
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From design to application: amphiphilic copolymers as antimicrobial materials
Zao
Cheng
and
Patrizio
Raffa
 *
*
Smart and Sustainable Polymeric Products, Engineering and Technology Institute Groningen, University of Groningen, Nijenborgh 4, 9747 AG, The Netherlands. E-mail: p.raffa@rug.nl
First published on 6th June 2025
Abstract
The rapid growth of harmful pathogens and their severe impact on health pose significant challenges to modern science. With comprehensive progress in human society, there is an increasing focus on personal health and quality of life, leading to a growing demand for antimicrobial materials. Amphiphilic copolymers can mimic the functions and molecular features of antimicrobial peptides (also known as host defense peptides), constituting a class of new antimicrobial materials often characterized by strongly cationic and hydrophobic domains. Due to their excellent and stable antimicrobial activity, low toxicity, and resistance prevention, amphiphilic antimicrobial copolymers have recently garnered considerable interest and attention in both academic and industrial sectors. This review outlines the latest advances of amphiphilic copolymers in antimicrobial applications, including their design strategies and current applications. Challenges and future directions of research in amphiphilic antimicrobial polymers are also discussed.
 From left to right: Zao Cheng and Patrizio Raffa | Patrizio Raffa (on the right) is associate professor in Smart and Sustainable Polymeric Products at the Faculty of Science and Engineering, University of Groningen, where he has been working since 2011. His current research aims at developing and synthesizing new multifunctional polymeric materials relevant for industrial and biomedical applications. In particular, he is interested in amphiphilic and stimuli-responsive polymers obtained via controlled polymerization methods, and in the use of bio-derived building blocks for the synthesis of sustainable polymeric products. His research group explores various applications for such materials as rheology modifiers, emulsifiers, dispersants, coatings, controlled release systems, pharmaceuticals & personal care products, and smart/multifunctional devices. Zao Cheng (on the left) started his PhD in Raffa's research group in November 2022. For his PhD project, he is developing novel amphiphilic polymers with antimicrobial properties, using bio-derived acrylic monomers, and controlling the polymer's architecture via RAFT polymerization. The goal of his research is to design the new generation materials for antimicrobial applications, with a particular focus on sustainability. |
1. Introduction
As the earliest life forms on earth, microorganisms have complex and diverse metabolic mechanisms and strong adaptability to various extreme environments.1,2 According to statistics, many human diseases and almost all infectious ones are related to microorganisms.3 Moreover, they can also cause corrosion and damage to various industrial equipment and raw materials, which causes huge waste of resources and economic losses.4 Therefore, how to inhibit the reproduction of harmful microorganisms, identify harmful bacteria, and reduce the impact of harmful microorganisms on human production and life has always been a matter of concern to humans.5,6 At the same time, the abuse of antibiotics has led to a significant increase in bacterial resistance, making the original antibacterial materials unable to meet people's needs.7–9 In recent years, researchers in related fields have been working hard to develop new materials, design various structures, and explore new ways to improve the antibacterial efficiency of materials, such as metal nanoparticles,10,11 antibacterial peptides12,13 and antibacterial polymers.14–18Amphiphilic copolymers, sometimes also referred to as polymeric surfactants,19 consist of two or more distinct moieties, hydrophilic and hydrophobic respectively, which are often incompatible, chemically bound together in a linear or branched arrangement. For the most common linear polymers, they can be divided into random copolymers, alternating copolymers and block copolymers according to the arrangement of the copolymer components.20 The amphiphilic copolymer chains exhibit self-assembling behavior in specific solvents. This unique property is a result of the hydrophilic–hydrophobic interactions among the polymer chains, similarly to low-molecular weight surfactants, leading to the formation of micellar structures in the nanometer and micrometer scales.21 This makes amphiphilic copolymers highly desirable for a wide range of applications, particularly in the biomedical field, such as tissue engineering,22 drug delivery,23 gene therapy,24 and as antibacterial agent.25
Some type of amphiphilic copolymer can mimic the functions and molecular characteristics of host defense peptides (HDPs), these typically have highly active cationic and hydrophobic chains and have shown strong antibacterial effects in many studies.26–28 The cationic and hydrophobic groups of these polymers are essential structures for selectively targeting and disrupting bacterial cell membranes. Achieving a fine-tuned balance in amphiphilic antimicrobial copolymers (the average quantity of hydrophobic and cationic residues present in polymer chains) is crucial for the design of highly selective antibacterial polymers.29 However, These synthetic amphiphilic polymers can only mimic the primary and secondary structures of HDPs, whereas the tertiary structure of HDPs endows them with a broader range of mechanisms of action. These include inhibiting RNA synthesis,30 suppressing protein synthesis,31 disrupting protein folding,32 interfering with lipid complexation,33 activating microbial autolysis systems,34 and neutralizing bacterial toxins.35 Increasing evidence has shown that HDPs can also function by activating the host immune system.36,37 This involves recruiting and activating macrophages and mast cells, inducing the production of chemokines. The immunomodulatory role of antimicrobial peptides has been established as one of the key features in determining their activity towards infectious diseases.38,39
HDPs are directly involved in antibacterial activity, endotoxin neutralization, chemotaxis, modulation of anti-inflammatory and pro-inflammatory immune responses, wound healing, tissue repair, angiogenesis, and apoptosis.40 This multifaceted functionality of HDPs further highlights the challenges in achieving clinical applicability for polymers designed only to mimic membrane-disruption mechanisms.
The outer surface of bacterial cell membranes generally carries a net negative charge, which is typically stabilized by the presence of divalent cations, such as Mg2+ and Ca2+. This stabilization involves the teichoic acids and polysaccharides in Gram-positive bacteria, the lipopolysaccharides in Gram-negative bacteria, and the cytoplasmic membrane itself (Fig. 1).41,42 Cationic antimicrobial agents often disrupt this stabilization by electrostatically replacing these divalent cations. Subsequently, the hydrophobic regions of these agents interact with the bacterial cell surface and integrate into the cytoplasmic membrane, leading to bacterial cell death.43 However, highly hydrophobic polymers may also bind to and penetrate human cell membranes independently of electrostatic interactions. This lack of specificity allows them to indiscriminately affect both human and microbial cells. To address this issue, achieving an appropriate balance between hydrophobic and cationic residues in antimicrobial polymers is critical for selectively targeting bacterial cells without harming human cells.44,45
 | ||
| Fig. 1 Schematic representation of the main differences between the cell membranes of mammalian cells and various microbial families. Reproduced with permission from ref. 41, copyright 2012, Elsevier. | ||
Thanks to recent advancements in polymerization technologies, particularly reversible deactivation radical polymerization (RDRP), such as atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain Transfer polymerization (RAFT),46–49 an enormous quantity of amphiphilic antimicrobial copolymers has been developed.50–56 In this review, we aim to focus on the recent advances of amphiphilic copolymers for antimicrobial applications, including the design strategies for amphiphilic antimicrobial copolymers. We also aim to highlight current application directions for amphiphilic antibacterial copolymers. Finally, the challenges and development directions of amphiphilic antimicrobial copolymers are pointed out.
2. Design strategies for amphiphilic antimicrobial copolymers
At present, the research of amphiphilic antibacterial polymers focuses mainly on macromolecules obtained through the copolymerization of compounds containing cations (mostly ammonium groups) and compounds containing hydrophobic alkyl side chains; the resulting cationic amphiphilic antimicrobial copolymers have strong membrane destruction ability and have been widely used in the field of antibacterial agents.57–59 Many quaternary amphiphilic disinfectants have been prepared using traditional polymers, including polyvinyl pyridine,60 polyvinyl alcohol,61 polyacrylate,62 and polystyrene.63 Studies have shown that the bactericidal mechanism of amphiphilic antibacterial agents involves the combination of electrostatic interactions with microbial cell membranes, followed by membrane penetration caused by the insertion of hydrophobic groups. By disrupting the cell membrane, the transmembrane potential is decomposed, causing the leakage of intracellular substances and ultimately leading to cell death.64–67 In such models, the hydrophobicity, amphiphilic character, and the abundance of cations are considered key determinants of antibacterial activity.This section will focus on recent researches on the impact of various components on the antibacterial activity of such amphiphilic copolymers (Fig. 2). Key aspects of localized polymer design include cationic groups,68,69 hydrophobic groups,70 end groups and their amphiphilic balance.71 Additionally, the global design of polymer chains is considered, encompassing molecular weight and structural characteristics relevant to antimicrobial activity. These structural designs range from linear and cyclic structures72 to more advanced architectures, such as star, hyper-branched,73 or brush polymers.74 A special case of advanced architecture can be considered the sequence-specific oligothioetheramides developed by Alabi's group, where control over the single hydrophobic units is achieved by a multi-step synthesis.75,76
 | ||
| Fig. 2 Systematic design principles of amphiphilic antibacterial polymers.77 | ||
2.1. Local polymer design
| Cation group | Structure | Proportions between components | Molecule weight | Antibacterial effects (MIC) | Cytotoxicity | Hemolytic for RBCs | Ref. |
|---|---|---|---|---|---|---|---|
| RBCs: human red blood cells; IC50: the maximum concentration of polymer less than 50% toxic to test cells: HC50 and HC10: the maximum concentration of polymers with less than 50% or 10% hemolytic activity on red blood cells; The polymer characterization data listed in the table are the polymers with the highest antibacterial activity in the corresponding article.a The values were calculated from analysis of the peak integration of the 1H NMR spectra.b The values were determined by GPC.c The values were theoretical. | |||||||
| Ammonium |

|
f = 0.12a | M n = 1600a | 24 μg mL−1 for E. coli | — | HC50 = 1600 μg mL−1 | 68 |
| n = 8.1a | |||||||
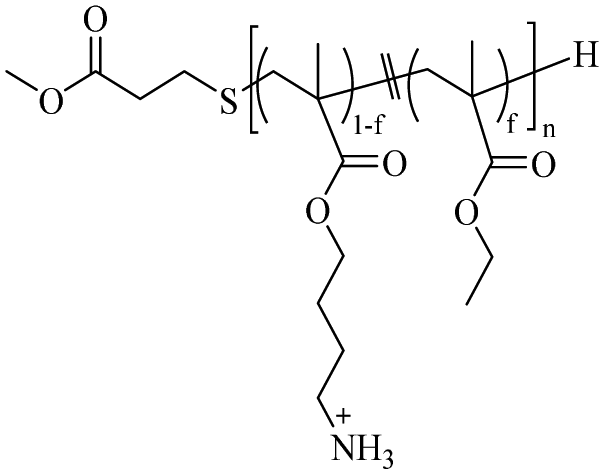
|
f = 0.29a | M n = 2900a | 21 μg mL−1 for E. coli | — | HC50 = 1300 μg mL−1 | 69 | |
| n = 12.5a | 63 μg mL−1 for S. aureus | ||||||
|
m = 20c | M n = 9200b | 4.3 μM for B. subtilis | — | HC50 > 54 μM | 91 | |
| PDI = 1.25b | |||||||
| n = 30 | 6.5 μM for S. aureus | ||||||
| 7.0 μM for MRSA | |||||||
| 10.8 μM for E. faecalis | |||||||
| 10.8 μM for C. neoformans | |||||||

|
n = 12a | M n = 1800a | 42–63 μg mL−1 for S. aureus | IC50 = 20 μg mL−1 for HEp-2 | HC50 > 1000 μg mL−1 | 92 | |
| 16 μg mL−1 for S. saprohyticus | IC50 = 55 μg mL−1 for COS-7 | ||||||

|
x = 0.69a | M n = 2600a | 15–60 μg mL−1 for S. aureus | IC50 > 1000 μg mL−1 for human fibroblasts cell | HC50 > 2000 μg mL−1 | 95 | |
| y = 0.31a | |||||||
| n = 16a | |||||||
| Ammonium |

|
x = 6.7a | M n = 2290a | 31 μg mL−1 for E. coli | — | HC50 = 740 μg mL−1 | 96 |
| y = 2.8a | |||||||
| z = 7.5a | |||||||
| n = 16.9a | |||||||
| M n = 3000b | |||||||
| PDI = 1.70b | 500 μg mL−1 for S. aureus | ||||||

|
x = 0.81a | 4900a | 6.5 μM for E. coli | IC50 > 8 MIC for human fibroblast cell | — | 72 | |
| y = 0.19a | 6.5 μM for S. aureus | ||||||
| n = 27a | |||||||

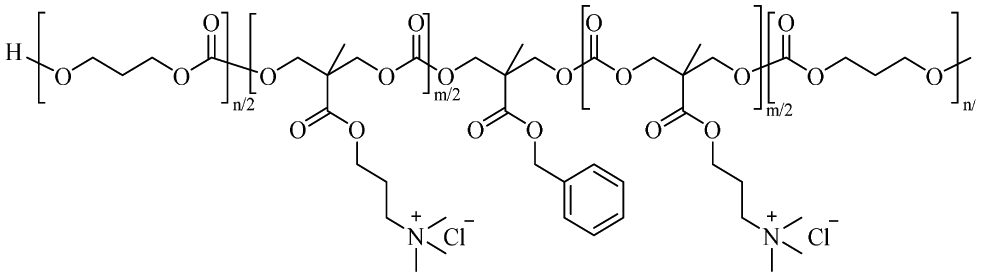
|
m = 41a | — | — | — | — | 80 | |
| n = 118a | |||||||
| Iminium |
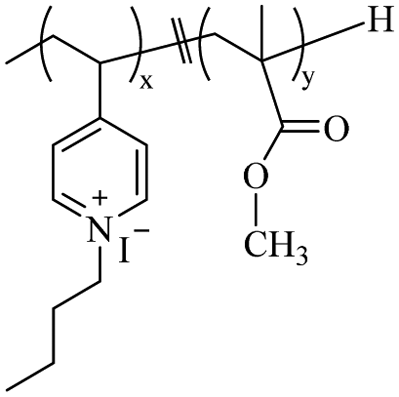
|
x = y = 0.50c |
M
n = 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–33000b 000–33000b |
50 μg mL−1 for E. coli | — | HC50 = 1245 μg mL−1 | 102 |
| 30 μg mL−1 for B. subtilis | |||||||

|
x = 0.4 | M n = 9326b | >250 μg mL−1 for E. coli in BM2MM | — | HC50 = 551.6 μg mL−1 | 106 | |
| y = 0.6 | 4 μg mL−1 for P. aeruginosa in BM2MM | ||||||
| 4 μg mL−1 for S. aureus in BM2MM | |||||||
| Iminium |

|
x = 0.79a |
M
n = 20400a |
312 μg mL−1S. aureus | — | HC50 = 2500 μg mL−1 | 104 |
| y = 0.21a |
M
n = 13000b |
5000 μg mL−1E. coli | |||||
| PDI = 1.14b | 312 μg mL−1S. epidermidis | ||||||
| 2500 μg mL−1P. aeruginosa | |||||||
| 312 μg mL−1C. parapsilosis | |||||||

|
n = 20c | — | 7.8 μg mL−1 for E. coli | — | — | 100 | |
| 7.8 μg mL−1 for K. pneumoniae | |||||||
| 15.6 μg mL−1 for P. aeruginosa | |||||||
| 15.6 μg mL−1 for C. albican | |||||||
| 3.9 μg mL−1 for S. aureus | |||||||

|
x = 6; molecular weight of n = 7000c and m = 3000c |
M
n = 11459a |
256 μg mL−1 for E. coli | — | HC50 > 1000 μg mL−1 | 103 | |
| M n = 5630b | 64 μg mL−1 for S. aureus | ||||||
| PDI = 3.24b | |||||||

|
— | M n = 6500b | 25 μg mL−1 for E. coli | — | HC50 = 740 μg mL−1 | 105 | |
| 50 μg mL−1 for MRSA | |||||||
| Iminium |
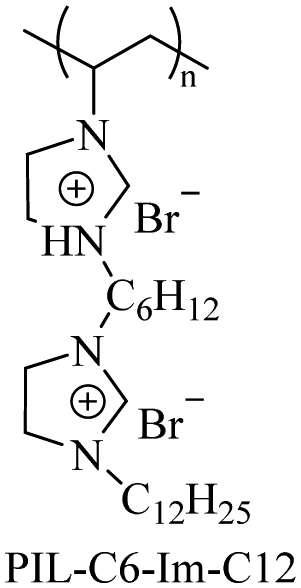
|
— |
M
n = 12000b |
0.018 μg mL−1 for E. coli | — | — | 107 |
| PDI = 1.02b | 0.009 μg mL−1 for S. aureus | ||||||

|
— | — | The antibacterial rate against E. coli and S. aureus >90% | — | Hemolytic activity (<4%) | 107 | |

|
— |
M
n = 26300b |
25 μg mL−1 for E. coli | IC50 > 100 μg mL−1 for NIH 3T3 | Hemolytic activity <5% at 100 μg mL−1 | 108 | |
| 25 μg mL−1 for S. aureus | IC50 > 100 μg mL−1 for 293T | ||||||
| PDI = 1.18b | |||||||
| Phosphonium |

|
— | M n = 6500b | 0.13 mM (MBC) for S. aureus and E. coli | — | NO hemolytic at 0.53 mM (sheep red blood cells) | 110 |
| PDI = 1.12b | |||||||
| Phosphonium |
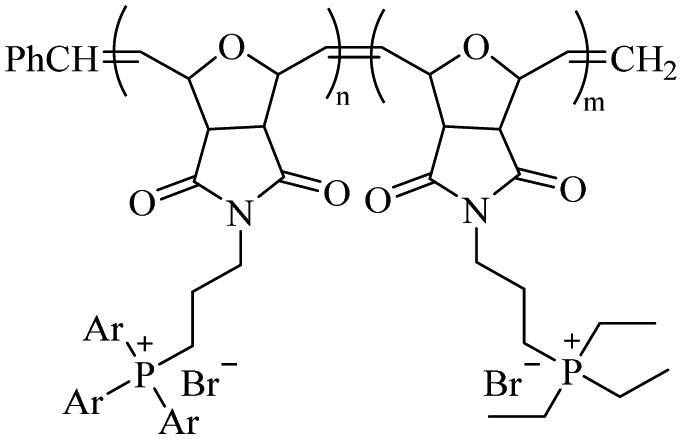
|
Phenyl:ethyl (5:5) |
M
n = 13131a |
128 μg mL−1 for E. coli | — | HC50 = 2048 μg mL−1 | 111 |
| m/n = 0.73a | M n = 6590b | 32 μg mL−1 for S. aureus | |||||
| PDI = 3.59b | |||||||
| Sulfonium |

|
— |
M
n = 10500b |
1.25 μg mL−1(MBC) for S. aureus | — | HC10 = 600 μg mL−1 | 87 |
| PDI = 1.53b | 1.25 μg mL−1(MBC) for MRSA | ||||||
| 10 μg mL−1(MBC) for E. faecalis | |||||||
| 2.5 μg mL−1(MBC) for E. coli | |||||||
| 20 μg mL−1(MBC) for P. aeruginosa | |||||||

|
x = 2 |
M
n = 94410b |
8 μg mL−1 for E. coli | — | HC10 = 600 32000 μg mL−1 |
79 | |
| n = 39b | PDI = 1.37b | 16 μg mL−1 for S. aureus | |||||

|
n = 28b | M n = 1270b | 10 μg mL−1 for E. coli | — | HC50 = 200 μg mL−1 (sheep red blood cells) | 88 | |
| PDI = 1.6b | 5 μg mL−1 for S. aureus | ||||||
| 10 μg mL−1 for M. smegmatis | |||||||
2.1.1.1. Ammonium group. The chemical structure of this class of antimicrobial agents is generally inspired by the functional and molecular characteristics of HDPs, so primary amines are the most common cationic groups, considering that lysine – with a primary amine functional group – is the main amino acid present in HDPs.90 Several examples described here are shown in Table 1. In 2005, Kuroda52 reported the use of N-(tert-butoxycarbonyl)aminoethyl methacrylate and butyl methacrylate to synthesize amphiphilic random polymethacrylate derivatives with a minimum inhibitory concentration (MIC) of 16 μg mL−1. In 2009, Kenichi's group prepared a library of amphiphilic random copolymers containing cationic and hydrophobic side chains by copolymerizing amine-functionalized methacrylate monomers with different proportions of alkyl methacrylates.68 The results showed that the copolymer series with primary and tertiary amine groups exhibited strong antibacterial activity and selectivity against Escherichia coli (E. coli) versus human red blood cells (RBCs). The copolymers containing quaternary ammonium groups show activity against E. coli only when the hydrophobic content of the polymer is higher. c and co-workers91 reported the synthesis of new biodegradable cationic amphiphilic polycarbonates. They reacted trimethylamine with a chlorinated polycarbonate to introduce quaternized ammonium cations; this polymer can easily self-assemble into cationic micellar nanoparticles by direct dissolution in water. It shows strong inhibition against Gram-positive bacteria (Enterococcus faecalis), methicillin-resistant S. aureus (MRSA), and fungi (Cryptococcus neoformans) and does not cause obvious hemolysis in a wide concentration range.
Kuroda69 reported structural modulation through cationic side chain spacer arms, including 2-aminoethylene, 4-aminobutylene, and 6-aminohexylene. The effect of amphiphilic random copolymers with cationic side chain spacer pairs of primary amines on the antibacterial and hemolytic activities was studied. The results showed that compared with copolymers with 2-aminoethylene spacer arms, for 4-aminobutene cationic side, The antibacterial activity of chain copolymers increased by an order of magnitude without causing undesirable hemolysis. When the spacer arm was further extended to hexene, the copolymer exhibited effective antibacterial and hemolytic activities. Then Kuroda's group92 reported the preparation of homopolymers with different Degree of Polymerization (DP = 7.7 to 12) from primary ammonium ethyl methacrylate. This polymer exhibits higher inhibitory effects against Gram-positive bacteria, including MRSA, than against Gram-negative bacteria; the authors suggested that this was related to the outer membrane structure on the bacterial surface, and the membrane disruption mode of such polymers. They further evaluated the activity of this polymer under physiological conditions, in the presence of fetal bovine serum (FBS). It also showed potent activity against S. aureus in the presence of FBS, while the activity of the antibiotic mupirocin was reduced under the same conditions. Studies have shown that the activity of HDPs is reduced in the presence of serum because serum salts inhibit the electrostatic binding of cationic peptides to anionic bacterial surfaces and serum proteins such as albumin bind to peptides nonspecifically.93,94 Moreover, they95 reported that an amphiphilic random copolymer composed of aminobutyl methacrylate (ABMA) and ethyl methacrylate (EMA) showed pH-dependent antibacterial activity. The polymer was effective at killing S. aureus at neutral pH but was inactive under acidic conditions (pH 5.5). In 2021, the same group also96 reported the synthesis of terpolymers containing primary ammonium cations and carboxylate anions, and hydrophobic side chain monomers. They kept the proportion of the hydrophobic monomer EMA in the polymer at 40 mol%, and synthesized a series of polymers with different total net charge (positive charge presence independent on the pH) by adjusting the percentages of amino-ethyl methacrylate (AEMA) and propanoic acid methacrylate (PAMA) side chains. The antibacterial and hemolytic activity of the copolymer is determined by a net charge of +3 or greater, and the anionic carboxylic acid groups have no significant effect on the antibacterial and hemolytic activity of the copolymer.
Quaternary ammonium has permanent cationic charges, regardless of the pH, and they are also commonly used in such amphiphilic antimicrobial polymers.97 Hedrick and Yang et al.98 reported the preparation of a series of antibacterial polyionenes by utilizing cations brought by two-sided quaternary amines, such as bis[2-(N,N-dimethylamino)ethyl] ether, through a simple addition polymerization reaction, in which the polymer formation reaction and charge installation occurred simultaneously. Tyagi and Mishra72 used aminopropyl methacrylamide (APMA), which remains protonated at physiological pH, thus providing a cationic charge, while benzylmethacrylamide (BMA) was chosen to provide hydrophobicity due to the presence of an aromatic hydrophobic benzyl group. Wang et al.80 synthesized a series of quaternized triblock copolymers that can self-assemble into reverse micelles (RM) in tetrahydrofuran (THF). The outer shell of the RM contains biocompatible poly(ε-caprolactone) (PCL) blocks to provide biosafety and responsiveness, and the core contains quaternary ammonium cationic blocks to provide antimicrobial activity. In the presence of bacterial lipase, the biodegradable PCL blocks are hydrolyzed, resulting in the responsive release of quaternary biocidal agents (QBA), thereby achieving self-sterilization. This provides a promising strategy for the development of bio-switchable self-sterilizing dressing materials.
2.1.1.2. Iminium group. Another example of nitrogen-based cationic center is the iminium group. This classification comprises several candidates, such as pyridinium, imidazolium, and guanidinium salts (Fig. 3). The iminium group exhibits uniformly distributed positive charges via π bonds and aromatic conjugated systems, potentially augmenting its affinity for negatively charged bacterial membranes and consequently enhancing antibacterial effectiveness.29
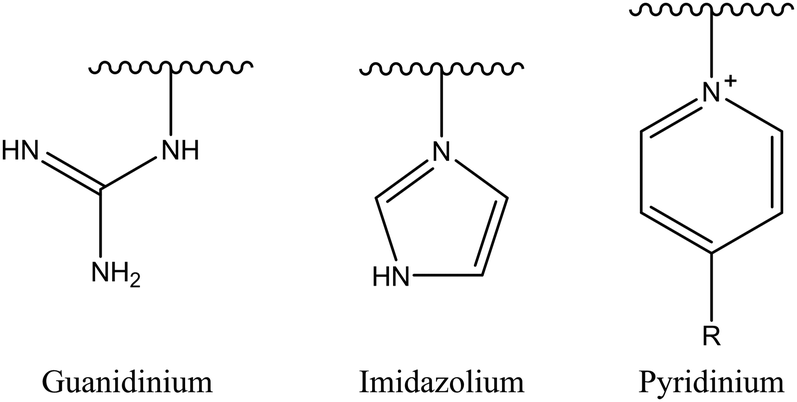 | ||
| Fig. 3 Schematic diagram of the iminium group structure. | ||
Among iminium groups, guanidinium salts are most widely used as cationic charge sources for such amphiphilic antimicrobial copolymers because of their close structural similarity to the natural amino acid arginine. Some studies have found that guanidine-functionalized polycarbonate can pass through bacterial cell membranes without damaging their integrity, and non-specifically binds to intracellular proteins and nucleic acids.99 Therefore, Leong et al. simply mixed quaternary ammonium homopolymer and guanidinium homopolymer to produce a more effective antibacterial behavior than a single mechanism, showing a synergistic or additive effect.100 An increased antibacterial activity for guanidine-containing oligothioetheramides, compared to other ammonium groups, was reported by Alabi's research group.101
Using a pyridine ring as a source of cations, Sambhy et al.102 investigated the effect of the spatial relationship of the charges and alkyl tails on the polymer backbone on membrane disruptive capacity. The results showed that placing the charges and tails in a spatially separated center resulted in higher membrane disruptive capacity, as evidenced by enhanced antimicrobial and hemolytic activities. Islam et al.103 synthesized random and block dendritic oxanorbornene polymers with different hydrophobicity/hydrophilicity and with pyridinium salt groups using ring-opening metathesis polymerization (ROMP), in which the copolymers with a six-carbon linker, a high charge density, and hexylpyridine functional groups per repeating unit, exhibited high antibacterial activity against Gram-positive bacteria (S. aureus). However, all polymers were inactive against Gram-negative bacteria (E. coli). Cuervo-Rodríguez et al.104 quaternized polymers containing thiazole ring monomers with butyl iodide or octyl iodide to obtain cationic amphiphilic antimicrobial copolymers containing thiazolium groups, and utilized the different alkyl groups chains length to adjust their hydrophobic/hydrophilic balance. The results showed that the more hydrophobic octanylated copolymer was more effective against all tested microorganisms. Hancock et al.105 synthesized N-methylpyridinium-fused norbornene monomers through ROMP, and successfully synthesized backbone cationic polymers with diverse structures, showing that these polymers are effective against Gram-negative (E. coli) and Gram-positive (S. aureus) bacteria, with MIC as low as 25 μg mL−1. In addition, polymers with a smaller DP showed increased selectivity for bacteria over human RBCs. Vishwakarma et al.106 reported the development of water-soluble synthetic peptide-mimicking polyurethanes incorporating arginine as a cationic functional group. The results demonstrated their ability to disrupt pre-formed biofilms of P. aeruginosa, S. aureus, and E. coli. Although these polyurethanes exhibited relatively weak antibacterial activity against planktonic bacteria, they effectively prevented surface adhesion and promoted bacterial surface motility. This dual action inhibited biofilm formation of both Gram-positive and Gram-negative bacteria at sub-inhibitory concentrations, while showing no toxicity toward mammalian cells.
Using imidazole groups as cation sources, Zheng et al.107 synthesized imidazolium-type ionic liquid monomers and their corresponding poly(ionic liquids) (PILs) membranes. By studying the impact of chemical on antibacterial activity, the results show that the antibacterial activity increases with the increase in charge density (double cation), while decreases with increasing carbon chain length. Shi et al.108 using ultrafast ring-opening polymerization (ROP) technology and “click” chemical reactions, prepared imidazolium-based block co-polypeptides with L configuration (PPGn-PILm); these can self-assemble into positively charged nanoscale micelles, and showed MIC against both Gram-positive (S. aureus) and Gram-negative (E. coli) of 25 μg mL−1.
2.1.1.3. Phosphonium group. Since the structure of phosphonium compounds is similar to that of quaternary ammonium ones, Kanazawa et al.109 first reported the use of phosphonium as a cationic charge source for amphiphilic antimicrobial copolymers in 1993. It was also shown that for polymers with the same structure but different cationic parts, the antibacterial activity of phosphates is two orders of magnitude higher than that of polymer ammonium salts. Hisey et al.110 connected phosphonium cations with different alkyl lengths to the ends of polyethylene glycol-polycaprolactone block copolymers and allowed the phosphonium-functionalized block copolymers to self-assemble in aqueous solution forming micro micelles to achieve selective antibacterial function. Kuday et al.111 synthesized five different aromatic side-chain phosphonium-based polymers via cyclopentene ROMP; the synthesized polymers showed higher antibacterial activity against Gram-positive strains than Gram-negative strains.
2.1.1.4. Sulfonium group. In 1993, Kanazawa et al.112 synthesized polymers with antibacterial activity using sulfur atoms as cation sources. In recent years, sulfonium compounds have attracted attention as sources of cationic charge for amphiphilic antimicrobial copolymers because they are similar in charge to quaternary ammonium groups and are also commonly used as therapeutic agents.79,87–89 Research results have shown that when the polymers contain sulfonium as a cationic moiety in the backbone, they can exhibit enhanced antimicrobial activity and selectivity compared to corresponding polymers with sulfonium as side chains. This makes the former potential candidates for combating antibiotic-resistant bacterial infections.87,88
Hu et al.87 developed a new class of AB alternating sequence main chain sulfonium-containing polymers. Broad-spectrum bactericidal properties against a group of clinically relevant bacteria and excellent biocompatibility were observed for the first time in a sulfonium-based macromolecule. This amphiphilic polymer showed excellent antibacterial activity against clinically relevant bacteria, including MRSA, with MIC in the range of 1.25–10 μg mL−1 and no hemolysis even at polymer concentrations as high as 10000 μg mL−1. Oh and Khan88 synthesized β-hydroxy-functionalized sulfonium polymers, which demonstrated structural stability and high efficacy against clinically significant Gram-positive bacterial strains, such as Mycobacterium smegmatis, which is crucial for combating tuberculosis infections. Furthermore, they are equally effective against challenging Gram-negative bacterial strains, such as E. coli. Additionally, the developed main-chain sulfonium polymers show similar efficacy to antibiotics like kanamycin and vancomycin.
In addition, Zhang et al.79 synthesized a series of star-shaped alkylated sulfonium amphiphilic antimicrobial copolymers by mimicking the structure unit of vitamin U via N-carboxyanhydrides (NCA) ring-opening reactions. The short hydrophobic moiety and highly cationic structure significantly enhance the hemocompatibility and bactericidal activity of G2-PM-1H+ (Table 1, HC10/MIC ratio as high as 16000). G2-PM-1H+ has broad-spectrum bactericidal activity (S. aureus: MIC = 4 μg mL−1, E. coli: MIC = 16 μg mL−1) and has excellent drug resistance compared with vancomycin.
In general, the full potential of sulfonium compounds as sources of cationic charge is still to be explored, especially sulfonium-based polymers with well-defined sequences.
 | ||
| Fig. 4 Schematic diagram illustrating the impact of cationic-hydrophobic balance on the cell selectivity of amphiphilic antimicrobial copolymers. Reproduced with permission from ref. 81, copyright 2017, American Chemical Society. | ||
The first design strategy involves controlling the hydrophobicity at the local level of the polymer by designing the structure of different hydrophobic monomers, thereby regulating the hydrophilic–hydrophobic balance of the polymer. So far, most studies on antimicrobial polymers have utilized hydrophobic alkyl chains as their hydrophobic groups. Therefore, the structural properties of the hydrophobic alkyl tails, such as the length of alkyl chains and the type of side chains, are crucial for determining the biological activity of the polymer. According to reports,56,81,86,104,113,114,117–119 extending the chain length within a certain range of carbon atoms in the side chains results in increased antibacterial activity. Hydrophobic structures can interact more strongly with the lipid bilayer of microbial membranes, thereby enhancing antibacterial activity. However, excessively long alkyl chains may lead to polymer over-hydrophobicity, potentially causing reduced water solubility and increased indiscriminate toxicity to pathogens and host cells, i.e., enhanced cytotoxicity. Therefore, optimizing the length of hydrophobic chains helps to regulate the balance between antibacterial efficacy and cytotoxicity, thereby enhancing the bioavailability of amphiphilic antimicrobial copolymers and maximizing their antibacterial activity.
In addition to hydrophobic chain length, the type of hydrophobic part also influences the bioactivity.50,113 Tan et al.120 synthesized a series of guanidinium-functionalized random polycarbonates through organocatalytic ring-opening polymerization (Fig. 5).
 | ||
| Fig. 5 Synthesis of guanidinium-functionalized random polycarbonates with different hydrophobic side chain lengths via organocatalytic ring-opening polymerization. Redrawn from ref. 120. | ||
By introducing various length of hydrophobic co-monomers (ethyl, propyl, isopropyl, benzyl, and hexyl) at different percentage contents, they modulated the hydrophobicity of the polymers. Overall, these polymers exhibited similar minimum inhibitory concentrations and minimum bactericidal concentrations against a broad spectrum of microbes. However, polymers with higher hydrophobicity showed a faster rate of bacterial elimination. At higher percentages (20 mol%), polymers containing a higher proportion of hydrophobic co-monomers displayed reduced selectivity due to their elevated hemolytic activity (Table 2).
| Polymer | R | x | y | MIC [μg mL−1]/selectivity (HC50/MIC) | HC50 [μg mL−1] | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gram-positive | Gram-negative | Fungus | ||||||||
| S. aureus | A. baumannii | E. coli | K. pneumoniae | P. aeruginosa | C. albicans | |||||
| a At 2000 μg mL−1, the hemolytic activity of P5 and P9 is ≈45% and ≈37%, respectively. | ||||||||||
| P1 | Bn | 17 | 2 | 7.8 (>256) | 7.8 (>256) | 7.8 (>256) | 15.6 (>128) | 15.6 (>128) | 31.3 (>64) | >2000 |
| P2 | Et | 18 | 2 | 7.8 (>256) | 7.8 (>256) | 7.8 (>256) | 15.6 (>128) | 15.6 (>128) | 31.3 (>64) | >2000 |
| P3 | iBu | 16 | 2 | 7.8 (>256) | 7.8 (>256) | 7.8 (>256) | 15.6 (>128) | 15.6 (>128) | 31.3 (>64) | >2000 |
| P4 | Bu | 18 | 2 | 7.8 (>256) | 7.8 (>256) | 7.8 (>256) | 15.6 (>128) | 15.6 (>128) | 31.3 (>64) | >2000 |
| P5 | Hex | 18 | 2 | 7.8 (>256) | 7.8 (>256) | 7.8 (>256) | 15.6 (>128) | 15.6 (>128) | 31.3 (>64) | >2000a |
| P6 | Bn | 15 | 4 | 7.8 (51) | 7.8 (51) | 7.8 (51) | 15.6 (26) | 15.6 (26) | 31.3 (13) | ≈400 |
| P7 | Et | 15 | 4 | 7.8 (>256) | 7.8 (>256) | 7.8 (>256) | 15.6 (>128) | 15.6 (>128) | 31.3 (>64) | >2000 |
| P8 | iBu | 14 | 4 | 7.8 (51) | 7.8 (51) | 7.8 (51) | 15.6 (29) | 15.6 (29) | 31.3 (14) | ≈450 |
| P9 | Bu | 13 | 5 | 7.8 (>256) | 7.8 (>256) | 7.8 (>256) | 15.6 (>128) | 15.6 (>128) | 31.3 (>64) | >2000a |
| P10 | Hex | 14 | 4 | 7.8 (22) | 15.6 (11) | 7.8 (22) | 15.6 (11) | 15.6 (11) | 31.3 (5) | ≈170 |
| P11 | — | 18 | — | 15.6 (>513) | 7.8 (>1026) | 15.6 (>513) | 31.3 (>256) | 15.6 (>513) | 31.3 (>256) | >8000121 |
A series of antimicrobial surfactant-like poly(ester polyurethane)s with different hydrophobic groups (short alkyl chain (PSA), aromatic group (PA) and long alkyl chain (PLA)) and cationic groups uniformly distributed along the polymer chain were studied (Fig. 6).122 Membrane permeability assays showed that all three antimicrobial polymers were able to permeabilize the outer (periplasm) and inner membrane (cytoplasm) of E. coli and that the S. aureus cell wall was unable to prevent these polymers from damaging the cytoplasmic membrane. PA and PLA caused significant damage to the plasma membrane of E. coli, whereas PSA caused slight damage to the integrity of the plasma membrane, but even this level of damage was able to dissipate the plasma membrane potential and lead to cell death (Table 3).
 | ||
| Fig. 6 Synthesis of antimicrobial surfactant-like poly(ester polyurethane)s with different hydrophobic pendant groups (PSA, PA, and PLA) and cationic groups distributed uniformly along the polymer chain. Redrawn from ref. 122. | ||
| Polymer | MIC | HC10 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gram-negative bacteria | Gram-positive bacteria | |||||||||
| E. coli | P. aeruginosa | S. aureus | S. epidermidis | |||||||
| μg mL−1 | μM | μg mL−1 | μM | μg mL−1 | μM | μg mL−1 | μM | μg mL−1 | μM | |
| PSA | 16 | 2.0 | 32 | 4.0 | 32 | 4.0 | 16 | 2.0 | 379.4 | 47.4 |
| PA | 8 | 1.6 | 16 | 3.1 | 16 | 3.1 | 4 | 0.8 | 19.8 | 3.8 |
| PLA | 8 | 0.9 | 16 | 1.8 | 16 | 1.8 | 16 | 1.8 | 3.5 | 0.4 |
| Ampicillin | 1 | 2.9 | 128 | 371.2 | 0.2 | 0.6 | 4 | 11.6 | — | — |
| Melittin | 25 | 8.8 | 50 | 17.6 | 25 | 8.8 | 0.8 | 0.3 | — | — |
The second and third design approaches focus more on the global polymer level, wherein the hydrophobic group influences bioactivity through its interaction with other comonomers present in the global amphiphilic balance of amphiphilic antimicrobial copolymers.
For example, Phuong et al.113 synthesized a library of 36 amphiphilic antimicrobial polymers using eight hydrophobic monomers (cyclic, aromatic, linear, or branched and classified into three categories, C5, C7, and C9, based on the carbon number (5, 7, or 9)) (Fig. 7) and screened them against four bacterial strains and sheep RBCs to assess their antimicrobial activity and blood compatibility. For polymers with saturated chains and similar DP and target component molar ratios, the antimicrobial activity decreases as the carbon number of the hydrophobic side chain groups increases (i.e., the MIC value for the C9 group is higher than those for C7 and C5). Within the same polymer group, copolymers containing branched monomers exhibit better antimicrobial activity than those with linear monomers. In the C5 group (see Fig. 7), the length of Im (6.2 Å) is slightly shorter than that of Pm (7.4 Å), and the MIC value of I40-1535 against Gram-negative bacteria is slightly lower than that of P40-1535. In the C7 group, the difference in chain length between branched (Pbm, 5 Å) and linear (Hm, 9.9 Å) monomers is significant, resulting in a substantial difference in antimicrobial activity (Table 4). Im monomer shows the highest compatibility with the selected cationic and hydrophilic monomers, as evidenced by polymers I40-1535 and I40-1040, which exhibit the highest antimicrobial activity among the tested polymers. In summary, it was found that linear and branched alkyl hydrophobic monomers exhibited better antimicrobial effects than cyclic and aromatic ones, and polymers with a Calculation logP (the partition constant of a compound between n-octanol (hydrophobic phase) and water (hydrophilic phase)) between 0 and 2 were most likely to achieve the optimal balance of high antimicrobial activity and low hemolytic activity.
 | ||
| Fig. 7 Synthesis of amphiphilic antimicrobial polymers using eight hydrophobic monomers (N-isopentylacrylamide (Im), N-pentylacrylamide (Pm), N-heptylacrylamide (Hm), N-butyl-N-propylacrylamide (Pbm), N-cycloheptylacrylamide (Cpm), N-(cyclohexylmethyl)acrylamide (Cxm), N-benzylacrylamide (Bm), and N-nonylacrylamide (Nm)). Redrawn from ref. 113. | ||
| Family of polymers | Hydrophobic monomer (C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) logP) logP) |
Polymer | Monomer ratio (x:y:z) |
ClogP of representative oligomer | MIC of polymer (μg mL−1) | |||
|---|---|---|---|---|---|---|---|---|
| PA01 | P. aeruginosa | E. coli | S. aureus | |||||
| I-family | Im(1.40) | I40-1040 | 50:10:40 |
2.70 | 16 | 16–32 | 16 | >256 |
| I40-1535 | 50:15:35 |
1.55 | 16 | 16–32 | 16 | >256 | ||
| I40-2030 | 50:20:30 |
0.39 | 16–32 | 32–64 | 32 | >256 | ||
| I40-2525 | 50:25:25 |
−0.77 | 256 | >256 | 128–256 | >256 | ||
| I40-3020 | 50:30:20 |
−1.93 | >256 | >256 | >256 | >256 | ||
| P-family | Pm(1.53) | P40-1040 | 50:10:40 |
3.22 | 32 | 32 | 16 | >256 |
| P40-1535 | 50:15:35 |
2.00 | 32 | 32 | 16 | >256 | ||
| P40-2030 | 50:20:30 |
0.78 | 32 | 32–64 | 32–64 | >256 | ||
| H-family | Hm(2.59) | H40-2030 | 50:20:30 |
3.95 | 64 | 128 | 64 | >256 |
| H40-2525 | 50:25:25 |
2.2 | 64 | 64 | 64 | >256 | ||
| H40-3020 | 50:30:20 |
0.45 | 64–128 | 128–256 | 128 | >256 | ||
| Pb-family | Pbm(2.37) | Pb40-2030 | 50:20:30 |
3.70 | 32 | 32–64 | 16–32 | >256 |
| Pb40-2525 | 50:25:25 |
2.00 | 32–64 | 32–64 | 32–64 | >256 | ||
| Pb40-3020 | 50:30:20 |
0.29 | 64 | 128 | 128–256 | >256 | ||
| Cp-family | Cpm(2.01) | Cp40-2030 | 50:20:30 |
2.20 | 32–64 | 64 | 32 | >256 |
| Cp40-2525 | 50:25:25 |
0.74 | 32–64 | 32–64 | 16–32 | >256 | ||
| Cx-family | Cxm(2.07) | Cx40-2030 | 50:20:30 |
2.38 | 32 | 32–64 | 32 | >256 |
| Cx40-2525 | 50:25:25 |
0.89 | 32–64 | 64 | 16–32 | >256 | ||
| B-family | Bm(1.18) | B40-1535 | 50:15:35 |
1.11 | 32–64 | 64 | 64–128 | >256 |
| B40-1040 | 50:10:40 |
2.23 | 16–32 | 32 | 32 | >256 | ||
| B40-2030 | 50:20:30 |
0.05 | 64 | 64–128 | 256 | >256 | ||
| N-family | Nm(3.65) | N40-2030 | 50:20:30 |
7.13 | >256 | >256 | >256 | >256 |
| N40-3020 | 50:30:20 |
2.57 | 128–256 | 256 | 128–256 | >256 | ||
| N40-3515 | 50:35:15 |
0.29 | 128–256 | 256 | 128–256 | >256 | ||
Another notable example comes from Mankoci et al.,123 which reported a series of water-soluble antimicrobial polyurethanes designed to mimic antimicrobial peptides (AMPs) (Fig. 8). These polyurethanes were synthesized by incorporating cationic side chains into one segment of the polymer chain, while functionalizing the other segment with various side-chain amino acids. Their antimicrobial activity and cytotoxicity were systematically compared. The findings revealed that the addition of hydrophobic, neutral polar, or anionic components did not significantly influence antibacterial activity. However, the introduction of hydrophobic groups enhanced cytoplasmic membrane disruption, and the proportion of cationic side-chain groups correlated with the degree of outer membrane disruption in Gram-negative bacteria. Meanwhile, neutral polar and anionic groups improved the compatibility of the polyurethanes with mammalian cells, highlighting the tunability of these materials for selective activity and reduced cytotoxicity.
 | ||
| Fig. 8 Chemical structures of the antimicrobial polyurethanes. mArg and mAsp (in green) represent the charged polar pendant groups; mVal, mAla, mTrp, and mPhe (in red) represent the hydrophobic pendant groups, and nPrDEA, cPrDEA, nBac, and mSer (in purple) were chosen as uncharged polar pendant groups for antimicrobial polyurethane design. Redrawn from ref. 123. | ||
Typical HDPs feature two functionalities, cationic groups and hydrophobic moieties. However, as mentioned above, an excess of cationic iminium82,86–88 and hydrophobic113,120,122 characteristics can lead to indiscriminate toxicity towards all cell types, including RBCs. This renders the optimization of binary polymer balance challenging. Consequently, a recent approach has been the introduction of neutral hydrophilic groups, resulting in a class of ternary polymers. The presence of neutral hydrophilic groups serves to reduce undesired protein complexation and hemolysis, thus maintaining polymer antimicrobial activity while imparting biocompatibility (Fig. 9).
 | ||
| Fig. 9 Proposed adoption of PEG group polymer and non-PEG group polymer in protein-rich media and in contact with bacterial and mammalian cell membranes. Reproduced with permission from ref. 70 and 124, copyright 2010, Royal Society of Chemistry. | ||
The role of the neutral hydrophilic group was reported first by Allison et al.125 They focus on quaternized poly(vinyl-pyridine) (PVP), which inherently possesses antibacterial properties but also exhibits high toxicity. They found that incorporation of polyethylene glycol comonomers could reduce hemolytic activity without compromising antimicrobial efficacy. This study paves the way for the development of new ternary polymer systems akin to HDPs. Since then, PEG has been commonly employed as a neutral hydrophilic residue.70,126–128 Additionally, various other neutral hydrophilic candidates exist for ternary polymer systems, such as hydroxyl substitutions (mimicking serine residues).45,129 Based on previous research, Pham et al.70 designed and developed a series of statistical amphiphilic ternary copolymers and systematically investigated the effects of each component group on antimicrobial activity and biocompatibility (Fig. 10).
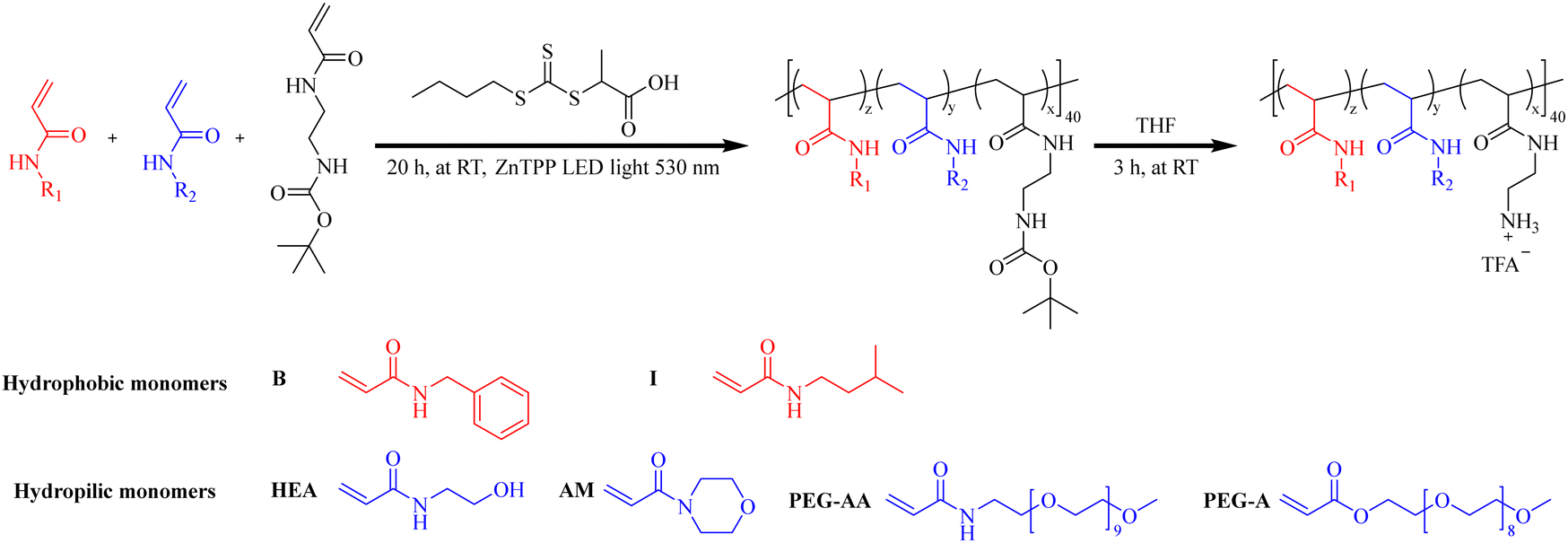 | ||
| Fig. 10 Synthesis of amphiphilic antimicrobial copolymers using different hydrophobic and hydrophilic monomers. R1, R2 is hydrophobic or hydrophilic chain, respectively. Hydrophilic monomer: hydroxyethyl acrylamide (HEA), 4-acryloylmorpholine (AM), poly(ethylene glycol) methyl ether acrylate (PEG-A), and PEG acrylamide(PEG-AA), hydrophobic monomers: N-isopentylacrylamide (I) and N-benzylacrylamide (B). Redrawn from ref. 70. | ||
The results indicate that, unlike hydrophobic moieties that directly damage cell membranes, hydrophilic groups indirectly but significantly influence bioactivity by modulating the hydrophobic/hydrophilic balance and overall hydrophobicity, leading to alterations in polymer solubility properties. The AM family (Fig. 10) exhibited the highest antibacterial activity against Gram-negative bacteria. The PEG-A and PEG-AA families, demonstrated antibacterial activity similar to the HEA family, except for polymers with a very low hydrophilic/hydrophobic ratio (PEG-A/PEG-AA/HEA-I-1040). Additionally, containing PEG as a neutral hydrophilic moiety exhibited significantly higher biocompatibility and selectivity compared to polymers with shorter, less hydrophilic, and less flexible chains (Table 5).
| Group | Family | Polymer | Monomer ratio (x:y:z) |
MIC of polymer (μg mL−1) | HC50 (μg mL−1) | IC50 against MEF (μg mL−1) | |||
|---|---|---|---|---|---|---|---|---|---|
| P. aeruginosa 01 | P. aeruginosa 37 | E. coli K12 | S. aureus ATCC29213 | ||||||
| Non-PEG group | HEA | HEA-I-1040 | 50:10:40 |
16 | — | 16 | >256 | 142 ± 6 | — |
| HEA-I-1535 | 50:15:35 |
16 | 32 | 16 | >256 | — | 29 ± 4 | ||
| HEA-I-2030 | 50:20:30 |
16–32 | 32–64 | 32 | >256 | >2000 | 122 ± 19 | ||
| HEA-I-3020 | 50:30:20 |
>256 | >256 | >256 | >256 | >2000 | — | ||
| HEA-B-1040 | 50:10:40 |
16–32 | — | 32 | 256 | 961 ± 425 | 59 ± 37 | ||
| HEA-B-1535 | 50:15:35 |
32–64 | — | 64–128 | >256 | >2000 | 43 ± 3 | ||
| AM | AM-I-1535 | 50:15:35 |
8–16 | 16–32 | 16 | >256 | 1825 | 56 ± 13 | |
| AM-I-2030 | 50:20:30 |
16 | 32 | 16 | >256 | 1505 ± 29 | 56 ± 15 | ||
| AM-I-3020 | 50:30:20 |
64 | 64 | 64–128 | >256 | >2000 | 326 ± 101 | ||
| AM-B-1040 | 50:10:40 |
32 | 32–64 | 32–64 | — | 878 ± 81 | 39 ± 25 | ||
| AM-B-1535 | 50:15:32 |
32 | 32–64 | 32–64 | — | 855 ± 212 | 36 ± 7 | ||
| PEG group | PEG-AA | PEG-AA-I-1040 | 50:10:40 |
8 | 16–32 | 8–16 | >256 | 390 ± 87 | 43 ± 10 |
| PEG-AA-I-1535 | 50:15:35 |
16 | 32 | 8–16 | >256 | >2000 | 166 ± 17 | ||
| PEG-AA-I-2030 | 50:20:30 |
16–32 | 32–64 | 32 | >256 | >2000 | ∼512 | ||
| PEG-A | PEG-A-I-1040 | 50:10:40 |
8 | 16–32 | 8–16 | >256 | 500 ± 343 | 39 ± 2 | |
| PEG-A-I-1535 | 50:15:35 |
16 | 32–64 | 8–16 | >256 | >2000 | 130 ± 30 | ||
| PEG-A-I-2030 | 50:20:30 |
16–32 | 64–128 | 32–64 | >256 | >2000 | ∼512 | ||
| PEG-A-I-3020 | 50:30:20 |
>256 | >256 | >256 | >256 | >2000 | >512 | ||
| PEG-A-B-1040 | 50:10:40 |
16–32 | 32–64 | 32–64 | — | 1563 ± 135 | 56 ± 7 | ||
| PEG-A-B-1535 | 50:15:35 |
32–64 | 64 | 64–128 | — | >2000 | 191 ± 30 | ||
In a similar approach, Mortazavian et al.45 adjusted the composition of cationic and hydrophobic monomers by introducing hydroxyethyl methacrylate (HEMA) monomer units to study the impact of each component on the antibacterial and hemolytic activities of the copolymer. The results showed that increasing the proportion of cationic groups in the copolymer beyond 30 mol% did not enhance its antibacterial efficacy against E. coli. They also proposed that the HEMA component (hydroxyl side chains) may have two active roles: (1) as a structural spacer to distribute the cationic and hydrophobic groups in monomer sequences and (2) as a modulator of copolymer chain insertion in bacterial and human cell membranes.
In summary, good neutral hydrophilic groups will maximize biocompatibility without negatively affecting the antibacterial efficacy of amphiphilic antimicrobial copolymers, which is of great significance for the design and synthesis of efficient and safe amphiphilic antimicrobial polymers.
000, so the impact of end groups on their biological activity cannot be ignored.71,130,131 Additionally, advancements in RAFT polymerization have made it easier to control the synthesis of polymer chains with different end groups (Fig. 11).
 | ||
| Fig. 11 Scheme of RAFT polymerization's main mechanism step.46 | ||
Nadres et al.130 used RAFT polymerization to produce a statistical methacrylate copolymer, followed by free radical-mediated conversion of the terminal groups successfully transformed the Z-end group of parent copolymer from dithiobenzoate to a cyanoisobutyl or aminoethyl cyanopentanoate group without any significant changes to the polymer molecular weight. The results showed that the parent copolymer with dithiobenzoate end groups exhibited the strongest antibacterial and hemolytic activity, while the ability of copolymers to destroy membranes does not depend on the end group structure. In a more in-depth research, Griesser's group131 used three different RAFT chain transfer agents to systematically study the effects of changing the R- and Z-RAFT end groups on the antibacterial activity and cytotoxicity of poly(methacrylate) polymers (Fig. 12). The results indicate that the R-group predominantly influences the toxicity of the polymers. Substituting the anionic cyanovaleric acid R-group (PA1) with the neutral isobutyronitrile group (PA3) led to over a 20-fold increase in the hemolytic activity of the polymers. However, the Z-group has a greater impact on the antimicrobial activity of the polymers against MRSA and C. albicans, where polymers with a long, lipophilic dodecylsulfanyl Z-group (PA1) were more effective than those with either an ethylsulfanyl or no Z group.
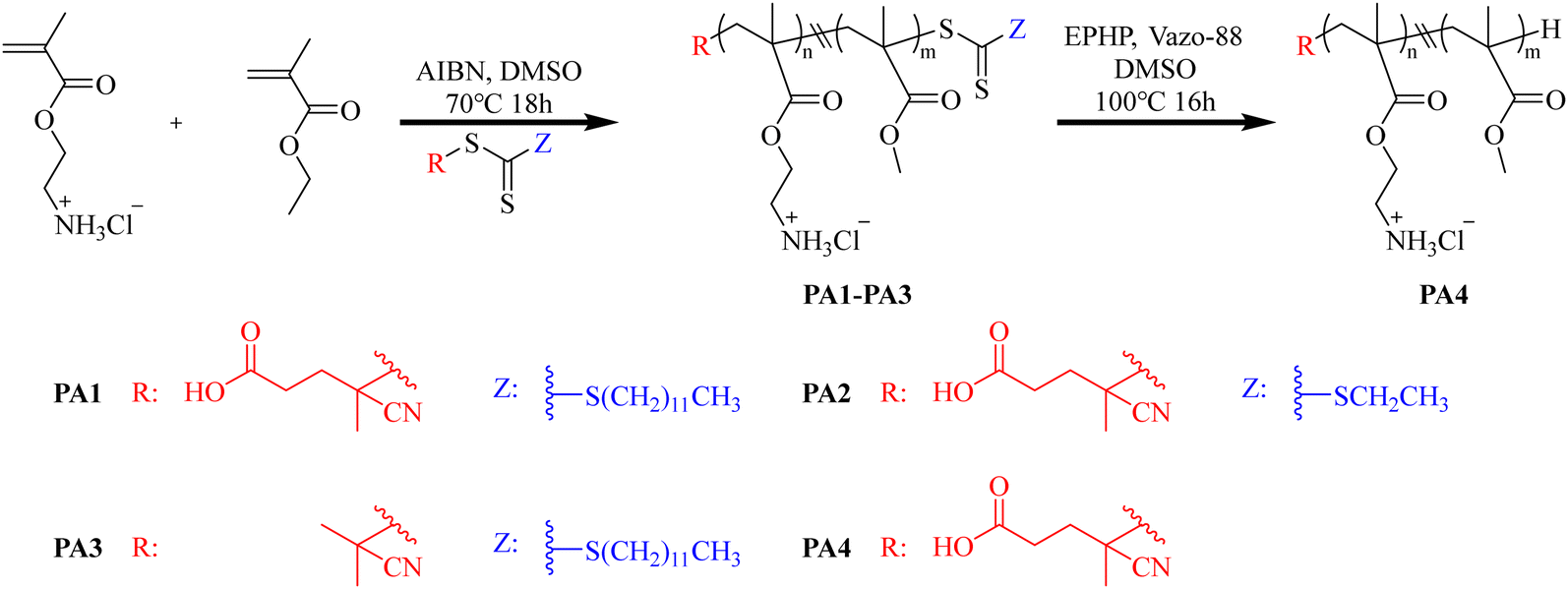 | ||
| Fig. 12 Synthesis of amphiphilic antimicrobial copolymers (PA1–PA4) by using RAFT agents to control the variation in the Z and R groups at the polymer ends. Redrawn from ref. 131. | ||
In conclusion, although the end group is not the main component of amphiphilic antimicrobial copolymers, modulating the end group should not be ignored to achieve optimal amphiphilic antimicrobial copolymers.
2.2. Global polymer design
| Polymer | M n/kDa | M w/kDa | PDI | MIC/μg mL−1 | HC50/μg mL−1 | IC50/μg mL−1 | ||
|---|---|---|---|---|---|---|---|---|
| E. coli | S. aureus | C. albicans | ||||||
| L-PTMI_0.8k | 0.80 | 0.91 | 1.14 | 256 | 128 | 32 | >2000 (2%) | 2220 |
| L-PTMI_1.2k | 1.2 | 1.4 | 1.22 | 16 | 16 | 16 | >2000 (4%) | 2570 |
| L-PTMI_3k | 2.8 | 3.7 | 1.33 | 2 | 4 | 16 | >2000 (3%) | 25.2 |
| L-PTMI_7k | 6.7 | 9.6 | 1.44 | 2 | 1 | 16 | 125 | 4.20 |
| L-PTMI_12k | 12.1 | 18.2 | 1.51 | 2 | 1 | 32 | 15.6 | 2.48 |
| L-PTMI_18k | 18.0 | 27.1 | 1.51 | 2 | 1 | 32 | 9.40 | 2.71 |
| L-PEI_4k | 4.0 | 5.3 | 1.30 | 16 | 16 | 32 | >2000 (39%) | 660 |
| Me-L-PTMI_7k | 3.6 | 5.4 | 1.53 | 16 | 16 | 16 | >2000 (2%) | 240 |
| MePTMI-co-Me2PTMI10% | 4.2 | 6.5 | 1.54 | 16 | 16 | 16 | >2000 (3%) | 420 |
| MePTMI-co-Me2PTMI20% | 3.7 | 5.5 | 1.49 | 16 | 16 | 16 | >2000 (3%) | 930 |
| Me-L-PEI_4k | 3.9 | 4.8 | 1.24 | 32 | 128 | 64 | >2000 (2%) | 600 |
| MePEI-co-Me2PEI10% | 3.6 | 4.6 | 1.27 | 32 | 64 | 32 | >2000 (3%) | 1370 |
| MePEI-co-Me2PEI20% | 3.6 | 4.7 | 1.31 | 16 | 64 | 16 | >2000 (2%) | 2000 |
 | ||
| Fig. 13 (a) Antimicrobial activities (MIC) and (b) hemotoxicity of guanidine and amine polymers as a function of polymer chain length (DP). Reproduced with permission from ref. 51, copyright 2013, American Chemical Society. | ||
Therefore, amphiphilic antimicrobial copolymers with different effects on Gram-positive and Gram-negative bacteria can be designed and synthesized by adjusting the molecular weight to meet the needs of different users.
2.2.2.1. Linear polymers. Linear polymers have been extensively studied due to their fundamental topological structure. An array of amphiphilic antimicrobial copolymers can be produced through diverse active polymerization techniques. The progress in controlled polymerization methodologies allows for precise adjustment of parameters like molecular weight, component composition ratios, and topological architectures, encompassing homopolymers, random, alternating, and block copolymers.21,138,139
2.2.2.1.1. Random polymers. Random polymers represent the simplest topology among copolymers. It is notable that numerous research reports have proven that random amphiphilic copolymers have efficient antibacterial effects.51,53,68,72,117,128,140–142 Due to their straightforward synthesis, devoid of any requirement for precise sequence control, the simplicity of the technology combined with favorable outcomes has made random copolymers an important part of the amphiphilic antimicrobial copolymers field.
However, the spatial distribution of components is bound to affect the secondary structure of the polymer, thereby affecting the overall biological efficacy by affecting its physicochemical properties and the conformation of the biological interface. Inspired by natural HDPs, researchers are dedicated to creating precisely defined sequence amphiphilic antimicrobial copolymers, manipulating the concentration of local structural domains within polymer chains, and mimicking the precise monomeric sequences and secondary sequences found in natural HDPs. For example, alternating or block copolymers are designed to enhance their biological activity and improve their selectivity between bacteria and mammalian cells.143–145
2.2.2.1.2. Block polymers. Block polymers are the most widely reported type of well-defined sequence.80,146–149 They are usually synthesized by living polymerization technologies, such as ATRP and RAFT polymerization.46,150
Many research groups142,151–153 have reported the preparation of random and diblock copolymers containing the same structure and composition and compared their biological activities. The results both showed that random and diblock copolymer series displayed similar antibacterial activity. But the random copolymer showed very high hemolytic activity and very low cell selectivity, while the diblock copolymer showed significantly lower hemolytic activity and significantly higher cell selectivity. A widely accepted hypothesis is that this is related to the difference in single chain conformation between block copolymers and random copolymers (Fig. 14). Block copolymers can form intramolecular aggregates surrounding a hydrophobic core of cationic segments in water. The cationic nature of these intramolecular aggregates increases their electrostatic binding to the anionic LPS on the bacterial surface and may displace the divalent cations that stabilize the outer membrane structure. Replacement of these divalent cations disrupts the membrane structure and increases the permeability of the outer membrane, thus facilitating the entry of polymers into the cell surface. As for the hemolytic activity, the hydrophobic segment was shielded by the cationic surface, which reduced the hydrophobic binding of the polymer to the cell membrane of RBCs and, therefore, did not cause significant hemolytic activity.153 On the other hand, the random copolymers, will adopt a random coil or slightly shrunk conformation because the hydrophobic groups randomly distributed along the polymer chain may not form a hydrophobic core if the hydrophobic content is low enough. These hydrophobic groups interact with the red blood cell membrane and cause cell lysis. In addition, the cationic functional groups of random copolymers can also enhance the binding of the polymer to bacterial cell membranes and cause membrane rupture.126,154
 | ||
| Fig. 14 Schematic illustration of the antibacterial and hemolytic activities of random and block amphiphilic copolymers. Reproduced with permission from ref. 151, copyright 2011, American Chemical Society. | ||
2.2.2.2. Cyclic polymers. Another possible structure for amphiphilic antimicrobial copolymer is a cyclic one. Cyclic polymers are linked end to end to form a ring structure and therefore have no end groups.155 Despite being inspired by many natural cyclic structures of amphiphilic antimicrobial copolymers,156 the utilization of cyclic amphiphilic polymers for antimicrobial applications remains relatively underexplored compared to other topological types. This may be attributed to the considerable difficulty in obtaining highly purified cyclic polymers.
For example, Xu and co-workers157 prepared cyclized cationic copolymers through ATRP (Fig. 15). These copolymers are more readily attracted to negatively charged bacterial cell membranes and cause membrane damage, exhibiting better antimicrobial performance compared to their linear counterparts. Additionally, the cytotoxicity of the cyclic cationic copolymers is slightly lower than that of the linear copolymers.
 | ||
| Fig. 15 Synthesis of cyclic cationic P(RMA-co-DMAEMA+) copolymers with different hydrophobic groups. Redrawn from ref. 157. | ||
Aquib et al.158 synthesized a functional RAFT agent containing benzaldehyde groups (Fig. 16(A)), which was subsequently utilized to prepare amphiphilic cationic statistical linear terpolymers via RAFT polymerization. These linear terpolymers were then cyclized into cyclic terpolymers through a Hetero-Diels–Alder click reaction (Fig. 16(B)). Compared to their linear counterparts, the cyclic terpolymers exhibited superior hemocompatibility and enhanced biocompatibility with mammalian macrophages, demonstrating improved safety profiles.
 | ||
| Fig. 16 (A) Synthesis scheme of RAFT agent. (B) Chemical structures of amphiphilic LPE, CPE, LPP, and CPP (X = percentage of cationic groups, Y = percentage of hydrophilic groups, and Z = percentage of hydrophobic groups). Redrawn from ref. 158. | ||
This indicates that cyclic polymers hold significant research value in the field of amphiphilic antimicrobial copolymers. It is anticipated that with innovations in the synthesis methods of cyclic polymers,159,160 more research on cyclic amphiphilic polymers will emerge in the future.
2.2.2.3. Advanced architecture design. Inspired by the precise and complex protein structure, many researchers tried to develop amphiphilic antimicrobial copolymers with higher structures. Along with a deeper understanding of amphiphilic antimicrobial copolymers, advances in polymer chemistry have made it possible to artificially synthesize a variety of advanced polymers with complex structures, such as hyperbranched, dendritic star or brush polymers.
For example, Santos et al. reported161 the synthesis of linear, 4-arm star and 6-arm star copolymers via supplemented activator and reducing agent atom transfer radical polymerization (SARA-ATRP) (Fig. 17). The results showed that 6-arm and 4-arm star polymers with similar molecular weights exhibited stronger antibacterial activity than linear polymers.
 | ||
| Fig. 17 Synthesis of the amphiphilic linear and linear, 4-arm star and 6-arm star shaped copolymers by SARA-ATRP. Redrawn from ref. 161. | ||
Aquib et al.162 synthesized linear polymers (LPs), hyperbranched polymers (HPs) and star polymers (SPs) via RAFT polymerization (Fig. 18). The results showed that the blood compatibility of HPs with the same ratio of hydrophobic to hydrophilic groups was significantly improved by 2 to 4 times compared with that of LPs; SPs with a low hydrophobic ratio are not toxic to red blood cells and retain similar potent antibacterial activity to linear polymers and hyperbranched polymers. Overall, SPs exhibit superior biological activity compared with HPs and LPs (SPs > HPs > LPs).
 | ||
| Fig. 18 Synthesis of LPs, HPs, and 4-arm SPs prepared by RAFT polymerization. Redrawn from ref. 162. | ||
Laroque et al.163 successfully utilized an arm-first synthesis strategy to create a library of amphiphilic cationic core-cross-linked star (CCS) copolymers through RAFT polymerization (Fig. 19). These polymers featured both diblock and homopolymer arms. The results indicated that transitioning from diblock to miktoarm CCS structures enhanced their antibacterial activity. Furthermore, while none of the synthesized polymers exhibited hemolytic activity in sheep red blood cells, there was a notable increase in the tendency for red blood cell aggregation with greater exposure of cationic units. Importantly, increasing the number of arms did not improve antibacterial activity compared to 4-arm polymers. The authors attributed this to the shielding effect of cationic units located in the polymer core, which hindered membrane interactions, although the miktoarm structures showed slight improvements in this regard.
 | ||
| Fig. 19 Planned synthesis and nomenclature of the “arm-first” core-cross-linked star (CCS) copolymer library. Linear polymers are labeled LP-X (LP = linear polymer, four different polymers numbered X = 1–4) and star polymers SP-D/M-Z (SP = star polymer, D = diblockarm, M = miktoarm, Z = DP of cross-linker). Redrawn from ref. 163. | ||
Islam et al.103 synthesized dendritic amphiphilic antimicrobial copolymers through ROMP, that showed strong cell selectivity, with high antibacterial activity against Gram-positive bacteria (S. aureus) (MIC = 32 μg mL−1) and non-toxicity to RBCs (HC50 >1000 μg mL−1). However, all polymers were inactive against Gram-negative bacteria (E. coli) (MIC > 256 μg mL−1). They hypothesize that dendritic copolymers, each containing three positive charges per dendritic unit, along with hexyl pyridinium functional groups, adhere to the lipopolysaccharide and outer membrane layers, resulting in diminished activity against E. coli.
Up to now, many studies have reported that amphiphilic antimicrobial copolymers with complex structures, including hyperbranched,162,164 dendritic star161,165 or brush polymers166,167 exhibit strong antibacterial activity and better biocompatibility than their linear counterparts. One potential explanation may be attributed to their intrinsic multivalent compact structures, where numerous end groups provide high local concentrations of active functional groups. These active moieties may synergistically enhance the initial adsorption and interaction between complex macromolecules and bacterial cells, subsequently leading to increased membrane disruption.
3. Antimicrobial applications of amphiphilic copolymers
In recent years, amphiphilic antimicrobial polymers have attracted much attention due to their low cost, simple preparation, effective antibacterial effect, easy modification, good biocompatibility, safety and non-toxicity. These properties makes amphiphilic antimicrobial copolymers highly desirable for a wide range of applications, particularly in the biomedical field, textiles, agriculture, food packaging, building materials, water treatment industry, public facilities, and medical facilities and equipment168,169 (Fig. 20). The next part of this review will discuss the application prospects of this type of amphiphilic antimicrobial polymers in different aspects, including coatings, hydrogels, vesicles, antibiotic substitutes, and more.170 | ||
| Fig. 20 Some applications of polymer antibacterial materials. | ||
3.1. Coatings
Material surfaces with antibacterial properties can prevent bacteria from adhering on and colonizing the surface of objects to form biofilms. The simplest way to give antibacterial functions to the surface of materials is to coat them. An ideal antimicrobial coating should meet the following main requirements: (a) be bactericidal against a broad spectrum of pathogenic microorganisms; (b) be active for a long time (preferably permanently); (c) be stable (should not decompose into toxic products); (d) be environmentally friendly; (e) be easy and cheap to synthesize; (f) be insoluble in water in many applications.171–173 Some type of synthetic amphiphilic antibacterial polymers meet these requirements.For example, Ma et al.174 designed a multifunctional environmentally adaptable polymer coating based on poly methacryloxyethyl dimethyl butyl ammonium bromide–polydimethylsiloxane (pMDBAB–PDMS) modified PET film (Fig. 21(a)). The soft polysiloxane chains and hydrophilic quaternary ammonium molecules enable the coating to have optimal anti-fog, antibacterial, antistatic, self-cleaning and self-lubricating properties in both water and air. At the same time, the cationic quaternary ammonium groups of the pMDBAB unit are covalently bonded in the coating network, making the coating highly antibacterial against Gram-positive S. aureus and Gram-negative E. coli. In addition, since the PDMS chains preferentially assemble in the air, the coating also exhibits good lubrication and oil adhesion resistance under dry air conditions.
 | ||
| Fig. 21 (a) Preparation process of amphiphilic multifunctional environmental adaptive coating; reproduced with permission from ref. 174, copyright 2022, American Chemical Society. (b) Synthesis of POSS-P(QAC-co-AEMA) and P(HEAA-co-GMA) copolymers for antibacterial and antifogging coatings; reproduced with permission from ref. 175, copyright 2020, American Chemical Society. (c) Schematic illustration for the fabrication of RM-coated GS and the mechanism of bioswitchable self-sterilization in bacterial infection site; reproduced with permission from ref. 80, copyright 2021, Wiley. (d) Preparation process of poly(DMA-G-MPC-MPA-N+) and dual-functional antimicrobial coating; reproduced with permission from ref. 176, copyright 2022, Elsevier. | ||
Bai et al.175 used synthetic polyhedral oligomeric silsesquioxane-poly(quaternary ammonium compound-co-2-aminoethyl methacrylate hydrochloride) (POSS-P(QAC-co-AEMA)) and poly(N-hydroxyethylacrylamide-co-glycidyl methacrylate) (P(HEAA-co-GMA)) to coat endoscopic lenses used in surgery (Fig. 21(b)). Due to the high water absorption capacity of HEAA and QAC, the coating has excellent anti-fogging properties under both in vitro and in vivo (mouse) fogging conditions. And by simply adjusting the mixing ratio of POSS-P (QAC-co-AEMA) and P(HEAA-co-GMA), the coating can achieve comprehensive bacteria-killing and bacteria-repelling properties, as well as a lower hemolysis rate.
Wang et al.80 synthesized a series of triblock copolymers with bactericidal quaternary ammonium groups at both ends of the polymer chain and bio-responsive PCL segments in the polymer chain (Fig. 21(c)). This triblock copolymer readily self-assembles to form RM with hydrophobic PCL segments in the shell and bactericidal quaternary ammonium groups in the core. Benefiting from the good biocompatibility of PCL, natural RM has little cytotoxicity. They demonstrated that RM can be easily impregnated into commercial gelatin sponge to fabricate an autonomously responsive antimicrobial coating (RM coated gelatin sponge) for the engineering design of self-sterilizing implantable medical dressings. In vivo experimental results in mice showed that when lipase emerges from released at the site of infection, PCL fragments are hydrolyzed, causing quaternary biocidal agents to be released in response, killing bacteria in the vicinity of the implant. In particular, S. aureus and B. subtilis were susceptible to quaternary biocidal agents released from RM-coated gelatin sponge.
Chen et al.176 employed a synthesized random terpolymer consisting of glycidyl methacrylate-derived quaternary ammonium cations (G-MPA-N+) as the antimicrobial component, zwitterionic 2-methacryloyloxyethyl phosphorylcholine (MPC) as the fouling-resistant component, and dopamine methacrylamide (DMA) as the anchoring moiety to modify the activated surface of PDMS elastomer, thereby constructing a dual-functional antibacterial coating (Fig. 21(d)). The resultant dual-functional antibacterial coating exhibits biocompatibility, effectively inhibiting the formation of pathogenic biofilms. Furthermore, it demonstrates minimal cytotoxicity towards mammalian cells concurrently. Of note, in an in vivo implantation model, the antibacterial coating can eradicate pathogenic biofilms within implants, preventing host tissue damage and inflammation.
These studies indicate that using this amphiphilic antimicrobial polymer as a coating has great application prospects, especially in the field of surgical medical devices, and it is believed that this research field will be further developed.
3.2. Hydrogels
Hydrogels are three-dimensional polymer networks that contain large amounts of water through hydrophilic groups or domains that hydrate in an aqueous environment.177 Chemical or physical cross-linking can render the network of homopolymers or copolymers resistant to dissolution in the aqueous phase. In addition to conventional properties such as high-water content, tunable mechanical properties, and lack of cytotoxic products after degradation, hydrogels also exhibit excellent biocompatibility and bioactivity.178,179 Due to the unique properties of antimicrobial hydrogels, they are widely used in biomedicine, tissue engineering, and smart textiles, including applications in drug delivery, regenerative medicine, wound dressings, and in vitro diagnostics. Antimicrobial hydrogels based on amphiphilic antimicrobial copolymers have also received significant attention because they combine the advantages of hydrogels and additives to exploit multiple properties on a single platform.180,181 For instance, the porous structure of hydrogels allows water/blood to flow in quickly, giving the material instant blood absorption capabilities and can also act as a barrier to prevent blood loss by forming a hemostatic plug.182,183 Therefore, the application of amphiphilic antimicrobial copolymers loaded hydrogels in wound healing dressings is a promising strategy.184–186 Particularly interesting are also injectable hydrogels, that can fill up wounds and cross-link in situ, combining antimicrobial activity with hemostatic capacity.173To address specific application scenarios, as illustrated below (Fig. 22(a)), a biodegradable amphiphilic block copolymer was utilized to design a temperature-responsive antimicrobial hydrogel.184 At room temperature, these systems exist as soluble micelles in aqueous solution. However, at physiological temperature (approximately 37 °C), they transform into gel-like materials with distinctive supramolecular fiber/ribbon-like structures and exhibit shear-thinning behavior. This hydrogel demonstrates broad-spectrum antimicrobial activity and excellent skin biocompatibility.
 | ||
| Fig. 22 (a) Preparation of temperature-sensitive mixed micellar solutions and gels containing PLLA-PEG-PLLA and PDLA-CPC-PDLA; reproduced with permission from ref. 184, copyright 2013, Wiley. (b) Schematic diagram of one-pot self-initiated polymerization to prepare highly adhesive and double-crosslinked antibacterial and antifouling gel dressing and its application in wound healing; reproduced with permission from ref. 185, copyright 2022, Elsevier. (c) Schematic diagram of the synthesis route of NPGHPs and their application in visual monitoring and antibacterial NPGHPs/SA gel; reproduced with permission from ref. 187, copyright 2022, Elsevier. | ||
Zhao et al.185 utilized the spontaneous radical polymerization of [2-(methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl) ammonium hydroxide (SBMA) to prepare an arginine-modified chitosan-oligosaccharide (COS-Arg) doped hydrogel system via a simple one-pot method (Fig. 22(b)). SBMA was covalently cross-linked with DMA and methacrylatoethyl trimethyl ammonium chloride (DMC) to form the hydrogel framework. The hydrogel exhibited excellent antibacterial activity and superior cytocompatibility. Additionally, it maintained a moist microenvironment at the wound site, promoted the production of vascular endothelial growth factor and hydroxyproline, reduced the formation of tumor necrosis factor-α, and effectively accelerated wound healing.
Additionally, Wang et al.187 encapsulated nanoengineered peptide-grafted hyperbranched polymers (NPGHP) in an anionic polyelectrolyte and, for the first time, constructed aggregation-induced emission (AIE) active fluorescent polymeric hydrogels (NPGHP/sodium alginate (SA) gels) using non-conjugated luminescent polymers (NCLPs) with AIE characteristics (Fig. 22(c)). The NPGHP/SA gel system, with good biocompatibility, exhibited significant luminescence changes during the release process, thereby enabling the visualization of dynamic drug release behavior. This system provides an advanced biomedical material with broad-spectrum antibacterial properties and offers a facile method for investigating drug release in hydrogel systems.
Distinct from common polymer hydrogels, Brahmachari et al.188 designed a novel amphiphilic hydrogelator utilizing physical cross-linking, such as hydrogen bonding between compounds. This design involved connecting a quaternary pyridinium unit to a hydrophobic long alkyl chain via an amide bond, resulting in a compound with a very simple synthesis. Efficient hydrogel formation was achieved through the interdigitated bilayer packing of the amphiphilic molecules, leading to the development of three-dimensional supramolecular aggregates. These hydrogelators exhibited high antibacterial activity against both Gram-positive and Gram-negative bacteria (MIC < 0.4 μg mL−1) while demonstrating very low cytotoxicity (IC50 > 100 μg mL−1).
There are also many studies that have investigated the incorporation of antimicrobial agents into hydrogels to achieve antimicrobial effects.189–192 However, wound dressings containing antimicrobial agents may lose their long-term bactericidal efficacy due to the leaching of these agents. Furthermore, certain nano-inorganic metal-based antimicrobial agents exhibit cytotoxicity, posing potential risks to biological systems.193–195 Additionally, traditional hydrogel systems often utilize initiators such as ammonium persulfate (APS) or 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (HHMP), which can leach from the hydrogel matrix. This leaching may irritate and corrode mucosal tissues and, with prolonged skin exposure, lead to allergic dermatitis.196 Consequently, the development of inherently antimicrobial hydrogels that do not rely on toxic initiators has emerged as a promising research direction. Such hydrogels offer the dual advantages of sustained antibacterial efficacy and excellent biocompatibility, addressing the limitations of traditional hydrogel systems.
3.3. Vesicles
Typically, polymer vesicles are nanoscale hollow spheres composed of three parts: an inner cavity, a hydrophobic membrane, and a hydrophilic corona, making them more complex than polymer micelles with simple core–shell nanostructures.197 Amphiphilic antimicrobial copolymers with cationic groups and hydrophobic chains (often as nanoparticles) have demonstrated excellent antibacterial performance against both Gram-positive and Gram-negative bacteria.198,199 However, strong positive charges can lead to severe cytotoxicity, such as hemolysis, which is a common side effect of cationic polymers. Therefore, forming nanostructured vesicles with amphiphilic antimicrobial copolymers that possess moderate positive charges may be a comprehensive strategy to balance antibacterial activity and cytotoxicity.91,147,151,200–203For instance, Rahman et al.198 selected bile acid and neutral PEG components to fabricate amphiphilic antimicrobial copolymers capable of forming nanoscale particles. These amphiphilic antimicrobial copolymers form spheres, rods, and vesicles (Fig. 23(a)). The nanoscale structures of these bile acid-based macromolecules preferentially interact with bacterial membranes, exhibiting broad-spectrum antibacterial activity against both Gram-positive and Gram-negative bacteria. The cholic acid-based copolymers with three QAC charges (P1–P5) demonstrated better efficacy compared to the deoxycholic acid-based copolymers with dual QAC charges (P6). Incorporating PEG into the copolymers not only enhanced the colloidal stability of the nanostructures but also increased their biocompatibility. Notably, Zhang and Zhu et al.147 reported water-soluble antibacterial polymer vesicles based on the novel thermo responsive di-block copolymer, which do not contain quaternary ammonium groups (amino groups were protected by Boc and not deprotected) or load any external antibiotics (Fig. 23(b)). Under physiological conditions, these polymer vesicles exhibited superior antibacterial efficacy against both Gram-negative and Gram-positive bacteria. The authors explained that the formation of vesicles increased the local concentration of cationic charges and polymer mass, resulting in stronger interactions between the polymer and the bacterial cell wall/membrane. This led to more effective antibacterial activity compared to individual, non-self-assembled polymer chains.
 | ||
| Fig. 23 (a) Schematic diagram of the antibacterial mechanism of the designed and synthesized cholic acid based gradient copolymers (P1–P4), diblock copolymer (P5), and deoxycholic acid based copolymer (P6) on the cell membrane of Gram-negative bacteria; reproduced with permission from ref. 198, copyright 2022, American Chemical Society. (b) Schematic diagram of antimicrobial vesicles formed by directly dissolving diblock copolymers in water; reproduced with permission from ref. 147, copyright 2013, Royal Society of Chemistry. (c) Synthesis of peptide-based vesicles with antibacterial activity and intrinsic blue fluorescence, and schematic diagram of real-time visualization of the antibacterial process; reproduced with permission from ref. 204, copyright 2021, Springer Nature. | ||
Recently, a vesicle capable of real-time visualization of antibacterial processes has been reported in the literature.204 Through modular design, Yang et al. integrated multiple functional segments to create vesicles with intrinsic blue fluorescence. The synthesized PCL25-b-PTrp2-b-P(Lys13-stat-Phe4) copolymer, featuring biocompatible and biodegradable PCL chains, forms hydrophobic vesicle membranes, while PTrp and P(Lys-stat-Phe) provide intrinsic fluorescence and broad-spectrum antibacterial activity (Fig. 23(c)). Additionally, during the self-assembly process, the aggregation of aromatic amino acids and formation of hydrogen bonds induce electron delocalization, shifting the fluorescence emission from the invisible ultraviolet range of the amino acids to the visible range (with a maximum emission at 436 nm). This transition enables the visualization of the antibacterial process targeting bacteria. Confocal fluorescence imaging of the vesicles with bacteria confirmed the specific adhesion of the vesicles to the bacteria and demonstrated bacterial death resulting from membrane disruption.
In summary, polymer vesicles typically exhibit superior antimicrobial activity compared to their linear counterparts or free antibiotics due to enhanced local charge density or increased delivery efficiency. However, most research on antimicrobial polymer vesicles has focused on those loaded with antibiotics,202,205–209 and there is limited work on the design of vesicles incorporating amphiphilic copolymers for antimicrobial purposes. Consequently, this field is still in a phase of rapid growth and discovery, and we believe that many challenges remain to be addressed in the future.
3.4. Antibiotic alternatives
The escalating rise in antibiotic resistance among bacteria and other pathogens has emerged as a new threat to global health.210 The emergence of this resistance phenomenon is attributed to various factors, including natural evolution under selective pressure, over prescription of antibiotics, and inappropriate use of antibiotics. Due to the development of resistance, there is a growing demand for novel antibiotics. Amphiphilic antimicrobial copolymers exhibit broad-spectrum antibacterial activity and low toxicity, distinct from HDPs, they can physically disrupt the cell membrane of organisms, thereby preventing the development of drug-resistant microorganisms and coupled with inherent advantages in synthetic methods. These attributes have piqued the interest of polymer chemists in exploring the potential of polymeric antibiotics.116,211,212For example, the amphiphilic antimicrobial copolymers poly[(APMA)-ran-(BMA)], synthesized by A. Tyagi and A. Mishra with approximately 20% hydrophobic benzylmethacrylamide units, were non hemolytic.72 Polymers with DP of 17 and 27 exhibited the optimal combination of high antibacterial efficiency and low cytotoxicity. In resistance studies, no increase in MIC values was observed for (AB-20)17 and (AB-20)27 polymers during serial passage over 25 and 30 days, respectively. However, when using ciprofloxacin (a fluoroquinolone antibiotic used as a control), the MIC values for E. coli increased by 256-fold, and for S. aureus, the MIC values increased by 64-fold (Table 7).
| Bacteria strain | Antibacterial agent | Number of passages | MIC (μM) | Fold change in MIC | |
|---|---|---|---|---|---|
| Initial | Final | ||||
| E. coli | (AB-20)17 | 25 | 12 | 12 | No resistance |
| Ciprofloxacin | 25 | 0.188 | 48.28 | 256 | |
| (AB-20)27 | 30 | 6.5 | 6.5 | No resistance | |
| Ciprofloxacin | 30 | 0.09 | 24.1 | 256 | |
| S. aureus | (AB-20)17 | 25 | 14 | 14 | No resistance |
| Ciprofloxacin | 25 | 0.75 | 48.28 | 64 | |
| (AB-20)27 | 30 | 6.5 | 6.5 | No resistance | |
| Ciprofloxacin | 30 | 0.75 | 24.1 | 32 | |
Despite extensive research by both academia and industry, the replacement of antibiotics with amphiphilic antimicrobial copolymers has yet to achieve industrial or clinical application. While these polymers exhibit broad-spectrum antimicrobial activity and a reduced risk of inducing resistance, MICs typically fall in the micromolar range—significantly higher than the nanomolar values of conventional antibiotics.213 This limits their applicability in systemic infection treatment and confines their current use to localized applications such as surface coatings, wound dressings, and medical device modifications.17,214 Moreover, balancing hydrophobicity and cationic charge to optimize antimicrobial efficacy without compromising biocompatibility remains a critical challenge. Meanwhile, commercial barriers to the use of amphiphilic antimicrobial copolymers as antibiotic alternatives remain high due to safety concerns, regulatory issues, and challenges in material development.215 However, some natural HDPs and their derivatives, such as daptomycin and colistin, have been clinically applied, and several synthetic antimicrobial peptides are currently undergoing clinical trials.216 We believe that amphiphilic antimicrobial copolymers will have potential applications as antibiotic alternatives in the future.
4. Conclusion and perspective
In summary, this review systematically summarizes the preparation strategies and wide applications of amphiphilic antibacterial polymers, as well as their advantages in the antibacterial field.In recent years, scientists have made exciting progress in elucidating the structure–activity relationships of antimicrobial polymers, providing unprecedented tools for the synthesis of amphiphilic antimicrobial copolymers. This has been achieved by chemists through the utilization of actively controlled methods to rigorously control structure, morphology, and topology. For instance, fine-tuning the amphiphilic antimicrobial copolymers balance can enhance selectivity and antimicrobial activity. Currently, the careful design and synthesis of amphiphilic antimicrobial copolymers, aimed at providing a clear understanding of structure/property relationships have become a reality. However, significant work still needs to be undertaken to address major challenges, such as: (1) enhancing antimicrobial activity while mitigating toxicity associated with certain strategies; (2) broadening the range of microbes effectively eliminated without inducing resistance; improving the long-term stability of antimicrobial polymers; (3) developing simple, cost-effective manufacturing solutions; (4) considering the biodegradability of amphiphilic antimicrobial copolymers in specific applications and (5) imbuing amphiphilic antimicrobial copolymers with targeted smart responses.
Moreover, despite the enormous potential applications of amphiphilic antimicrobial copolymers, particularly in biomedical materials such as antibiofilm, wound healing, and tissue engineering, they are still in a rapid growth and discovery phase. Therefore, we believe that there are still many challenges worth attention in the future:
(1) How to design and synthesize amphiphilic antimicrobial copolymers to meet specific application requirements? In certain specific scenarios, antimicrobial agents need to be active against only one or a few microorganisms. This requires a deep understanding of microbial structures and antibacterial mechanisms. Synthesizing such specialized amphiphilic antimicrobial copolymers has consistently been highly challenging.
(2) How to increase the selectivity toward bacteria and mammalian cells? Currently, side effects can be reduced by developing precisely sequence-controlled and highly structured polymers rather than simpler models. Recent studies have found that incorporating neutral hydrophilic groups into traditional binary polymers to create novel ternary antimicrobial polymers can significantly enhance their biocompatibility with host cells. However, our ongoing goal is to further increase antimicrobial activity while reducing the cytotoxicity of antimicrobial polymers.
(3) How to achieve antibacterial and anticancer activities simultaneously? In clinical settings, bacterial infections often accompany tumor sites due to the decreased immunity of patients. Therefore, achieving simultaneous antibacterial and anticancer effects in biomedical materials holds special significance. This could bring new insights into the field of cancer treatment.
Abbreviations
| ABMA | Aminobutyl methacrylate |
| AEMA | Amino-ethyl methacrylate |
| AIE | Aggregation-induced emission |
| AM | 4-Acryloylmorpholine |
| APMA | Aminopropyl methacrylamide |
| APS | Ammonium persulfate |
| ATRP | Atom transfer radical polymerization |
| B | N-Benzylacrylamide |
| Bm | N-Benzylacrylamide |
| BMA | Benzylmethacrylamide |
| CCS | Core-cross-linked star |
| CPC | Polycarbonate |
| Cpm | N-Cycloheptylacrylamide |
| Cxm | N-(Cyclohexylmethyl)acrylamide |
| DMA | Dopamine methacrylamide |
| DMC | Methacrylatoethyl trimethyl ammonium chloride |
| DP | Degree of polymerization |
| EMA | Ethyl methacrylate |
| HC50 and HC10 | The maximum concentration of polymers with less than 50% or 10% hemolytic activity on RBCs |
| HEA | Hydroxyethyl acrylamide |
| HEMA | Hydroxyethyl methacrylate |
| HHMP | 2-Hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone |
| Hm | N-Heptylacrylamide |
| HPs | Hyperbranched polymers |
| I | N-Isopentylacrylamide |
| IC50 | The maximum concentration of polymer less than 50% toxic to test cells |
| IL | Ionic liquid |
| Im | N-isopentylacrylamide |
| G-MPA-N+ | Glycidyl methacrylate quaternary ammonium cations |
| HDPs | Host defense peptides |
| LPs | Linear polymers |
| LPS | lipopolysaccharide |
| L-PTMI | Linear polytrimethyleneimine |
| MIC | Minimum inhibitory concentration |
| MPC | Methacryloyloxyethyl phosphorylcholine |
| MRSA | Methicillin-resistant S. aureus |
| NCA | N-Carboxyanhydrides |
| Nm | N-Nonylacrylamide |
| NPGHP | Nanoengineered peptide-grafted hyperbranched polymers |
| PA | Poly(ester polyurethane)s with aromatic group as hydrophobic group |
| PAMA | Propanoic acid methacrylate |
| Pbm | N-Butyl-N-propylacrylamide |
| PCL | Poly(ε-caprolactone) |
| PDLA | Poly(D-lactide) |
| PDMS | Poly dimethylsiloxane |
| PEG | Poly ethylene glycol |
| PEG-A | Poly(ethylene glycol)methyl ether acrylate |
| PEG-AA | Poly(ethylene glycol)acrylamide |
| P(HEAA-co-GMA) | poly(N-hydroxyethylacrylamide-co-glycidyl methacrylate) |
| PILs | Poly(ionic liquids) |
| PLA | Poly(ester polyurethane)s with long alkyl chain as hydrophobic group |
| PLLA | Poly(L-lactide) |
| Pm | N-Pentylacrylamide |
| PM | Poly(methionine) |
| PMDBAB | Poly methacryloxyethyl dimethyl butyl ammonium bromide |
| POSS-P(QAC-co-AEMA) | polyhedral oligomeric silsesquioxane-poly(quaternary ammonium compound-co-2-aminoethyl methacrylate hydrochloride) |
| PPGn-PILm | Imidazolium-based block copolypeptides with L configuration |
| PSA | Poly(ester polyurethane)s with short alkyl chain as hydrophobic group |
| PVP | Poly(vinyl-pyridine) |
| QAC | Quaternary ammonium compound |
| QBA | Quaternary biocidal agents |
| RAFT | Reversible addition–fragmentation chain transfer polymerization |
| RBCs | Red blood cells |
| RDRP | Reversible deactivation radical polymerization |
| RM | Reverse micelles |
| ROMP | Ring-opening metathesis polymerization |
| SA | Sodium alginate |
| ROP | Ring-opening polymerization |
| SARA-ATRP | Supplemented activator and reducing agent atom transfer radical polymerization |
| SBMA | [2-(Methacryloyloxy)ethyl]dimethyl-(3-sulfopropyl) ammonium hydroxide |
| SPs | Star polymers |
| THF | Tetrahydrofuran |
Data availability
This is a review article, therefore it does not contain original data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The first author of this work was financially supported by the China Scholarship Council (CSC) under grant number 202204910075.References
- L. J. Rothschild and R. L. Mancinelli, Nature, 2001, 409, 1092–1101 CrossRef CAS.
- G. Antranikian, C. E. Vorgias and C. Bertoldo, Adv. Biochem. Eng./Biotechnol., 2005, 96, 219–262 CrossRef CAS PubMed.
- P. Sarmah, M. M. Dan, D. Adapa and T. Sarangi, Electron. J. Biol., 2018, 14, 50–58 Search PubMed.
- M. Lavanya, J. Bio-Tribo-Corros., 2021, 7, 125 CrossRef.
- G. McDonnell and A. D. Russell, Clin. Microbiol. Rev., 1999, 12, 147–179 CrossRef CAS PubMed.
- C. Marambio-Jones and E. M. Hoek, J. Nanopart. Res., 2010, 12, 1531–1551 CrossRef CAS.
- B. K. English and A. H. Gaur, Hot Topics in Infection and Immunity in Children VI, 2010, pp. 73–82 Search PubMed.
- R. J. Fair and Y. Tor, Perspect. Med. Chem., 2014, 6, 25–64 Search PubMed.
- G. Mancuso, A. Midiri, E. Gerace and C. Biondo, Pathogens, 2021, 10, 1310 CrossRef CAS PubMed.
- O. O. Adeniji, M. O. Ojemaye and A. I. Okoh, ACS Appl. Bio Mater., 2022, 5, 4814–4826 CrossRef CAS PubMed.
- R. Rawashdeh and Y. Haik, Dyn. Biochem. Process Biotechnol. Mol. Biol., 2009, 3, 12–20 Search PubMed.
- Y. Yan, Y. Li, Z. Zhang, X. Wang, Y. Niu, S. Zhang, W. Xu and C. Ren, Colloids Surf., B, 2021, 202, 111682 CrossRef CAS PubMed.
- J. K. Boparai and P. K. Sharma, Protein Pept. Lett., 2020, 27, 4–16 CrossRef CAS.
- S. Duan, R. Wu, Y. Xiong, H. Ren, C. Lei, Y. Zhao, X. Zhang and F. Xu, Prog. Mater. Sci., 2022, 125, 100887 CrossRef CAS.
- K. Huang, C. Yang, S. Huang, C. Chen, Y. Lu and Y. Lin, Int. J. Mol. Sci., 2016, 17, 1578 CrossRef.
- L. Timofeeva and N. Kleshcheva, Appl. Microbiol. Biotechnol., 2011, 89, 475–492 CrossRef CAS PubMed.
- J. Chen, F. Wang, Q. Liu and J. Du, Chem. Commun., 2014, 50, 14482–14493 RSC.
- A. Jain, L. S. Duvvuri, S. Farah, N. Beyth, A. J. Domb and W. Khan, Adv. Healthcare Mater., 2014, 3, 1969–1985 CrossRef CAS PubMed.
- P. Raffa, D. A. Z. Wever, F. Picchioni and A. A. Broekhuis, Chem. Rev., 2015, 115, 8504–8563 CrossRef CAS PubMed.
- L. C. Klein and A. B. Wojcik, Encyclopedia of Materials: Science and Technology, Elsevier, Oxford, 2001, pp. 7577–7584 Search PubMed.
- F. Perin, A. Motta and D. Maniglio, Mater. Sci. Eng., C, 2021, 123, 111952 CrossRef CAS PubMed.
- A. B. Kutikov and J. Song, ACS Biomater. Sci. Eng., 2015, 1, 463–480 CrossRef CAS.
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- X. Xiong, Z. Binkhathlan, O. Molavi and A. Lavasanifar, Acta Biomater., 2012, 8, 2017–2033 CrossRef CAS.
- Z. Si, W. Zheng, D. Prananty, J. Li, C. H. Koh, E. T. Kang, K. Pethe and M. B. Chan-Park, Chem. Sci., 2022, 13, 345–364 RSC.
- N. Mookherjee, M. A. Anderson, H. P. Haagsman and D. J. Davidson, Nat. Rev. Drug Discovery, 2020, 19, 311–332 CrossRef CAS PubMed.
- E. F. Palermo, K. Lienkamp, E. R. Gillies and P. J. Ragogna, Angew. Chem., Int. Ed., 2019, 58, 3690–3693 CrossRef CAS PubMed.
- K. Kuroda and G. A. Caputo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2013, 5, 49–66 CAS.
- Y. Yang, Z. Cai, Z. Huang, X. Tang and X. Zhang, Polym. J., 2018, 50, 33–44 CrossRef CAS.
- A. Patrzykat, C. L. Friedrich, L. Zhang, V. Mendoza and R. E. Hancock, Antimicrob. Agents Chemother., 2002, 46, 605–614 CrossRef CAS PubMed.
- A. Krizsan, D. Volke, S. Weinert, N. Sträter, D. Knappe and R. Hoffmann, Angew. Chem., Int. Ed., 2014, 53, 12236–12239 CrossRef CAS PubMed.
- G. Kragol, S. Lovas, G. Varadi, B. A. Condie, R. Hoffmann and L. Otvos, Jr., Biochemistry, 2001, 40, 3016–3026 CrossRef CAS PubMed.
- D. G. McCafferty, P. Cudic, M. K. Yu, D. C. Behenna and R. Kruger, Curr. Opin. Chem. Biol., 1999, 3, 672–680 CrossRef CAS PubMed.
- I. Ginsburg, Med. Hypotheses, 2004, 62, 367–374 CrossRef CAS PubMed.
- L. Otvos, Jr., H. Flick-Smith, M. Fox, E. Ostorhazi, R. M. Dawson and J. D. Wade, Protein Pept. Lett., 2014, 21, 374–381 CrossRef PubMed.
- Y. Lai and R. L. Gallo, Trends Immunol., 2009, 30, 131–141 CrossRef CAS PubMed.
- D. J. Davidson, A. J. Currie, G. S. Reid, D. M. Bowdish, K. L. MacDonald, R. C. Ma, R. E. Hancock and D. P. Speert, J. Immunol., 2004, 172, 1146–1156 CrossRef CAS PubMed.
- S. V. Guryanova and T. V. Ovchinnikova, Int. J. Mol. Sci., 2022, 23, 2499 CrossRef CAS PubMed.
- H. Li, J. Niu, X. Wang, M. Niu and C. Liao, Pharmaceutics, 2023, 15, 2278 CrossRef CAS.
- L. Brandenburg, J. Merres, L. Albrecht, D. Varoga and T. Pufe, Polymers, 2012, 4, 539–560 CrossRef.
- A. C. Engler, N. Wiradharma, Z. Ong, D. J. Coady, J. L. Hedrick and Y. Yang, Nano Today, 2012, 7, 201–222 CrossRef CAS.
- K. Matsuzaki, Biochimica et Biophysica Acta, 2009, 1788, 1687–1692 CrossRef CAS PubMed.
- P. Gilbert and L. E. Moore, J. Appl. Microbiol., 2005, 99, 703–715 CrossRef CAS PubMed.
- M. F. Ilker, K. Nüsslein, G. N. Tew and E. B. Coughlin, J. Am. Chem. Soc., 2004, 126, 15870–15875 CrossRef CAS PubMed.
- H. Mortazavian, L. L. Foster, R. Bhat, S. Patel and K. Kuroda, Biomacromolecules, 2018, 19, 4370–4378 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Chem. – Asian J., 2013, 8, 1634–1644 CrossRef CAS PubMed.
- F. L. Hatton, Polym. Chem., 2020, 11, 220–229 RSC.
- N. P. Truong, G. R. Jones, K. G. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem., 2021, 5, 859–869 CrossRef CAS PubMed.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- D. S. Uppu, S. Samaddar, C. Ghosh, K. Paramanandham, B. R. Shome and J. Haldar, Biomaterials, 2016, 74, 131–143 CrossRef CAS PubMed.
- K. E. Locock, T. D. Michl, J. D. Valentin, K. Vasilev, J. D. Hayball, Y. Qu, A. Traven, H. J. Griesser, L. Meagher and M. Haeussler, Biomacromolecules, 2013, 14, 4021–4031 CrossRef CAS PubMed.
- K. Kuroda and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 4128–4129 CrossRef CAS PubMed.
- K. Kuroda, G. A. Caputo and W. F. DeGrado, Chem. – Eur. J., 2009, 15, 1123–1133 CrossRef CAS PubMed.
- K. Lienkamp and G. N. Tew, Chem. – Eur. J., 2009, 15, 11784–11800 CrossRef CAS PubMed.
- K. Lienkamp, A. E. Madkour, A. Musante, C. F. Nelson, K. Nusslein and G. N. Tew, J. Am. Chem. Soc., 2008, 130, 9836–9843 CrossRef CAS PubMed.
- G. J. Gabriel, J. A. Maegerlein, C. F. Nelson, J. M. Dabkowski, T. Eren, K. Nüsslein and G. N. Tew, Chem. – Eur. J., 2009, 15, 433–439 CrossRef CAS PubMed.
- B. Dizman, M. O. Elasri and L. J. Mathias, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5965–5973 CrossRef CAS.
- E. R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- T. Tashiro, Macromol. Mater. Eng., 2001, 286, 63–87 CrossRef CAS.
- J. C. Tiller, C. J. Liao, K. Lewis and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5981–5985 CrossRef CAS PubMed.
- F. Baudrion, A. Perichaud and S. Coen, J. Appl. Polym. Sci., 1998, 70, 2657–2666 CrossRef CAS.
- E. R. Kenawy, F. I. Abdel-Hay, A. El-Shanshoury and M. H. El-Newehy, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2384–2393 CrossRef CAS.
- M. A. Gelman, B. Weisblum, D. M. Lynn and S. H. Gellman, Org. Lett., 2004, 6, 557–560 CrossRef CAS PubMed.
- N. M. Milovic, J. Wang, K. Lewis and A. M. Klibanov, Biotechnol. Bioeng., 2005, 90, 715–722 CrossRef CAS PubMed.
- R. F. Epand, B. P. Mowery, S. E. Lee, S. S. Stahl, R. I. Lehrer, S. H. Gellman and R. M. Epand, J. Mol. Biol., 2008, 379, 38–50 CrossRef CAS PubMed.
- J. Y. Huang, R. R. Koepsel, H. Murata, W. Wu, S. B. Lee, T. Kowalewski, A. J. Russell and K. Matyjaszewski, Langmuir, 2008, 24, 6785–6795 CrossRef CAS PubMed.
- H. Murata, R. R. Koepsel, K. Matyjaszewski and A. J. Russell, Biomaterials, 2007, 28, 4870–4879 CrossRef CAS PubMed.
- E. F. Palermo and K. Kuroda, Biomacromolecules, 2009, 10, 1416–1428 CrossRef CAS PubMed.
- E. F. Palermo, S. Vemparala and K. Kuroda, Biomacromolecules, 2012, 13, 1632–1641 CrossRef CAS PubMed.
- P. Pham, S. Oliver, E. H. Wong and C. Boyer, Polym. Chem., 2021, 12, 5689–5703 RSC.
- X. Yang, K. Hu, G. Hu, D. Shi, Y. Jiang, L. Hui, R. Zhu, Y. Xie and L. Yang, Biomacromolecules, 2014, 15, 3267–3277 CrossRef CAS PubMed.
- A. Tyagi and A. Mishra, ACS Omega, 2021, 6, 34724–34735 CrossRef CAS PubMed.
- W. Yang, K. G. Neoh, E. T. Kang, S. L. M. Teo and D. Rittschof, Polym. Chem., 2013, 4, 3105–3115 RSC.
- W. Zheng, M. Anzaldua, A. Arora, Y. Jiang, K. McIntyre, M. Doerfert, T. Winter, A. Mishra, H. Ma and H. Liang, Biomacromolecules, 2020, 21, 2187–2198 CrossRef CAS PubMed.
- J. S. Brown, Z. J. Mohamed, C. M. Artim, D. N. Thornlow, J. F. Hassler, V. P. Rigoglioso, S. Daniel and C. A. Alabi, Commun. Biol., 2018, 1, 220 CrossRef CAS PubMed.
- M. Porel, D. N. Thornlow, C. M. Artim and C. A. Alabi, ACS Chem. Biol., 2017, 12, 715–723 CrossRef CAS PubMed.
- P. Pham, S. Oliver and C. Boyer, Macromol. Chem. Phys., 2022, 224, 2200226 CrossRef.
- A. M. Carmona-Ribeiro and L. D. de Melo Carrasco, Int. J. Mol. Sci., 2013, 14, 9906–9946 CrossRef PubMed.
- Z. Zhang, X. Wang, J. Liu, H. Yang, H. Tang, J. Li, S. Luan, J. Yin, L. Wang and H. Shi, Angew. Chem., Int. Ed., 2024, 63, e202318011 CrossRef CAS PubMed.
- A. Wang, S. Duan, X. Ding, N. Zhao, Y. Hu, X. Ding and F. Xu, Adv. Funct. Mater., 2021, 31, 2011165 CrossRef CAS.
- H. Takahashi, G. A. Caputo, S. Vemparala and K. Kuroda, Bioconjugate Chem., 2017, 28, 1340–1350 Search PubMed.
- L. Liu, K. Xu, H. Wang, P. K. Tan, W. Fan, S. Venkatraman, L. Li and Y. Yang, Nat. Nanotechnol., 2009, 4, 457–463 CrossRef CAS PubMed.
- A. King, S. Chakrabarty, W. Zhang, X. Zeng, D. E. Ohman, L. F. Wood, S. Abraham, R. Rao and K. J. Wynne, Biomacromolecules, 2014, 15, 456–467 CrossRef CAS PubMed.
- I. Mukherjee, A. Ghosh, P. Bhadury and P. De, ACS Omega, 2017, 2, 1633–1644 CrossRef CAS PubMed.
- S. Mankoci, R. L. Kaiser, N. Sahai, H. A. Barton and A. Joy, ACS Biomater. Sci. Eng., 2017, 3, 2588–2597 CrossRef CAS PubMed.
- T. Eren, A. Som, J. R. Rennie, C. F. Nelson, Y. Urgina, K. Nüsslein, E. B. Coughlin and G. N. Tew, Macromol. Chem. Phys., 2008, 209, 516–524 CrossRef CAS.
- Y. Hu, J. Zhao, J. Zhang, Z. Zhu and J. Rao, ACS Macro Lett., 2021, 10, 990–995 CrossRef CAS PubMed.
- J. Oh and A. Khan, Biomacromolecules, 2021, 22, 3534–3542 CrossRef CAS PubMed.
- Y. Zhao, X. Wang, Y. Hu, J. Zhao, M. Sun, M. Yang, H. Xuan, X. Wang, J. Zhang, Z. Zhu and J. Rao, ACS Appl. Polym. Mater., 2023, 5, 4437–4447 CrossRef CAS.
- K. J. Cutrona, B. A. Kaufman, D. M. Figueroa and D. E. Elmore, FEBS Lett., 2015, 589, 3915–3920 CrossRef CAS PubMed.
- F. Nederberg, Y. Zhang, J. P. Tan, K. Xu, H. Wang, C. Yang, S. Gao, X. Guo, K. Fukushima and L. Li, Nat. Chem., 2011, 3, 409–414 CrossRef CAS PubMed.
- L. M. Thoma, B. R. Boles and K. Kuroda, Biomacromolecules, 2014, 15, 2933–2943 CrossRef CAS PubMed.
- A. K. Marr, W. J. Gooderham and R. E. Hancock, Curr. Opin. Pharmacol., 2006, 6, 468–472 CrossRef CAS PubMed.
- G. Maisetta, M. Di Luca, S. Esin, W. Florio, F. L. Brancatisano, D. Bottai, M. Campa and G. Batoni, Peptides, 2008, 29, 1–6 CrossRef CAS PubMed.
- S. Hong, H. Takahashi, E. T. Nadres, H. Mortazavian, G. A. Caputo, J. G. Younger and K. Kuroda, PLoS One, 2017, 12, e0169262 CrossRef PubMed.
- R. Bhat, L. L. Foster, G. Rani, S. Vemparala and K. Kuroda, RSC Adv., 2021, 11, 22044–22056 RSC.
- S. Buffet-Bataillon, P. Tattevin, M. Bonnaure-Mallet and A. Jolivet-Gougeon, Int. J. Antimicrob. Agents, 2012, 39, 381–389 CrossRef CAS PubMed.
- S. Liu, R. J. Ono, H. Wu, J. Y. Teo, Z. C. Liang, K. Xu, M. Zhang, G. Zhong, J. P. Tan, M. Ng, C. Yang, J. Chan, Z. Ji, C. Bao, K. Kumar, S. Gao, A. Lee, M. Fevre, H. Dong, J. Y. Ying, L. Li, W. Fan, J. L. Hedrick and Y. Y. Yang, Biomaterials, 2017, 127, 36–48 CrossRef CAS PubMed.
- W. Chin, G. Zhong, Q. Pu, C. Yang, W. Lou, P. F. De Sessions, B. Periaswamy, A. Lee, Z. Liang, X. Ding, S. Gao, C. W. Chu, S. Bianco, C. Bao, Y. Tong, W. Fan, M. Wu, J. Hedrick and Y. Yang, Nat. Commun., 2018, 9, 917 CrossRef PubMed.
- J. Leong, C. Yang, J. Tan, B. Tan, S. Hor, J. L. Hedrick and Y. Yang, Biomater. Sci., 2020, 8, 6920–6929 RSC.
- J. S. Brown, Z. J. Mohamed, C. M. Artim, D. N. Thornlow, J. F. Hassler, V. P. Rigoglioso, S. Daniel and C. A. Alabi, Commun. Biol., 2018, 1, 220 Search PubMed.
- V. Sambhy, B. R. Peterson and A. Sen, Angew. Chem., Int. Ed., 2008, 47, 1250–1254 CrossRef CAS PubMed.
- M. N. Islam, B. Aksu, M. Güncü, M. Gallei, M. Tulu and T. Eren, J. Polym. Sci., 2020, 58, 872–884 CrossRef CAS.
- R. Cuervo-Rodríguez, A. Muñoz-Bonilla, F. López-Fabal and M. Fernández-García, Polymers, 2020, 12, 972 CrossRef PubMed.
- S. N. Hancock, N. Yuntawattana, E. Diep, A. Maity, A. Tran, J. D. Schiffman and Q. Michaudel, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2311396120 CrossRef CAS PubMed.
- A. Vishwakarma, F. Dang, A. Ferrell, H. A. Barton and A. Joy, J. Am. Chem. Soc., 2021, 143, 9440–9449 Search PubMed.
- Z. Zheng, Q. Xu, J. Guo, J. Qin, H. Mao, B. Wang and F. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 12684–12692 CrossRef CAS PubMed.
- Z. Shi, X. Zhang, Z. Yu, F. Yang, H. Liu, R. Xue, S. Luan and H. Tang, Biomacromolecules, 2021, 22, 2373–2381 CrossRef CAS PubMed.
- A. Kanazawa, T. Ikeda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 335–343 CrossRef CAS.
- B. Hisey, P. J. Ragogna and E. R. Gillies, Biomacromolecules, 2017, 18, 914–923 CrossRef CAS PubMed.
- H. Kuday, N. C. Süer, A. Bayır, B. Aksu, A. Hatipoğlu, M. M. Güncü, İ. Acaroğlu Degitz, M. Gallei and T. Eren, ACS Appl. Polym. Mater., 2021, 3, 6524–6538 CrossRef CAS.
- A. Kanazawa, T. Ikeda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2873–2876 Search PubMed.
- P. T. Phuong, S. Oliver, J. He, E. H. H. Wong, R. T. Mathers and C. Boyer, Biomacromolecules, 2020, 21, 5241–5255 Search PubMed.
- E. F. Palermo and K. Kuroda, Appl. Microbiol. Biotechnol., 2010, 87, 1605–1615 CrossRef CAS.
- C. Ergene, K. Yasuhara and E. F. Palermo, Polym. Chem., 2018, 9, 2407–2427 RSC.
- H. Takahashi, G. A. Caputo and K. Kuroda, Biomater. Sci., 2021, 9, 2758–2767 RSC.
- E. F. Palermo, I. Sovadinova and K. Kuroda, Biomacromolecules, 2009, 10, 3098–3107 CrossRef CAS PubMed.
- I. Sovadinova, E. F. Palermo, M. Urban, P. Mpiga, G. A. Caputo and K. Kuroda, Polymers, 2011, 3, 1512–1532 CrossRef CAS.
- M. Ward, M. Sanchez, M. O. Elasri and A. B. Lowe, J. Appl. Polym. Sci., 2006, 101, 1036–1041 CrossRef CAS.
- J. Tan, Y. Zhao, J. L. Hedrick and Y. Yang, Adv. Healthcare Mater., 2022, 11, 2100482 CrossRef CAS PubMed.
- W. Chin, G. Zhong, Q. Pu, C. Yang, W. Lou, P. F. De Sessions, B. Periaswamy, A. Lee, Z. C. Liang, X. Ding, S. Gao, C. W. Chu, S. Bianco, C. Bao, Y. W. Tong, W. Fan, M. Wu, J. L. Hedrick and Y. Y. Yang, Nat. Commun., 2018, 9, 917 Search PubMed.
- C. Peng, A. Vishwakarma, S. Mankoci, H. A. Barton and A. Joy, Biomacromolecules, 2019, 20, 1675–1682 CrossRef CAS PubMed.
- S. Mankoci, J. Ewing, P. Dalai, N. Sahai, H. A. Barton and A. Joy, Biomacromolecules, 2019, 20, 4096–4106 CrossRef CAS PubMed.
- M. A. Rahman, M. Bam, E. Luat, M. S. Jui, M. S. Ganewatta, T. Shokfai, M. Nagarkatti, A. W. Decho and C. Tang, Nat. Commun., 2018, 9, 5231 Search PubMed.
- B. C. Allison, B. M. Applegate and J. P. Youngblood, Biomacromolecules, 2007, 8, 2995–2999 CrossRef CAS PubMed.
- R. Namivandi-Zangeneh, R. J. Kwan, T.-K. Nguyen, J. Yeow, F. L. Byrne, S. H. Oehlers, E. H. Wong and C. Boyer, Polym. Chem., 2018, 9, 1735–1744 RSC.
- C. Ergene and E. F. Palermo, J. Mater. Chem. B, 2018, 6, 7217–7229 RSC.
- S. Schaefer, D. Melodia, C. Pracey, N. Corrigan, M. D. Lenardon and C. Boyer, Biomacromolecules, 2024, 25, 871–889 CrossRef CAS PubMed.
- P. Pham, S. Oliver, D. T. Nguyen and C. Boyer, Macromol. Rapid Commun., 2022, 43, 2200377 CrossRef CAS.
- E. T. Nadres, H. Takahashi and K. Kuroda, J. Polym. Sci., Part A: Polym. Chem., 2016, 55, 304–312 CrossRef.
- T. D. Michl, K. E. S. Locock, N. E. Stevens, J. D. Hayball, K. Vasilev, A. Postma, Y. Qu, A. Traven, M. Haeussler, L. Meagher and H. J. Griesser, Polym. Chem., 2014, 5, 5813–5822 RSC.
- W. Chin, C. Yang, V. W. L. Ng, Y. Huang, J. Cheng, Y. W. Tong, D. J. Coady, W. Fan, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2013, 46, 8797–8807 CrossRef CAS.
- K. Lienkamp, K. N. Kumar, A. Som, K. Nüsslein and G. N. Tew, Chem. – Eur. J., 2009, 15, 11710–11714 CrossRef CAS PubMed.
- J. Pachla, R. J. Kopiasz, G. Marek, W. Tomaszewski, A. Głogowska, K. Drężek, S. Kowalczyk, R. Podgórski, B. Butruk-Raszeja, T. Ciach, J. Mierzejewska, A. Plichta, E. Augustynowicz-Kopeć and D. Jańczewski, Biomacromolecules, 2023, 24, 2237–2249 CrossRef CAS PubMed.
- Y. Qiao, C. Yang, D. J. Coady, Z. Ong, J. L. Hedrick and Y. Yang, Biomaterials, 2012, 33, 1146–1153 CrossRef CAS PubMed.
- S. Oliver, L. Zhao, A. J. Gormley, R. Chapman and C. Boyer, Macromolecules, 2019, 52, 3–23 CrossRef CAS.
- D. A. Tomalia, Prog. Polym. Sci., 2005, 30, 294–324 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- B. D. Fairbanks, P. A. Gunatillake and L. Meagher, Adv. Drug Delivery Rev., 2015, 91, 141–152 CrossRef CAS PubMed.
- H. Takahashi, E. T. Nadres and K. Kuroda, Biomacromolecules, 2017, 18, 257–265 CrossRef CAS PubMed.
- S. Zhao, W. Huang, C. Wang, Y. Wang, Y. Zhang, Z. Ye, J. Zhang, L. Deng and A. Dong, Biomacromolecules, 2020, 21, 5269–5281 CrossRef CAS PubMed.
- Y. Wang, J. Xu, Y. Zhang, H. Yan and K. Liu, Macromol. Biosci., 2011, 11, 1499–1504 CrossRef CAS PubMed.
- A. Kuroki, P. Sangwan, Y. Qu, R. Peltier, C. Sanchez-Cano, J. Moat, C. G. Dowson, E. G. Williams, K. E. Locock and M. Hartlieb, ACS Appl. Mater. Interfaces, 2017, 9, 40117–40126 CrossRef CAS PubMed.
- P. R. Judzewitsch, T.-K. Nguyen, S. Shanmugam, E. H. H. Wong and C. Boyer, Angew. Chem., Int. Ed., 2018, 57, 4559–4564 CrossRef CAS PubMed.
- H. Takahashi, I. Sovadinova, K. Yasuhara, S. Vemparala, G. A. Caputo and K. Kuroda, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2023, 15, e1866 CAS.
- K. Zhang, Y. Du, Z. Si, Y. Liu, M. E. Turvey, C. Raju, D. Keogh, L. Ruan, S. L. Jothy and S. Reghu, Nat. Commun., 2019, 10, 4792 CrossRef PubMed.
- C. Zhang, Y. Zhu, C. Zhou, W. Yuan and J. Du, Polym. Chem., 2013, 4, 255–259 RSC.
- W. Yuan, J. Wei, H. Lu, L. Fan and J. Du, Chem. Commun., 2012, 48, 6857–6859 RSC.
- C. Chang, C. Chang, Y. Yang, H. Chen, S. Yang, W. Yao and C. Chao, Polymers, 2022, 14, 250 CrossRef CAS PubMed.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- Y. Oda, S. Kanaoka, T. Sato, S. Aoshima and K. J. B. Kuroda, Biomacromolecules, 2011, 12, 3581–3591 CrossRef CAS PubMed.
- Y. Oda, K. Yasuhara, S. Kanaoka, T. Sato, S. Aoshima and K. Kuroda, Polymers, 2018, 10, 93 CrossRef PubMed.
- M. Casanova, H. Olleik, S. Hdiouech, C. Roblin, J.-F. Cavalier, V. Point, K. Jeannot, B. Caron, J. Perrier, S. Charriau, M. Lafond, Y. Guillaneuf, S. Canaan, C. Lefay and M. Maresca, Antibiotics, 2023, 12, 120 CrossRef CAS PubMed.
- Y. Tominaga, M. Mizuse, A. Hashidzume, Y. Morishima and T. Sato, J. Phys. Chem. B, 2010, 114, 11403–11408 CrossRef CAS PubMed.
- C. Chen and T. Weil, Nanoscale Horiz., 2022, 7, 1121–1135 RSC.
- J. Koehbach and D. J. Craik, Trends Pharmacol. Sci., 2019, 40, 517–528 CrossRef CAS PubMed.
- J. Xu, L. Pu, J. Ma, S. K. Kumar and H. Duan, Polym. Chem., 2020, 11, 6632–6639 RSC.
- M. Aquib, W. Yang, L. Yu, V. K. Kannaujiya, Y. Zhang, P. Li, A. Whittaker, C. Fu and C. Boyer, Chem. Sci., 2024, 15, 19057–19069 RSC.
- R. Liénard, J. De Winter and O. Coulembier, J. Polym. Sci., 2020, 58, 1481–1502 CrossRef.
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 CrossRef CAS PubMed.
- M. R. E. Santos, P. V. Mendonça, M. C. Almeida, R. Branco, A. C. Serra, P. V. Morais and J. F. J. Coelho, Biomacromolecules, 2019, 20, 1146–1156 CrossRef CAS PubMed.
- M. Aquib, S. Schaefer, H. Gmedhin, N. Corrigan, V. A. Bobrin and C. Boyer, Eur. Polym. J., 2024, 205, 112698 CrossRef CAS.
- S. Laroque, K. E. S. Locock and S. Perrier, Biomacromolecules, 2024, 26, 190–200 CrossRef PubMed.
- K. A. Gibney, I. Sovadinova, A. I. Lopez, M. Urban, Z. Ridgway, G. A. Caputo and K. Kuroda, Macromol. Biosci., 2012, 12, 1279–1289 CrossRef CAS PubMed.
- H. Liu, X. Zhang, Z. Zhao, F. Yang, R. Xue, L. Yin, Z. Song, J. Cheng, S. Luan and H. Tang, Biomater. Sci., 2021, 9, 2721–2731 RSC.
- N. Hadjesfandiari, K. Yu, Y. Mei and J. N. Kizhakkedathu, J. Mater. Chem. B, 2014, 2, 4968–4978 RSC.
- T. Chen, H. Yang, X. Wu, D. Yu, A. Ma, X. He, K. Sun and J. Wang, Langmuir, 2019, 35, 3031–3037 CrossRef CAS PubMed.
- H. Luo, X. Q. Yin, P. F. Tan, Z. P. Gu, Z. M. Liu and L. Tan, J. Mater. Chem. B, 2021, 9, 2802–2815 RSC.
- M. R. E. Santos, A. C. Fonseca, P. V. Mendonca, R. Branco, A. C. Serra, P. V. Morais and J. F. J. Coelho, Materials, 2016, 9, 599 CrossRef PubMed.
- M. M. Konai, B. Bhattacharjee, S. Ghosh and J. Haldar, Biomacromolecules, 2018, 19, 1888–1917 CrossRef CAS PubMed.
- M. Cloutier, D. Mantovani and F. Rosei, Trends Biotechnol., 2015, 33, 637–652 CrossRef CAS PubMed.
- S. Arora, V. Yadav, P. Kumar, R. Gupta and D. Kumar, Int. J. Pharm. Sci. Rev. Res., 2013, 23, 279–290 Search PubMed.
- J. Hoque, S. Ghosh, K. Paramanandham and J. Haldar, ACS Appl. Mater. Interfaces, 2019, 11, 39150–39162 CrossRef CAS PubMed.
- Z. Ma, Y. Liu, K. Feng, J. Wei, J. Liu, Y. Wu, X. Pei, B. Yu, M. Cai and F. Zhou, ACS Appl. Mater. Interfaces, 2022, 14, 18901–18909 CrossRef CAS PubMed.
- S. Bai, X. Li, Y. Zhao, L. Ren and X. Yuan, ACS Appl. Mater. Interfaces, 2020, 12, 12305–12316 CrossRef CAS PubMed.
- C. Chen, Z. Li, X. Li, C. Kuang, X. Liu, Z. Song, H. Liu and Y. Shan, Composites, Part B, 2022, 232, 109623 CrossRef CAS.
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef.
- H. Chang, C. Li, R. Huang, R. Su, W. Qi and Z. He, J. Mater. Chem. B, 2019, 7, 2899–2910 RSC.
- H. Qu, Q. Yao, T. Chen, H. Wu, Y. Liu, C. Wang and A. Dong, Adv. Colloid Interface Sci., 2024, 325, 103099 CrossRef CAS PubMed.
- C. M. González-Henríquez, M. A. Sarabia-Vallejos and J. Rodriguez-Hernandez, Materials, 2017, 10, 232 CrossRef PubMed.
- M. Malmsten, Soft Matter, 2011, 7, 8725–8736 RSC.
- X. Qin, K. Labuda, J. Chen, V. Hruschka, A. Khadem, R. Liska, H. Redl and P. Slezak, Adv. Funct. Mater., 2015, 25, 6606–6617 CrossRef CAS.
- X. Zhao, H. Wu, B. Guo, R. Dong, Y. Qiu and P. X. Ma, Biomaterials, 2017, 122, 34–47 CrossRef CAS PubMed.
- Y. Li, K. Fukushima, D. J. Coady, A. C. Engler, S. Liu, Y. Huang, J. S. Cho, Y. Guo, L. S. Miller and J. P. Tan, Angew. Chem., Int. Ed., 2013, 52, 674–678 CrossRef CAS PubMed.
- N. Zhao and W. Yuan, Composites, Part B, 2022, 230, 109525 CrossRef CAS.
- Y. Hong, Y. Xi, J. Zhang, D. Wang, H. Zhang, N. Yan, S. He and J. Du, J. Mater. Chem. B, 2018, 6, 6311–6321 RSC.
- Y. Wang, J. Zhao, Z. Dong, C. Wang, H. Meng, Y. Li, H. Jin and C. Wang, Mater. Today Chem., 2021, 21, 100537 Search PubMed.
- S. Brahmachari, S. Debnath, S. Dutta and P. K. Das, Beilstein J. Org. Chem., 2010, 6, 859–868 CrossRef CAS PubMed.
- S. Li, S. Dong, W. Xu, S. Tu, L. Yan, C. Zhao, J. Ding and X. Chen, Adv. Sci., 2018, 5, 1700527 CrossRef PubMed.
- S. Vidovic, J. Stojkovska, M. Stevanovic, B. Balanc, M. Vukasinovic-Sekulic, A. Marinkovic and B. Obradovic, R. Soc. Open Sci., 2022, 9, 211517 CrossRef CAS PubMed.
- K. Neibert, V. Gopishetty, A. Grigoryev, I. Tokarev, N. Al-Hajaj, J. Vorstenbosch, A. Philip, S. Minko and D. Maysinger, Adv. Healthcare Mater., 2012, 1, 621–630 CrossRef CAS PubMed.
- C. T. Gustafson, F. Boakye-Agyeman, C. L. Brinkman, J. M. Reid, R. Patel, Z. Bajzer, M. Dadsetan and M. J. Yaszemski, PLoS One, 2016, 11, e0146401 CrossRef PubMed.
- S. C. Bondy, Curr. Opin. Toxicol., 2021, 26, 8–13 CrossRef CAS.
- K. Gwon, I. Han, S. Lee, Y. Kim and D. N. Lee, ACS Appl. Mater. Interfaces, 2020, 12, 20234–20242 Search PubMed.
- D. Han, Y. Li, X. Liu, B. Li, Y. Han, Y. Zheng, K. W. K. Yeung, C. Li, Z. Cui, Y. Liang, Z. Li, S. Zhu, X. Wang and S. Wu, Chem. Eng. J., 2020, 396, 125194 Search PubMed.
- B. Zeng, Z. Cai, J. Lalevée, Q. Yang, H. Lai, P. Xiao, J. Liu and F. Xing, Toxicol. In Vitro, 2021, 72, 105103 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- M. A. Rahman, M. S. Jui, M. Bam, Y. Cha, E. Luat, A. Alabresm, M. Nagarkatti, A. W. Decho and C. Tang, ACS Appl. Mater. Interfaces, 2020, 12, 21221–21230 Search PubMed.
- Z. Shi, K. G. Neoh, E. T. Kang and W. Wang, Biomaterials, 2006, 27, 2440–2449 CrossRef CAS PubMed.
- H. Zhu, Q. Geng, W. Chen, Y. Zhu, J. Chen and J. Du, J. Mater. Chem. B, 2013, 1, 5496–5504 Search PubMed.
- K. Lienkamp, K. N. Kumar, A. Som, K. Nüsslein and G. N. Tew, Chemistry, 2009, 15, 11710–11714 Search PubMed.
- L. D. Blackman, Z. Y. Oo, Y. Qu, P. A. Gunatillake, P. Cass and K. E. S. Locock, ACS Appl. Mater. Interfaces, 2020, 12, 11353–11362 CrossRef CAS PubMed.
- N. J. Bassous and T. J. Webster, Small, 2019, 15, 1804247 Search PubMed.
- Y. Yang, L. Chen, M. Sun, C. Wang, Z. Fan and J. Du, Chin. J. Polym. Sci., 2021, 39, 1412–1420 CrossRef CAS.
- Y. Zhu, B. Yang, S. Chen and J. Du, Prog. Polym. Sci., 2017, 64, 1–22 CrossRef CAS.
- H. Sun, Y. Wang and J. Song, Polymers, 2021, 13, 2903 Search PubMed.
- Y. Chen, Y. Huang and Q. Jin, Macromol. Chem. Phys., 2022, 223, 2100440 Search PubMed.
- T. Wang, Y. Li, E. J. Cornel, C. Li and J. Du, ACS Nano, 2021, 15, 9027–9038 Search PubMed.
- Y. Li, G. Liu, X. Wang, J. Hu and S. Liu, Angew. Chem., Int. Ed., 2016, 55, 1760–1764 CrossRef CAS PubMed.
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao and E. Wool, Lancet, 2022, 399, 629–655 Search PubMed.
- S. Alfei and A. M. Schito, Polymers, 2020, 12, 1195 CrossRef CAS PubMed.
- H. Etayash and R. E. W. Hancock, Pharmaceutics, 2021, 13, 1820 CrossRef CAS PubMed.
- N. F. Kamaruzzaman, L. P. Tan, R. H. Hamdan, S. S. Choong, W. K. Wong, A. J. Gibson, A. Chivu and M. d F. Pina, Int. J. Mol. Sci., 2019, 20, 2747 Search PubMed.
- K. Malhotra and Y. Singh, Racing for the Surface: Pathogenesis of Implant Infection and Advanced Antimicrobial Strategies, 2020, pp. 543–581 Search PubMed.
- R. Namivandi-Zangeneh, E. H. H. Wong and C. Boyer, ACS Infect. Dis., 2021, 7, 215–253 Search PubMed.
- C. H. Chen and T. K. Lu, Antibiotics, 2020, 9, 24 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |