
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of lanthanide-based metal–organic frameworks for CO2 capture using computational modeling†
Zeynep Pinar
Haslak‡
,
Hasan Can
Gulbalkan‡
 and
Seda
Keskin
*
and
Seda
Keskin
*
Department of Chemical and Biological Engineering, Koc University, Rumelifeneri Yolu, Sariyer, 34450 Istanbul, Turkey. E-mail: skeskin@ku.edu.tr; Tel: +90 (212) 338-1362
First published on 24th March 2025
Abstract
Metal organic frameworks (MOFs) have emerged as promising materials in the context of CO2 capture and separation. Thanks to their tunable nature, various functionalities can be introduced to improve their separation performances. Lanthanide MOFs (Ln-MOFs) with high coordination numbers offer a promising space for the design of new high-performing and stable adsorbents for gas adsorption and separation. In this study, we combined molecular simulations with quantum mechanical (QM) calculations for designing new hypothetical materials offering superior CO2/N2 separation performances. An Ln-MOF having high CO2/N2 selectivity and working capacity was originally selected and its linkers were exchanged with five different types of linkers and its metal atom was exchanged with 12 different Ln3+ metals to generate 77 different types of hypothetic Ln-MOFs. Following the initial geometry optimizations at the molecular mechanics (MM) level, these structures were studied for CO2/N2 separation by performing grand canonical Monte Carlo (GCMC) simulations. Five MOFs were found to outperform the original Ln-MOF structure and they were optimized at the QM level to obtain geometries with minimized total energy, which finally led to two hypothetic Ln-MOFs offering superior CO2/N2 separation performance. The computational work that we described in this study will be useful for the rational design of new Ln-based MOFs with improved CO2 separation properties.
1. Introduction
Despite the massive investments and incitements in research to achieve efficient CO2 capture and to reduce greenhouse gas emissions, global CO2 emissions continue to rise.1 Currently, absorption/scrubbing, adsorption, cryogenic distillation, and membrane-based separation are the leading technologies for CO2/N2 separation. The most widely used technology in the industry for CO2 separation is amine-based scrubbing, despite its several drawbacks, such as the large energy penalty and high solvent volatility.2–4 Adsorption-based CO2 separation with porous materials has emerged as an alternative since it is reversible and requires less energy than scrubbing.3Metal organic frameworks (MOFs), materials in which inorganic nodes are connected through organic linkers to create highly porous structures, have been considered as a versatile platform for gas adsorption, separation, catalysis, and sensing.5,6 The greatest advantage of MOFs over conventional porous materials is that by varying the combination of inorganic nodes and organic linkers, a wide range of MOFs with various pore shapes/sizes and chemical functions can be synthesized. MOFs having desirable functionalities can be generated by modulating the choice of the parent MOF, also known as the genotype MOF, through introducing functional groups to the linker scaffolds,7–10 linker exchange,11–13 or metal exchange.14–16 However, it is not always guaranteed that the designed MOF will be stable after synthesis, its porosity will be permanent, or it will perform better than the parent MOF. Due to the large number of linkers and metal atoms that can be used to design a MOF, synthesis, and performance evaluation of thousands of structures through purely experimental methods is not efficient. Computational methods offer great advantages for the design of new MOFs with high CO2 selectivities at an affordable cost.
Computational studies focusing on HKUST-1,17 IRMOFs,18 and ZnO-MOFs19 reported the significance of metal-exchange in achieving increased CO2 adsorption capacity. The impact of ligand functionalization in MOFs was also computationally evaluated for MOF-74, MIL-53, UiO-66, UiO-67,20 and multivariate MOFs,21 and results showed that ligand functionalization can improve the CO2 affinity of the MOFs. On the other hand, linker exchange and its impact on the gas affinity of MOFs have not been widely studied: MIL-88 analogues were computationally constructed from larger dicarboxylic acid moieties to extend its pore size,22 however, when the CO2 capture capacity of MIL-88 analogues was computationally investigated, they were reported to underperform the parent MIL-88.23 Benzene-1,4-dicarboxylic acid linker of MOF-5 and 2,5-dioxobenzene-1,4-dicarboxylate (DOBDC) linker of MOF-74 were replaced with various dicarboxylic acids to enhance their CH4 and CO2 uptakes.24,25 Hypothetical MOFs with Zr nodes and different topologies were generated by combining linker-exchange and functionalization approaches and these MOFs achieve high CH4 deliverable capacities.26
Lanthanide MOFs (Ln-MOFs, also known as lanthanide organic frameworks-LOFs) are multi-functional materials having remarkable structural diversity with interesting chemical and physical properties that enable them to be used in various applications such as luminescence sensing, catalysis, magnetism, gas storage, and separation applications.27–32 Due to the high coordination numbers of Ln-metals and their flexible coordination environment, modification and construction of Ln-MOFs and the ease of solvent removal from their framework without any permanent pore collapse offer a promising space for the design and synthesis of new high-performing and stable Ln-MOFs for gas adsorption and separation.33–36 However, CO2/N2 separation studies using Ln-MOFs are limited in the literature.
Experimental studies only focused on a few Ln-MOFs, which have relatively low CO2/N2 selectivities up to 19.2,37 22.4,38 and 29.039 at 273 K. Large-scale computational screening studies revealed that Ln-MOFs with distinct structural properties are very promising for CO2/N2 separation among thousands of MOFs. For example, Qiao et al.31 calculated the adsorption selectivity, working capacity, and regenerability of 4764 computation-ready experimental (CoRE) MOFs for CO2/N2 separation and results showed that 13 out of 15 top-performing MOFs contain Ln as the metal. CATART03, SEVWEV, and MUFJAZ01 were identified as the most promising MOFs, which contain Ca, Nd, and Eu metals, respectively. Our group computed CO2/N2 selectivities and CO2 permeabilities of 3806 MOFs obtained from the Cambridge Structural Database (CSD) and showed that three top-performing Ln-MOFs are NURVAZ, PEXSAO, and SAJFEO with La, Gd, and La metals, respectively.40 The promising Ln-based MOFs for membrane-based CO2/N2 separation were found to possess narrow pores, low porosities, and low surface areas. As this literature review demonstrates, existing studies either focus on experimental measurement of the CO2 adsorption and separation performance of a particular Ln-MOF or merely mention the importance of Ln-MOFs for CO2/N2 separation through computational screening of many MOFs. To better understand the potential of Ln-MOFs for CO2 capture, studying these structures at the atomic-level and establishing useful structure–performance relationships based on molecular-insights is strongly required.
In this work, we used both atomic- and molecular-level calculations to tune the CO2/N2 separation performances of an Ln-MOF by linker- and metal-exchange methods. We first searched the CoRE MOF database to identify 2288 Ln-MOFs and then performed grand canonical Monte Carlo (GCMC) simulations to compute their CO2/N2 mixture adsorption and separation performance metrics such as selectivities, working capacities, adsorbent performance scores, and regenerabilities. One of the top-performing Ln-MOFs was selected as the genotype MOF for metal and linker exchange. The linkers of the selected Ln-MOF were exchanged with 5 different linkers and its metal atom was exchanged with 12 different Ln metals. In this way, we generated 77 different types of hypothetical Ln-MOFs, constructed from a total of 6 linkers and 13 Ln-metals, excluding the parent MOF. These structures were first optimized at the molecular mechanics (MM)-level, and then their CO2/N2 mixture adsorption and separation performances were studied by GCMC simulations. Computer-generated Ln-based MOFs, which outperformed the genotype Ln-MOF in terms of selectivity and adsorbent performance score, were identified and subjected to quantum mechanics (QM)-level optimizations. The relationships between the separation performance metrics of hypothetical Ln-MOFs and their physical and chemical properties were investigated in detail to understand which molecular features are critical to achieve high-performance CO2 capture. The computational approach we described and followed in this study is expected to guide future studies for the rational design and discovery of novel functional materials with desirable gas separation properties.
2. Computational details
The computational protocol that we followed for the selection of a genotype Ln-MOF among 2288 Ln-MOFs and the modification of the genotype Ln-MOF with the aim of designing a superior hypothetical material for CO2/N2 separation is displayed in Fig. 1. We used the most recent computation-ready experimental, all-solvents-removed MOF database (CoRE MOF 2019 ASR), which contains 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 143 different types of non-disordered MOFs.41 2288 MOFs containing lanthanides (La3+, Ce3+, Pr3+, Nd3+, Sm3+, Eu3+, Gd3+, Tb3+, Dy3+, Ho3+, Er3+, Tm3+, Yb3+, and Lu3+) were initially selected from this database. Structural properties of these Ln-based MOFs, such as pore limiting diameter (PLD), the largest cavity diameter (LCD), N2 accessible surface area (Sacc), and porosity (ϕ) were computed using Zeo++ software.42 MOFs with nonzero gravimetric surface areas and PLDs greater than 3.64 Å were considered for further investigation to ensure that both CO2 (3.30 Å) and N2 (3.64 Å) molecules could be adsorbed into the pores of the adsorbents.
143 different types of non-disordered MOFs.41 2288 MOFs containing lanthanides (La3+, Ce3+, Pr3+, Nd3+, Sm3+, Eu3+, Gd3+, Tb3+, Dy3+, Ho3+, Er3+, Tm3+, Yb3+, and Lu3+) were initially selected from this database. Structural properties of these Ln-based MOFs, such as pore limiting diameter (PLD), the largest cavity diameter (LCD), N2 accessible surface area (Sacc), and porosity (ϕ) were computed using Zeo++ software.42 MOFs with nonzero gravimetric surface areas and PLDs greater than 3.64 Å were considered for further investigation to ensure that both CO2 (3.30 Å) and N2 (3.64 Å) molecules could be adsorbed into the pores of the adsorbents.
 | ||
| Fig. 1 Schematic representation of the computational workflow, together with the number of Ln-MOFs passed to the next level of calculations in each step. | ||
To compute the CO2/N2 mixture adsorption in Ln-MOFs, electrostatic interactions between the gas molecules and MOFs should be considered and hence the charge equilibration method (Qeq), as implemented in the RASPA43 simulation code, was used to assign partial point charges to Ln-MOF atoms.44,45 Any MOF with a partial point charge larger than 5.0 or smaller than −5.0 was eliminated. The Ln-MOFs for which Henry's constant of CO2 (KCO2) was computed to be higher than 10−2 mol kg−1 Pa−1 were not considered since the regenerability of a material with a very high Henry's constant might be challenging due to the presence of strong adsorbate–adsorbent interactions.46,47 After these eliminations, 487 Ln-MOFs were found to be eligible for CO2/N2 separation application. Fig. S1 (ESI†) shows the distribution of metals in 487 Ln-MOFs evaluated in this study: Gd-MOFs exhibit the largest group (101 MOFs), whereas Tm-MOFs constitute the group with the lowest number of MOFs in the distribution (5 MOFs). There is no MOF constructed from Pm3+ metal in the CoRE MOF database, and Nd3+-based MOFs were eliminated due to the filters described above.
GCMC simulations were performed using the RASPA43 simulation code version 2.0.37 to obtain the CO2/N2 mixture adsorption data of 487 different types of Ln-MOFs. To represent dry flue gas, we specified the composition of the CO2/N2 mixture as 15/85. The temperature was fixed to the ambient temperature (298 K). The Lennard-Jones (LJ) potential was used to compute the non-bonded interactions, and a cut-off distance of 13 Å was set for the truncation of these interactions. The simulation cell lengths were set to at least twice of the cut-off distance. The universal force field (UFF) was used for the potential parameters of MOF atoms.48 Potential parameters for CO2 molecule were taken from the TraPPE force field.49 CO2 was modeled as a three-site linear, rigid molecule with partial point charges located at the center of each site. For N2 molecule, a three-site model was used with N atoms at the two sites, and the third site was the center of mass with partial charges.50 The Lorentz–Berthelot mixing rules were applied to obtain pair potentials between dissimilar atoms. The Ewald summation was used to consider the long-range electrostatic interactions.51
CO2/N2 mixture adsorption performances of 487 Ln-MOFs were calculated under vacuum swing adsorption (VSA) and pressure swing adsorption (PSA) process conditions, where adsorption (desorption) pressure was set to 1 (0.1) and 10 (1) bar, respectively, for VSA and PSA processes. GCMC simulations for CO2/N2 mixture were performed with 60000 cycles (10000 cycles for initialization, 50000 cycles for taking ensemble averages). To further understand how cyclic adsorption processes affect the gas separation performances; mixture selectivities (Sads,CO2/N2), working capacities (ΔNCO2), and adsorbent performance scores (APS) of MOFs were computed at each process condition using the formulas given in Table S1 (ESI†). The ratio of the composition of the adsorbed gases divided by the bulk composition of the mixture was used to calculate mixture adsorption selectivity. Working capacity was calculated as the difference of the CO2 uptake computed at the adsorption and desorption pressures. APS was calculated as the multiplication of selectivity and working capacity. The isosteric heat of adsorption values for CO2 and N2 were obtained from the mixture GCMC simulations conducted at 1 bar and 10 bar. KCO2 and KN2 were calculated using the Widom particle insertion method52 at infinite dilution with a total of 60000 cycles where 10000 cycles were used for the initialization.
were obtained from the mixture GCMC simulations conducted at 1 bar and 10 bar. KCO2 and KN2 were calculated using the Widom particle insertion method52 at infinite dilution with a total of 60000 cycles where 10000 cycles were used for the initialization.
487 Ln-MOFs were then ranked with respect to their calculated APSs under VSA condition, and one of the top-performing Ln-MOFs, YUSWIU, was selected as the genotype MOF. Original metal and linker atoms of this selected Ln-MOF, Dy3+ and furan-2,5-dicarboxylate, respectively, were exchanged using the “edit structure” tool implemented in Mercury software.53 77 hypothetical Ln-MOFs were generated. Since QM level optimizations of this large number of structures are computationally very demanding and time consuming, initial geometry optimizations of these structures were performed by using the Forcite module of Materials Studio54 with the aim to relax all atomic coordinates and cell parameters for an efficient pre-screening of the generated Ln-MOFs. The convergence tolerance quality of ultra-fine (energy tolerance value of 2 × 10−5 kcal mol−1, displacement tolerance value of 10−5 Å, and force tolerance value of 10−3 kcal) and smart algorithm, which uses the steepest descent, adjusted basis set Newton-Raphson and quasi-Newton methods to find the lowest energy conformation with maximum iteration numbers of 10000 were employed, and the cell parameters were allowed to be optimized. The potential parameters were taken from the UFF for MOFs, and the partial point charges were assigned by the Qeq method, which offers an affordable computational cost with high accuracy. After this initial geometry optimization at the MM-level, 37 hypothetical Ln-MOFs were identified to meet the eligibility requirements discussed above (PLD > 3.64 Å, Sacc > 0 m2 g−1, K0 < 10−2 mol kg−1 Pa−1). GCMC simulations were performed to assess their CO2/N2 mixture adsorption and separation performances, and they were ranked with respect to their selectivities and APSs computed under VSA and PSA conditions. Five of them were identified to outperform the parent Ln-MOF both in VSA and PSA conditions.
To obtain MOF geometries closer to expected real structures, these five hypothetical Ln-MOFs were further subjected to geometry optimizations at the QM-level. QM-level optimizations of the one-unit cell of the Ln-MOFs were conducted by the DMol3 module55,56 implemented in the Materials Studio program package. With the aim of testing the capability of the most commonly employed density functional theory (DFT) methodologies for reproducing the experimental structural properties, the generalized gradient approximation (GGA) Perdew–Burke–Ernzerhof (PBE) functional,57 hybrid-GGA exchange–correlation Becke-3-parameter-Lee–Yang–Parr (B3LYP) functional58,59 together with double numerical plus d-function (DND) and double numerical plus polarization function (DNP) basis sets were used. A spin-unrestricted formalism was employed for the treatment of spin polarization. The Monkhorst–Pack scheme k-points grid sampling was set as 1 × 3 × 1. The convergence tolerance quality of fine (energy tolerance value of 10−5 Ha, displacement tolerance value of 5 × 10−5 Å, force tolerance value of 2 × 10−3 Ha Å−1, and SCF tolerance value of 10−6) was set, in which the cell parameters were allowed to be optimized. GCMC simulations were then performed to assess the CO2/N2 separation performance of QM-level optimized Ln-MOFs. In these simulations, partial atomic charges in metal–organic frameworks (PACMOF)60 charges which are based on the density derived electrostatic and chemical (DDEC) charges61 were assigned to QM-optimized Ln-MOF structures for the highest accuracy.
3. Results and discussions
Identification of the MOF genotype with desired properties is the principal step in materials design which is followed by the modification of its organic linkers and inorganic nodes. While linker replacement provides the freedom for the incorporation of chemical moieties of interest, metal exchange expands the structural and compositional diversity of a material. With the aim of finding the most suitable MOF as the genotype, adsorption-based CO2/N2 separation performances of 487 Ln-MOFs were initially examined to find the top-performing materials offering high Sads,CO2/N2 and ΔNCO2 with a high R%.
Fig. 2(a) and (b) show the simulated Sads,CO2/N2 and ΔNCO2 values of 487 Ln-MOFs computed at VSA and PSA conditions. At VSA (PSA) condition, Sads,CO2/N2 and ΔNCO2 values varied between 3–15508 and 0.03–3.9 mol kg−1 (3–19284 and 0.03–8.1 mol kg−1), respectively. For an efficient and cost-effective gas separation process, an adsorbent should have both a high working capacity and a high selectivity. APS, which is the multiplication of adsorption selectivity and working capacity, was employed to evaluate the performance of Ln-MOFs for CO2/N2 separation. Ln-MOFs that are represented with blue and red points in Fig. 2 correspond to materials offering moderate and high APSs, respectively. Under VSA condition, 11 Ln-MOFs out of 487 have moderate to high APSs (553–14193 mol kg−1) because of their very high Sads,CO2/N2 (734–15508), although they have considerably low ΔNCO2 (< 1 mol kg−1), as shown in Fig. 2(a). 10 Ln-MOFs with moderate Sads,CO2/N2 (140–362) and high ΔNCO2 (> 3 mol kg−1) also achieve a range of moderately high APSs (452–1400 mol kg−1). Similarly, at PSA condition, Ln-MOFs with very high Sads,CO2/N2 (1136–19234) and low ΔNCO2 (< 1 mol kg−1) have moderate to high APSs (141–3513 mol kg−1) and Ln-MOFs with moderate Sads,CO2/N2 (108–208) and high ΔNCO2 (>3 mol kg−1) have APSs in the range of 426–1100 mol kg−1, as shown in Fig. 2(b). Since the VSA process yielded higher APSs compared to the PSA process, the top promising 25 Ln-MOFs were identified for CO2/N2 separation, considering the highest APS for the VSA process with R% > 75%. All calculated adsorbent performance metrics of the top 25 Ln-MOFs, together with their structural properties, were listed in Table S2 (ESI†) and results show that the top Ln-MOFs having R% > 75% also have moderate APSs (145–843 mol kg−1) and Sads,CO2/N2 (73–325).
 | ||
| Fig. 2 Simulated Sads,CO2/N2 and ΔNCO2 values for 487 Ln-MOFs for CO2/N2 separation at (a) VSA and (b) PSA conditions. Green, blue, and red points represent MOFs with (a) APS < 100 mol kg−1, 100 < APS < 2000 mol kg−1, and APS > 2000 mol kg−1, respectively, (b) APS < 100 mol kg−1, 100 < APS < 1100 mol kg−1, and APS > 1100 mol kg−1, respectively. | ||
The top 25 Ln-MOFs are mainly composed of benzene- and pyridine-derived organic linkers, three of them have cyclohexane (COWMIL, KULRIT, and COWMAR), one contains oxalato (OJONOQ), and two have furan scaffolds (YUSWIU and QUZVEO). Linkers connected to the inorganic nodes of Ln-MOFs through carboxylate fragments were reported to implement porous and robust character to the framework.33,35 Besides, rigid and linear ligands preserve the rigidity and topological network of the crystal structure. Among the top performing 25 Ln-MOFs listed in Table S2 (ESI†), modification of YUSWIU, which offers moderate APSs (146 and 200 mol kg−1 at VSA and PSA conditions), was interesting since furan-2,5-dicarboxylate linkers can be replaced with various rigid and commercially available 5-membered heterocyclic-dicarboxylic acids. Therefore, we focused on both the linker and metal-exchange of the MOF named YUSWIU.
Dy3+ atoms in YUSWIU bond to six carboxylate O atoms of furan-2,5-dicarboxylic acid, one O atom is coordinated to dimethylformamide (DMF) solvent, and one O atom is coordinated to methanol solvent.60 Four carboxylate groups bridge the two Dy3+ centers. The furan-2,5-dicarboxylate linkers of YUSWIU were replaced with 5 different types of linkers which are: pyrrole-2,5-dicarboxylate (hereafter denoted as Ln-pyrrole), 4-amino-1,2,4-triazole-3,5-dicarboxylate (Ln-aminetriazole), 4-methyl-1,2,4-triazole-3,5-dicarboxylate (Ln-methyltriazole), isoindoline-1,3-dicarboxylate (Ln-isoindoline) and 1,3,4-oxadiazole-2,5-dicarboxylate (Ln-oxadiazole). Then, Dy3+ metal of every linker-exchanged YUSWIU structure and the original YUSWIU were replaced with 12 Ln-metals; La3+, Ce3+, Pr3+, Sm3+, Eu3+, Gd3+, Tb3+, Ho3+, Er3+, Tm3+, Yb3+, Lu3+ to generate a total of 77 YUSWIU-derived Ln-MOFs as shown in Fig. 3.
 | ||
| Fig. 3 Proposed linkers and Ln metals to be exchanged with the linker and metal atom of the genotype Ln-MOF, YUSWIU. (a) Pyrrole-2,5-dicarboxylic acid, (b) 1,3,4-oxadiazole-2,5-dicarboxylic acid, (c) 4-methyl-1,2,4-triazole-3,5-dicarboxylic acid, (d) 4-amino-1,2,4-triazole-3,5-dicarboxylic acid, (e) isoindoline-1,3-dicarboxylic acid. | ||
Initial conformations of all generated structures were subjected to periodic geometry optimizations at the MM-level with the Forcite module of the Materials Studio program package to attain a MOF geometry with minimized total energy. Although generic force fields such as UFF may have limitations in the representation of interactions, optimizations at the MM-level were shown to perform well in predicting the geometric parameters for MOFs.18 We note that lanthanides have unique electronic properties, and UFF parameterization might not capture them well. In this study, UFF-based optimization was used for pre-screening the materials before the computationally demanding QM-level optimizations. To test the reliability of our method, the crystal structure of YUSWIU was optimized with the parameters described in the methodology section. The Dy–O and Dy–Dy bond lengths of YUSWIU were reported to be 2.256–2.594 Å (shortest–longest) and 4.122 Å, respectively.62 The corresponding bond distances in the optimized YUSWIU were observed to be fairly close to the reported values (rDy–O = 2.220–2.388 Å, rDy–Dy = 4.317 Å). The unit cell parameters of the optimized YUSWIU deviated from the experimental data by less than 4.7%, which gives an acceptable degree of confidence in terms of the quality of the geometry for the initial screening of the generated Ln-MOFs.
When the Dy3+ atoms of YUSWIU (Dy-furan) were exchanged with 12 Ln-metals, optimized Ln–Ln distances decreased gradually from 57La3+ (4.606 Å) to 71Lu3+ (2.701 Å) in accordance with the lanthanide contraction concept; the atomic radius of lanthanide elements decreases as the atomic number increases,63 with only exceptions in Eu–Eu (4.508 Å) and Yb–Yb (4.946 Å) distances, as given in Table S3 (ESI†). Eu has an electronic configuration of [Xe] 6s2 4f6 (half-filled f subshell), and Yb has an electronic configuration of [Xe] 6s2 4f14 (full-filled f subshell), and thus they can access +2 oxidation state instead of +3 which makes them relatively more stable. Eu and Yb have larger metallic radii compared to other trivalent Ln-metals, consistent with the divalent nature of these two Ln-metals. As a result, they behave exceptionally in the Ln–Ln bond length trend. Ln–O bond lengths also show a rough decrease from 57La3+ to 71Lu3+ as observed in Ln–Ln distances, with large deviations in Eu–O and Yb–O bonds. When the furan ring in YUSWIU (Dy-furan) was exchanged with 5-membered heterocycles, Dy–Dy and the shortest Dy–O distances were observed to be slightly longer in Dy-aminetriazole, Dy-oxadiazole, and Dy-methyltriazole than YUSWIU, whereas Dy–Dy and the largest Dy–O bond lengths were calculated to be slightly shorter in Dy-pyrrole and Dy-isoindoline, as in Table S4 (ESI†).
After the geometry optimizations of YUSWIU-derived MOFs, their structural properties (PLD, LCD, Sacc, ϕ) were computed. 40 generated Ln-MOFs were eliminated due to the previously applied filters such that if an Ln-MOF has PLD < 3.64 Å or KCO2 > 10−2 mol kg−1 Pa−1. As a general trend, none of the Ln-MOFs constructed from isoindoline-1,3-dicarboxylate (Ln-isoindoline) and 4-methyl-1,2,4-triazole-3,5-dicarboxylate (Ln-methyltriazole) could give PLD > 3.64 Å because of the steric effects caused by the large molecular volumes of these moieties. Thus, we ended up with 37 YUSWIU-derived structures. Table S5 (ESI†) represents the structural properties of these 37 Ln-MOFs, having pore sizes of 4.87 Å ≤ LCD ≤ 6.41 Å and 3.74 Å ≤ PLD ≤ 5.74 Å. MOFs generated from 4-amino-1,2,4-triazole-3,5-dicarboxylate linkers (Ln-aminetriazole) have the lowest Sacc, varying between 125–559 m2 g−1, whereas the MOFs generated from furan-2,5-dicarboxylate linkers (Ln-furan) generally have the highest Sacc, varying between 1043–1332 m2 g−1 (excluding Lu-furan), followed by Ln-pyrroles and Ln-oxadiazoles, in accordance with the molecular volumes of the linkers. On the other hand, when the metal atoms are considered, calculated Sacc values generally decreased with the increased atomic size of the Ln-metal; Lu-MOFs have the lowest Sacc, varying between 323–431 m2 g−1 and Yb- and La-MOFs have the largest Sacc, varying between 1074–1332 m2 g−1, excluding aminetriazoles.
Fig. 4 shows the Sads,CO2/N2 and ΔNCO2 values computed from the results of molecular simulations of 37 hypothetical Ln-MOFs that we generated by linker and metal-exchange, together with 487 synthesized Ln-MOFs for which the Sads,CO2/N2 and ΔNCO2 results were previously presented in Fig. 2. As a general trend, Sads,CO2/N2 and ΔNCO2 values of hypothetical YUSWIU analogues are cumulated amid the Sads,CO2/N2 and ΔNCO2 values of the synthesized Ln-MOFs. Under VSA condition, the range of Sads,CO2/N2 and ΔNCO2 for hypothetical YUSWIU-derived MOFs were computed as 16–595 and 0.5–2.4 mol kg−1, respectively. Among 37 YUSWIU derived-MOFs, 8 of them have higher Sads,CO2/N2 (117–595) and APS (236–1023 mol kg−1) than YUSWIU (80 and 146 mol kg−1, respectively) at VSA process. The range of Sads,CO2/N2 and ΔNCO2 values for YUSWIU-derived MOFs under PSA condition were computed between 27–198 and 0.7–3.3 mol kg−1, respectively. Six YUSWIU-analogues were identified to have higher Sads,CO2/N2 (91–198) and APS (202–500 mol kg−1) than the parent Ln-MOF (88 and 200 mol kg−1, respectively), whereas three of them have higher APS (237–260 mol kg−1) but slightly lower Sads,CO2/N2 (79–84 mol kg−1) at PSA process.
 | ||
| Fig. 4 Simulated Sads,CO2/N2 and ΔNCO2 for 37 different types of computer-generated YUSWIU-derived MOFs for CO2/N2 separation at (a) VSA and (b) PSA conditions. Blue circle represents the performance of YUSWIU. Green, blue, and red points represent the YUSWIU-derived MOFs with (a) APS < 100 mol kg−1, 100 < APS < 5000 mol kg−1, and APS > 500 mol kg−1, respectively, (b) APS < 100 mol kg−1, 100 < APS < 300 mol kg−1, and APS > 300 mol kg−1, respectively. Simulated data for 487 synthesized Ln-MOFs were shown by grey in the back for comparison. | ||
Fig. 4 also shows interesting trends in the MOFs generated from Ln metals. For instance, Ln-aminetriazoles were calculated to have low porosities (0.46–0.52) and low Sacc (125–559 m2 g−1), which limits their gas uptakes and APSs. On the other hand, no hypothetic Ln-MOF with Dy, Er, Gd, Ho, Lu, Tb or Yb metal could outperform YUSWIU in terms of selectivity and APS. We observed a linear trend between the Qeq charges assigned to Ln-metals and their CO2 uptakes at 1 bar, when MOFs have similar porosities. 19 out of 37 Ln-MOFs listed in Table S5 (ESI†) have porosities between 0.57–0.59 and Ln-MOFs with more positively charged lanthanides appear to have stronger CO2 uptake, as depicted in Fig. S2 (ESI†). While partial charges for Dy, Er, Gd, Ho, Tb, and Yb are scattered between 1.16–1.87e, partial charges of Ce, Eu, La, Lu, Pr, Sm, and Tm span a wide range between 2.11–3.93e. Although Lu metal has a partial positive charge of ∼2.65e, the narrow porosity of Lu-furan and Lu-pyrrole (0.49) permits less CO2 uptake than Ln-MOFs with higher Ln charges, but higher uptake than those with lower charges. Since lanthanides are electropositive elements, electrons of the Ln metals are withdrawn by the electronegative coordinating oxygens of the linkers. We also observed that carboxylate oxygens of Ce, Eu, La, Lu, Pr, Sm, and Tm-MOFs are more negatively charged compared to the carboxylate oxygens of Dy, Er, Gd, Ho, Tb, and Yb-MOFs, and the more electron-rich oxygens attract Lewis acidic (electron acceptor) CO2 stronger. Further examination of 19 Ln-MOFs having similar porosities for the effect of linker type on CO2 uptake reveals that oxadiazole linkers almost double the CO2 uptake compared to furan, which adsorbs CO2 stronger than pyrrole at 1 bar. Furan and pyrrole-based linkers were observed to adsorb N2 strongly compared to oxadiazole-based linkers. Oxadiazole linkers were previously shown to have high CO2/N2 selectivity due to the presence of two nitrogen atoms (bearing lone pairs) in its heterocyclic ring, which attract CO2.64 Five YUSWIU derived-MOFs common in VSA and PSA conditions were identified to outperform the original YUSWIU structure in terms of selectivity and APS. Their calculated properties are given in Table S6 (ESI†).
In the next step, these promising five materials identified to have higher selectivity and APS than the original YUSWIU structure were considered for the computationally more demanding QM-level geometry optimizations. For the benchmarking study of the DFT method, PBE/DNP, PBE/DND, and B3LYP/DNP level theories were tested. The crystal structure of YUSWIU was subjected to geometry optimizations by employing the three DFT functional and basis set combinations. When the geometrical parameters including the bond lengths and unit cell parameters of the crystal structure of YUSWIU were compared with the three optimized geometries, B3LYP/DNP was found to give markedly longer Dy–O and Dy–Dy bonds (5–7%) and larger cell volume (20.9%), whereas the corresponding deviations were smaller than 2.9% in bond lengths and 7.4% larger in cell volume when YUSWIU was optimized with PBE/DND. On the other hand, when PBE/DNP level of theory was used, the largest and the shortest Dy–O bonds of optimized YUSWIU (2.779 Å and 2.454 Å) were slightly overestimated and underestimated, respectively, compared to the original crystal structure (2.256 and 2.594 Å), and Dy–Dy bond (4.092 Å) was slightly underestimated with respect to the crystal structure (4.122 Å), whereas the unit cell parameters and cell volume deviate from the experimental data by less than 2.7% and 5.3%, respectively. Besides, geometry optimization at the PBE/DNP level resulted in closer unit cell parameters compared to the MM-level optimization (less than 2.7% deviation for QM and less than 4.7% deviation for MM-level optimized YUSWIU). Thus, five promising hypothetical Ln-MOFs were further optimized by using the PBE/DNP level of theory. Calculated structural properties, LCD, PLD, Sacc, and ϕ of QM-level optimized Ln-MOFs presented in Table S7 (ESI†) were observed to be different from the calculated properties for their MM-optimized geometries. MOFs containing furan linkers (La-furan and Tm-furan) were computed to have slightly lower LCD and PLDs (less than 0.12 Å) in QM-level optimized structures, whereas higher LCD and PLDs were computed for QM-level optimized structures of YUSWIU-derived MOFs with oxadiazole (Pr-oxadiazole, Sm-oxadiazole and Tm-oxadiazole) linkers (between 0.37–0.97 Å) compared to their MM-optimized counterparts. These changes are considered within the acceptable limits according to a previous computational study, where 90% of 879 MOFs experienced changes in their LCDs less than 1 Å upon QM-level geometry optimization.65
Adsorbent performance metrics were computed from scratch for the QM-level optimized YUSWIU analogues by assigning PACMOF charges. To ensure consistency of the performance comparisons between YUSWIU and its hypothetic analogues, PACMOF charges were also assigned to YUSWIU. Comparisons of the simulated NCO2 and Sads,CO2/N2 values of MM- and QM-level optimized Ln-MOFs are shown in Fig. 5. The calculated metrics for YUSWIU and five hypothetic YUSWIU-derived MOFs were found to be different when two different methodologies (MM-level optimization with Qeq charges vs. QM-level optimization with PACMOF charges) were used. MM-level optimized and Qeq charge-assigned hypothetical Ln-MOFs exhibit equal or higher CO2 uptake at 1 bar than QM-level optimized and PACMOF charge-assigned counterparts (Fig. 5(a)), whereas the reverse trend is observed at 10 bar (Fig. 5(b)) and for YUSWIU at 1 bar and 10 bar. For N2 uptakes, while similar trends are observed for hypothetical Ln-MOFs, YUSWIU presents a reverse trend (Fig. S3, ESI†). Nazarian et al.65 showed that the CO2 uptake of 82% of 879 MOFs changed more than 5% after the geometry optimization, indicating that gas adsorption properties can differ depending on the free energy landscape of the MOFs. Thus, the changes in geometrical parameters upon optimization at the QM-level result in different CO2 and N2 uptakes. On the other hand, when we considered the charge effect, CO2 uptake of PACMOF-charged YUSWIU at 1 bar was computed to be 17% more than the Qeq-charged YUSWIU (Fig. 5(a)), while at 10 bar, the assigned charges have little impact on the CO2 uptake as expected due to the decreased impact of adsorbate–adsorbent interactions (Fig. 5(b)).
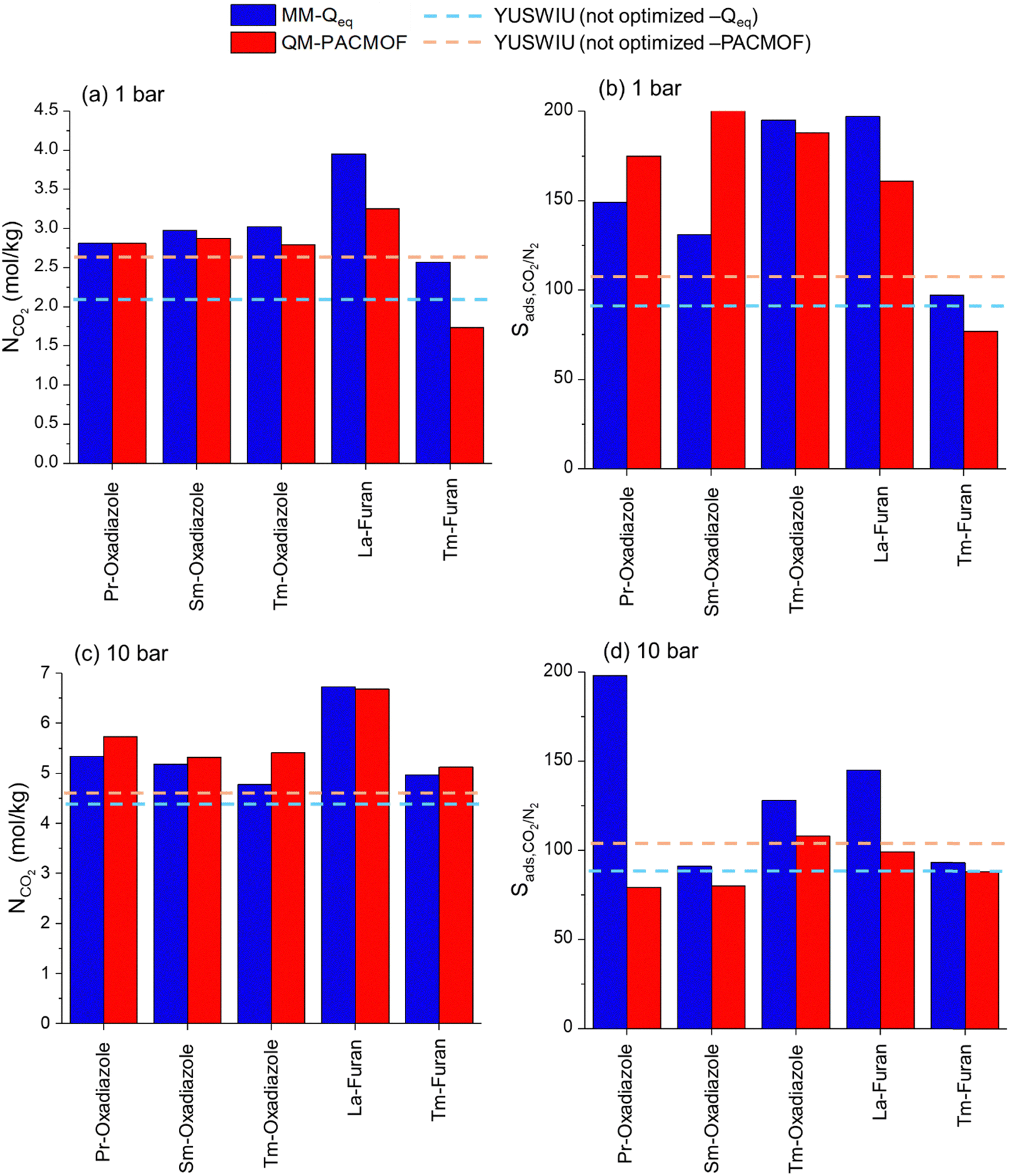 | ||
| Fig. 5 Calculated CO2 uptakes and Sads,CO2/N2 at 1 bar (a) and (b), and at 10 bar (c) and (d) for original YUSWIU and YUSWIU-derived hypothetical MOFs. | ||
At 1 bar, Pr-oxadiazole, Sm-oxadiazole, Tm-oxadiazole and La-furan were calculated to possess higher CO2 uptakes (Fig. 5(a)), whereas five hypothetic Ln-MOFs have lower N2 uptakes than YUSWIU (Fig. S3, ESI†) which results in higher Sads,CO2/N2 for Pr-oxadiazole, Sm-oxadiazole, Tm-oxadiazole, and La-furan compared to YUSWIU when the two methodologies were considered (Fig. 5(b)). However, at 10 bar, although all the hypothetical Ln-MOFs have higher CO2 uptakes than YUSWIU (Fig. 5(c)), only Tm-oxadiazole exhibits a higher CO2 selectivity assessed by both methodologies (Fig. 5(d)), because of the increased N2 uptakes in Pr-oxadiazole, Sm-oxadiazole, La-furan and Tm-furan (Fig. S3, ESI†). We observed a large gap between the calculated Sads,CO2/N2 of MM- and QM-level optimized Pr-oxadiazole at 10 bar which is due to the drastically higher N2 uptake calculated for the QM-level optimized, PACMOF-charged structure than the MM-level optimized, Qeq-charged structure (Fig. S3, ESI†).
With the aim to see how the gas uptakes change with our proposed methodology, we tested the combined effect of charge and the optimization method on the gas uptakes by performing the simulations on the MM-level optimized, Qeq charge-assigned YUSWIU and the QM-level optimized, PACMOF charge-assigned YUSWIU. Results showed that CO2 uptake of the QM-level optimized, PACMOF charge-assigned YUSWIU at 1 (10) bar is 12% (7%) higher than the MM-level optimized, Qeq charge-assigned YUSWIU. While charge alone did not have a significant impact on N2 uptake (Fig. S3, ESI†), MM-level optimized, Qeq charge-assigned YUSWIU have ∼30% higher N2 uptakes than QM-level optimized, PACMOF charge-assigned YUSWIU at 1 bar and 10 bar. These results point out the importance of the choice of the methodology to be applied in the computational design of materials.
Table S8 (ESI†) shows the comparison between the highest positive and the lowest negative charges when Qeq and PACMOF charges were assigned to the QM- and MM-level optimized geometries of five promising structures. For some MOFs, notable differences were observed, particularly for the metal centers. For instance, in Tm-furan, the highest positive charge for the QM-level optimized geometry is +2.10 (Qeq) vs. 1.73 (PACMOF) for Tm atom, while the lowest negative charge is −0.47 (Qeq) vs. −0.70 (PACMOF) for O atom, which resulted in higher KCO2 for the PACMOF charge-assigned QM-level optimized geometry (1.4 × 10−4 mol kg−1 Pa−1) than the Qeq charge-assigned one (6.2 × 10−5 mol kg−1 Pa−1). In the case of the MM-level optimized geometry of Pr-oxadiazole, the highest positive charge for the Qeq charge-assigned structure (+3.87) is higher than the PACMOF charge-assigned one (+2.28), which resulted in higher KCO2 (4.0 × 10−4 for Qeq charges and 4.6 × 10−5 mol kg−1 Pa−1 for PACMOF charges). Despite these variations, we observed that the differences between the selectivities obtained from the ratio of Henry's constants of gases (KCO2/KN2) remained within an order of magnitude across different charge schemes, supporting the applicability of the Qeq method for high-throughput screening while acknowledging its limitations in the selectivity predictions.
To gain deeper insights into the adsorption dynamics of the Ln-MOFs, CO2/N2 mixture adsorption snapshots in YUSWIU and five promising hypothetical Ln-MOFs (Pr-oxadiazole, Sm-oxadiazole, Tm-oxadiazole, La-furan, and Tm-furan) were generated. Fig. 6 shows that CO2 and N2 are gathered primarily around the carboxylate oxygens (red) in all Ln-MOFs. While CO2 is also cumulated around furan and oxadiazole rings, N2 is only attracted by the furan ring since ring oxygens are weakly attractive for N2, while nitrogen sites in oxadiazole repel N2 molecules, decreasing their uptakes by the MOF. This behavior of N2 molecules can be quantified by the isosteric heat of adsorption (Qst) values, which shows the strength of the interactions between an adsorbate and an adsorbent. According to the Qst values calculated at 1 bar (Table S9, ESI†), Pr-oxadiazole, Sm-oxadiazole, Tm-oxadiazole, La-furan, Tm-furan, and YUSWIU have a very high affinity for CO2 and moderate affinity for N2
and moderate affinity for N2 . As a general trend, Ln-MOFs constructed from furan rings (Ln-furans) display slightly higher affinity for N2, whereas Ln-oxadiazoles present a slightly lower affinity. On the other hand, we observe that Tm-oxadiazole has a much higher affinity for CO2 (36.27 kJ mol−1) than Tm-furan (32.64 kJ mol−1), for which the node atoms are the same, but the linkers are different. These results indicate that 1,3,4-oxadiazole-2,5-dicarboxylate linkers dramatically increase the affinity for CO2 and decrease the affinity for N2 when replaced with furan-2,5-dicarboxylate leading to increased CO2 selectivity. Both 1,3,4-oxadiazole and furan are aromatic rings, the latter more than the former. According to the previous DFT calculations, lone pair electrons on the oxygen and nitrogen atoms of the linker rings, which act as Lewis bases (electron-donor), are very good CO2 adsorption sites that act as Lewis acid (electron-acceptor).66,67 N2, as a weak Lewis base, is also bound to oxygen sites, but more weakly compared to CO2.
. As a general trend, Ln-MOFs constructed from furan rings (Ln-furans) display slightly higher affinity for N2, whereas Ln-oxadiazoles present a slightly lower affinity. On the other hand, we observe that Tm-oxadiazole has a much higher affinity for CO2 (36.27 kJ mol−1) than Tm-furan (32.64 kJ mol−1), for which the node atoms are the same, but the linkers are different. These results indicate that 1,3,4-oxadiazole-2,5-dicarboxylate linkers dramatically increase the affinity for CO2 and decrease the affinity for N2 when replaced with furan-2,5-dicarboxylate leading to increased CO2 selectivity. Both 1,3,4-oxadiazole and furan are aromatic rings, the latter more than the former. According to the previous DFT calculations, lone pair electrons on the oxygen and nitrogen atoms of the linker rings, which act as Lewis bases (electron-donor), are very good CO2 adsorption sites that act as Lewis acid (electron-acceptor).66,67 N2, as a weak Lewis base, is also bound to oxygen sites, but more weakly compared to CO2.
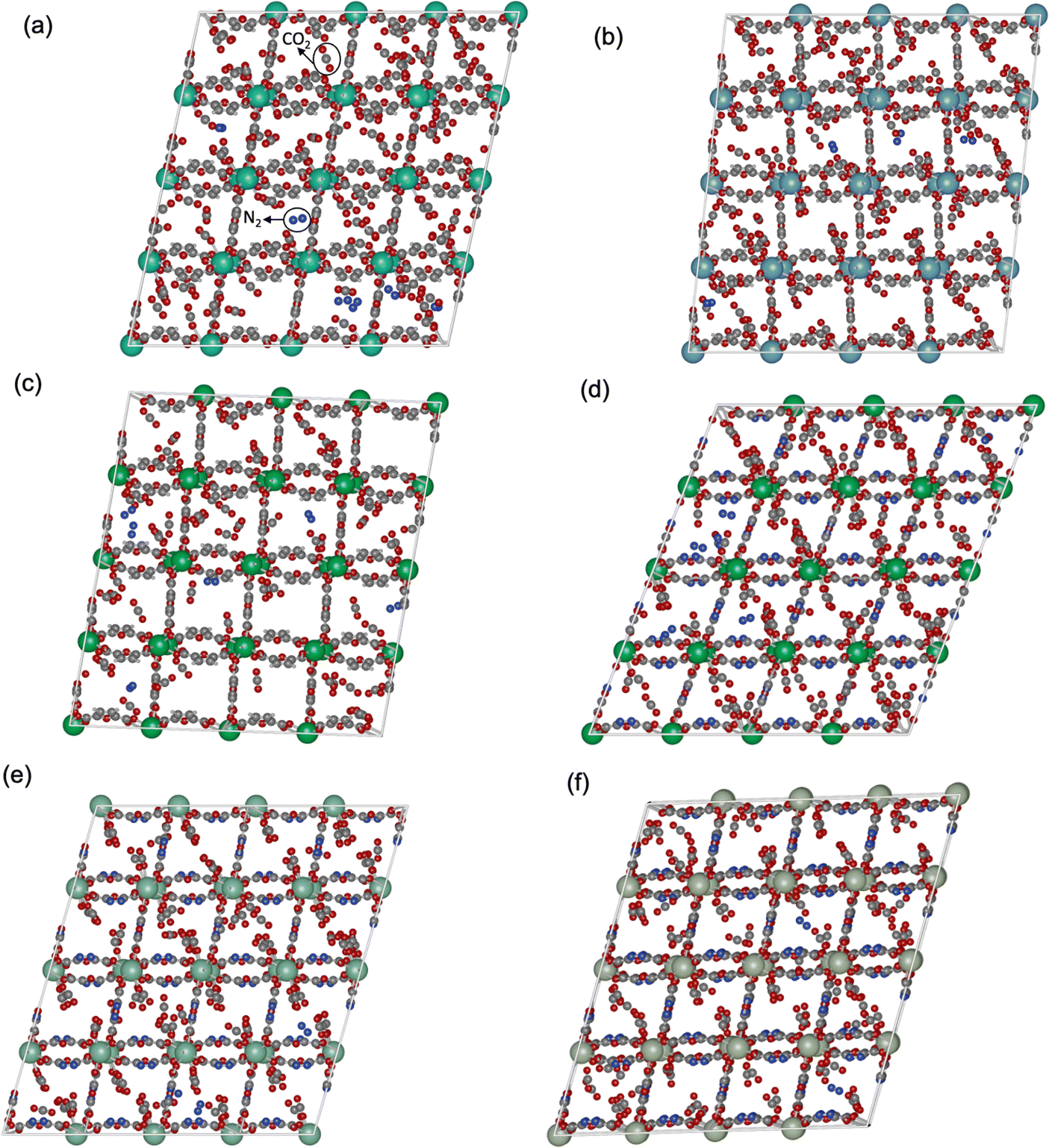 | ||
| Fig. 6 Snapshots for CO2 and N2 adsorption in (a) YUSWIU (Dy-furan), (b) La-furan, (c) Tm-furan, (d) Tm-oxadiazole, (e) Sm-oxadiazole, and (f) Pr-oxadiazole at 1 bar, 298 K. Dy, La, Tm, Sm, C, O, H and N atoms are represented with cyan, light blue, dark green, light green, gray, red, white and dark blue, respectively. | ||
Selectivity is an interplay between various properties such as pore size, inorganic node, and linker type of the adsorbent, adsorbate polarizability and its affinity for the adsorbent. Higher CO2 uptake and lower N2 uptake in Tm-oxadiazole, thus higher Sads,CO2/N2 compared Tm-furan, can be both attributed to the linker affinities for CO2 and N2, and to the larger pores of Tm-oxadiazole where more gas molecules can penetrate. On the other hand, although exchanging the Dy3+ nodes in YUSWIU (Dy-furan) with La3+ (La-furan) decreases the affinity of CO2 ( for YUSWIU and 32.68 kJ mol−1 for La-furan) and N2 (
for YUSWIU and 32.68 kJ mol−1 for La-furan) and N2 (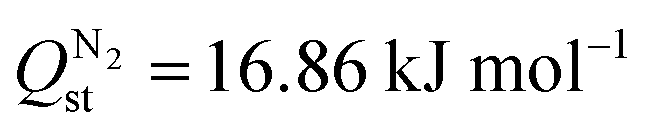 for YUSWIU and 14.47 kJ mol−1 for La-furan), larger porosity of La-furan allows higher CO2 uptake, while N2 uptake is slightly lower. These findings pointed out the significance of furan-2,5-dicarboxylic acid and 1,3,4-oxadiazole-2,5-dicarboxylic acid in CO2/N2 separation performance of the material when incorporated into the MOFs as linkers and using Ln-metals as nodes which have electropositive character, willing to donate their electrons to coordinating oxygens, which in turn becomes more attractive for CO2.
for YUSWIU and 14.47 kJ mol−1 for La-furan), larger porosity of La-furan allows higher CO2 uptake, while N2 uptake is slightly lower. These findings pointed out the significance of furan-2,5-dicarboxylic acid and 1,3,4-oxadiazole-2,5-dicarboxylic acid in CO2/N2 separation performance of the material when incorporated into the MOFs as linkers and using Ln-metals as nodes which have electropositive character, willing to donate their electrons to coordinating oxygens, which in turn becomes more attractive for CO2.
At 10 bar, the CO2 affinities of YUSWIU, Pr-oxadiazole, Sm-oxadiazole, and Tm-oxadiazole were slightly lower ( , respectively), whereas La-furan and Tm-furan had slightly higher CO2 affinities (
, respectively), whereas La-furan and Tm-furan had slightly higher CO2 affinities ( , respectively) compared to their corresponding values at 1 bar (Table S9, ESI†). In addition, the N2 affinities of all Ln-MOFs were calculated to be profoundly higher
, respectively) compared to their corresponding values at 1 bar (Table S9, ESI†). In addition, the N2 affinities of all Ln-MOFs were calculated to be profoundly higher  , resulted in lower CO2 selectivities at 10 bar (Sads,CO2/N2 varying between 79 and 108).
, resulted in lower CO2 selectivities at 10 bar (Sads,CO2/N2 varying between 79 and 108).
The comparison of APS values of these five promising QM-optimized Ln-MOFs (Pr-oxadiazole, Sm-oxadiazole, Tm-oxadiazole, La-furan, Tm-furan) and YUSWIU is presented in Fig. 7. Under VSA condition, four hypothetical Ln-MOFs offer higher APSs than their original structure YUSWIU (248 mol kg−1), except Tm-furan (Table S7, ESI†). At PSA condition, La-furan, Tm-furan, Pr-oxadiazole, and Tm-oxadiazole have higher APSs (339, 298, 231, and 284 mol kg−1, respectively) than YUSWIU (208 mol kg−1). Overall, considering all the calculated performance metrics obtained using two different methodologies, and considering the notable difference between calculated Sads,CO2/N2 at 10 bar for Pr-oxadiazole, we identified La-furan and Tm-oxadiazole as novel Ln-MOF candidates with improved APS values both at VSA and PSA conditions.
 | ||
| Fig. 7 APS of promising Ln-MOF candidates at VSA and PSA conditions. Geometries were optimized at QM-level and PACMOF charges were assigned. Data provided for YUSWIU were taken from the calculations performed for the PACMOF-assigned crystal structure. | ||
We finally compared the CO2/N2 separation performances of the Ln-MOFs that we generated in this work with the promising MOFs and COFs reported in the literature. The CO2/N2 selectivities of Zn-MOF-74, Ti-MIL-91, Cu-TDPAT, UTSA-16, and SIFSIX-3-Ni for the separation of CO2/N2: 15/85 mixture were computed as 63, 92, 214, 246, and 285, respectively, using the results of GCMC simulations.68 Similarly, simulated CO2/N2 selectivities of the top-performing COFs for the CO2/N2: 15/85 mixture were computed as 14–40 and 6–19 at 1 bar and 10 bar, respectively.69 In experiments, single-component gas adsorption measurements were performed for Mg-MOF-74,70 SMOF-SIFSIX-1,71 MIL-53(Al),72 Ni-AG5,73 MOF-505,74 and MIL-101(Cr)72 and CO2/N2 selectivities for the separation of CO2/N2: 15/85 mixture were predicted by the ideal adsorbed solution theory (IAST) as 150, 132, 64, 52, 37, and 8 at 1 bar, respectively. Our results showed that the modified Ln-MOFs could achieve CO2/N2 mixture selectivities up to 200 both at 1 and 10 bar, which are comparable to or better than those reported in the literature.
We proposed a computational workflow (Fig. 1) as an ideal scheme for the selection of promising Ln-MOFs for CO2 capture and the design of better performing materials by the linker and metal node modification. Since screening thousands of MOFs with high precision is computationally very demanding, we used several assumptions, and our results should be evaluated under the validity of these assumptions. Ln-MOFs were assumed to be rigid in our molecular simulations, a widely used assumption in the literature, to save computational time for studying a large number of materials. Recent studies showed that molecular adsorption is only weakly affected by the inclusion of MOFs’ flexibility and considering the framework flexibility primarily affects the diffusion properties of gas molecules in MOFs.46,75,76 For more realistic simulations, MOFs should be modeled as flexible frameworks in simulations in future studies.
Another assumption is related to the assignment of the partial atomic charges since the accuracy of the electrostatic interactions is strongly dependent on the charges of the atoms that make up the system. We note that partial atomic charges are not physically observable and there are several atomic charge assignment schemes. The charges can be derived from the electron wave function, electron density, electrostatic potential, or semi-empirical approaches.77 Although DFT-derived charges are more accurate than the semi-empirical charge methods, it is not realistic to screen thousands of MOFs by computationally demanding DFT-derived charges. Thus, in this work, we used semi-empirically derived Qeq charges to initially screen 2288 Ln-MOFs to obtain fast and reliable results, and then we performed a more detailed analysis for the 5 Ln-MOFs which appeared as the most promising materials for CO2/N2 separation, by assigning computationally demanding DFT-derived PACMOF charges to obtain more accurate results. The gas uptakes and adsorption selectivities of MOFs were shown to vary with the charge method used,78,79 and in this study, we observed the presence of notable differences between CO2 and N2 uptakes of Qeq and PACMOF-charged Ln-MOFs in some cases.
Another assumption we used in this study concerns the level of theory to be used in geometry optimizations of generated Ln-MOFs. Following the computational generation of a hypothetical MOF, the geometry should be optimized to obtain a more realistic geometry close to a local minima on the potential energy surface. Depending on the choice of the level of theory (such as force field, DFT functional/basis set, smearing procedure, convergence tolerance quality, etc.), the self-consistent field (SCF) convergence may appear as a difficult task to achieve. Especially when the material includes lanthanide atoms, filling electrons in 4f-orbitals makes the calculations very complicated, and thus, increasing the SCF iterations up to tens of thousands may be needed,80 which in turn significantly increases the computational time. Since the optimization of a large number of generated Ln-MOFs at the QM level appears to be unrealistic, we optimized the generated 77 Ln-MOFs at a faster MM-level for the initial screening in this work. The geometric parameters of MOFs were predicted well by this method in previous studies.18 Then 5 Ln-MOFs, identified as the most promising materials, were optimized at the QM-level to obtain more reasonable geometrical parameters. The validities of our geometry optimization methods were checked by comparing the results with the experimentally reported structural parameters of YUSWIU. While the unit cell parameters of the MM-level optimized YUSWIU deviated less than 4.7%, QM-level optimized YUSWIU deviated to a maximum of 2.7%. Overall, our proposed workflow given in Fig. 1 works well within the limits of the assumptions that we discussed. We believe that this methodology can be useful for future studies, helping to identify key challenges and guide improvements in the screening and design of novel materials.
We finally note that it is crucial to address the synthesizability and stability of the top-performing adsorbents, as these Ln-MOFs are currently hypothetical. Recent studies have demonstrated that hypothetical MOFs can be synthesized since they are constructed using linkers similar to those available in the experimentally realized MOFs.25,81 Future experimental studies are required to show the synthesizability and stability of the hypothetical Ln-MOFs that we computationally generated.
4. Conclusions
In this study, we presented a multi-level computational approach for generating high-performing hypothetical Ln-MOFs and testing them for adsorption-based CO2/N2 separation. A promising Ln-MOF, YUSWIU, having a moderate CO2/N2 selectivity was initially identified from the MOF database based on the results of molecular simulations, and then modified by linker and metal exchanges to generate 77 new hypothetical Ln-MOFs. After MM-level geometry optimization of these materials, 5 hypothetic Ln-MOFs which outperform the original YUSWIU in terms of selectivity and/or APS under VSA and PSA conditions were further optimized at QM-level and separation performances of these computer-generated MOFs were examined. La-furan and Tm-oxadiazole were identified as novel materials for CO2/N2 separation with very high APSs (334 and 409 mol kg−1 for VSA, 339 and 284 mol kg−1 for PSA, respectively) and selectivities (161 and 188 at 1 bar, 99 and 108 at 10 bar, respectively). Calculated Qst values and adsorption snapshots obtained from molecular simulations indicated that oxygen atoms on the carboxylate ends, furan, and oxadiazole rings are the most attractive sites for both gases, whereas nitrogen atoms on oxadiazole rings were found to be attractive for only CO2. The repulsive oxadiazole rings for N2 decrease the affinity of N2, allowing Ln-MOFs generated from 1,3,4-oxadiazole-2,5-dicarboxylate linkers to be more CO2 selective than furan-2,5-dicarboxylate linkers. Thus, hypothetical Pr-oxadiazole, Sm-oxadiazole, and Tm-oxadiazole were found to outperform the parent MOF, YUSWIU (Dy-furan), in terms of selectivity and APS, especially under VSA condition, because of their CO2 attractive and N2 repulsive 1,3,4-oxadiazole-2,5-dicarboxylate linkers. On the other hand, Ln nodes were observed to alter the electronic nature of the coordinating oxygens, allowing them to be richer in electrons, and vary the porosity of the MOF. When the Dy3+ nodes in YUSWIU were changed with La3+ (La-furan), the larger porosity of La-furan allowed more CO2 molecules to be adsorbed, making La-furan more CO2/N2 selective than YUSWIU. Considering the robustness of the results obtained from MM and QM-level calculations, Tm-oxadiazole and La-furan were proposed as promising adsorbents for CO2/N2 separation under VSA and PSA conditions.Author contributions
ZPH: methodology, software, validation, investigation, conceptualization, writing – original draft, writing – review & editing. HCG: methodology, software, validation, investigation, conceptualization, writing – original draft, writing – review & editing. SK: conceptualization, supervision, methodology, writing – original draft, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI† and five cifs are available at Zenodo at https://doi.org/10.5281/zenodo.14616175.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. K. acknowledges funding by the European Union (ERC, STARLET, 101124002). Views and opinions expressed are, however, those of the authors only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them.References
- G. P. Peters, R. M. Andrew, J. G. Canadell, P. Friedlingstein, R. B. Jackson, J. I. Korsbakken, C. Le Quéré and A. Peregon, Nat. Clim. Change, 2020, 10, 3–6 CrossRef
.
- J. F. Brennecke and B. E. Gurkan, J. Phys. Chem. Lett., 2010, 1, 3459–3464 CrossRef CAS
- M. Oschatz and M. Antonietti, Energy Environ. Sci., 2018, 11, 57–70 RSC
- L. B. Hamdy, C. Goel, J. A. Rudd, A. R. Barron and E. Andreoli, Mater. Adv., 2021, 2, 5843–5880 RSC
- S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC
- B. Yeskendir, J.-P. Dacquin, Y. Lorgouilloux, C. Courtois, S. Royer and J. Dhainaut, Mater. Adv., 2021, 2, 7139–7186 RSC
- R. Das, D. Muthukumar, R. S. Pillai and C. M. Nagaraja, Chem. – Eur. J., 2020, 26, 17445–17454 CrossRef CAS PubMed
- B. Arstad, H. Fjellvåg, K. O. Kongshaug, O. Swang and R. Blom, Adsorption, 2008, 14, 755–762 CrossRef CAS
- H. Li, K. Wang, D. Feng, Y. P. Chen, W. Verdegaal and H. C. Zhou, ChemSusChem, 2016, 9, 2832–2840 CAS
- Z. Lu, J. Bai, C. Hang, F. Meng, W. Liu, Y. Pan and X. You, Chem. – Eur. J., 2016, 22, 6277–6285 CAS
- S. Wang, H. Reinsch, N. Heymans, M. Wahiduzzaman, C. Martineau-Corcos, G. De Weireld, G. Maurin and C. Serre, Matter, 2020, 2, 440–450 CAS
- D. H. Hong and M. P. Suh, Chem. – Eur. J., 2014, 20, 426–434 CAS
- Y. W. Abraha, C.-W. Tsai, J. H. Niemantsverdriet and E. H. Langner, ACS Omega, 2021, 6, 21850–21860 CAS
- X. Wu, Z. Bao, B. Yuan, J. Wang, Y. Sun, H. Luo and S. Deng, Microporous Mesoporous Mater., 2013, 180, 114–122 CAS
- C. R. Wade and M. Dincă, Dalton Trans., 2012, 41, 7931–7938 CAS
- S. Waitschat, D. Fröhlich, H. Reinsch, H. Terraschke, K. Lomachenko, C. Lamberti, H. Kummer, T. Helling, M. Baumgartner and S. Henninger, Dalton Trans., 2018, 47, 1062–1070 CAS
- G. Avci, S. Velioglu and S. Keskin, J. Phys. Chem. C, 2019, 123, 28255–28265 CrossRef CAS
- J. Borycz, D. Tiana, E. Haldoupis, J. C. Sung, O. K. Farha, J. I. Siepmann and L. Gagliardi, J. Phys. Chem. C, 2016, 120, 12819–12830 CrossRef CAS
- G. Avci, C. Altintas and S. Keskin, J. Phys. Chem. C, 2021, 125, 17311–17322 CrossRef CAS PubMed
- Z. Qiao, N. Wang, J. Jiang and J. Zhou, Chem. Commun., 2016, 52, 974–977 RSC
- S. Li, Y. G. Chung, C. M. Simon and R. Q. Snurr, J. Phys. Chem. Lett., 2017, 8, 6135–6141 CAS
- S. Surblé, C. Serre, C. Mellot-Draznieks, F. Millange and G. Férey, Chem. Commun., 2006, 284–286 Search PubMed
- N. T. X. Huynh, O. K. Le, T. P. Dung and V. Chihaia, RSC Adv., 2023, 13, 15606–15615 CAS
- R. L. Martin, L.-C. Lin, K. Jariwala, B. Smit and M. Haranczyk, J. Phys. Chem. C, 2013, 117, 12159–12167 CAS
- M. Witman, S. Ling, S. Anderson, L. Tong, K. C. Stylianou, B. Slater, B. Smit and M. Haranczyk, Chem. Sci., 2016, 7, 6263–6272 CAS
- D. A. Gomez-Gualdron, O. V. Gutov, V. Krungleviciute, B. Borah, J. E. Mondloch, J. T. Hupp, T. Yildirim, O. K. Farha and R. Q. Snurr, Chem. Mater., 2014, 26, 5632–5639 CAS
- X. Wang, L. Zhang, J. Yang, F. Liu, F. Dai, R. Wang and D. Sun, J. Mater. Chem. A, 2015, 3, 12777–12785 CAS
- Y. Cui, B. Chen and G. Qian, Coord. Chem. Rev., 2014, 273, 76–86 Search PubMed
- B. Yan, Inorg. Chem. Front., 2021, 8, 201–233 CAS
- Y. Zhang, S. Liu, Z.-S. Zhao, Z. Wang, R. Zhang, L. Liu and Z.-B. Han, Inorg. Chem. Front., 2021, 8, 590–619 CAS
- Z. Qiao, K. Zhang and J. Jiang, J. Mater. Chem. A, 2016, 4, 2105–2114 CAS
- B.-B. Xing, Y.-S. Wang, T. Zhang, J.-Y. Liu, H. Jiao and L. Xu, Mater. Adv., 2025, 6, 756–765 CAS
- A. Zulys, F. Yulia, A. Buhori, N. Muhadzib, M. Ghiyats and B. B. Saha, J. Mater. Res. Technol., 2020, 9, 7409–7417 Search PubMed
- Q. Yao, A. Bermejo Gómez, J. Su, V. Pascanu, Y. Yun, H. Zheng, H. Chen, L. Liu, H. N. Abdelhamid and B. N. Martín-Matute, Chem. Mater., 2015, 27, 5332–5339 CAS
- Y. Chen and S. Ma, Rev. Inorg. Chem., 2012, 32, 81–100 CAS
- D. H. Le, R. P. Loughan, A. Gładysiak, N. Rampal, I. A. Brooks, A.-H. A. Park, D. Fairen-Jimenez and K. C. Stylianou, J. Mater. Chem. A, 2022, 10, 1442–1450 RSC
- Z. Lin, R. Zou, J. Liang, W. Xia, D. Xia, Y. Wang, J. Lin, T. Hu, Q. Chen and X. Wang, J. Mater. Chem., 2012, 22, 7813–7818 RSC
- S. Pal, A. Bhunia, P. P. Jana, S. Dey, J. Möllmer, C. Janiak and H. P. Nayek, Chem. – Eur. J., 2015, 21, 2789–2792 CrossRef CAS PubMed
- W. Mu, X. Huang, R. Zhong, W. Xia, J. Liu and R. Zou, CrystEngComm, 2015, 17, 1637–1645 RSC
- H. Daglar and S. Keskin, J. Phys. Chem. C, 2018, 122, 17347–17357 CrossRef CAS PubMed
- Y. G. Chung, E. Haldoupis, B. J. Bucior, M. Haranczyk, S. Lee, H. Zhang, K. D. Vogiatzis, M. Milisavljevic, S. Ling and J. S. Camp, J. Chem. Eng. Data, 2019, 64, 5985–5998 CrossRef CAS
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CrossRef CAS
- D. Dubbeldam, S. Calero, D. E. Ellis and R. Q. Snurr, Mol. Simul., 2016, 42, 81–101 CrossRef CAS
- A. K. Rappe and W. A. Goddard III, J. Phys. Chem., 1991, 95, 3358–3363 CrossRef CAS
- C. E. Wilmer and R. Q. Snurr, Chem. Eng. J., 2011, 171, 775–781 CrossRef CAS
- C. Altintas and S. Keskin, ACS Sustainable Chem. Eng., 2018, 7, 2739–2750 CrossRef PubMed
- Z. Qiao, C. Peng, J. Zhou and J. Jiang, J. Mater. Chem. A, 2016, 41, 15904–15912 RSC
- A. K. Rappé, C. J. Casewit, K. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef
- B. L. Eggimann, A. J. Sunnarborg, H. D. Stern, A. P. Bliss and J. I. Siepmann, Mol. Simul., 2014, 40, 101–105 CrossRef CAS
- C. Murthy, K. Singer, M. Klein and I. McDonald, Mol. Phys., 1980, 41, 1387–1399 CrossRef CAS
- P. P. Ewald, Ann. Phys., 1921, 369, 253–287 Search PubMed
- T. Vlugt, E. García-Pérez, D. Dubbeldam, S. Ban and S. Calero, J. Chem. Theory Comput., 2008, 4, 1107–1118 CrossRef CAS PubMed
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens and M. Towler, Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed
- D. S. Biovia, Dassault Systèmes: San Diego, BW, Release, 2017.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS
- A. D. Becke, Phys. Rev. A:At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed
- S. Kancharlapalli, A. Gopalan, M. Haranczyk and R. Q. Snurr, J. Chem. Theory Comput., 2021, 17, 3052–3064 CrossRef CAS PubMed
- T. A. Manz and D. S. Sholl, J. Chem. Theory Comput., 2010, 6, 2455–2468 CrossRef CAS PubMed
- K. Liu, H. Li, X. Zhang, W. Shi and P. Cheng, Inorg. Chem., 2015, 54, 10224–10231 CAS
- S. C. Bart, Inorg. Chem., 2023, 62(9), 3713–3714 CAS
- D. Ko, H. A. Patel and C. T. Yavuz, Chem. Commun., 2015, 51, 2915–2917 CAS
- D. Nazarian, J. S. Camp, Y. G. Chung, R. Q. Snurr and D. S. Sholl, Chem. Mater., 2017, 29, 2521–2528 CAS
- G. O. Aksu, I. Erucar, Z. P. Haslak and S. Keskin, J. CO2 Util., 2022, 62, 102077 CAS
- H. C. Gulbalkan, Z. P. Haslak, C. Altintas, A. Uzun and S. Keskin, Sep. Purif. Technol., 2022, 287, 120578 CAS
- D. Yancy-Caballero, K. T. Leperi, B. J. Bucior, R. K. Richardson, T. Islamoglu, O. K. Farha, F. You and R. Q. Snurr, Mol. Syst. Des. Eng., 2020, 5, 1205–1218 CAS
- O. F. Altundal, C. Altintas and S. Keskin, J. Mater. Chem. A, 2020, 8, 14609–14623 RSC
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030–3040 RSC
- J. Dai, D. Xie, Y. Liu, Z. Zhang, Y. Yang, Q. Yang, Q. Ren and Z. Bao, Ind. Eng. Chem. Res., 2020, 59, 7866–7874 CrossRef CAS
- N. Singh, S. Dalakoti, A. Sharma, R. Chauhan, R. S. Murali, S. Divekar and S. Dasgupta, Sep. Purif. Technol., 2024, 341, 126820 CrossRef CAS
- L. Asgharnejad, A. Abbasi and A. Shakeri, Microporous Mesoporous Mater., 2018, 262, 227–234 CAS
- Y. Chen, D. Lv, J. Wu, J. Xiao, H. Xi, Q. Xia and Z. Li, Chem. Eng. J., 2017, 308, 1065–1072 CAS
- I. Erucar and S. Keskin, J. Membr. Sci., 2016, 514, 313–321 CAS
- E. Haldoupis, T. Watanabe, S. Nair and D. S. Sholl, ChemPhysChem, 2012, 13, 3449–3452 CAS
- M. Cho, N. Sylvetsky, S. Eshafi, G. Santra, I. Efremenko and J. M. Martin, ChemPhysChem, 2020, 21, 688–696 CAS
- C. Cleeton, F. L. de Oliveira, R. F. Neumann, A. H. Farmahini, B. Luan, M. Steiner and L. Sarkisov, Energy Environ. Sci., 2023, 16, 3899–3918 CAS
- C. Altintas and S. Keskin, Mol. Syst. Des. Eng., 2020, 5, 532–543 CAS
- V. A. Basiuk, Comput. Theor. Chem., 2025, 115170 CAS
- P. G. Boyd, A. Chidambaram, E. García-Díez, C. P. Ireland, T. D. Daff, R. Bounds, A. Gładysiak, P. Schouwink, S. M. Moosavi and M. M. Maroto-Valer, Nature, 2019, 576, 253–256 CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ma00017c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |