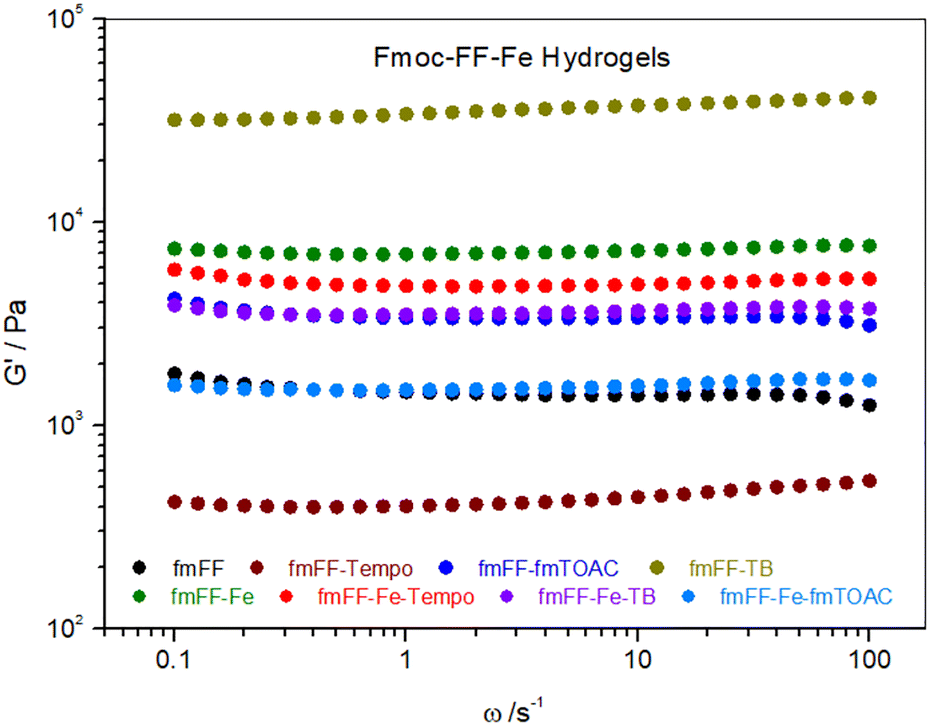 | ||
| Fig. 7 Frequency-dependent rheological characteristics (storage modulus) of spin probed Fmoc–FF-hydrogels in combinations with Fe3+ and the three spin probes used in this study. In the figure legend, Fmoc is abbreviated as fm, for the sake of brevity. | ||
Adding spin probes to Fmoc–FF-Fe gels clearly changed their viscoelastic properties. The effect of the spin probes was particularly pronounced for TB and Fmoc-TOAC, both of which showed slightly decreased elastic properties compared to the Fmoc–FF-Fe-TEMPO sample. This suggests that the steric hindrance effect for these two spin probes outweighs the potential π–π-interactions and H-bonding interactions (possibly driven by the NO moiety in TEMPO), which play a key role in the solvation of metal cations in aqueous media and, consequently, in the formation of hydrogels.37,71
3. IR spectroscopy of MCHs
The IR spectra of Fmoc–FF (reference) and metal-containing gels are shown in Fig. 8. All presented spectra are corrected for the water contribution. Key features of the Fmoc–FF hydrogel spectra are the different carbonyl vibration, of which, according to the molecular structure (see Fig. 1(A)), three different types can be recognized: (i) the carbamate linker between the Fmoc and the Phe group, (ii) the amide bond between the two Phe residues and the terminal carboxylic acid. The carbamate vibration gives rise to a strong vibrational band at 1693 cm−1,72 which is most prominent in the Fmoc–FF gel and detected for the MCHs at the same position but in reduced intensity. The amide group vibrations give rise to a group of prominent bands, which includes the amide I band (mainly C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) centered at 1652 cm−1 (see Fig. 8(B)). This position is typical for the vibration of unstructured proteins or peptide. In addition, clear amide A (3302 cm−1), amide II (1536 cm−1), and amide III (1256 cm−1) bands are visible in the spectra. While the amide A mode is due to the N–H stretching vibration, amide II and III are dominated by N–H bending modes.73 The positions and intensities of these bands are strongly influenced by transition dipole coupling and the hydrogen bonding pattern in structures formed by the peptides. Here, all amide band intensities are reduced by the addition of the trivalent metal cations, with the amide A vibration (Fig. 8(A)) being not discernible at all in the MHC spectra. This drastic intensity reduction of all amide modes indicates the involvement of the amide groups in the coordination of the metal ions.74,75 Presumably this metal coordination disturbs the vibrational coupling though inter-molecular hydrogen bonds, that are, in absence of metal ions, formed between N–H and CO groups. Such reduction in vibrational band intensity is also observed when Fmoc-dipeptide gels are compared to monomeric Fmoc-dipeptides vibrations,72 indicating that the intensity is largely influenced by peptide aggregation. The CO stretching vibration of the terminal carboxylic acid group is visible at 1747 cm−1 in all three spectra. Also this vibration is reduced in intensity upon metal addition. However, both the carboxylic acid and the carbamate bands are less affected than the amide bands. In general, the intensity reduction is more pronounced for Fe3+-containing gels than for Al3+-containing gels, indicating a stronger complexation of Fe3+ over Al3+. Other vibrations, like the C–H stretching vibrations of the aromatic fluorenyl groups at 3016 cm−1 the ring aromatic ring stretching and aromatic C–H bending modes between 1300 and 1500 cm−1, and the C–O and C–N stretching modes (not shown) are not influenced by the addition of the metal cations, indicating that the integrity of the molecules is maintained. Moreover, these bands could be used as internal standard for normalization of the spectra in order to eliminate concentration differences between the samples. Hence, all discussed differences in band intensities can clearly be attributed to reduced dipole strengths by metal complexation.
O stretching) centered at 1652 cm−1 (see Fig. 8(B)). This position is typical for the vibration of unstructured proteins or peptide. In addition, clear amide A (3302 cm−1), amide II (1536 cm−1), and amide III (1256 cm−1) bands are visible in the spectra. While the amide A mode is due to the N–H stretching vibration, amide II and III are dominated by N–H bending modes.73 The positions and intensities of these bands are strongly influenced by transition dipole coupling and the hydrogen bonding pattern in structures formed by the peptides. Here, all amide band intensities are reduced by the addition of the trivalent metal cations, with the amide A vibration (Fig. 8(A)) being not discernible at all in the MHC spectra. This drastic intensity reduction of all amide modes indicates the involvement of the amide groups in the coordination of the metal ions.74,75 Presumably this metal coordination disturbs the vibrational coupling though inter-molecular hydrogen bonds, that are, in absence of metal ions, formed between N–H and CO groups. Such reduction in vibrational band intensity is also observed when Fmoc-dipeptide gels are compared to monomeric Fmoc-dipeptides vibrations,72 indicating that the intensity is largely influenced by peptide aggregation. The CO stretching vibration of the terminal carboxylic acid group is visible at 1747 cm−1 in all three spectra. Also this vibration is reduced in intensity upon metal addition. However, both the carboxylic acid and the carbamate bands are less affected than the amide bands. In general, the intensity reduction is more pronounced for Fe3+-containing gels than for Al3+-containing gels, indicating a stronger complexation of Fe3+ over Al3+. Other vibrations, like the C–H stretching vibrations of the aromatic fluorenyl groups at 3016 cm−1 the ring aromatic ring stretching and aromatic C–H bending modes between 1300 and 1500 cm−1, and the C–O and C–N stretching modes (not shown) are not influenced by the addition of the metal cations, indicating that the integrity of the molecules is maintained. Moreover, these bands could be used as internal standard for normalization of the spectra in order to eliminate concentration differences between the samples. Hence, all discussed differences in band intensities can clearly be attributed to reduced dipole strengths by metal complexation.
 | ||
| Fig. 8 ATR-IR spectra for metal containing Fmoc–FF gels. The spectra in panels (A) and (B) correspond to high and low wave number regions. | ||
4. Spin probing EPR of MCHs
EPR spectroscopy together with spin probing is an indirect but straightforward approach to gather information about the surrounding environment and rotational dynamics of small paramagnetic molecules (spin probes). We used TEMPO as an amphiphilic probe, enabling us to simultaneously monitor environments with different polarities around the probe. TB was selected to further explore the effects of steric hindrance and separation of bulky aromatic groups on gelation and the driving forces behind it. Fmoc-TOAC, a synthetic amino acid spin probe, provides insights into various aspects: conformational changes due to the incorporation of a new amino acid into the gel network, steric hindrances caused by the protective Fmoc group, and the enhancement of aromatic π–π interactions from this group. The two key properties of isotropic hyperfine coupling (Aiso) and rotational correlation time (τc) report on the polarity of the environment around the spin probe and its dynamics on the picosecond to nanosecond timescale, respectively.We initially measured CW-EPR spectra of the prepared samples at room temperature at X-band frequency (∼9.4 GHz). All spectra revealed the presence of an aqueous medium around the spin probes, reflected in their Aiso values (∼48 MHz). The exception was the Fmoc-TOAC probed samples, which had Aiso ∼45 MHz, indicating a strong hydrophobic medium, compared to Aiso ∼ 47 MHz of spin probe in water (see Table S2, ESI†). Interestingly, even at room temperature, not all spectra displayed isotropic rotational behavior, as indicated by varying peak intensities. This effect was particularly evident in the samples containing Fmoc-TOAC (see Fig. S7, ESI†). To better resolve the dynamic and anisotropic behavior of the spin-probed hydrogels, we measured the samples at higher field/frequency (Q-band, 34 GHz) and hence higher resolution.
Experimental and simulated Q-band EPR spectra of the TEMPO-containing samples are shown in Fig. 9(A). The TEMPO spin probe dissolved in water displays a two-component spectrum: a dominant hydrophobic component (Aiso ∼44 MHz), which contributes ∼80% of the spectrum, and a second, more hydrophilic component, which additionally shows much faster rotational dynamics. TEMPO is a well-known amphiphilic probe capable of altering the structure of the solvent/water shell into aqueous lower polarity solvation shells, or ALPSS, a phenomenon first described by us.76 We find the ALPSS around a variety of different amphiphilic radicals (e.g. di-tert-butyl nitroxide, DTBN, see Fig. S8, ESI†) and different aqueous solvent mixtures, which is currently being explored in more detail. Here, the basic idea is that TEMPO and in particular the amount of ALPSS and aqueous component, respectively, is a very sensitive probe of the polarity of the environment.
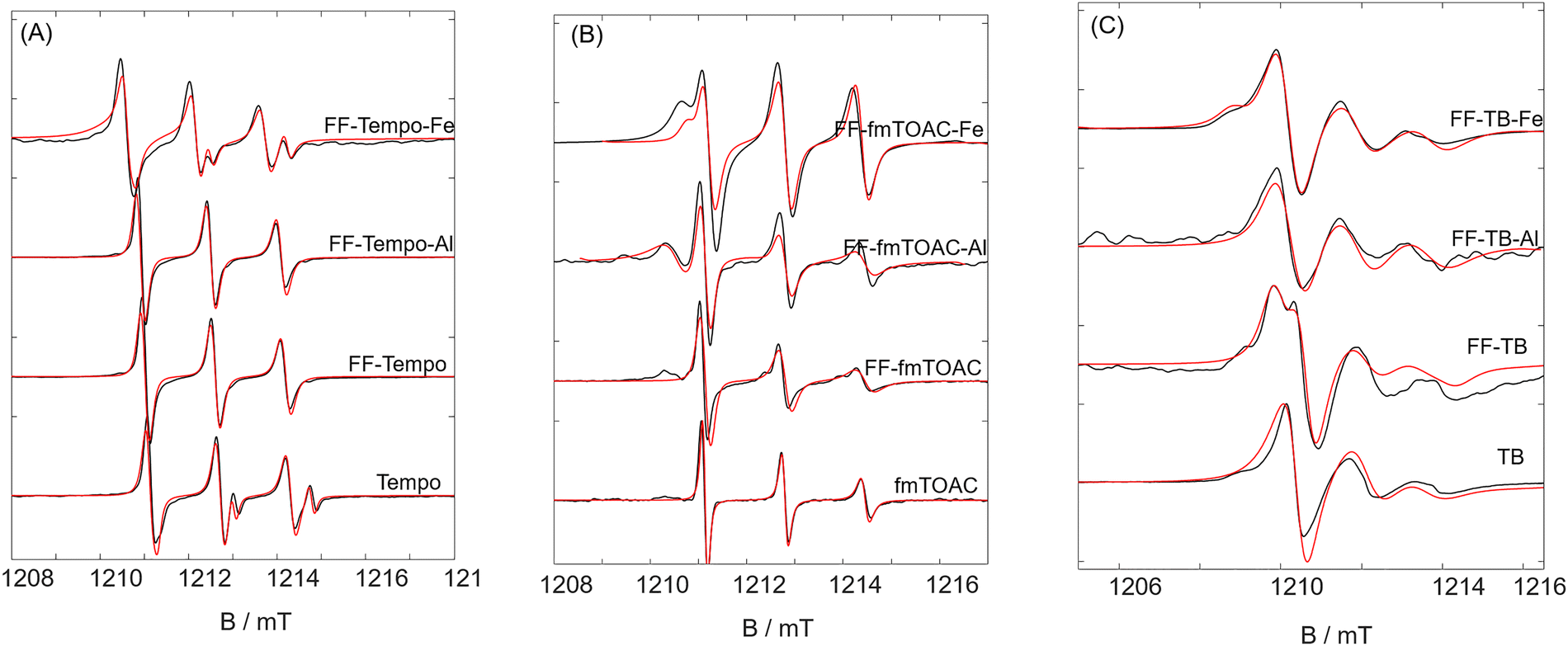 | ||
| Fig. 9 Experimental room temperature EPR spectra (in black) and corresponding simulations (in red) of (A) TEMPO, (B) Fmoc-TOAC, and (C) TB-containing metal-hydrogels. The dashed lines denote the position of the high-field peak of the ALPSS species indicating non-polar solvation. | ||
When TEMPO is added to Fmoc–FF gels, spectroscopically only thehydrophobic ALPSS component is detected (see dashed lines), so that only TEMPO spin probes in adequately low-polar environment are found, with no purely water-solvated species being present any more. In view of the hydrophobic nature of Fmoc–FF, this result is not surprising. Spectral simulations also support these findings, reflected in decreased Aiso and increased giso (see Table S1, ESI†). The presence of aluminium cations does not significantly alter this behavior, with hydrophobic environments within the gel system still being detected by TEMPO. However, the addition of iron cations leads to a breakdown of hydrophobicity for a large portion of the TEMPO molecules. Not only is the bulk-type, hydrophilic hydration shell around TEMPO partially restored (∼20% of TEMPO), but a third hydrophilic component (indicated by an Aiso of ∼48 MHz) also emerges, also featuring spectral broadenings that indicate spin exchange interactions. These observations suggest that a higher local concentration of TEMPO resides in the more water-rich sections of the hydrogel or that hydrogen bonds exist between water molecules in the solvation shell or between the hydration shell of iron and bulk water.
Experimental and theoretical studies on ferric ions in aqueous solutions have shown that the first hydration shell around Fe3+ complexes is highly ordered, with six water molecules surrounding the metal center. The second hydration shell forms hydrogen bonds with the first shell and acts as a transient medium for proton exchange with bulk water, occurring very rapidly (∼10−10 s),77–79 in contrast to the slower water exchange in hydrated Al3+ cations. The presence of spin exchange couplings (interactions) in Fe3+-containing Fmoc–FF-TEMPO hydrogels, as evidenced by EPR spectral simulations, is an indirect indication of the fast exchange rate of hydrogen-bonded TEMPO probes80 in the aqueous parts of the hydrogel. However, we cannot yet specify whether this exchange occurs between the water shells of hydrated metal cations or between these water shells and bulk water. This can be examined implementing pulse EPR techniques of hyperfine spectroscopy and isotope labeling, e.g. through use of D2O and is currently planned in our lab for future research.
Experimental spectra and corresponding simulations of Fmoc-TOAC gels are shown in Fig. 9(B). In this case, we observed a situation quite different from that of TEMPO. The spin probe itself was found to be solvated in a more hydrophilic environment (Fig. 9(B)), and there is only one type of hydrophilic solvation in the hydration shell and no ALPSS is observed. Note that the rotational motion is fast but anisotropic as evidenced from the three lines with different line intensities. This can be attributed to the rather large Fmoc group. Upon adding Fmoc–FF, the hydrophilic nature of the hydrogel was not only preserved but also slightly increased (by about 3 MHz, see Table S2, ESI†). However, the dynamics of the spin probe clearly become restricted, which could be due to the interaction between the aromatic Fmoc group of the probe and the Fmoc–FF gel network, or the probe itself being directly influenced and rotationally hampered by the gel structure.
The presence of aluminium cations induces a minor population of low-polarity radical species (∼20%), but the majority of the spin probes still resides in hydrophilic (water-rich) environments. Similar to the effect observed with TEMPO, the addition of iron results in the detection of three distinct spin species for Fmoc-TOAC. However, in contrast to TEMPO, the low-polarity solvation and spin exchange interactions (∼5 MHz) are more dominant. The polarity sensed by the third species (∼20%) is increased significantly, with a ∼7 MHz difference from the two other components.
The spectral shapes of TB spin probe species in the gels are more anisotropic compared to the other two spin probes (see Fig. 9(C)). The spin probe itself resides in a highly polar environment, as indicated by the Aiso of ∼53 MHz. As expected, TB exhibits slower dynamics, which can be attributed to the rather rigid attachment of the TEMPO-based radical esterified to a benzoic acid group in the TB molecules. This iss reflected in the broad and anisotropic EPR spectra of all TB-containing hydrogels. The rotational dynamics of TB-containing samples are also slower compared to those of the other two spin probes (see Tables S1–S3, ESI†), which for Fmoc-TOAC may seem surprising, as Fmoc is much larger/heavier and more rigid than the benzoic acid part of TB. When inspecting Fig. 1, it becomes clear that the chemical attachment of the NO-containing part to the Fmoc region is through several single bonds or bonds with only partial double-bond character, so that in TB the NO-containing part is more rigidly attached. In the presence of Fmoc–FF, the hydrophilic nature of the spin probe is thus maintained, but its rotational motion significantly slowed down (∼24 ns). A component with slower dynamic (low-field peak) also becomes visible more clearly. This suggests that TB molecules reside in water-rich regions that are confined by the Fmoc–FF gel network, similar to the behavior observed for Fmoc-TOAC. Adding aluminium cations to Fmoc–FF gels containing TB induces a highly non-polar environment around the spin probe, as indicated by a sharp decrease in Aiso (∼12 MHz, from ∼53 MHz for Fmoc–FF-TB to 41 MHz, see Fig. 9(C) and Table S3, ESI†). Additionally, the spectral component with the more strongly restricted rotational motion seems reduced in intensity.
Both observations contrast with the behavior observed for Fmoc-TOAC in Fmoc–FF gels with Al3+, which highlights the presence of TB in a confined hydrophobic surroundings (possibly deep inside the gel network) in the presence of aluminium cations. The addition of iron cations to Fmoc–FF-TB gels also led to a decrease in the polarity of the environment with an Aiso of ∼46 MHz, accompanied by very slow dynamics that are present again, similar to the Fmoc–FF-TB system.
Among the spin probes used, only TEMPO induces hydrophobicity in its surrounding water shell by forming a lower-polarity solvation shell. This effect was also observed in the presence of Fe3+. The other two spin probes, TB and Fmoc-TOAC, exhibited fully hydrophilic environments around the probes (in reference samples without hydrogels). Spectral simulations also revealed different solvation tendencies between the MCHs. Spin probing of Fe3+-containing hydrogels shows that iron induces higher-polarity solvation shells around the spin probes, while the overall gel network remains relatively hydrophobic, with Aiso values between 41–44 MHz (see Tables S1–S3, ESI†).
This suggests that Fe3+ promotes the formation of more water-rich areas within the gel system. Iron ions do, however, also lead to a reduction of rotational motion for a fraction of the spin probes, in particular visible for the two probes containing aromatic residues. The situation is quite different in Al3+-containing gels, in particular visible for the two probes containing aromatic residues. In these gels, the effect of adding the Al3+ cations changes the interplay between two intermolecular weak forces—hydrogen bonding and π–π interactions—differently. Also, all spin probes seem to exhibit strongly hydrophobic surroundings, with Aiso values between 41–43 MHz. When considering that the IR-spectroscopic findings indicate a stronger complexation of amides (and to a lesser degree of carbamates) with Al3+ over Fe3+, one may conclude that Fe3+ ions tend to dominate and strengthen the solvated and hydrogen-bonded amide/carboxylic acid regions, effectively restricting residence times of the amphiphilic spin probes (around their NO groups) in these strongly polar regions. Al3+ions do not complex the amide/carbamates as strongly as Fe3+, however it benefits from a wide range of non-covalent interactions which may translate into more loosely packed highly polar regions that can then be probed mainly by the amphiphilic parts of the spin probes. A graphical summary of these observation is depicted in Scheme 1.
 | ||
| Scheme 1 A schematic cross-section of different types of solvation shells based on EPR data is shown. Hydrophilicity and hydrophobicity are color-coded as blue and red, respectively. (A) TEMPO exhibits the ALPSS effect, while (B) the TEMPO moiety in TEMPO-benzoate (TB), even in the presence of Fmoc–FF, resides in fully hydrophilic regions. (C) Hydrophobic effect and strong solvated, metal-dominated regions govern aluminium-containing samples with TB, leading to strongly hydrophobic NO environments. (D) Addition of iron to Fmoc-TOAC-FF gels increases the contribution of a lower-polarity solvation shell. | ||
Conclusion
In materials science, functionality of the material is of prime interest. Yet, a deep understanding of the molecular mechanisms underlying the materials’ functions is not only of fundamental scientific interest but also needed for the ability to establish correlations between the “bottom-up” features of materials, predict their performance, and ultimately improve and design new materials. In this report, we studied how the incorporation of trivalent metal cations, aluminium and iron, into short peptide-based protected diphenylalanine hydrogels influences their nanoscale and macroscopic properties, using ATR-IR and EPR spectroscopies, along with rheological characterizations. The addition of metal cations, although in a very small quantities significantly increased the toughness of the gels, by almost an order of magnitude. IR spectra suggest for complexation between the peptide amide groups and metal cations, as evidenced by shifts in the amide A and carbonyl stretching bands. Among the two employed cations, Fe3+ was complexed more strongly by the amino acids than Al3+, which confirms observations made by dynamic moduli measurements, indicating that the toughness of the metal-containing hydrogels may be attributed to the formation of ligand biding between the metal cations and the peptide, in synergy with non-covalent hydrogen bonding including differential solvation shells and π–π interactions.TEM images reveal distinct fibril formation mechanisms, including helical structures and superstructures. In the presence of Al3+ cations, we observed larger droplet sizes, suggesting the development of liquid–liquid phase separation and the emergence of solute-rich droplets. This observation is further supported by spin-probing EPR spectroscopy. Aluminium tends to induce lower-polarity hydration shells and, creating a hydrophobic environment for co-dissolved spin probe molecules. In contrast, iron complexation forms more robust solvated complexes with the amide groups of the dipeptide and promotes more water-rich regions within the hydrogel, which remain accessible to spin probes while still maintaining strong mechanical properties.
Altogether, we were able to develop a detailed model of the interactions occurring at the nanoscale with notably different interaction tendencies between the two metal cations studied.
Since biological systems often function in buffered solutions, we also prepared buffered hydrogels to investigate the effect of electrostatic interactions on the self-assembly process. Our results suggest that the potential for microgel formation should be taken into account, and that better gelation does not necessarily correlate with higher stability of the formed gels. TEM micrographs reveal helical superstructures and stiff ribbons in all three used buffers and water. In contrast, the MCHs do not show the stiff ribbon structures, a discrepancy that will be further elucidated in following reports.
Author contributions
M. Kemesies: buffered hydrogels, rheological characterization and data curation, writing original draft. V. Jerschabek: IR spectroscopy, data curation, writing original draft. C. E. Fidelis: metal containing and spin probed-gel samples preparations, rheology and data curation A. Meister: TEM imaging, data curation, writing original draft. J. Volmer: conceptualization, Editing and data curation. A. F. Roth: EPR spectroscopy, data curation. C. Schwieger: IR spectroscopy, data curation, writing original draft H. H. Haeri: conceptualization, Investigation, supervision, EPR measurements and spectral simulations, writing original draft, review and editing. D. Hinderberger: conceptualization, supervision, project administration and funding acquisition, writing, review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
The authors thank Annekatrin Schiebel and Heike Schimm for their assistance with chemicals and instruments. Financial support by the DFG (436494874-GRK 2670, Beyond Amphiphilicity BEAM) is gratefully acknowledged. We acknowledge the Core Facility Microscopy, Biozentrum, Martin Luther University Halle-Wittenberg for providing equipment, expertise, and assistance.References
- M. Reches and E. Gazit, Science, 2003, 300, 625 CrossRef CAS PubMed
.
- M. Reches and E. Gazit, Nat. Nanotechnol., 2006, 1, 195 CrossRef CAS PubMed
- E. Gaziz, Chem. Soc. Rev., 2007, 36, 1263 RSC
- C. H. Goerbitz, Chem. Commun., 2006, 2332 RSC
- A. Mishra, K. H. Chan, M. R. Reithofer and C. A. E. Hauser, RSC Adv., 2013, 3, 9985 RSC
- P. Makam and E. Gazit, Chem. Soc. Rev., 2018, 47, 3406 RSC
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611 CrossRef CAS
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37 CrossRef CAS
- C. H. Görbitz, Chem. – Eur. J., 2007, 13, 1022–1031 CrossRef PubMed
- E. Fuentes, K. Boháčová, A. M. Fuentes-Caparrós, R. Schweins, E. R. Draper, D. J. Adams, S. Pujals and L. Albertazzi, Chem. – Eur. J., 2020, 26, 9869–9873 CrossRef CAS PubMed
- A. Nadernezhad, L. Forster, F. Netti, L. Adler-Abramovich, J. Teßmar and J. Groll, Polym. J., 2020, 52, 1007–1012 CrossRef CAS
- E. Rosa, P. Pellegrino, M. Cascione, R. Rinaldi, E. Gianolio, C. Edwards-Gayle, I. W. Hamley, G. Morelli, A. Accardo and C. Diaferia, ACS Appl. Polym. Mater., 2024, 6(12), 7197–7208 CrossRef CAS
- J. Li, R. Xiang, S. Bai and X. Yan, Soft Matter, 2019, 15, 1704 RSC
- C. A. Haueser, S. Maurer-Stroh and I. C. Martins, Chem. Soc. Rev., 2014, 43, 5326 RSC
- M. J. Ford, Y. Ohm, K. Chin and C. Majidi, J. Mater. Res., 2022, 37, 2–24 CrossRef CAS
- H. Li, P. Yang, P. Pageni and C. Tang, Macromol. Rapid Commun., 2017, 38(14), 10 CrossRef PubMed
- T. Zhu, Y. Sha, J. Yan, P. Pageni, Md. A. Rahman, Y. Yan and C. Tang, Nat. Commun., 2018, 9, 4329 CrossRef PubMed
- Y. Miller, B. Ma and R. Nussinov, Coord. Chem. Rev., 2012, 256, 2245 CrossRef CAS
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684 CrossRef CAS PubMed
- B. Xing, M. F. Choi and B. Xu, Chem. – Eur. J., 2002, 8, 5028 CrossRef CAS PubMed
- M. Ajmal, M. Siddiq, H. Al-Lohedan and N. Sahiner, RSC Adv., 2014, 4, 59562 RSC
- K. Gayen, K. Basu, D. Bairagi, V. Castelletto, I. W. Hamley and A. Banerjee, ACS Appl. Bio. Mater., 2018, 1, 1717 CrossRef CAS PubMed
- A. M. Garcia, M. Kurbasic, S. Kralj, M. Melchionna and S. Marchesan, Chem. Commun., 2017, 53, 8110 RSC
- Y. Liu, Y. Zheng, X. H. Chen, J. A. Yang, H. Pan, D. Chen, L. Wang, J. Zhang, D. Zhu, S. Wu, K. W. K. Yeung, R. Zeng, Y. Han and S. Guan, Adv. Funct. Mater., 2019, 29, 1805402 CrossRef
- N. Kashimura, Y. Suzuki, T. Nonoyama and J. P. Gong, Chem. Mater., 2024, 36, 2944–2952 CrossRef CAS
- M. Vitale, C. Ligorio, B. McAvan, N. W. Hodson, C. Allan, S. M. Richardson, J. A. Hoyland and J. Bella, Acta Biomater., 2022, 138, 144 CrossRef CAS PubMed
- Z. Y. Li, Y. L. Su, B. Q. Xie, H. L. Wang, T. Wen, C. C. He, H. Shen, D. C. Wu and D. J. Wang, J. Mater. Chem. B, 2013, 1, 1755 RSC
- M. Rivas, L. J. Del Valle, C. Alemán and J. Puiggali, Gels, 2019, 5(1), 14 CrossRef CAS PubMed
- Y. Wang, T. Xu, S. Jin, J. Y. Wang, Q. Yuan, H. Liu, Y. Tang, S. Zhang, W. Yan, Y. Jiao and G. Li, Chem. – Eur. J., 2024, 30, e202400005 CrossRef CAS PubMed
- M. C. Mañas-Torres, C. Gila-Vilchez, J. A. González-Vera, F. Conejero-Lara, V. Blanco, J. M. Cuerva, M. T. Lopez-Lopez, A. Orte and L. Álvarez de Cienfuegos, Mater. Chem. Front., 2021, 5, 5452 RSC
- M. C. Mañas-Torres, G. B. Ramírez-Rodríguez, J. I. García-Peiro, B. Parra-Torrejón, J. M. Cuerva, M. T. Lopez-Lopez, L. Álvarez de Cienfuegos and J. M. Delgado-López, Inorg. Chem. Front., 2022, 9, 743–752 RSC
- G. Domingos da Silveira, C. Izabelle, B. Saubamea, A. Varenne and F. d’Orlyé, Int. J. Pharm., 2023, 648, 123559 CrossRef PubMed
- T. Shao, N. Falcone and H. B. Kraatz, ACS Omega, 2020, 5(3), 1312–1317 CrossRef CAS PubMed
- S. Anjum, P. Gurave, M. V. Badiger, A. Torris, N. Tiwari and B. Gupta, Polymer, 2017, 126, 196 CrossRef CAS
- D. Liang, G. Zhou, Y. Hu, C. Zhao and C. Chen, J. Mater. Sci., 2021, 56, 14531 CrossRef CAS
- W. Ji, C. Yuan, S. Zilberzwige-Tal, R. Xing, P. Chakraborty, K. Tao, S. Gilead, X. Yan and E. Gazit, ACS Nano, 2019, 13(6), 7300 CrossRef CAS PubMed
- D. M. Roquero, A. Othman, A. Melman and E. Katz, Mater. Adv., 2022, 3, 1849 RSC
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49(24), 9637 CrossRef CAS
- B. Strachota, A. Strachota, G. Gąsior and M. Šlouf, J. Polym. Res., 2021, 28, 211 CrossRef CAS
- D. F. Wallace, Clin. Biochem. Rev., 2016, 37(2), 51–62 Search PubMed
- J. M. Connorton, J. Balk and J. Rodríguez-Celma, Metallomics, 2017, 9(7), 813 CrossRef CAS PubMed
- M. R. Will and J. Savory, Lancet, 1983, 2(8340), 29–34 CrossRef PubMed
- Z. Wei, J. He, T. Liang, H. Oh, J. Athas, Z. Tong, C. Wang and Z. Nie, Polym. Chem., 2013, 4, 4601 RSC
- T. J. Auletta, G. J. LeDonne, K. G. Gronborg, C. D. Ladd, H. Liu, W. W. Clark and T. Y. Meyer, Macromolecules, 2015, 48, 1736–1747 CrossRef
- Y. Hu, Z. Du, X. Deng, T. Wang, Z. Yang, W. Zhou and C. Wang, Macromolecules, 2016, 49(15), 5660–5668 CrossRef CAS
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49(24), 9637–9646 CrossRef CAS
- R. Xing, C. Yuan, S. Li, J. Song, J. Li and X. Yan, Angew. Chem., Int. Ed., 2018, 57(6), 1537 CrossRef CAS PubMed
- M. C. Gonzalez, M. I. Peñas, F. Barbault, A. J. Müller, F. Boulmedais and R. Hernández, Nanoscale, 2024, 16, 9887 RSC
- M. M. Nguyen, K. M. Eckes and L. J. Suggs, Soft Matter, 2014, 10, 2693 RSC
- S. Basak, I. Singh, A. Banerjee and H. B. Kraatz, RSC Adv., 2017, 7, 14461 RSC
- Y. Huang, Z. Qiu, Y. Xu, J. Shi, H. Lin and Y. Zhang, Org. Biomol. Chem., 2011, 9(7), 2149–2155 RSC
- M. Ikeda, T. Tanida, T. Yoshii and I. Hamachi, Adv. Mater., 2011, 23(25), 2819–2822 CrossRef CAS PubMed
- C. Diaferia, E. Rosa, G. Morelli and A. Accardo, Pharmaceuticals, 2022, 15, 1048 CrossRef CAS PubMed
- C. Tang, A. Am Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25(16), 9447 CrossRef CAS PubMed
- S. H. Arabi, B. Aghelnejad, C. Schwieger, A. Meister, A. Kerth and D. Hinderberger, Biomater. Sci., 2018, 6, 478 RSC
- S. H. Arabi, B. Aghelnejad, J. Volmer and D. Hinderberger, Protein Sci., 2020, 29(12), 2459 CrossRef CAS PubMed
- H. H. Haeri, V. Jerschabek, A. Sadeghi and D. Hinderberger, Macromol. Chem. Phys., 2020, 2000262 CrossRef
- N. Sanaeifar, K. Mäder and D. Hinderberger, Macromol. Biosci., 2020, 2000126 CrossRef CAS PubMed
- K. Laass, F. Garcia Quiroz, J. Hunold, S. Roberts, A. Chilkoti and D. Hinderberger, Biomacromolecules, 2021, 22(2), 1015 CrossRef CAS PubMed
- J. Raeburn, C. Mendoza-Cuenca, B. N. Cattoz, M. A. Little, A. E. Terry, A. Z. Cardoso, P. C. Griffiths and D. J. Adams, Soft Matter, 2015, 11(5), 927–935 RSC
-
MATLAB version: 9.13.0(R2022b) Natick, The MathWorks Inc., Massachusetts, 2022, https://www.mathworks.com Search PubMed
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178(1), 42 CrossRef CAS PubMed
- C. Yuan, A. Levin, W. Chen, R. Xing, Q. Zou, T. W. Herling, P. K. Challa, T. P. J. Knowles and X. Yan, Angew. Chem., Int. Ed., 2019, 58, 18116–18123 CrossRef CAS PubMed
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932 CrossRef CAS PubMed
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, Y. Zhao, A. B. Ihsan, H. L. Guo and X. F. Li, Macromolecules, 2014, 47, 6037 CrossRef CAS
- S. Sato and M. Kobayashi, Molecules, 2023, 28, 3555 CrossRef CAS PubMed
- W. W. Rudolph, R. Mason and C. Pye, Phys. Chem. Chem. Phys., 2000, 2, 5030 RSC
- E. J. Bylaska, M. Valiev, J. R. Rustad and J. H. Weare, J. Chem. Phys., 2007, 126(10), 104505 CrossRef PubMed
- I. Waluyo, C. Huang, D. Nordlund, U. Bergmann, T. M. Weiss, L. G. M. Pettersson and A. Nilsson, J. Chem. Phys., 2011, 134(6), 064513 CrossRef PubMed
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32(5), 751 CrossRef
- I. Persson, Dalton Trans., 2024, 53, 15517 RSC
- S. Fleming, P. W. Frederix, I. Ramos Sasselli, N. T. Hunt, R. V. Ulijn and T. Tuttle, Langmuir, 2013, 29, 9510 CrossRef CAS PubMed
- S. Krimm and J. Bandekar, Adv. Protein Chem., 1986, 38, 181 CrossRef CAS PubMed
- A. Barth, Biochem. Biophys. Acta, 2007, 1767, 1073 CAS
-
N. V. Chukanov and A. D. Chervonnyi, Infrared Spectroscopy of Minerals and Related Compounds, Springer Mineralogy, 2016, ISBN 978-3-319-25349-7 (eBook) Search PubMed
- J. Hunold, J. Eisermann, M. Brehm and D. Hinderberger, J. Phys. Chem. B, 2020, 124(39), 8601 CrossRef CAS PubMed
- L. Degreve and C. Quintale, J. Electroanal. Chem., 1996, 409, 25 CrossRef
- T. Remsungnen and B. M. Rode, Chem. Phys. Lett., 2004, 385, 491 CrossRef CAS
- C. W. Bock, G. D. Markham, A. K. Katz and J. P. Glusker, Inorg. Chem., 2003, 42(5), 1538 CrossRef CAS PubMed
- V. V. Khramtsov, A. A. Bobko, M. Tseytlin and B. Driesschaert, Anal. Chem., 2017, 89, 4758 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00339c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |