
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi–NiCr nanoparticles incorporated carbon nanofibers as robust electrocatalysts for efficient glycerol oxidation†
Nasser A. M.
Barakat
 *a,
Shimaa
Hamada
a,
Ibrahim
Mustafa
b and
Hesham
Alhumade
bc
*a,
Shimaa
Hamada
a,
Ibrahim
Mustafa
b and
Hesham
Alhumade
bc
aChemical Engineering Department, Minia University, El-Minia, 61516, Egypt. E-mail: nasbarakat@mu.edu.eg; Fax: +20862364420; Tel: +20862348005
bDepartment of Chemical and Materials Engineering, Faculty of Engineering, King Abdulaziz University, Jeddah 21589, Saudi Arabia
cCenter of Excellence in Desalination Technology, King Abdulaziz University, Jeddah 21589, Saudi Arabia
First published on 5th June 2025
Abstract
The development of cost-effective and durable electrocatalysts for glycerol oxidation is essential for advancing energy conversion technologies and chemical production. In this study, we synthesized Ni–NiCr alloy nanoparticles incorporated into a graphitic carbon nanofiber (CNF) matrix using an electrospinning technique followed by thermal treatment. The structural and electrochemical properties of the resulting Ni–NiCr-CNFs were systematically investigated. X-ray diffraction (XRD) confirmed the formation of a Ni–NiCr alloy phase, while scanning and transmission electron microscopy (SEM/TEM) revealed a uniform nanofiber morphology with embedded crystalline nanoparticles. Cyclic voltammetry (CV) and chronoamperometry demonstrated the excellent electrocatalytic activity and stability of the Ni–NiCr-CNFs toward glycerol oxidation in alkaline media. The optimized electrocatalyst, prepared with 15 wt% chromium acetate precursor, exhibited a maximum current density of 102.7 mA cm−2 in 0.5 M glycerol at 1.0 M KOH, surpassing many reported precious metal-based catalysts. The temperature dependence study indicated negligible impact on electrocatalytic activity, underscoring the robustness of the catalyst under varying conditions. Chronoamperometry further confirmed the stability and durability of the catalyst, with consistent current densities across different applied voltages. These findings highlight the synergistic effects of the Ni–NiCr alloy and the graphitic CNF matrix in enhancing catalytic performance, making the Ni–NiCr-CNFs a promising candidate for sustainable and efficient glycerol oxidation.
1. Introduction
Glycerol, a renewable and non-toxic byproduct of biodiesel production, has gained significant attention in recent years as a valuable feedstock for sustainable energy conversion and chemical production. The biodiesel industry, which has rapidly expanded to meet the demand for renewable energy, generates approximately 10 wt% glycerol as a byproduct during the transesterification of triglycerides. This oversupply has led to a substantial drop in glycerol's market value, creating an urgent need to valorize this resource into high-value chemicals and energy sources. Consequently, the development of efficient strategies for glycerol utilization is not only economically advantageous but also environmentally significant.1–3Electrochemical glycerol oxidation, commonly referred to as the glycerol oxidation reaction (GOR), represents a promising and sustainable approach to glycerol valorization. GOR offers dual benefits: it converts glycerol into high-value chemicals, such as glyceric acid, tartronic acid, dihydroxyacetone, mesoxalic acid, and formic acid, while simultaneously generating electrical energy in direct glycerol fuel cells (DGFCs). These products have broad industrial applications in pharmaceuticals, cosmetics, food additives, and polymer precursors. Furthermore, GOR can be carried out under mild reaction conditions, utilizing renewable electricity as the energy input, making it a highly attractive process for green energy technologies.3–5
The efficiency of glycerol oxidation is critically dependent on the electrocatalyst employed. Precious metals such as platinum (Pt), palladium (Pd), and gold (Au) have been extensively studied for GOR due to their high activity and selectivity. However, these catalysts suffer from several drawbacks, including high cost, limited availability, and susceptibility to poisoning by intermediates such as carbon monoxide. To address these challenges, research efforts have focused on developing non-precious metal-based electrocatalysts that are cost-effective, abundant, and efficient.6–9
Nickel (Ni)-based electrocatalysts have emerged as promising candidates for GOR in alkaline media. Nickel's ability to form Ni(OH)2/NiOOH redox species is a key factor in its catalytic activity, enabling the oxidation of glycerol and other alcohols.10,11 The incorporation of electrocatalysts into carbon-based supports has further advanced the field of GOR. Carbon nanofibers (CNFs) are particularly attractive as catalyst supports due to their high surface area, excellent electrical conductivity, and structural stability. The integration of metal nanoparticles into CNFs offers several advantages. CNFs enable uniform distribution of active metal sites, reducing agglomeration and increasing the availability of catalytic surfaces.12,13 The high conductivity of graphitic CNFs facilitates efficient electron transport during electrochemical reactions.14 Moreover, CNFs provide robust support, ensuring long-term stability under operating conditions.
Additionally, alloying nickel with other metals, such as cobalt (Co), copper (Cu), and iron (Fe), has been a common approach to enhance catalytic activity and stability.15–17 These alloying elements are known to modify the electronic structure of nickel, improve the adsorption of glycerol and hydroxyl ions, and increase resistance to poisoning by reaction intermediates. However, to the best of our knowledge, the incorporation of chromium (Cr) as a co-catalyst with nickel for glycerol electrooxidation has not been previously reported in the literature.
Chromium is widely recognized for its ability to enhance the electronic properties of metallic alloys and improve corrosion resistance in harsh environments. In electrochemical systems, Cr is often used as a stabilizing agent or a dopant to improve the structural and catalytic performance of various materials.18 However, its role as a co-catalyst for glycerol oxidation in combination with nickel represents a novel approach. In this study, the synergistic effects of Ni and Cr in the Ni–NiCr alloy are demonstrated to provide several advantages: (1) enhanced electronic interactions that optimize the adsorption and activation of glycerol molecules and hydroxyl ions, (2) improved durability and resistance to deactivation, and (3) stabilization of the active sites during prolonged electrochemical operation. These unique properties distinguish the Ni–Cr alloy from conventional Ni-based or bimetallic catalysts.
The novelty of this work lies in introducing Cr as a co-catalyst with Ni to form Ni–NiCr alloy nanoparticles incorporated into a graphitic carbon nanofiber matrix. This combination leverages the electronic and structural advantages of Ni–Cr alloys while benefiting from the high surface area, excellent electrical conductivity, and robust mechanical stability of the CNF support. The results presented in this study indicate that the Ni–NiCr-CNFs catalyst outperforms many reported electrocatalysts, including precious metal-based ones, in terms of activity, stability, and cost-effectiveness for glycerol oxidation. To the best of our knowledge, this is the first report in the literature that investigates and highlights the potential of Cr as a co-catalyst with Ni for glycerol electrooxidation, paving the way for the development of a new class of non-precious metal-based electrocatalysts for sustainable energy and chemical applications. Electrochemical testing demonstrated the exceptional performance of the optimized electrocatalyst, particularly the formulation containing 15 wt% chromium acetate precursor. Furthermore, chronoamperometric analysis confirmed the excellent stability of the catalyst, with negligible performance degradation over extended operation. Temperature-dependent studies showed that the catalyst maintained consistent activity across a wide thermal range, highlighting its robustness under diverse operational conditions. This work underscores the potential of Ni–NiCr-CNFs as a cost-effective and high-performance electrocatalyst for glycerol oxidation, paving the way for the development of sustainable energy systems and value-added chemical production.
2. Experimental section
2.1. Chemicals and reagents
All chemicals used in this study were of analytical grade and used without further purification. Nickel(II) acetate tetrahydrate (Ni(CH3COO)2·4H2O, ≥99% purity), chromium(II) acetate hydrate (Cr(CH3COO)3·xH2O, ≥99% purity), and poly(vinyl alcohol) (PVA, molecular weight 89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–98000, 99% hydrolyzed) were purchased from Sigma-Aldrich (USA). These chemicals were used as precursors for the synthesis of the Ni–NiCr alloy nanoparticles-incorporated carbon nanofibers (Ni–NiCr-CNFs).
000–98000, 99% hydrolyzed) were purchased from Sigma-Aldrich (USA). These chemicals were used as precursors for the synthesis of the Ni–NiCr alloy nanoparticles-incorporated carbon nanofibers (Ni–NiCr-CNFs).
Potassium hydroxide (KOH, ≥85% purity) was obtained from Merck (Germany) and used as the supporting electrolyte in all electrochemical experiments. Glycerol (C3H8O3, ≥99% purity) was supplied by Alfa Aesar (USA) and used as the reactant in glycerol oxidation studies. Deionized (DI) water (resistivity >18.2 MΩ cm) was used in all solution preparations and experimental procedures. All solvents and additional reagents used were of analytical grade and were obtained from local suppliers.
2.2. Catalyst preparation
The Ni–NiCr alloy nanoparticles-incorporated carbon nanofibers (Ni–NiCr-CNFs) were synthesized via an electrospinning process followed by thermal treatment. To prepare the electrospinning solutions, 1 g of nickel acetate was dissolved in 5 mL of deionized water (DI), and the desired amounts of chromium acetate were added to achieve chromium contents of 5, 10, 15, and 35 wt% relative to nickel acetate. The salt solution was then mixed with 15 mL of a 10 wt% aqueous PVA solution. The mixture was stirred at 50 °C for 5 hours to ensure complete dissolution and homogeneity. The resulting solution was subjected to electrospinning under the following parameters: an applied voltage of 20 kV, a tip-to-collector distance of 15 cm, and a feeding rate of 0.07 mL h−1 using syringe pumps. The electrospun nanofiber mats were collected on aluminum foil and vacuously dried at 60 °C overnight.The dried nanofiber mats were calcined under vacuum at 850 °C with a heating rate of 2 °C min−1 and a holding time of 5 hours. This calcination process facilitated the removal of the polymer template and the formation of the Ni–NiCr alloy phase within the carbon nanofiber matrix. The resulting material was cooled to room temperature under vacuum conditions, yielding the final Ni–NiCr-CNFs catalyst.
2.3. Characterizations
The structural and morphological properties of the synthesized Ni–NiCr-CNFs were analyzed using various advanced techniques. The surface morphology and fiber structure of the electrospun nanofibers were examined using scanning electron microscope (SEM, JEOL JSM-7610F). High-resolution imaging and structural analysis were conducted using a FEI Tecnai G2 F20 TEM, operated at an accelerating voltage of 200 kV. TEM analysis was used to determine the particle size of the Ni–NiCr alloy nanoparticles and their dispersion within the nanofibers. The inter-planar spacings and crystallinity of the alloy were further examined using high-resolution TEM (HR-TEM). The crystalline structure and phase composition of the Ni–NiCr-CNFs were characterized using a Rigaku diffractometer equipped with Cu Kα radiation (λ = 1.5406 Å). XRD patterns were recorded over a 2θ range of 10°–80° at a scan rate of 0.02° s−1. The diffraction peaks corresponding to the Ni–NiCr alloy and graphitic carbon phases were identified and indexed using standard ICCD reference patterns.The electrochemical performance of the Ni–NiCr-CNFs for glycerol oxidation was evaluated using cyclic voltammetry (CV), chronoamperometry (CA), and other electrochemical techniques. All experiments were performed at room temperature in a three-electrode electrochemical cell using a CHI660E electrochemical workstation (CH Instruments, USA). The working electrode was prepared by depositing 60 μL of an ink solution onto a glassy carbon electrode (GCE, diameter: 3 mm) and allowing it to dry under ambient conditions. The ink was composed of 2 mg of Ni–NiCr-CNFs, 20 μL of Nafion solution (5 wt% in isopropanol), and 400 μL of isopropanol. The mixture was ultrasonicated for 30 minutes to ensure uniform dispersion of the nanofibers before deposition.
A graphite rod was used as the counter electrode, while an Ag/AgCl (3 M KCl) electrode served as the reference electrode. The electrolyte for all measurements was 1.0 M KOH solution containing different concentration of glycerol, depending on the experimental requirements. CV experiments were conducted over a potential range of 0.2–1.0 V (vs. Ag/AgCl) at scan rates ranging from 5 to 100 mV s−1. These measurements provided insights into the catalytic activity, redox behavior, and charge transfer characteristics of the electrocatalyst. Long-term stability and durability of the electrocatalyst were evaluated using chronoamperometry at various fixed potentials. The current densities were monitored as a function of time to assess the catalyst's resistance to deactivation under operational conditions.
3. Results and discussion
3.1. Electrocatalyst characterizations
 | ||
| Fig. 1 XRD pattern for the prepared Ni–NiCr NPs-incorporated CNFs from electrospun solution containing 15 wt% CrAc and calcined at 850 °C. | ||
• The Cr (200) peak, which is typically located at 2θ = 64.55° (ICCD #85-1336), shifted to approximately 64.64°.
• The Ni (200) peak, which is usually observed at 2θ = 51.85°, shifted to around 52.2°.
These shifts in the diffraction peaks suggest the formation of a solid solution or an alloy phase.19 The observed shifts can be attributed to the incorporation of Cr atoms into the Ni lattice, which induces lattice distortions and changes in the interatomic spacing. This is consistent with previous studies on Ni–Cr systems, where the formation of alloys or solid solutions leads to similar peak shifts.20
As it was reported, in inert atmosphere, NiAc decomposes completely to zero-valent nickel under an inert atmosphere, rather than forming nickel oxides. This decomposition releases reducing gases such as carbon monoxide and hydrogen, which assist in the formation of pure nickel. The reactions involved in this process include.21,22
| Ni(CH3COO)2·4H2O → 0.86Ni(CH3COO)2·0.14Ni(OH)2 + 0.28CH3COOH + 3.72H2O | (1) |
| 3(0.86 Ni(CH3COO)2·0.14Ni(OH)2) + 0.28H2O → 3NiCO3 + 2.44CH3COCH3 + 1.12H2 | (2) |
| NiCO3 → NiO + CO2 | (3) |
| NiO + CO/H2 → Ni + CO2/H2O | (4) |
Analogy to NiAc, CrAc behave similar behavior to form zero-valent metal. Then, these two metals combined to form the NiCr alloy as it was concluded by XRD results. The calcination process at high temperature under an inert atmosphere provides the necessary thermal energy for diffusion and intermixing of Ni and Cr atoms. During this process: Cr atoms diffuse into the Ni lattice, forming a solid solution. The incorporation of Cr atoms induces lattice distortions, resulting in the observed peak shifts in the XRD pattern. Extended calcination stabilizes the NiCr alloy phase, as evidenced by the uniform shifts in the XRD peaks. The behavior observed in this study aligns with findings from the structural evolution of Ni-20Cr alloys during ball milling and heat treatments.19 Studies have shown that the incorporation of Cr into Ni leads to the broadening and shifting of XRD peaks, indicative of alloy or solid solution formation. These phenomena are attributed to lattice strain and the high defect density introduced during alloy formation.
Poly(vinyl alcohol) (PVA) has a relatively high carbon content compared to other vinyl polymers, which makes it a suitable precursor for crafting carbon nanofibers. However, challenges arise due to its low melting point and its decomposition into volatile, low-molecular-weight compounds during the heat treatment process.23,24 To overcome these limitations, researchers have employed two primary strategies: pre-treatment before carbonization and the use of catalysts to enhance the graphitization process. These methods involve structural modifications to the PVA chain, which result in compounds with higher melting points, thereby facilitating efficient graphitization.25,26
During the calcination process, techniques such as dehydration and dehydrogenation are used to extract aromatic carbon from PVA. This enhances its carbon yield and contributes to the stability of the nanofiber structure. A schematic illustration in Fig. S1 in the ESI† outlines the optimal breakdown process of PVA to maximize yield.27,28
Beside Ni and NiCr representative peaks, there is a broad peak at two theta value around 26°.29 This peak can be assigned to graphite counterpart in the prepared materials. Therefore, it can claimed that the formed zero-valent metals served as catalyst for graphitization of the PVA polymer which was also concluded in our previous study.30 Accordingly, it can be concluded that calcination of NiAc/CrAc/PVA electrospun nanofibers results in formation of Ni–NiCr-graphite composite material. It is noteworthy mentioning that the other prepared samples revealed similar XRD patterns.
 | ||
| Fig. 2 Low; (A) and high; (B) SEM magnifications images for the prepared Ni-NiCr NPs-incorporated CNFs from electrospun solution containing 15 wt% CrAc and calcined at 850 °C. | ||
The preservation of the nanofibrous structure during calcination can be attributed to the polycondensation tendency of the acetate precursors (nickel acetate and chromium acetate). Typically, acetate precursors exhibits a polycondensation tendency, which helps maintain the nanofibrous morphology during calcination.31,32 This reaction also promotes the formation of a cohesive gel with the polymer during the electrospinning process, ensuring uniform nanofiber production. This polycondensation reaction can be expressed as follows.27
 | (5) |
During heat treatment, these precursors undergo a series of thermal decomposition reactions, leading to the gradual release of volatile by-products such as acetic acid, acetone, and water. This controlled release prevents the collapse or agglomeration of the nanofibers, allowing the structure to remain intact.
Nickel acetate, in particular, facilitates the formation of a cohesive matrix by forming a gel-like intermediate during decomposition. This gel-like phase stabilizes the fiber morphology, ensuring that the structure remains intact even at elevated temperatures. Studies have shown that the decomposition of acetate-based precursors proceeds through intermediate carbonate or hydroxide phases, which also contribute to maintaining the structural integrity of nanofibers.33,34
The polycondensation reactions of acetate precursors result in the formation of interconnected networks that prevent fiber breakage or deformation. This process is particularly effective in stabilizing the nanofibrous structure during the calcination phase. The gradual thermal decomposition of acetate groups avoids abrupt material loss, minimizing stress and deformation within the fibers. This controlled process ensures that the nanofibers maintain their smooth and continuous morphology.35 The presence of nickel and chromium also aids in stabilizing the fiber structure. These metals act as catalysts for cross-linking reactions during the decomposition of the organic matrix, further supporting the structural integrity of the nanofibers.36
The TEM image (Fig. 3A) reveals the internal structure of the prepared Ni/NiCr NPs-incorporated carbon nanofibers (CNFs). The observed structure comprises amorphous nanofiber matrix. The nanofiber matrix is largely amorphous, which can be attributed to graphitic carbon. This conclusion aligns with the XRD results (broad peak at ∼2θ = 26°), indicative of disordered or turbostratic graphite.
 | ||
| Fig. 3 Normal TEM image; (A) and HR-TEM image; (B) for the prepared NiCr NPs-incorporated CNFs from electrospun solution containing 15 wt% CrAc and calcined at 850 °C. The inset displays SAED pattern for the crystalline nanoparticles. | ||
Embedded crystalline Ni–NiCr nanoparticles distributing throughout the matrix are highly crystalline metallic nanoparticles. These nanoparticles exhibit a wide size distribution, ranging from a few nanometers to approximately 50 nm. The estimated average diameter of the nanofibers (∼150 nm) matches the measurements from SEM analysis (Fig. 2), confirming the consistency of the electrospinning and calcination processes.
The HR-TEM image (Fig. 3B) provides insight into the crystal structure of the embedded nanoparticles. The interplanar spacing measured at 0.3nm is consistent with the crystal lattice of the NiCr alloy. For nickel (Ni), the standard interplanar spacing (dd) for the (111) plane is approximately 0.203nm, and for the (200) plane, 0.176nm.37 For chromium (Cr), the standard interplanar spacing for the (110) plane is 0.203nm. In a NiCr alloy, the lattice parameter shifts slightly due to atomic intermixing. The observed spacing of 0.3nm likely corresponds to a specific alloy plane, further confirming the successful formation of a NiCr alloy.38 Studies have demonstrated that interplanar spacings in Ni-based alloys vary due to the incorporation of other elements like chromium, creating lattice distortions that are detectable in HR-TEM.39
The crystallinity observed in HR-TEM suggests effective thermal treatment and alloying, producing well-defined NiCr phases even at the nanoscale. The TEM and HR-TEM results are consistent with observations from SEM and XRD. SEM analysis confirmed the uniform nanofiber diameter and morphology. XRD analysis revealed characteristic peaks of the NiCr alloy, corresponding to the crystalline nature of the nanoparticles embedded in the CNF matrix. Amorphous carbon matrices embedding metal nanoparticles have been extensively studied, with turbostratic graphite forming due to the thermal decomposition of carbon-rich precursors like poly(vinyl alcohol).13,40
The inset of Fig. 3A displays the SAED pattern for the crystalline nanoparticles embedded in the CNFs. The ring pattern observed in the SAED image is characteristic of polycrystalline materials and provides crucial insights into the structural properties of the NiCr alloy nanoparticles. The concentric rings in the SAED pattern indicate the presence of randomly oriented crystallites within the sample. This confirms the polycrystalline nature of the embedded NiCr nanoparticles. The observed rings correspond to specific crystallographic planes of the NiCr alloy. For example, the innermost ring likely represents the (111) plane, while the subsequent rings correspond to higher-order planes such as (200) and (220). This interpretation is consistent with the XRD findings. The polycrystalline nature of the nanoparticles can be attributed to the alloying process during calcination, where the atomic diffusion and intermixing of Ni and Cr create crystalline domains of varying orientations.
3.2. Electrochemical properties
Nickel-based electrocatalysts exhibit remarkable electrocatalytic activity, primarily attributed to the formation of a Ni(OOH) active layer on their surface. This transformation occurs during cyclic voltammetry (CV) sweeps in an alkaline medium and involves several distinct stages: Initial NiO Layer Formation: Due to the inherent activity of nickel, a thin layer of nickel oxide (NiO) naturally forms on the surface upon exposure to air or moisture. This passive oxide layer serves as the precursor for subsequent electrochemical transformations in an alkaline environment.
In alkaline media, the naturally formed NiO layer undergoes hydration, converting to Ni(OH)2. Despite this transformation, the corresponding redox peaks are often undetectable in CV profiles. This can be attributed to the rapid kinetics of the NiO to Ni(OH)2 transition, coupled with the overlapping of signals from other electrochemical processes, which collectively obscure the detection of distinct peaks for this step.43,44
Upon further sweeping, Ni(OH)2 undergoes oxidation to form Ni(OOH). This reaction is marked by the emergence of distinct redox peaks in the CV profile. The intensity of these peaks typically increases with repeated CV cycles as more Ni(OOH) species are generated on the electrode surface, enhancing the active catalytic area activity reactions.45–47
Fig. 4A illustrates the CV analysis for the synthesized NiCr NPs-incorporated CNFs in 1.0 M KOH for 50 successive cycles at 50 mV s−1. The redox peaks observed in the CV curves correspond to the Ni(OH)2 to Ni(OOH) are consistent with established findings for nickel-based catalysts in alkaline environments. Contrary to the typical behavior of increasing peak intensity with successive cycles, the CV analysis of the Ni–NiCr NPs-loaded CNFs shows a decline in the intensity of the Ni(OH)2/Ni(OOH) redox peaks with an increasing number of cycles (Fig. 4B). This paradoxical behavior can be attributed to the formation of a self-limiting oxide layer.
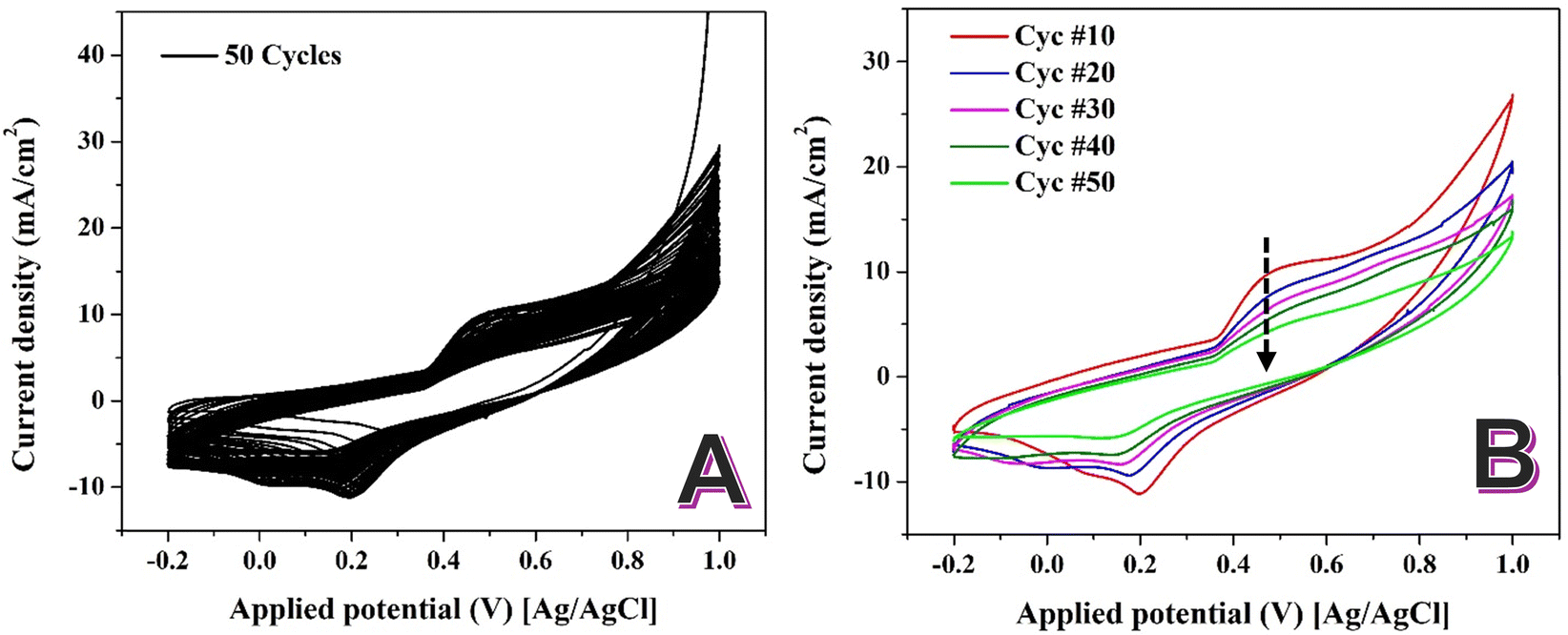 | ||
| Fig. 4 Sweeping of the prepared NiCr NPs-incorporated carbon nanofibers in 1.0 M KOH at scan rate of 50 mV s−1 at 25 °C for 50 successive cycles; (A) and selected cycles; (B). | ||
In NiCr alloys, repeated CV cycling in alkaline media induces the formation of a dense, self-limiting oxide layer. This layer, approximately 2 nm thick, forms due to the high chemical stability of the NiCr alloy and its ability to resist further oxidation beyond this critical thickness.48,49
The self-limiting nature of this oxide film prevents the continuous formation of new Ni(OOH) species. As a result, the active sites available for redox reactions are progressively reduced, leading to a decline in the intensity of the Ni(OH)2/Ni(OOH) redox peaks with successive cycles. The formation of the self-limiting oxide layer in NiCr alloys can be explained through its unique chemical and structural properties. Chromium in the alloy preferentially oxidizes to form Cr2O3, which integrates with the Ni oxide to create a compact, mixed oxide film. This layer enhances the stability of the material but limits further electrochemical reactions. The dense oxide film reduces the number of electrochemically active nickel sites, leading to a decrease in the redox peak intensity over time. This behavior contrasts with pure nickel-based catalysts, where the absence of chromium allows for sustained formation of Ni(OOH).
It should be noted that while the decrease in Ni(OH)2/NiOOH redox peak intensity with cycling is consistent with the expected formation of a self-limiting passive oxide layer, this conclusion is based on indirect electrochemical evidence. No direct surface characterization (e.g., XPS or TEM-EDS) was conducted in this study to confirm the composition or thickness of the oxide layer, and such investigations will be the focus of our future work.
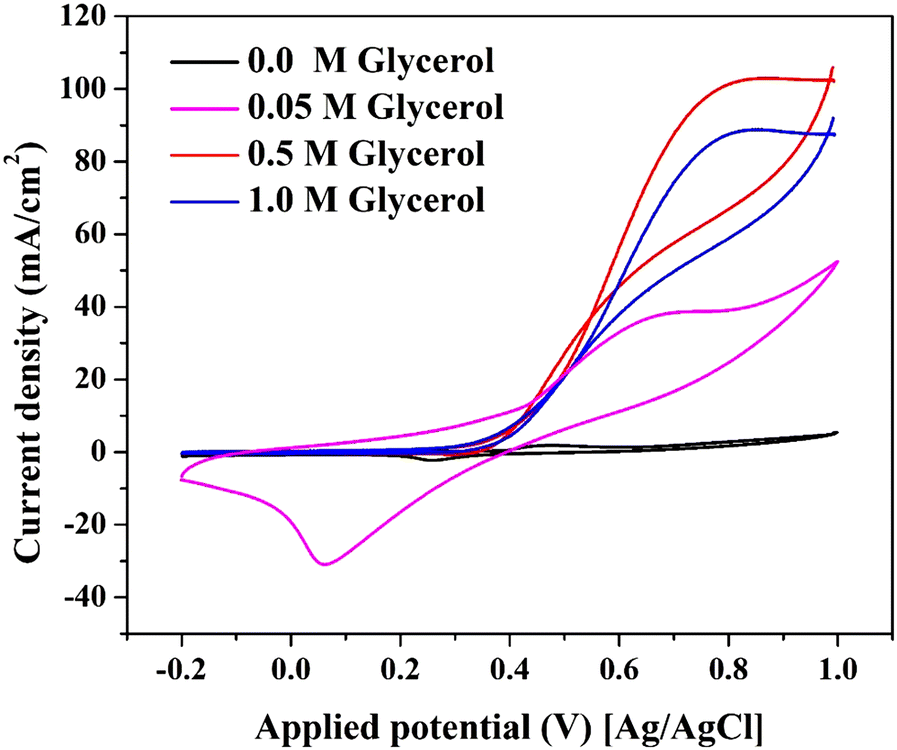 | ||
| Fig. 5 Electrooxidation of glycerol using the prepared Ni–NiCr NPs-incorporated CNFs (15 wt% sample) at different glycerol concentration in 1.0 M KOH solution at 50 mV s−1 scan rate and 25 °C. | ||
Table S1 (ESI†) highlights the electrocatalytic activity of various materials for glycerol oxidation. The synthesized Ni–NiCr-CNFs exhibit a maximum current density of 102.7 mA cm−2 in 1.0 M KOH with 0.5 M glycerol, surpassing even precious metal-based catalysts like Pd, Au, and Pt.
The superior performance of the NiCr-CNFs, despite not forming Ni(OOH) active species (as previously discussed in Fig. 4), suggests a distinct catalytic mechanism driven by their unique electronic structure and the adsorption properties of the graphite nanofiber matrix. The incorporation of Cr into the Ni matrix modifies the electronic structure of the catalyst's surface. This modification enhances the density of states near the Fermi level, optimizing the adsorption and activation of glycerol molecules and hydroxyl ions (OH−) in alkaline media.
Cr atoms contribute to enhanced charge transfer by acting as secondary adsorption sites for OH−, a critical step in breaking the C–C bonds in glycerol molecules. This synergistic interaction is characteristic of high-performance electrocatalysts. The graphitic carbon matrix plays a pivotal role in the mechanism of glycerol oxidation. Graphite has a well-documented ability to adsorb organic molecules, including glycerol. This facilitates the pre-concentration of glycerol molecules on the catalyst surface, increasing the availability of reactants for oxidation.51,52 The high conductivity of the graphitic matrix ensures efficient electron transport between the NiCr active sites and the electrode, minimizing resistance and enhancing catalytic performance.53 Moreover, the carbon matrix prevents agglomeration of the NiCr nanoparticles, maintaining a high surface area and active site availability throughout the reaction.
Conventional Ni-based catalysts derive their activity from Ni(OH)2/Ni(OOH) redox transitions, which facilitate the adsorption and oxidation of glycerol. However, the NiCr-CNFs exhibit remarkable activity without forming Ni(OOH). This deviation can be attributed to the catalytically active alloy surface. The NiCr alloy surface provides energetically favorable sites for glycerol oxidation, bypassing the need for Ni(OOH) intermediates. Moreover, the Cr-enriched oxide layer stabilizes the alloy surface, preventing over-oxidation and maintaining catalytic activity. In other words, there is dual-site catalytic mechanism: primary sites: Ni atoms act as active centers for the adsorption of glycerol molecules, enabling their dehydrogenation and oxidation. Secondary sites: Cr atoms facilitate the adsorption of OH− ions, enhancing the oxidation process through synergistic charge transfer. This dual-site mechanism ensures efficient catalysis even at high current densities.
The graphitic matrix further amplifies this mechanism by ensuring the uniform distribution of active sites and improving glycerol interaction at the interface.54
Unlike Pd, Pt, or Au, which rely on scarce and expensive metals, the NiCr-CNFs are composed of abundant and cost-effective elements, making them a viable alternative for large-scale applications. Paradoxically, compared to the precious metals, the introduced nanofibers reveal better electrocatalytic activity toward glycerol oxidation. The NiCr-CNFs exhibit superior electrocatalytic performance for glycerol oxidation due to their unique electronic structure, synergistic interaction between Ni and Cr, and the adsorption capacity of the graphitic nanofiber matrix. These findings underscore the potential of NiCr-CNFs as a sustainable and high-performance alternative to precious metal-based catalysts in energy and chemical conversion applications.
 | ||
| Fig. 6 Effect of the activation process on the electrocatalytic activity of the proposed NiCr-CNFs (15 wt% sample) at 0.05 M glycerol in 1.0 M KOH, scan rate 50 mV s−1. | ||
Indeed, it was proved Ni(OOH) species reveals good activity toward electrooxidation of several compounds including urea, methanol, ethanol… etc.55–57 However, based on literature, Ni-based electrocatalysts don’t show distinct catalytic activity toward glycerol oxidation which indicates that Ni(OOH) does not have enough ability to dissociate the glycerol molecules.
Therefore, it can be claimed that, the formation of a NiOOH layer, during activation of the introduced nanofiber, can block active sites on the NiCr alloy surface, hindering the adsorption and oxidation of glycerol molecules. This phenomenon has been observed in studies where the presence of NiOOH did not enhance, and in some cases, even reduced the electrocatalytic activity for glycerol oxidation.58
The presence of NiOOH may alter the reaction pathway, leading to less efficient oxidation processes. For instance, in certain nickel-based catalysts, the formation of NiOOH did not correspond to improved glycerol oxidation rates, indicating that NiOOH might not be the active species in all nickel-containing electrocatalysts.58
The superior performance of the non-activated Ni–NiCr-CNFs suggests that the intrinsic properties of the NiCr alloy, in combination with the conductive and supportive CNF matrix, are sufficient to facilitate efficient glycerol oxidation. The formation of NiOOH during the activation process appears to obstruct active sites and impede the reaction, highlighting the importance of maintaining a NiOOH-free surface for optimal electrocatalytic activity in this specific catalyst system.
 | ||
| Fig. 7 Effect of the CrAc content in the NiAc/CrAc/PVA electrospun solution on the electrocatalytic activity of the proposed NiCr-CNFs toward glycerol at 0.05 M glycerol in 1.0 M KOH, scan rate 50 mV s−1. | ||
The observed minimum current density (21 mA cm−2) corresponding to the lowest Cr content (5 wt%), indicates insufficient chromium content to enhance activity. At 10 wt% chromium acetate, moderate current density (36.5 mA cm−2), showing some improvement due to partial alloy formation. At 15 wt% chromium acetate, the observed maximum current density (102.7 mA cm−2), suggests optimal synergy between Ni and Cr in the alloy. However, increasing the Cr precursor more than the optimum threshold shows negative impact on the electrocatalytic activity. At 35% chromium acetate: Decrease in current density (48.1 mA cm−2), likely due to excess chromium disrupting the catalytic balance. Moreover, the onset potential for glycerol oxidation shifts positively with increasing chromium content beyond 15%, suggesting reduced catalytic efficiency at higher chromium levels.
Chromium incorporation into the Ni matrix alters the electronic density of states near the Fermi level, optimizing the adsorption of glycerol and OH− ions. This synergy is maximized at 15% Cr content, resulting in enhanced activity.59 Ni atoms provide active sites for glycerol oxidation, while Cr atoms facilitate OH− adsorption, promoting the catalytic reaction. Excess Cr (e.g., 35%) can dilute the active Ni sites, reducing activity. At 15%, the Ni–Cr alloy achieves a balance between active site density and electronic enhancement, maximizing glycerol adsorption and oxidation efficiency. This composition likely corresponds to the formation of a homogenous alloy structure with minimal lattice strain.
Excess Cr disrupts the alloy structure, reducing the availability of Ni active sites and increasing the likelihood of inactive Cr oxides on the surface. The synergistic interaction between Ni and Cr diminishes as the Cr content increases beyond the optimal range, reducing the catalytic efficiency.
The study highlights the critical role of chromium content in determining the electrocatalytic performance of Ni–NiCr-CNFs. The optimal Cr content (15%) provides the best balance between active site density, electronic structure enhancement, and catalytic synergy, resulting in superior glycerol oxidation performance. Excess Cr disrupts this balance, reducing activity. These findings emphasize the importance of precise compositional control in developing high-performance electrocatalysts.
 | ||
| Fig. 8 Cyclic voltammetry curves of the Ni–NiCr-CNFs (15 wt% sample) electrode at different scan rates (5–100 mV s−1) in 0.05 M glycerol with 1.0 M KOH as the supporting electrolyte; (A), and the relationship between the anodic and cathodic peak current densities and the square root of the scan rate; (B). | ||
The broadening of peaks with increasing scan rates reflects the influence of mass transport limitations, as higher scan rates reduce the time for diffusion of the reactants to the electrode surface. The shifts in peak potentials suggest that the reaction involves a quasi-reversible electron transfer mechanism. This means that while electron transfer is relatively fast, it is not entirely reversible under the experimental conditions. The potential shifts observed in the CV profiles are consistent with findings in bimetallic catalysts such as PdAu and NiCo alloys, which exhibit quasi-reversible behavior under alkaline conditions.15,62 The CV curves exhibit stable and consistent profiles across all scan rates, indicating that the catalyst maintains its activity and structural integrity during repeated cycling. This stability is crucial for practical applications in energy and chemical conversion systems.
Fig. 8B represents the relationship between the cathodic and anodic peak current densities and the square root of the scan rate for a given electrochemical system. This analysis provides critical insights into the mechanism and kinetics of the electrochemical process. The linear relationship between the peak current density and the square root of the scan rate indicates that the electrochemical process is diffusion-controlled. This behavior follows the Randles–Ševčík equation for reversible redox systems, suggesting that the electron transfer rate is faster than the diffusion of reactants to the electrode surface.63
| ip = (2.69 × 105)n1.5AD0.5Cv0.5 | (6) |
The high R2 values (0.997 for anodic peaks and 0.995 for cathodic peaks) confirm the excellent linearity of the plot, supporting the conclusion that the process is predominantly diffusion-controlled. The symmetry between the anodic and cathodic trends suggests a reversible redox reaction, meaning that the forward (oxidation) and reverse (reduction) reactions occur without significant kinetic limitations.
From the slope of the lines, the diffusion coefficients for the electroactive species can be calculated using the Randles–Ševčík equation. By comparing the slopes of the anodic and cathodic lines, any discrepancies in diffusion rates for the oxidized and reduced species can be analyzed. The diffusion control indicates that the process relies heavily on the transport of glycerol and hydroxyl ions to the electrode surface. This behavior suggests that enhancing mass transport (e.g., through stirring or using a flow cell) could improve the catalytic activity.
The linear relationship indicates that the catalyst provides a stable surface for the redox reaction without significant kinetic barriers. This reinforces the suitability of the catalyst for diffusion-controlled electrochemical processes. The consistent trends for anodic and cathodic peaks suggest that the redox couple involved in the reaction maintains reversibility under the experimental conditions.
All electrochemical measurements were performed without applying iR compensation. The potential values presented in this study reflect the uncompensated behavior, including the contribution from the solution and contact resistances. This approach was chosen to evaluate the practical performance of the catalyst under realistic conditions. However, it should be noted that at higher current densities, the influence of ohmic drop could slightly shift the measured potential. In future studies, iR compensation will be considered to more accurately determine the intrinsic electrocatalytic activity of the developed materials.
 | ||
| Fig. 9 Effect of reaction temperature on the electrocatalytic activity of the Ni–NiCr-CNFs nanofiber electrode using 0.05 M glycerol solution at 50 mV s−1 scan rate. | ||
The negligible differences in the anodic and cathodic peak currents across the temperature range suggest that the electrocatalytic activity of the Ni–NiCr-CNFs is relatively insensitive to temperature. This stability can be attributed to robust catalyst design. The Ni–NiCr alloy nanoparticles and the graphitic carbon nanofiber matrix provide a stable and efficient platform for glycerol oxidation, minimizing the effect of temperature variations. Moreover, the glycerol oxidation reaction over the Ni–NiCr-CNFs may operate with sufficient kinetic favorability across the tested temperatures, maintaining a consistent performance.
The lack of a distinct trend with temperature highlights the versatility of the Ni–NiCr-CNFs catalyst, which can maintain stable performance across a wide range of operating temperatures. This temperature insensitivity is advantageous for practical applications, allowing the catalyst to perform effectively without the need for precise thermal control.63
Unlike many electrocatalysts, where performance improves or declines significantly with temperature due to changes in mass transport or reaction kinetics,63,64 the Ni–NiCr-CNFs exhibit consistent behavior. This distinguishes the catalyst as a thermally stable system, ideal for scalable and robust applications. This thermal stability underscores the robustness of the Ni–NiCr-CNFs catalyst and its suitability for practical applications under varying thermal conditions.
 | ||
| Fig. 10 Chronoamperometric analysis of the Ni–NiCr-CNFs (15 wt%) electrocatalyst at different applied voltages (0.4–1.0 V vs. Ag/AgCl) in 0.05 M glycerol with 1.0 M KOH. | ||
The steady-state current density at each voltage remains stable over the measurement period, demonstrating the catalyst's durability and ability to maintain activity without significant degradation. The steady-state current density increases proportionally with applied voltage, suggesting an enhanced rate of glycerol oxidation with increasing overpotential.
The sharp increase in current density immediately after each voltage step is indicative of a rapid electrochemical reaction at the electrode surface. This initial spike corresponds to the oxidation of glycerol molecules adsorbed on the catalyst surface and the activation of reaction intermediates. The subsequent decrease and stabilization in current density are characteristic of: Mass transport limitations: As the reaction progresses, the diffusion of glycerol and hydroxyl ions to the catalyst surface becomes the rate-limiting step. The accumulation of partially oxidized intermediates, such as glyceraldehyde or dihydroxyacetone, can block active sites temporarily, reducing the current density before stabilization.
The linear increase in steady-state current density with applied voltage suggests that the Ni–NiCr-CNFs catalyst is highly responsive to overpotential, enabling efficient glycerol oxidation at higher voltages. This behavior is consistent with electrocatalysts exhibiting strong adsorption and activation of reactants.
The steady-state current density remains consistent throughout the chronoamperometric test at each voltage step, indicating excellent stability of the Ni–NiCr-CNFs catalyst. This stability can be attributed to robust catalyst design. The combination of Ni–NiCr alloy nanoparticles and the graphitic CNF matrix provides structural integrity and resistance to deactivation. Moreover, the alloy's surface resists passivation or fouling by intermediates, maintaining catalytic activity over time.
The consistent performance across a range of voltages underscores the potential of the Ni–NiCr-CNFs electrocatalyst for practical applications in energy conversion and electrochemical systems, where stability and durability are critical. The observed decay in current density after each voltage step suggests that improving mass transport (e.g., by enhancing electrolyte convection or optimizing electrode design) could further enhance catalytic performance.
In summary, the chronoamperometry analysis demonstrates that the Ni–NiCr-CNFs electrocatalyst exhibits excellent stability and durability for glycerol oxidation. The catalyst maintains consistent activity across various applied voltages, with stable steady-state current densities at each step. These findings highlight the robustness of the Ni–NiCr-CNFs catalyst, making it a promising candidate for applications in electrochemical energy conversion and alcohol oxidation processes.
To further evaluate the structural stability of the synthesized Ni–NiCr-CNFs electrocatalyst after the electrochemical glycerol oxidation tests, post-reaction SEM analysis was performed, as shown in Fig. 11. The SEM images at two magnifications confirm that the nanofibrous morphology of the catalyst was largely preserved after the electrocatalytic operation, indicating the robust mechanical and structural stability of the material under the applied electrochemical conditions. It is important to note that compared to the original SEM images (before use), some additional fine threads and small-scale structures were observed on the surface of the nanofibers. These newly appeared features are attributed to the Nafion binder and residual traces of isopropanol solvent, which were used during the electrode preparation step to ensure proper adhesion of the catalyst onto the glassy carbon substrate. Similar morphological features arising from polymeric binders and solvents after electrochemical testing have been previously reported in the literature and are known not to negatively impact the intrinsic catalytic performance of the active material itself.
 | ||
| Fig. 11 Low; (A) and high; (B) SEM magnifications images for the used Ni-NiCr-CNFs (15 wt% sample) after conducting the electrochemical measurement. | ||
Moreover, to further confirm the structural integrity of the catalyst after electrochemical glycerol oxidation, XRD analysis was conducted on the Ni–NiCr-CNFs (15 wt%) after use, as shown in Fig. S3 (ESI†). The XRD pattern shows the characteristic peaks corresponding to the Ni–NiCr alloy and graphitic carbon matrix, which are consistent with the pattern observed before the reaction. Notably, no new crystalline phases, such as nickel or chromium oxides, were detected, and no significant peak broadening was observed. This confirms that the Ni–NiCr alloy phase and the carbon nanofiber structure remained stable during the electrochemical testing, further supporting the robustness and durability of the developed electrocatalyst under the applied conditions.
4. Conclusions
Ni–NiCr alloy nanoparticles-incorporated graphitic carbon nanofiber can be exploited as effective electrocatalyst for glycerol oxidation in alkaline media. The synthesis process, involving electrospinning followed by thermal treatment, resulted in a robust and efficient electrocatalyst with well-dispersed Ni–NiCr nanoparticles embedded in a conductive CNF matrix. The optimized electrocatalyst, prepared with 15 wt% chromium acetate precursor, demonstrated a maximum current density of 102.7 mA cm−2 in 0.5 M glycerol with 1.0 M KOH, outperforming many reported precious metal-based catalysts. The temperature-dependent study revealed that the catalytic activity of the Ni–NiCr-CNFs was largely unaffected by thermal variations, underscoring the robustness and versatility of the material. Additionally, chronoamperometric analysis indicated excellent stability and durability, with the electrocatalyst maintaining consistent performance across a wide range of applied voltages. The exceptional electrocatalytic activity of the Ni–NiCr-CNFs is attributed to the synergistic effects of the Ni and Cr components, which enhance electronic properties, optimize reactant adsorption, and stabilize the active sites. The graphitic CNF matrix further contributed by improving electron transfer efficiency, providing a high surface area, and preventing nanoparticle agglomeration during electrochemical reactions. This work demonstrates that Ni–NiCr-CNFs are a cost-effective, high-performance alternative to precious metal-based catalysts for glycerol oxidation. The catalyst's stability, scalability, and compatibility with alkaline media make it a promising candidate for practical applications in direct glycerol fuel cells and the production of value-added chemicals. These findings contribute to advancing the development of non-precious metal electrocatalysts, supporting the transition to sustainable energy and chemical technologies. Future studies will focus on further optimizing the composition and structure of the catalyst to enhance its activity and selectivity for specific glycerol oxidation products, as well as exploring its applicability in other electrochemical energy conversion processes. Additionally, although the current study focused primarily on the electrocatalytic activity and stability of the developed Ni–NiCr-CNFs catalyst, future work will aim to perform a comprehensive analysis of the glycerol oxidation products using larger-scale electrolysis setups, enabling detailed insight into the reaction pathways and product selectivity.Author contributions
Nasser A. M. Barakat: conceptualization; methodology; writing – original draft; supervision; project administration. Shimaa Hamads: investigation; formal analysis. Ibrahim Mustafa: data curation; visualization. Hesham Alhumade: supervision; writing – review & editing.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no conflict of interest.References
- M. Braun, C. S. Santana, A. C. Garcia and C. Andronescu, Curr. Opin. Green Sustainable Chem., 2023, 41, 100829 CrossRef CAS.
- P. A. Alaba, C. S. Lee, F. Abnisa, M. K. Aroua, P. Cognet, Y. Pérès and W. M. A. Wan Daud, Rev. Chem. Eng., 2021, 37, 779–811 CrossRef CAS.
- M. S. Ahmad, M. H. Ab Rahim, T. M. Alqahtani, T. Witoon, J.-W. Lim and C. K. Cheng, Chemosphere, 2021, 276, 130128 CrossRef CAS PubMed.
- H. Wan, C. Dai, L. Jin, S. Luo, F. Meng, G. Chen, Y. Duan, C. Liu, Q. Xu and J. Lu, ACS Appl. Mater. Interfaces, 2022, 14, 14293–14301 CrossRef CAS PubMed.
- M. Oliveira, A. Ramos, E. Monteiro and A. Rouboa, Sustainability, 2022, 14, 1747 CrossRef CAS.
- T. Li and D. A. Harrington, ChemSusChem, 2021, 14, 1472–1495 CrossRef CAS PubMed.
- M. Simões, S. Baranton and C. Coutanceau, Appl. Catal., B, 2011, 110, 40–49 CrossRef.
- W.-Y. Zhang, X.-Y. Ma, S.-Z. Zou and W.-B. Cai, J. Electrochem., 2021, 27, 7 Search PubMed.
- Y. Zhou, Y. Shen and J. Piao, ChemElectroChem, 2018, 5, 1636–1643 CrossRef CAS.
- M. S. Houache, E. Cossar, S. Ntais and E. A. Baranova, J. Power Sources, 2018, 375, 310–319 CrossRef CAS.
- V. Oliveira, C. Morais, K. Servat, T. Napporn, G. Tremiliosi-Filho and K. Kokoh, Electrochim. Acta, 2014, 117, 255–262 CrossRef CAS.
- N. A. Barakat, M. A. Abdelkareem, M. El-Newehy and H. Y. Kim, Nanoscale Res. Lett., 2013, 8, 1–6 CrossRef PubMed.
- N. A. Barakat, M. El-Newehy, S. S. Al-Deyab and H. Y. Kim, Nanoscale Res. Lett., 2014, 9, 1–10 CrossRef CAS PubMed.
- N. A. Barakat, H. E. Gomaa, K. Khalil, F. S. Al-Mubaddel and M. M. Abdel-Aty, PLoS One, 2023, 18(10), e0308365 CrossRef PubMed.
- B. Habibi and N. Delnavaz, RSC Adv., 2016, 6, 31797–31806 RSC.
- Z. Zhong, M. Li, J. Wang, J. Lin, J. Pan, S. Jiang, A. Xie and S. Luo, Int. J. Hydrogen Energy, 2022, 47, 13933–13945 CrossRef CAS.
- K. Fernández-Caso, M. Molera, T. Andreu, J. Solla-Gullón, V. Montiel, G. Díaz-Sainz, M. Álvarez-Guerra and A. Irabien, Chem. Eng. J., 2024, 480, 147908 CrossRef.
- H. Nady, M. El-Rabiei, M. Samy, M. Deyab and G. M. Abd El-Hafez, Int. J. Hydrogen Energy, 2021, 46, 34749–34766 CrossRef CAS.
- I. Lopez-Baez, E. Martinez-Franco, H. Zoz and L. Trapaga-Martinez, Rev. Mex. Fis., 2011, 57, 176–183 CAS.
- I. Ban, J. Stergar, M. Drofenik, G. Ferk and D. Makovec, Acta Chim. Slov., 2013, 60, 750–755 CAS.
- J. C. De Jesus, I. González, A. Quevedo and T. Puerta, J. Mol. Catal. A: Chem., 2005, 228, 283–291 CrossRef CAS.
- N. A. Barakat, K. A. Khalil, I. H. Mahmoud, M. A. Kanjwal, F. A. Sheikh and H. Y. Kim, J. Phys. Chem. C, 2010, 114, 15589–15593 CrossRef CAS.
- S. Gea, B. Attaurrazaq, S. A. Situmorang, A. F. R. Piliang, S. Hendrana and S. Goutianos, Polymers, 2022, 14, 446 CrossRef CAS PubMed.
- U. K. Fatema, A. J. Uddin, K. Uemura and Y. Gotoh, Text. Res. J., 2011, 81, 659–672 CrossRef CAS.
- N. A. Barakat, B. Kim, S. Park, Y. Jo, M.-H. Jung and H. Y. Kim, J. Mater. Chem., 2009, 19, 7371–7378 RSC.
- N. A. Barakat, A. M. Tayeb, R. Hamad, M. Hashem, H. Fouad, H. Y. Kim and R. A. Hefny, RSC Adv., 2024, 14, 34904–34917 RSC.
- N. A. Barakat, K. A. Khalil and H. Y. Kim, Mater. Res. Bull., 2012, 47, 2140–2147 CrossRef CAS.
- N. A. Barakat and M. A. Ali, Sci. Rep., 2022, 12, 22574 CrossRef CAS PubMed.
- R. Phillips, K. Jolley, Y. Zhou and R. Smith, Carbon Trends, 2021, 5, 100124 CrossRef CAS.
- N. A. Barakat, M. Motlak, A. A. Elzatahry, K. A. Khalil and E. A. Abdelghani, Int. J. Hydrogen Energy, 2014, 39, 305–316 CrossRef CAS.
- M. M. Abdel-Aty, H. E. Gomaa, H. M. Abdu, R. A. Almasri, O. M. Irfan and N. A. Barakat, Polymers, 2023, 15, 2430 CrossRef CAS PubMed.
- W. Song, K. Wang, X. Lian, F. Zheng, C. Xu and H. Niu, Inorg. Chem. Front., 2024, 11, 7437–7450 RSC.
- Z. Liu, Z. Ma, X. Pang, M. Ahmad, Y. Zhao, N. Su, J. Liu, Y. Zhang, H. Sun and F. Subhan, Chem. Eng. J., 2023, 478, 147254 CrossRef CAS.
- J. Lu, H. Wan, T. Ju, Z. Ying, W. Zhang, B. Li and Y. Zhang, J. Alloys Compd., 2019, 774, 593–600 CrossRef CAS.
- Q. Fan, C. Ma, L. Wu, C. Wei, H. Wang, Y. Song and J. Shi, RSC Adv., 2019, 9, 6419–6428 RSC.
- A. Yousef, I. M. Maafa, A. Abutaleb, S. M. Matar, A. A. Alamir and M. El-Halwany, Polymers, 2024, 16, 3541 CrossRef CAS PubMed.
- B. D. Cullity and R. Smoluchowski, Phys. Today, 1957, 10, 50 CrossRef.
- F. Abe and T. Tanabe, Int. J. Mater. Res., 1985, 76, 420–425 CrossRef CAS.
- E. Aschauer, S. Sackl, T. Schachinger, T. Wojcik, H. Bolvardi, M. Arndt, P. Polcik, H. Riedl and P. H. Mayrhofer, Vacuum, 2018, 157, 173–179 CrossRef CAS.
- N. A. Barakat, M. Motlak, B.-S. Kim, A. G. El-Deen, S. S. Al-Deyab and A. Hamza, J. Mol. Catal. A: Chem., 2014, 394, 177–187 CrossRef CAS.
- X. Jiang, X. Ma, Y. Yang, Y. Liu, Y. Liu, L. Zhao, P. Wang, Y. Zhang, Y. Lin and Y. Wei, Nano-Micro Lett., 2024, 16, 275 CrossRef CAS PubMed.
- S. Jiang, T. Xiao, C. Xu, S. Wang, H.-Q. Peng, W. Zhang, B. Liu and Y.-F. Song, Small, 2023, 19, 2208027 CrossRef CAS PubMed.
- S. Bhakta, D. Macdonald, B. Pound and M. Urquidi-Macdonald, J. Electrochem. Soc., 1991, 138, 1353 CrossRef CAS.
- A. G. Oshchepkov, G. Braesch, A. Bonnefont, E. R. Savinova and M. Chatenet, ACS Catal., 2020, 10, 7043–7068 CrossRef CAS.
- F. Hahn, B. Beden, M. Croissant and C. Lamy, Electrochim. Acta, 1986, 31, 335–342 CrossRef CAS.
- M. Vuković, J. Appl. Electrochem., 1994, 24, 878–882 CrossRef.
- A. Rahim, R. Abdel Hameed and M. Khalil, J. Power Sources, 2004, 134, 160–169 CrossRef.
- M. Danışman and N. Cansever, J. Alloys Compd., 2010, 493, 649–653 CrossRef.
- P. Franklin, A reliability assessment of bipolar PROMs 14th International Reliability Physics Symposium, IEEE, 1976, pp. 207–218.
- L. Fan, B. Liu, X. Liu, N. Senthilkumar, G. Wang and Z. Wen, Energy Technol., 2021, 9, 2000804 CrossRef CAS.
- M. N. Jackson and Y. Surendranath, Acc. Chem. Res., 2019, 52, 3432–3441 CrossRef CAS PubMed.
- N. A. Barakat, M. A. Abdelkareem and H. Y. Kim, Appl. Catal., A, 2013, 455, 193–198 CrossRef CAS.
- K. Wakabayashi, Y. Takane, M. Yamamoto and M. Sigrist, New J. Phys., 2009, 11, 095016 CrossRef.
- M. Zhou, W. Kong, M. Xue, H. Li, M. A. Khan, B. Liu, F. Lu and X. Zeng, Catal. Sci. Technol., 2023, 13, 4615–4634 RSC.
- S. Akkari, V. Vivier and C. M. Sánchez-Sánchez, Electrochim. Acta, 2024, 474, 143526 CrossRef CAS.
- V. T. T. Phan, Q. P. Nguyen, B. Wang and I. J. Burgess, J. Am. Chem. Soc., 2024, 146, 4830–4841 CrossRef CAS PubMed.
- S.-J. Li, W. Guo, B.-Q. Yuan, D.-J. Zhang, Z.-Q. Feng and J.-M. Du, Sens. Actuators, B, 2017, 240, 398–407 CrossRef CAS.
- A. C. Brix, D. M. Morales, M. Braun, D. Jambrec, J. R. Junqueira, S. Cychy, S. Seisel, J. Masa, M. Muhler and C. Andronescu, ChemElectroChem, 2021, 8, 2336–2342 CrossRef CAS.
- S. P. Babu and A. Falch, ChemCatChem, 2022, 14, e202200364 CrossRef CAS.
- M. Z. Iqbal, S. Zakar, M. Tayyab, S. S. Haider, M. Alzaid, A. M. Afzal and S. Aftab, Appl. Nanosci., 2020, 10, 3999–4011 CrossRef CAS.
- M. D. Levi and D. Aurbach, J. Electroanal. Chem., 1997, 421, 79–88 CrossRef CAS.
- L. Chen, N. Chen, Y. Hou, Z. Wang, S. Lv, T. Fujita, J. Jiang, A. Hirata and M. Chen, ACS Catal., 2013, 3, 1220–1230 CrossRef CAS.
- N. Neghmouche, A. Khelef and T. Lanez, J. Fundam. Appl. Sci., 2009, 1, 23–30 CrossRef.
- B. Chi, J. Li, Y. Han and Y. Chen, Int. J. Hydrogen Energy, 2004, 29, 605–610 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00295h |
| This journal is © The Royal Society of Chemistry 2025 |