
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extraordinary U(VI) sorption capacity of high surface area super-oxidized carbons†
Tamuna
Bakhiia
 a,
Bartosz
Gurzeda
b,
Gui
Li
b,
Nicolas
Boulanger
b,
Konstantin
Maslakov
a,
Anna
Krot
ac,
Alexander
Trigub
ad,
Anna Yu.
Romanchuk
*a,
Stepan N.
Kalmykov
a and
Alexandr V.
Talyzin
*b
a,
Bartosz
Gurzeda
b,
Gui
Li
b,
Nicolas
Boulanger
b,
Konstantin
Maslakov
a,
Anna
Krot
ac,
Alexander
Trigub
ad,
Anna Yu.
Romanchuk
*a,
Stepan N.
Kalmykov
a and
Alexandr V.
Talyzin
*b
aDepartment of Chemistry, Lomonosov Moscow State University, Leninskie Gory, Moscow 119991, Russia. E-mail: romanchuk.anna@gmail.com
bDepartment of Physics, Umeå University, Umeå S-90187, Sweden. E-mail: alexandr.talyzin@umu.se
cA.N. Frumkin Institute of Physical Chemistry and Electrochemistry RAS, 31/4, Leninsky pr., Moscow, 119071, Russia
dNational Research Centre “Kurchatov Institute”, Akademika Kurchatova pl. 1, 123182 Moscow, Russia
First published on 28th April 2025
Abstract
Porous carbons based on activated reduced graphene oxide (rGO) have been demonstrated as excellent sorbents for U(VI), with their sorption capacity correlating with the degree of their oxidation. Herein, we demonstrate an extraordinarily high U(VI) sorption of ∼7050 μmol g−1 for super-oxidized porous carbon (SOPC) with a specific surface area (SSA) of ∼970 m2 g−1 and an extremely high degree of oxidation (C/O = 2.1), similar to graphene oxide. The SOPC materials were prepared using an oxidation treatment applied to activated carbon produced from spruce cones. The extremely high SSA of the precursor activated carbon (∼3400 m2 g−1) as well as its microporous structure and mild oxidation treatment allowed for the preservation of a significant part of the surface area, providing materials with rather narrow pore size distribution (∼7.5 Å). The SOPC prepared from spruce cone biochar is similar to defective graphene oxide but with a significantly higher surface area, resulting in superior U(VI) sorption. Analysis of EXAFS and XPS data shows that U(VI) likely binds to carboxylic groups on the opposite sides of the micropores. The small size of the micropores and irregular pore wall structure are the main factors affecting pore sorption. The spruce-cone biochar has a strong advantage compared with earlier used rGO as a precursor for the preparation of SOPC.
Introduction
Environmental pollution due to radionuclides, particularly U(VI), mostly originates from mining, nuclear plant incidents, waste storage and other industrial processes.1,2 Moreover, a large amount of waste solutions contaminated with radionuclides is produced during nuclear waste reprocessing and the decontamination of construction parts of nuclear plants.3 Therefore, the removal of radionuclides, including U(VI), from aqueous solutions is an important industrial and environmental issue that requires the design of new materials with high sorption capacity.4–6Porous carbon materials are excellent sorbents for the removal of certain contaminants.7,8 A very high specific surface area (SSA) is a key property that enables the application of porous carbon as a sorbent. It is well known that porous carbons with an SSA of up to ∼2000–3300 m2 g−1 can be prepared using various activation procedures.9–19 These carbons can be further chemically modified for the removal of different pollutants.20,21
Our recent studies have demonstrated that almost any pure carbon precursor can be used for producing activated carbons with an SSA exceeding ∼3000 m2 g−1 using the optimized KOH activation procedure. This treatment was successfully applied to reduced graphene oxide, wood and cone-derived biochar and spent mushroom planting substrates.15–17,22,23 An extremely high BET surface area of these ACs exceeds the theoretical value for graphene (∼2600 m2 g−1), indicating that pore walls are composed of extremely defective monolayers of carbon atoms. The pore wall structure of ACs is completely disordered, most likely curved and somewhat uncertain, in contrast to graphene, which has a rather regular 2D structure.24–26
The three-dimensional structure of activated carbons is an advantage for sorption applications compared to 2D materials such as graphene and graphene oxide.18 2D materials must be dispersed in solutions to expose the entire surface area, which is not always possible. In contrast, the 3D structure of ACs exhibits the entire surface for any atom or molecule capable of penetrating their pores. However, the hydrophobic nature of AC is a limiting parameter for the sorption of many contaminants, such as radionuclides, due to the lack of suitable sorption sites. Our recent studies demonstrate that porous carbon can be converted into hydrophilic materials via strong surface oxidation.18,27
Oxidized carbon materials show improved sorption of radionuclides and in particular for U(VI). For instance, graphene oxide (GO) is an effective sorbent for the removal of actinides from solutions.28–30 The sorption capacity for U(VI) correlates with an increased number of hole and vacancy defects.31,32 The defects facilitate U(VI) sorption due to the termination of hole edges with carboxylic groups, which are absent in defect-free GO. Carboxylic groups are known to bind metal cations, as reported in several previous studies.33,34 The number of defects in GO cannot be increased infinitely due to the breakup of 2D sheets into extremely small fragments.32 Moreover, the SSA of GO in aqueous dispersions is much smaller (700–800 m2 g−1) compared to the theoretical value of about 2400 m2 g−1 for single-layered GO.35,36
An alternative approach to increase radionuclide sorption is the surface oxidation of carbons, which has been tested with, e.g., carbon nanotubes,37,38 activated carbons39 and natural carbon materials.38 However, oxidized materials often have relatively small SSA38 and a degree of oxidation that is much smaller than those of GO.
Our previous studies demonstrated that materials with a surface oxidation degree similar to that of GO can be prepared using activated reduced graphene oxide (arGO), which is alternatively called “activated graphene”. Mild but prolonged oxidation treatment with ammonia persulphate allowed us to produce materials with a C/O ratio similar to that of standard graphene oxides (C/O = 2.4–3.3) with relatively high BET SSA up to ∼800 m2 g−1.18,27
Strongly oxidized arGO exhibited exceptionally high sorption of U(VI) (∼5400 μmol g−1 at pH = 5.1). Moreover, a correlation between the degree of oxidation and the sorption of U(VI) has been reported.18 Following this trend, the theoretical possibility of a further 2–3 fold increase in U(VI) sorption has been suggested for hypothetical materials with similarly high degrees of oxidation and SSA close to the theoretical value of GO.18 Notably, oxidation treatment resulted in a 3–4-fold decrease in SSA due to the collapse of mesopores.18 The stronger the oxidation, the more likely some pores will collapse because some C–C bonds break up due to a reaction with oxygen.18,27 Therefore, a further increase of sorption could be achieved if a smaller sacrifice of surface area during the oxidation treatment is combined with a higher oxidation degree.18
However, graphene oxide is not an attractive precursor for the industrial production of SOPC due to its high cost, complicated and environmentally unfriendly synthesis involving strong acids. Attempts to oxidize commercial activated carbon (Kuraray) were not successful due to the collapse of porous structure evident from a strong decrease in BET SSA (∼2500 m2 g−1 to ∼40 m2 g−1).18,27 Further studies revealed that microporous carbons activated the same way as arGO can be oxidized to the same degree while preserving relatively high SSA.40 The material previously reported as “activated graphene”41,42 appeared to be nearly identical to activated carbons synthesized from wood- and cone-based biochar subjected to the identical activation treatments.15
Pine and spruce cones, wood chips, bark15 and spent mushroom substrate22 precursors were used successfully to produce microporous activated carbons with SSA exceeding 3000 m2 g−1.15,40 Surface oxidation treatment applied to porous carbons has been demonstrated in our earlier study to result in improvement of Cu(II) sorption.40 However, super-oxidized activated carbons have never been tested for radionuclide sorption.
Here, we report the extremely high sorption of U(VI) by super-oxidized activated carbons produced from spruce cones. The procedure included three steps: pyrolysis of the bio-precursor (spruce cone scales) to prepare biochar, then KOH activation to synthesize microporous activated carbons with SSA up to ∼3300 m2 g−1 and prolonged oxidation treatment, which resulted in SOPC with C/O = 2.1 and SSA of ∼970 m2 g−1. The characterization of SOPCs shows that the degree of oxidation and type of oxygen functionalities in these materials are similar to those of defect-rich GO. Our experiments revealed exceptionally high sorption of U(VI) of SOPC (∼7000 μmol g−1), exceeding the sorption capacity of all previously reported sorbent materials. The mechanism of U(VI) sorption by super-oxidized activated carbons was studied using EXAFS data and XPS analysis, revealing a correlation between sorption and a high abundance of carboxylic groups on the surface of SOPC. The low cost of activated carbons produced from wood industry waste is a strong advantage compared to the expensive rGO precursor, which opens the road to the mass production of SOPC for sorbent applications.
Experimental
Materials
The sample was heated inside a tube furnace under argon flow to 200 °C at a heating rate of 5 °C min−1 to remove water and other volatile impurities and then kept at this temperature for 30 min. The temperature was then increased to 850 °C at a heating rate of 4 °C min−1 and maintained at this temperature for 3 h, and then cooled down. Washed overnight with 1 L of acetic acid solution (10 vol%) under stirring was applied to the activated sample to remove acid soluble impurities. Final washing was performed using 4 L of deionized (DI) water, which was passed over the sample using a vacuum filtration setup. The samples were dried in a vacuum oven at 60 °C overnight.
Oxidation of carbon materials
The surface oxidation treatment applied to the SCAC sample was optimized in our earlier studies.15,40 A new batch was prepared for the U(VI) sorption study presented here using the same procedure as in ref. 40. Ammonium persulfate, (NH4)2S2O8 (>99%) was obtained from Sigma-Aldrich (Germany). Carbon materials were oxidized using the following standard procedure: 30 mL of saturated ammonium persulfate solution (∼0.85 g of (NH4)2S2O8 per 1 mL of H2O) was added to 0.5 g of carbon material in a glass beaker and covered with plastic foil to reduce water evaporation. The mixture was stirred for 14 days at ambient temperature. After oxidation, the SOPC materials were washed by passing DI water through the sample in a vacuum filtration setup. Finally, the samples were dried in a vacuum oven at 60 °C for 24 hours.Sorption
Plastic vials were used in all sorption experiments to minimize water retention on the walls. A mixture of 232,233U and natural uranium was used in sorption experiments to record isotherms. The solid-phase concentration was 0.5 g L−1. A 0.1 M NaClO4 electrolyte solution was used to maintain constant ionic strength. The pH was monitored using a combined glass pH electrode (InLab Expert Pro, Mettler Toledo) connected to a pH meter (SevenEasy pH S20-K, Mettler Toledo). The pH was adjusted using small amounts of diluted HClO4 or NaOH. The pH was monitored over several days to ensure that it remained stable at 5.1 ± 0.1. Sorption was measured after equilibration under constant pH conditions. The solid phase was separated from the solution using centrifuging of the suspension aliquots at 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 20 minutes (Allegra 64R, Beckman Coulter). Sorption was calculated as the difference between the initial radionuclide activity and the activity in the solution after centrifugation. Liquid scintillation spectroscopy was used to measure the radionuclide activity using Quantulus-6220 (PerkinElmer). The experimental uncertainty in the sorption experiment was assessed based on the systematic uncertainty in measuring the uranium content, weighing the carbon-contained sample, and taking aliquots. In the above experiments, this value was determined to be 4%.
000g for 20 minutes (Allegra 64R, Beckman Coulter). Sorption was calculated as the difference between the initial radionuclide activity and the activity in the solution after centrifugation. Liquid scintillation spectroscopy was used to measure the radionuclide activity using Quantulus-6220 (PerkinElmer). The experimental uncertainty in the sorption experiment was assessed based on the systematic uncertainty in measuring the uranium content, weighing the carbon-contained sample, and taking aliquots. In the above experiments, this value was determined to be 4%.
Characterization
The N2 sorption isotherms were recorded using an Autosorb iQ XR sorptometer (Quantachrome) at 77 K. The interval of relative pressure (P/P0) for the evaluation of BET surface area was selected using a procedure based on Rouquerol parameters43 specifically designed for microporous materials. This procedure was also verified using BETSI software,44 thereby providing agreement within 2–3 m2 g−1.18 The cumulative surface area, pore volume and pore size distribution were evaluated using the slit-pore QSDFT equilibrium model from the Quantachrome software package. Fourier transform infrared (FTIR) spectra were recorded using a Vertex 80v spectrometer in attenuated total reflectance (ATR) mode equipped with a diamond crystal under vacuum conditions. The Mettler Toledo TGA/DSC1 STARe System was used for the TGA scans. Typically, a heating rate of 5 °C min−1 was used for scans performed under a nitrogen flow with a rate of 40 mL min−1. The error in the estimation of the surface area of the samples characterized in this study was limited to ±5 m2 g−1 and pore volume to ±0.01 cm3 g−1.X-ray photoelectron spectroscopy (XPS) spectra were acquired using an Axis Ultra DLD spectrometer (Kratos Analytical, UK) with a monochromatic AlKα radiation source (hν = 1486.7 eV, 150 W). The pass energies of the analyzer were 160 eV and 40 eV for the survey spectra and high-resolution scans, respectively. Double-sided nonconductive adhesive tape was used to support the powder samples. The Kratos charge neutralizer system was used because of its charging effects. The spectra were referenced to take into account charging issues by setting the C–C component of the C1s spectra to 285.0 eV. CasaXPS software was used to process all spectra. FTIR spectra of the samples after sorption were recorded using the FT-801 IR Fourier spectrometer (Simex, Russia) equipped with a diamond attenuated total internal reflection detector. Fifty scans were collected for each spectrum using a range of 4000–600 cm−1 with a spectral resolution of 4 cm−1. The errors in the element concentrations provided by XPS for our samples are within ±0.1 at%.
The Structural Materials Science beamline of the Kurchatov Synchrotron Radiation Source (Moscow, Russia) was used to collect the XAFS spectra.45 The beamline was connected to the storage ring with an electron beam energy of 2.5 GeV and a current in the range of 80–100 mA. XAFS was recorded using a monochromatic X-ray beam produced with a Si (220) channel-cut monochromator, which provided an energy resolution of ΔE/E ≈ 2 × 10−4. The damping of the higher-energy harmonics was achieved via distortion of the monochromator geometry. Energy calibration was performed using the XAFS spectrum of UO2. FEFF8.5L and IFEFFIT 1.2.11 software were used for fitting experimental spectra.46
Results and discussion
Characterization of activated carbons before and after oxidation
Surface oxidation treatment was applied to activated carbons with extremely high BET SSA values, which exceeded 3000 m2 g−1. These activated carbons were synthesized in two steps, starting from the pyrolysis of spruce cone scales, followed by KOH activation of biochar. The optimization of the synthesis procedure for achieving maximum SSA of SCAC was performed in our earlier studies.15,17,40 The most important optimized parameters are the loading ratio of KOH/biochar, temperature and duration of activation. The SCAC material prepared using an identical procedure was characterized in detail before and after a 14-day oxidation treatment in our previous publication using XPS, TGA, XRD, FTIR, and analysis of N2 sorption isotherms.15,40 Characterization of two new batches (denoted as SCAC1 and SCAC2) used here for U(VI) sorption experiments confirmed the reproducibility of the material synthesis, demonstrating only some slight batch-to-batch variations. The BET SSA values of SCAC1 and SCAC2 batches evaluated from the N2 sorption isotherms were 3280 and 3560 m2 g−1 (Table 1). These values are among the highest values reported in the literature for activated carbons. Surface oxidation of SCAC was performed using a saturated solution of ammonium persulfate (APS) following the procedure optimized in our earlier studies.15,40 The treatment was performed at ambient temperature for 5, 10 and 14 days.| Sample | Duration of oxidation, days | BET SSA, m2 g−1 | Cumulative SSA areaa, m2 g−1 | Cumulative pore volumeb, cm3 g−1 | C/O ratioc |
|---|---|---|---|---|---|
| a Cumulative SSA by QSDFT method. b Cumulative pore volume by QSDFT method. c Carbon/oxygen ratio calculated based on at% determined using XPS (Fig. 2). | |||||
| SCAC1 | 0 | 3560 | 2640 | 1.90 | 17.60 |
| SCAC2 | 0 | 3280 | 2375 | 2.10 | 18.14 |
| 5D-SCAC1 | 5 | 1820 | 1510 | 0.89 | 2.63 |
| 10D-SCAC1 | 10 | 1120 | 1084 | 0.47 | 2.51 |
| 14D-SCAC2 | 14 | 970 | 950 | 0.41 | 2.08 |
The surface area and pore size distribution of SCAC before and after oxidation treatment were analyzed using nitrogen sorption isotherms (Fig. 1). Two main features are found in the plot of the volumetric pore size distribution of the precursor SCAC materials: a narrow peak corresponding to ∼8.5 Å micropores and a broader peak corresponding mostly to mesopores (15–35 Å). The oxidation treatment resulted in almost complete disappearance of all mesopores, while the narrow peak of micropores shifted to a lower width of ∼7 Å. It can be concluded that mesopores are not stable and collapse during oxidation treatment, whereas micropores are likely narrow due to oxygen functionalization (Fig. 1).
 | ||
| Fig. 1 Volumetric pore size distributions of SCAC before and after oxidation treatment: (a) SCAC1 before and after 10 days of oxidation; (b) SCAC2 before and after 14 days of oxidation. | ||
The strong surface oxidation of ammonium persulfate-treated SCACs was confirmed by XPS analysis of the SCAC1 and SCAC2 samples before and after oxidation for 10 and 14 days, respectively. The C/O ratio calculated from the XPS spectra correlates with the duration of oxidation treatment. Moreover, a very high oxidation degree (C/O = 2.08) was achieved for the sample 14D-SCAC2 while preserving a relatively high BET SSA value (970 m2 g−1). The oxidation degree and surface area of this sample significantly exceed those of the oxidized arGO samples previously tested for U(VI) sorption in our earlier studies.18 Therefore, a high sorption capacity for U(VI) is expected for oxidized 14D-SCAC2 (C/O = 2.08 and SSA = 970 m2 g−1). The 10D-SCAC1 sample exhibited a lower oxidation degree (C/O = 2.51) and higher SSA = 1120 m2 g−1.
Analysis of C1s XPS spectra (Fig. 2 and Table 2) show that strongly oxidized SCAC is similar to graphene oxides in terms of the type of oxygen functionalization. Therefore, the spectra were deconvoluted using the recommendations in ref. 47 for oxidized carbons. The C1s spectra were interpreted using three components: the first was assigned to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of non-oxidized carbons (285.0 eV), and the second was assigned to single bonded carbons (hydroxyl, epoxide, ether) (286.9 eV). Finally, the component assigned to double-bonded oxygen CO groups (including both carbonyls and carboxyl) is found at 288.9 eV. The 290.8 eV feature corresponds to π–π* satellites (Fig. 2).47,48 The relative amounts of functional groups are provided in Table 2 as the % of carbon atoms in each of the three main deconvolution components. The data demonstrate that the sample subjected to the longest oxidation also showed the maximal relative number of carbons in double-bonded oxygen groups (21.9%). These types of oxygen functional groups bind U(VI) in graphene oxides.31
C of non-oxidized carbons (285.0 eV), and the second was assigned to single bonded carbons (hydroxyl, epoxide, ether) (286.9 eV). Finally, the component assigned to double-bonded oxygen CO groups (including both carbonyls and carboxyl) is found at 288.9 eV. The 290.8 eV feature corresponds to π–π* satellites (Fig. 2).47,48 The relative amounts of functional groups are provided in Table 2 as the % of carbon atoms in each of the three main deconvolution components. The data demonstrate that the sample subjected to the longest oxidation also showed the maximal relative number of carbons in double-bonded oxygen groups (21.9%). These types of oxygen functional groups bind U(VI) in graphene oxides.31
 | ||
| Fig. 2 C1s XPS spectra of SCAC before (a) and (b) and after oxidation treatment (c) and (d) for 10 days (10D-SCAC1) and 14 days (14-SCAC2). | ||
| Sample name | C–C/CC (%) |
C–O–C, C–OH (%) | COOH, CO (%) |
|---|---|---|---|
| SCAC1 | 71.8 | 18.7 | 9.5 |
| SCAC2 | 70.0 | 24.3 | 5.7 |
| 5D-SCAC1 | 58.1 | 27.6 | 14.3 |
| 10D-SCAC1 | 57.7 | 25.1 | 17.2 |
| 14D-SCAC2 | 48.2 | 29.9 | 21.9 |
Remarkably, sulfur impurity was found either as rather small traces (0.1%) or not at all. In contrast, the sulphate anions attached to graphene layers are typically found in graphene oxide due to H2SO4 used in the Hummers oxidation procedure.49 Using ammonia persulfate does not lead to the functionalization of the carbon surface with sulfate groups, as confirmed by the FTIR data (see below).
The similarity between super-oxidized ACs (10D-SCAC1 and 14D-SCAC2) and graphene oxides was also confirmed by analysis of the FTIR spectra and TGA scans.
Several strong peaks appeared in the spectra of the SCAC samples after oxidation treatment, whereas the precursor spectra were almost featureless (Fig. S2 in the ESI† file). The FTIR spectra were recorded under vacuum to reduce the amount of water typically physisorbed by hydrophilic SOPC at ambient conditions. Despite the dynamic vacuum, the 1618 cm−1 peak originating from water was still observed in all three oxidized SCAC samples (Fig. 3), indicating the presence of water strongly bound to the material surface. This peak overlaps the signal arising from C–C bonds (1568 cm−1). The main features found in the spectra of oxidized SCAC are assigned here to CO (1714 cm−1), C–OH (1134 cm−1, 1348 cm−1 and 1421 cm−1) and C–O–C (1027 cm−1 and 940 cm−1) bonds.49 The peak at 1230 cm−1 is more intense in the stronger oxidized samples and likely originated from carboxylic and epoxy groups. A similar type of functionalization with oxygen-containing groups was reported in our earlier studies of persulphate-oxidized arGO.18 The FTIR spectra of super-oxidized SCAC also show a certain similarity to the spectra of graphene oxide.49–52
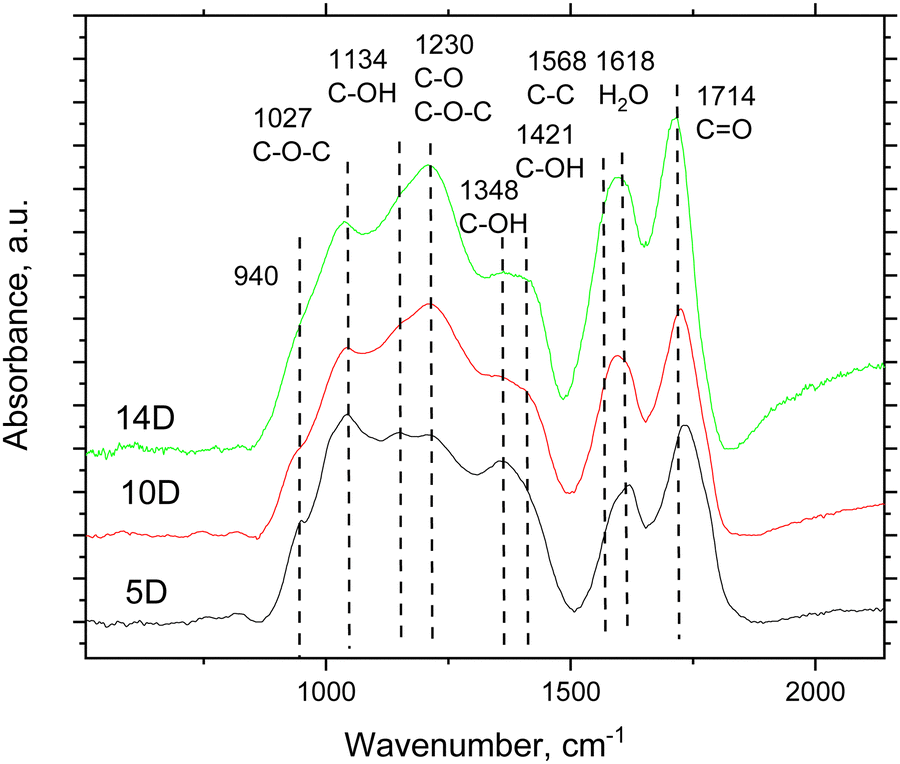 | ||
| Fig. 3 Part of the FTIR spectra of SCAC samples oxidized for 5 days (5D-SCAC1), 10 days (10D-SCAC1) and 14 days (14D-SCAC2) (some background subtracted). The original spectra (no background subtraction) of the pristine (non-oxidized) SCAC2 and 14D-SCAC2 samples are shown in the ESI† file (Fig. S2). The positions of the main features in the 14D-SCAC2 spectrum are shown for deconvolution. | ||
TGA scans recorded from 10D-SCAC1 and 14D-SCAC2 samples (Fig. 4) reveal three distinct temperature regions similar to earlier published TGA of super-oxidized arGO and graphene oxides.18,50,53 The low-temperature region (<110 °C) is due to evaporation of water (∼20 wt%) sorbed by hydrophilic carbons at ambient humidity,54 the 110–260 °C region (∼20 wt%) is assigned to deoxygenation with the breakup of C–OH and C–O–C functional groups.
 | ||
| Fig. 4 TGA data for the 10D-SCAC1 and 14D-SCAC2 samples: weight loss curves and weight loss derivative plots. | ||
A gradual weight loss at temperatures above 260 °C is related to the removal of remaining CO groups.50 The overall weight loss observed for the 14D-SCAC2 sample was approximately 58%, confirming a very high degree of oxidation. Note that the weight loss recorded in TGA scans cannot be directly used for the estimation of the oxygen content of super-oxidized carbons and graphene oxides since it is known that deoxygenation proceeds with the formation of carbon oxides (CO and CO2).52,55 Notably, the TGA data recorded here for SCAC samples are nearly identical to the data previously reported for super-oxidized arGO with a similar degree of oxidation (Fig. S1 in ESI† file). The TGA data also provide evidence of the hydrophilic nature of super-oxidized SCAC that absorbs a significant amount of water at ambient humidity.
Summarizing this section, the oxidation treatment of high specific surface area (>3000 m2 g−1) SCAC allowed us to produce samples with an extremely high degree of oxidation while preserving high SSA (C/O = 2.1 and 970 m2 g−1 for the most oxidized sample). The detailed characterization of these two SOPC batches is in agreement with our earlier study on surface oxidation applied to several types of porous carbons.39 The surface oxidation of SCAC, which produces a high amount of CO functional groups (carboxyls and carbonyls), was expected to result in an extremely large sorption capacity for U(VI) following earlier reported trends.
Sorption of U(VI)
The sorption of U(VI) was studied for the two samples with the highest degrees of oxidation (10D-SCAC1 and 14D-SCAC2). The sorption isotherms of these samples were obtained at pH 5.1 (Fig. 5). The resulting isotherms exhibit a pattern similar to those previously obtained for samples of oxidized “activated graphene”: a linear increase in sorption at low uranium concentrations, followed by a gradual flattening of the curve, indicating surface saturation with uranium. This saturation allows us to calculate the maximum sorption capacity using the Langmuir equation: Csorb = Qmax × KLa × Csolution/(1 + KLa × Csolution), where Csorb is the equilibrium concentration of sorbed radionuclides and Csolution is the equilibrium concentration of radionuclides in aqueous solution. The obtained isotherms and their modeling (Table 3) indicate that the sorption capacity of the SCAC1 sample is very close to that of the previously studied AP14D sample, amounting to 5430 μmol g−1. For the 14D-SCAC2 sample, the capacity was even higher, reaching 7040 μmol g−1. | ||
| Fig. 5 Isotherms of U(VI) sorption onto 10D-SCAC1 and 14D-SCAC2 in comparison with the previously studied arGO material before and after 14 days of ammonium persulfate oxidation (AP14D).18 Error bars are smaller than the size of symbols (due to log scale). | ||
| Sample | BET SSA, m2 g−1 | Cumulative pore vol., cm3 g−1 | C/O ratio | Formula | U(VI) sorption at pH 5.1, μmol g−1 | Ref. |
|---|---|---|---|---|---|---|
| 14D-SCAC2 | 970 | 0.46 | 2.08 | CO0.48 | 7040 ± 520 | This work |
| 10D-SCAC1 | 1120 | 0.47 | 2.5 | CO0.4 | 5530 ± 250 | |
| AP14D* | 690 | 0.31 | 2.30 | CO0.43 | 5430 ± 321 | 18 |
| arGO* | 2680 | 2.27 | 19.64 | CO0.05 | 644 ± 78 |
This value is significantly higher than U(VI) sorption reported for other types of materials (e.g., graphene oxides, activated carbons, carbon nanotubes, etc), see summary table in our earlier publication.18
Thanks to the combination of extremely strong surface oxidation (C/O = 2.08) and high specific surface area (970 m2 g−1), the 14D-SCAC2 sample showed a record high sorption capacity of 7040 μmol g−1 for U(VI). The results of sorption experiments presented in Fig. 5 and Table 3 confirm the trend reported in our previous study.18
Higher sorption of U(VI) is found for materials with a stronger degree of oxidation, even when the specific surface area decreases because of mesopore collapsing (Fig. 6). This trend indicates that the sorption of U(VI) is mostly limited to micropores, which survive after oxidation treatment.
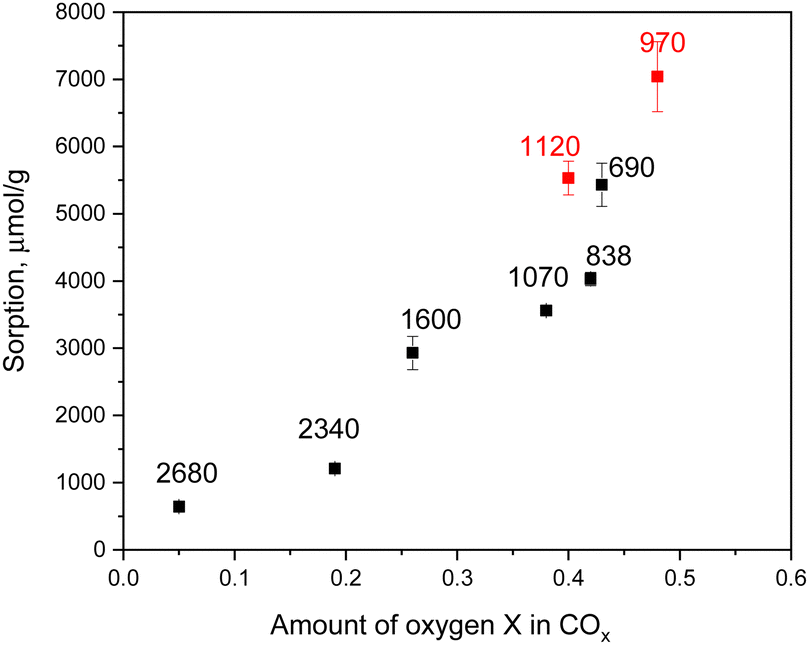 | ||
| Fig. 6 Sorption of U(VI) (at pH = 5.1) vs. oxydation degree determined using XPS. Red symbols are data from this study for super-oxidized SCAC materials, and black symbols are for earlier published super-oxidized arGO.18 The values of BET surface area (m2 g−1) are provided for each material. | ||
To determine the species in which uranium was sorbed on the studied samples, a combination of the EXAFS, FTIR, and XPS methods was used. The U4f XPS spectra of the samples (Fig. 7a) were characteristic of hexavalent uranium56 in terms of both the U4f7/2 binding energy (381.8 eV) and satellite positions, confirming the absence of redox reactions during sorption. The spectra show that the intense satellites are shifted by approximately 3.5 eV from the main peaks and that the less intense satellite is shifted by 10.5 eV from the U4f5/2 peak. Some differences in the position and intensity of the satellites are observed, indicating variations in the uranium environment within the samples.
 | ||
| Fig. 7 (a) U4f XPS spectra of the oxidized SCAC samples after U(VI) sorption, (b) FTIR of 14D-SCAC2 before and after U(VI) sorption. | ||
The strong changes in the FTIR spectra of the 14D-SCAC2 sample exposed to U(VI) sorption are rather obvious. The new peak found at ∼920 cm−1 is assigned to UO22+, providing evidence of significant sorption. The decreased intensity of the CO peak (∼1720 cm−1) and the nearly complete disappearance of the broad 1230 cm−1 feature assigned to C–O from carboxylic groups indicate that sorption of U(VI) is likely to occur by the interaction of uranyl cations with double-bonded CO groups on the surface of SCAC. A similar mechanism of sorption was revealed in our studies of sorption by graphene oxides, suggesting that carboxylic groups are capable of binding U(VI). However, some differences between the sorption of U(VI) by SCAC and graphene oxide are unavoidable because of the differences in the structures of these materials. Graphene oxides were dispersed in solution for the sorption tests, thus exhibiting the entire surface of 2D flakes to U(VI). Oxidized activated carbons are materials with a 3D porous structure.
Previously, we demonstrated that uranium binds predominantly to carboxyl groups at the edges of GO sheets or within defects.31 However, in the case of microporous materials, an important question remains: does uranium penetrate the micropores? To address this question, the EXAFS method was used because it allows the determination of the local environment.
Several structural models were used to describe the recorded EXAFS spectrum, considering uranium sorption both inside and outside the pores, including interactions with carboxyl groups. The best result was obtained with the structure shown in Fig. 8c. The reverse Monte Carlo method with the evolutionary algorithm (RMC/EA) was used to optimize the structural parameters of the proposed structural model based on the EXAFS spectroscopy data. This approach allows the extraction of three-dimensional atomic structures from the local atomic environment. The fitting was performed using the EvAX program.57
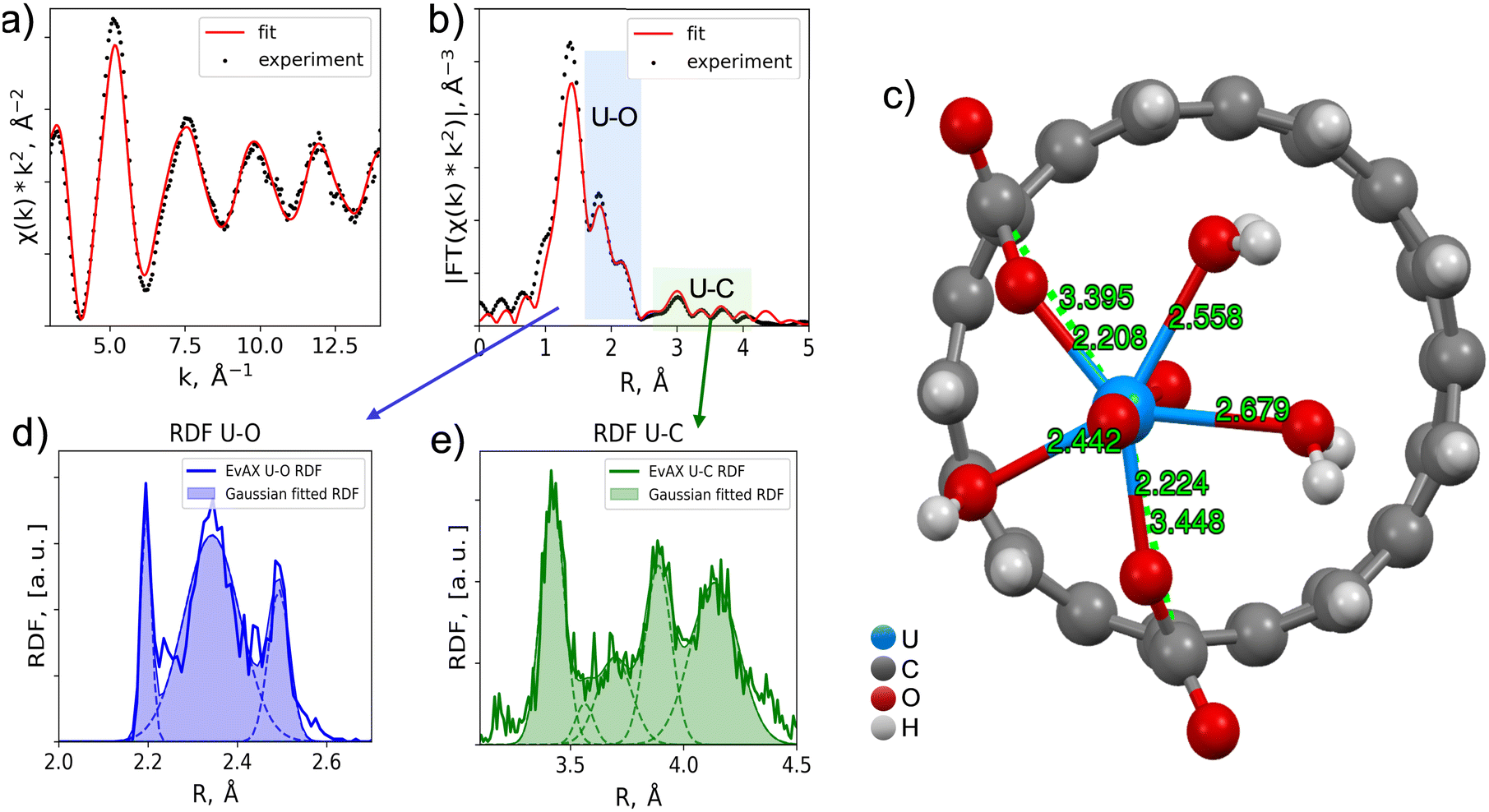 | ||
| Fig. 8 Comparison of experimental and calculated EXAFS spectra of U(VI) sorbed onto 10D-SCAC2: (a) the oscillating part of the EXAFS spectrum, and (b) its Fourier transform modules; (c) structural model of U(VI) sorption onto SCAC; the radial distribution functions of (d) oxygen and (e) carbon around uranium. | ||
A key advantage of the RMC method is its automatic handling of multiple scattering paths. Including multiple scattering paths in a standard EXAFS fitting procedure typically leads to a significant increase in the number of variable parameters, often making the results unreliable. In EvAX, the EXAFS spectrum is averaged over a set of χ(k) signals calculated using the FEFF8 multiple scattering code. Experimental and theoretical spectra were compared in wavelet transform (WT) space. The minimization procedure was performed in WT space with a k2 weighting factor using a k-range of 3–14 Å−1 and an R-range of 1–5 Å.
The initial geometry proposed included a uranyl complex positioned above a fragment of a carbon nanotube with a diameter of 7 Å, simulating a pore in the studied material. The RMC-EXAFS procedure was applied to a supercell of size 4 × 4 × 4 without considering periodic boundary conditions. The supercell contained 64 uranium atoms. In all calculations, the amplitude reduction factor S02 was fixed at 0.9. At each RMC iteration, new atomic configurations were generated by randomly displacing all atoms, with a maximum displacement of 0.4 Å from their initial positions. Eight simulations were performed using different random initial seeds to obtain the RDF functions and their corresponding statistics. The number of atomic configurations (supercells) simultaneously considered in the EA algorithm was 32.57 All simulations included 1000 iterations to ensure convergence. The final EXAFS and RDF fitting curves were calculated by averaging all such computations.
To distinguish individual coordination spheres, the resulting radial density function was deconvoluted into separate Gaussian components. The deconvolution results are presented in Table 4. The nearest oxygen atoms to uranium are axial and belong to the uranyl complex structure. Next are the oxygen atoms at characteristic distances for water molecules in close proximity to uranium. Oxygen atoms at distances of 2.4–2.5 Å represent functional groups through which uranyl binds to the carbon material. Carbon atoms are at distances characteristic of monodentate binding of uranium with carboxyl groups and are at the edge of the micropore. Thus, the presence of micropores plays a significant role, as their edges allow for the formation of carboxyl groups, which facilitate the binding process. Considering that activated carbon pores have a highly irregular shape with many bends (unlike straight carbon nanotube pores), a high number of edge-like defects are likely present inside each pore, providing additional sorption sites.
| Shell | Coordination number N | Distance R, Å | Debye–Waller factor σ2, Å2 |
|---|---|---|---|
| U-O | 2.00 | 1.78 | 0.001 |
| U-O | 0.7 | 2.20 | 0.01 |
| U-O | 3.3 | 2.34 | 0.063 |
| U-O | 1.0 | 2.49 | 0.024 |
| U-C | 2.6 | 3.42 | 0.052 |
| U-C | 0.3 | 3.56 | 0.040 |
| U-C | 1.4 | 3.70 | 0.076 |
| U-C | 2.3 | 3.89 | 0.058 |
Oxidized SCAC maintains a three-dimensional porous structure, and sorption occurs inside irregularly shaped pores, in contrast to sorption of U(VI) on the planes or inside hole defects of 2D sheets of graphene oxide. Moreover, the atomically thin walls of SCAC are very different from the relatively regular structure of the graphene skeleton of GO. In an oversimplified representation, the pores of SCAC can be considered approximately cylindrical and composed of extremely irregular carbon walls. Therefore, a difference in the local environment of U(VI) sorbed on graphene oxide and SCAC is expected due to the small size of pores (0.7–0.8 nm) in SCAC and the broad variety of sorption sites related to oxygen functional groups attached to rather irregular carbon pore walls. It is difficult to model the structure of disordered materials and even more difficult to model the sorption of U(VI) in materials with complex and disordered structures. Nevertheless, the main features of the EXAFS spectra recorded from SCAC after U(VI) sorption were found to agree with the model, suggesting nanotube-like pores and the binding of U(VI) to the opposite sides of the pore, as shown in Fig. 8.
In summary, in this section, we assigned extremely high sorption of U(VI) to the high abundance of carboxylic groups attached to the walls of SOPC pores. The most essential parameters that affect sorption and provide a high abundance of carboxylic groups are the rather strong oxidation degree of our materials, along with a high specific surface area. It is also crucial that the size of micropores is not limiting the sorption and is sufficiently large to accommodate U(VI).
Conclusions
Extending the previously revealed trend correlating a stronger oxidation degree of porous carbons to higher sorption of U(VI),58 super-oxidized activated carbons with a record high oxidation degree were produced, and superior sorption properties of these materials were demonstrated. Oxidation treatment with ammonium persulfate over prolonged periods was used here to convert hydrophobic activated carbons with SSA exceeding 3000 m2 g−1 into their hydrophilic oxidized analogues, while keeping a significant part of the surface area preserved. The main results of this study can be summarized as follows:- Unlike commercial activated carbons (Kuraray in ref. 18), activated carbons produced from spruce cone biochar survive prolonged oxidation treatment without a loss of porous structure. Mesopores collapse during prolonged oxidation, resulting in microporous SOPC.
- Surface oxidation of the atomically thin pore walls of SCAC results in the addition of a variety of oxygen functional groups (epoxide, hydroxyl, carboxyl and carbonyl). These materials are very similar to graphene oxide in many properties, e.g., types of oxygen functionalization, thermal stability and hydrophilicity.
- Extremely high oxidation degree (C/O = 2.08) and BET SSA (∼970 m2 g−1) of the best surface oxidized SCAC material resulted in an extraordinarily high U(VI) sorption capacity of ∼7050 μmol g−1. The sorption of U(VI) by super-oxidized SCAC exceeds that previously reported for other materials, including super-oxidized arGO reported in our earlier study (5400 μmol g−1 (ref. 58)).
- This study confirms and extends the previously reported general correlation between sorption capacity and degree of carbon oxidation. However, replacing exotic arGO with activated carbon is a big advantage for scaling up the production of inexpensive sorbent materials.
- The observed trend predicts a possible increase in sorption capacity up to ∼15000 μmol g−1 for materials with an oxidation degree of C/O = 2.1 if their specific surface area could be increased up to a realistic value of about 2000 m2 g−1.
- Analysis of the sorption mechanism indicates that a narrow pore size distribution and small pore diameter enable the bonding of U(VI) to the opposite sides of the pore walls.
- The microporous nature of SCACs with atomically thin carbon walls enables the formation of carboxyl groups at their defects and pore edges during oxidation. The high abundance of carboxylic groups compared to other oxidized carbon materials enables exceptionally high cation sorption.
Our results demonstrate that SOPCs with an extremely high oxidation degree and large surface area can be prepared using relatively cheap activated carbons produced by the KOH activation of biochar. Scaling up the production of SOPC starting from abundant and nearly cost-free wood waste (cones, bark, wood chips) is much easier than synthesis based on an rGO precursor. The production of significant amounts of rGO requires first the strong oxidation of graphite (which involves strong acids), followed by the thermal reduction of GO. In contrast, biochar is produced in a single step by pyrolysis of a carbon-rich bioprecursor. Using biochar instead of rGO will resolve environmental issues related to the use of wood industry waste. The properties of porous carbon produced from rGO and biochar are nearly identical (SSA, pore size distribution), and both materials exhibit similar resistance to oxidation treatment by ammonia persulfate, related mostly due to their microporous nature. Therefore, it is highly beneficial to use activated carbon for producing SOPC as it opens the way to mass production and practical sorbent applications of these materials in water filtration, actinide recovery from waste solutions and waste wood recycling.
Author contributions
Tamuna Bakhiia: methodology and investigation. Bartosz Gurzęda: methodology, investigation, writing – original draft, and visualization. Nicolas Boulanger, Gui Li: investigation, writing – review and editing. Konstantin Maslakov: investigation, writing – review and editing. Anna Krot: investigation. Alexander Trigub: investigation, formal analysis, and visualization. Anna Yu. Romanchuk: conceptualization, methodology, visualization, writing – review and editing. Stepan N. Kalmykov: supervision and funding acquisition. Alexandr V. Talyzin: conceptualization, methodology, resources, writing – review and editing, supervision, project administration, and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
AT acknowledges the support from Åforsk grant N 23-275 and Carl Tryggers Stiftelse. AT and BG thank Kempestiftelserna for funding BG stipend. The Vibrational Spectroscopy Platform and Umeå Core Facility Electron Microscopy (UCEM) at Umeå University are acknowledged by AT. A. Shchukarev is acknowledged for support with recording and interpreting XPS data. AYR, AK and SNK acknowledge funding from the Russian Science Foundation (grant 23-73-30006) for supporting U(VI) sorption studies. The experimental studies were partially performed using equipment acquired with funding from the M.V. Lomonosov Moscow State University Program of Development. FTIR measurements with uranium were performed using the equipment of the Center of shared use of IPCE RAS.References
- K. M. Campbell, R. K. Kukkadapu, N. P. Qafoku, A. D. Peacock, E. Lesher, K. H. Williams, J. R. Bargar, M. J. Wilkins, L. Figueroa, J. Ranville, J. A. Davis and P. E. Long, Appl. Geochem., 2012, 27, 1499–1511 CrossRef CAS.
- Y. L. Lu, J. J. Yuan, D. Du, B. Sun and X. J. Yi, Geogr. Sustain., 2021, 2, 95–98 Search PubMed.
- A. R. Lang, D. L. Engelberg, C. Walther, M. Weiss, H. Bosco, A. Jenkins, F. R. Livens and G. T. W. Law, ACS Omega, 2019, 4, 14420–14429 CrossRef CAS PubMed.
- C. Madic, M. Lecomte, P. Baron and B. Boullis, Cell Rep. Phys. Sci., 2002, 3, 797–811 CAS.
- L. Wang, L. Y. Yuan, K. Chen, Y. J. Zhang, Q. H. Deng, S. Y. Du, Q. Huang, L. R. Zheng, J. Zhang, Z. F. Chai, M. W. Barsoum, X. K. Wang and W. Q. Shi, ACS Appl. Mater. Interfaces, 2016, 8, 16396–16403 CrossRef CAS PubMed.
- X. Y. Zhang and Y. Liu, Environ. Sci.:Nano, 2020, 7, 1008–1040 RSC.
- I. I. Salame and T. J. Bandosz, J. Colloid Interface Sci., 2001, 240, 252–258 CrossRef CAS PubMed.
- M. Suzuki, Carbon, 1994, 32, 577–586 CrossRef CAS.
- T. Otowa, Y. Nojima and T. Miyazaki, Carbon, 1997, 35, 1315–1319 CrossRef CAS.
- Y. H. Lu, S. L. Zhang, J. M. Yin, C. C. Bai, J. H. Zhang, Y. X. Li, Y. Yang, Z. Ge, M. Zhang, L. Wei, M. X. Ma, Y. F. Ma and Y. S. Chen, Carbon, 2017, 124, 64–71 CrossRef CAS.
- T. Otowa, R. Tanibata and M. Itoh, Gas Sep. Purif., 1993, 7, 241–245 CrossRef CAS.
- N. M. Keppetipola, M. Dissanayake, P. Dissanayake, B. Karunarathne, M. A. Dourges, D. Talaga, L. Servant, C. Olivier, T. Toupance, S. Uchida, K. Tennakone, G. R. A. Kumara and L. Cojocaru, RSC Adv., 2021, 11, 2854–2865 RSC.
- Y. J. Zhang, Z. J. Xing, Z. K. Duan, M. Li and Y. Wang, Appl. Surf. Sci., 2014, 315, 279–286 CrossRef CAS.
- M. MolinaSabio, M. T. Gonzalez, F. RodriguezReinoso and A. SepulvedaEscribano, Carbon, 1996, 34, 505–509 CrossRef CAS.
- G. Li, A. Iakunkov, N. Boulanger, O. A. Lazar, M. Enachescu, A. Grimm and A. V. Talyzin, RSC Adv., 2023, 13, 14543–14553 RSC.
- A. Nordenström, N. Boulanger, A. Iakunkov, G. Li, R. Mysyk, G. Bracciale, P. Bondavalli and A. V. Talyzin, Nanoscale Adv., 2022, 4, 4689–4700 RSC.
- A. Iakunkov, V. Skrypnychuk, A. Nordenström, E. A. Shilayeva, M. Korobov, M. Prodana, M. Enachescu, S. H. Larsson and A. V. Talyzin, Phys. Chem. Chem. Phys., 2019, 21, 17901–17912 RSC.
- N. Boulanger, G. Li, T. Bakhiia, K. I. Maslakov, A. Y. Romanchuk, S. N. Kalmykov and A. Talyzin, J. Hazard. Mater., 2023, 457 Search PubMed.
- T. Otowa, Y. Nojima and M. Itoh, Activation mechanism, surface properties and adsorption characteristics of KOH activated high surface area carbon, in Fundamentals of Adsorption: Proceedings of the Fifth International Conference on Fundamentals of Adsorption, Springer, 1996, 709–716 Search PubMed.
- G. P. Rao, C. Lu and F. Su, Sep. Purif. Technol., 2007, 58, 224–231 CrossRef CAS.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Orfao, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- N. Boulanger, A. V. Talyzin, S. J. Xiong, M. Hultberg and A. Grimm, Colloids Surf., A, 2024, 680, 132684 CrossRef CAS.
- A. Klechikov, G. Mercier, T. Sharifi, I. A. Baburin, G. Seifert and A. V. Talyzin, Chem. Commun., 2015, 51, 15280–15283 RSC.
- Y. Huang, F. S. Cannon, J. K. Watson, B. Reznik and J. P. Mathews, Carbon, 2015, 83, 1–14 CrossRef CAS.
- P. J. F. Harris, Z. Liu and K. Suenaga, J. Phys.:Condens. Matter, 2008, 20, 362201 CrossRef.
- J. C. Palmer, J. K. Brennan, M. M. Hurley, A. Balboa and K. E. Gubbins, Carbon, 2009, 47, 2904–2913 CrossRef CAS.
- N. Boulanger, A. S. Kuzenkova, A. Iakunkov, A. Nordenström, A. Y. Romanchuk, A. L. Trigub, P. V. Zasimov, M. Prodana, M. Enachescu, S. Bauters, L. Amidani, K. O. Kvashnina, S. N. Kalmykov and A. V. Talyzin, Adv. Mater. Interfaces, 2022, 9, 2200510 CrossRef CAS.
- A. Y. Romanchuk, A. S. Slesarev, S. N. Kalmykov, D. V. Kosynkin and J. M. Tour, Phys. Chem. Chem. Phys., 2013, 15, 2321–2327 RSC.
- Z. J. Li, F. Chen, L. Y. Yuan, Y. L. Liu, Y. L. Zhao, Z. F. Chai and W. Q. Shi, Chem. Eng. J., 2012, 210, 539–546 CrossRef CAS.
- Y. Xie and B. A. Powell, ACS Appl. Mater. Interfaces, 2018, 10, 32086–32092 CrossRef CAS PubMed.
- A. S. Kuzenkova, A. Y. Romanchuk, A. L. Trigub, K. I. Maslakov, A. V. Egorov, L. Amidani, C. Kittrell, K. O. Kvashnina, J. M. Tour, A. V. Talyzin and S. N. Kalmykov, Carbon, 2020, 158, 291–302 CrossRef CAS.
- N. Boulanger, A. S. Kuzenkova, A. Iakunkov, A. Y. Romanchuk, A. L. Trigub, A. V. Egorov, S. Bauters, L. Amidani, M. Retegan, K. O. Kvashnina, S. N. Kalmykov and A. V. Talyzin, ACS Appl. Mater. Interfaces, 2020, 12, 45122–45135 CrossRef CAS PubMed.
- R. R. Amirov, J. Shayimova, Z. Nasirova, A. Solodov and A. M. Dimiev, Phys. Chem. Chem. Phys., 2018, 20, 2320–2329 RSC.
- R. R. Amirov, J. Shayimova, Z. Nasirova and A. M. Dimiev, Carbon, 2017, 116, 356–365 CrossRef CAS.
- H. P. Boehm, A. Clauss, G. Fischer and C. Hofmann, Proceedings of the Fifth Conference on Carbon, Oxford, Pergamon, 1962, 73–80 Search PubMed.
- P. Montes-Navajas, N. G. Asenjo, R. Santamaria, R. Menendez, A. Corma and H. Garcia, Langmuir, 2013, 29, 13443–13448 CrossRef CAS PubMed.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS.
- A. Khannanov, V. V. Nekljudov, B. Gareev, A. Kiiamov, J. M. Tour and A. M. Dimiev, Carbon, 2017, 115, 394–401 CrossRef CAS.
- H. Guedidi, L. Reinert, J. M. Leveque, Y. Soneda, N. Bellakhal and L. Duclaux, Carbon, 2013, 54, 432–443 CrossRef CAS.
- B. G. Gurzeda, N. Boulanger, L. B. Enache, M. Enachescu and A. V. Talyzin, Microporous Mesoporous Mater., 2024, 378, 113259 CrossRef CAS.
- T. Kim, G. Jung, S. Yoo, K. S. Suh and R. S. Ruoff, ACS Nano, 2013, 7, 6899–6905 CrossRef CAS PubMed.
- Y. W. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2006, 160, 49–56 CrossRef.
- J. W. M. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider and R. Ameloot, et al. , Adv. Mater., 2022, 34 Search PubMed.
- A. A. Chernyshov, A. A. Veligzhanin and Y. V. Zubavichus, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 603, 95–98 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 96–100 CrossRef CAS PubMed.
- T. R. Gengenbach, G. H. Major, M. R. Linford and C. D. Easton, J. Vac. Sci. Technol., A, 2021, 39, 013204 CrossRef CAS.
- C. A. Amadei and C. D. Vecitis, J. Phys. Chem. Lett., 2016, 7, 3791–3797 CrossRef CAS PubMed.
- V. Brusko, A. Khannanov, A. Rakhmatullin and A. M. Dimiev, Carbon, 2024, 229, 119507 CrossRef CAS.
- A. V. Talyzin, G. Mercier, A. Klechikov, M. Hedenström, D. Johnels, D. Wei, D. Cotton, A. Opitz and E. Moons, Carbon, 2017, 115, 430–440 CrossRef CAS.
- T. Szabó, O. Berkesi and I. Dékány, Carbon, 2005, 43, 3186–3189 CrossRef.
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef.
- N. Boulanger, A. S. Kuzenkova, A. Iakunkov, A. Nordenström, A. Y. Romanchuk, A. L. Trigub, P. V. Zasimov, M. Prodana, M. Enachescu, S. Bauters, L. Amidani, K. O. Kvashnina, S. N. Kalmykov and A. V. Talyzin, Adv. Mater. Interfaces, 2022, 9, 2200510 CrossRef CAS.
- A. Lerf, A. Buchsteiner, J. Pieper, S. Schöttl, I. Dekany, T. Szabo and H. P. Boehm, J. Phys. Chem. Solids, 2006, 67, 1106–1110 CrossRef CAS.
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- E. S. Ilton and P. S. Bagus, Surf. Interface Anal., 2011, 43, 1549–1560 CrossRef CAS.
- J. Timoshenko, A. Kuzmin and J. Purans, J. Phys.:Condens. Matter, 2014, 26, 055401 CrossRef CAS PubMed.
- N. Boulanger, G. Li, T. Bakhiia, K. I. Maslakov, A. Y. Romanchuk, S. N. Kalmykov and A. V. Talyzin, J. Hazard. Mater., 2023, 457, 131817 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00277j |
| This journal is © The Royal Society of Chemistry 2025 |