
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decoupling the role of lignin, cellulose/hemi-cellulose, and ash on ZnCl2-activated carbon pore structure†
Chengjun
Wu
,
Graham W.
Tindall
,
Carter L.
Fitzgerald
,
Mark C.
Thies
and
Mark E.
Roberts
*
Department of Chemical and Biomolecular Engineering, 127 Earle Hall, Clemson University, Clemson, South Carolina 29634-0909, USA. E-mail: mrober9@clemson.edu; Tel: +1 864 656-6307
First published on 21st January 2025
Abstract
Activated carbon (AC), generally synthesized from fossil fuels or biomass waste, is a crucial form of porous carbon used for the purification of gases and liquids. Its key performance metrics vary widely when produced from biomass because of the differing amounts of cellulose, hemicellulose, lignin, and mineral/ash content. In this study, we adapted the Aqueous Lignin Purification using Hot Agents (ALPHA) process, originally developed for purifying lignin-rich waste streams, to control the sugars (as cellulose/hemicellulose) and mineral/ash content of a given biomass. Biomass samples having a wide range of sugars (0.01–56 wt%) and mineral/ash compositions (0.01–7.1 wt%) were generated from a single, hybrid poplar cultivar and used to create AC using ZnCl2-impregnation and low-temperature carbonization. Strong correlations were developed between the biomass sugars and mineral/ash composition and the AC surface area, pore size, and pore distribution, with the maximum surface area of 2500 m2 g−1 being obtained from the precursor with the highest level (56 wt%) of sugars. These findings may provide a path to predicting the properties of AC from biomasses encompassing a wide range of compositions, and furthermore, select AC precursors for target applications.
Introduction
Porous carbon materials, specifically activated carbon (AC) or active charcoal, are important for a wide range of applications, from water and gas purification1–3 to heterogeneous catalysis4 and electronics.5–7 AC is generally synthesized from carbon-rich sources, such as coal,8 biomass,9–11 wood,12 or decomposed organic matter,13via chemical or physical activation14 followed by carbonization (pyrolysis). The surface area and pore structure, two key metrics of the performance of AC, can be tailored by adjusting the activation method and conditions, along with the carbonization time and temperature.11–13,15,16 The ability to control these properties via various synthetic methods is important for optimizing performance, while simultaneously reducing production costs.Although coal has been a predominant precursor for AC,8 the use of biomass waste (e.g., coconut shells and rice husks) has been steadily increasing, as it provides a low-cost, renewable alternative.9–11 Biomass comprises three key biopolymers: cellulose, hemicellulose, and lignin, typically with a mass fraction of 40–50% cellulose, 15–30% hemicellulose, and 15–30% lignin.17–20 Because of the disparities in chemical composition and functionality of these natural polymers, it would be expected that biopolymer percentage would impact the surface area and pore structure of the resulting AC product; however, no clear correlations are available, and the properties of AC from these different materials can vary dramatically. Isolated lignin has also been proposed21 as a starting material for AC because of the substantial volume generated as a waste product from lignocellulosic biofuel production. When alkaline pretreatment of biomass is employed in a lignocellulosic biorefinery, two byproduct streams containing significant amounts of lignin are produced during the conversion of biomass to ethanol,22 namely, black liquor (a liquid) and a residual solid from enzymatic digestion, known as lignin cake. The US Department of Energy23,24 has substantial interest in isolating the lignin in these byproducts for applications requiring carbon-rich precursors, including AC, in order to add value to the biorefinery process. However, the use of isolated bulk lignin has shown limited performance metrics, with the highest surface areas reported to be 1500–1700 m2 g−1.21
The relationship between the biomass composition and AC surface area and pore structure is not well-understood. Previous work has shown that surface area and pore structure are dependent upon both the activation process (either physical or chemical) and biomass composition. For example, waste materials such as coconut shells and rice husks, which comprise 10 to 50 wt% lignin, can yield AC surface areas ranging from 166 to 3302 m2 g−1.3,10,25,26 However, even with relatively high performance values, further optimization is limited by a lack of control for key variables (particularly variation in both biopolymer and impurity content) when evaluating alternative activation agents and/or carbonization conditions. Limited research has addressed how biomass composition affects the properties of the AC, but the results are generally inconsistent due to unidentified variables.
For example, previous work with ZnCl2 activation27 reported that the surface area of AC gradually increased with the percent cellulose in the precursor, with a maximum of 1078 m2 g−1 obtained for 100% cellulose. However, the overall yield and absorption capacity did not correlate to cellulose content in a similar manner. The authors concluded that carbonization of cellulose leads to the mesoporous structure, while lignin contributes to the microporous structure. Unfortunately, this is contrary to our understanding of pore formation and development. In another study, which used potassium hydroxide (KOH) activation and low-temperature carbonization,28 researchers found that the highest surface area in AC was achieved with cellulose as the precursor, followed by lignin and then xylan. However, when the cellulose was mixed with the other biopolymers, adsorption properties decreased, and no correlation could be developed between the percentage of biopolymer and pore structure.
Phosphoric acid (H3PO4) activation work29 has indicated that the mineral (and thus ash) content of lignocellulosic biomass affects pore properties, resulting in a decrease in the AC surface area. The authors also found that the micropore volume of the natural biomass increased with the addition of a lignin, whereas the mesopore volume increased with the addition of cellulose and hemicellulose, which supports previous research27 that concludes how each biopolymer affects pore size.
When both physical activation (steam) and high-temperature pyrolysis were used, the microstructure of the AC was predicted based on the percentage of each biopolymer – cellulose, hemicellulose, and lignin – in the starting material.30 The authors concluded that lignin is the major contributor to char and carbon in final ACs. In another study31 that used pyrolysis with no chemical activating agent, the high level of oxygen-containing functional groups in cellulose and hemicellulose were shown to be eliminated as H2O, CO2, and CO to produce micropores, whereas lignin produced only nonporous materials. Even when KHCO3 was added before carbonization, the char yield was found to be essentially proportional to the mass ratio of lignin, but there was no obvious trend for the surface area of the char from the mixtures.31
With no clear relationship and often contradictory results on how biomass composition and the mineral/ash content of biomass affect AC surface area and pore structure, we have adapted a process originally developed for the fractionation and purification of lignin (i.e., aqueous lignin purification with hot agents (ALPHA))32–35 – and are now using it to control the composition of biomass-based, AC precursors in a well-defined manner. The essence of ALPHA is as follows: when a hot renewable solvent that forms one phase with water (e.g., acetic acid or ethanol) is contacted with a raw bulk (i.e., impure) lignin, two liquid phases are created, with the three biomass biopolymers and mineral/ash impurities all distributing between these two phases. Partitioning of these components between the two phases can vary dramatically, depending on the biopolymer type and impurity, and can be controlled by organic solvent/water ratio, total solvent/lignin ratio, and temperature.35,36 Although the ALPHA process was developed for recovering lignins of very high purity and molecular weight, in this work we ironically use ALPHA to control, that is, both decrease and increase, the biopolymer (e.g., cellulose, hemi-cellulose, and lignin) and mineral/ash content in the final biomass sample.
The starting biomass for this study was hybrid poplar wood chips derived from a single cultivar. Two diverse biomass feedstocks were subsequently isolated from the wood chips, following an alkaline pretreatment step: (1) a low-purity lignin containing significant levels of all three biopolymers plus mineral/ash and (2) a cellulose-rich lignin cake. By applying ALPHA to these feedstocks, biomass samples encompassing a wide range of sugars and mineral/ash compositions were isolated and used to synthesize AC using ZnCl2 impregnation and low-temperature carbonization. As a result, we have been able to develop clear relationships between biomass composition (i.e., cellulose, hemicellulose, lignin, and mineral/ash content) and the key performance metrics of AC (surface area, pore volume, pore size, and fractional conversion of carbon). Our findings may provide a path to predicting the properties of AC from diverse biomass material; and furthermore, these correlations may eventually help guide the selection of AC precursors for target applications.
Experimental
Biomass sample preparation (see Fig. 1)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 liquor-to-wood ratio by weight. The reactor vessel was heated to 150 °C and held for 3 h, after which the vessel was cooled to <100 °C prior to opening. The resulting alkaline black liquor was decanted from the residual solids. The pH of the liquor was adjusted to 2.0 through the addition of 96.6 wt% sulfuric acid, and the bulk lignin feed (BLF; ∼95 wt% lignin) solids precipitated from the black liquor. The resulting slurry was centrifuged for 2 h at 8000 rpm, the supernatant was decanted, and DI water was added (lignin:water ratio of 1:5 by mass). The process of centrifuging and washing was repeated, and the BLF solids were dried under vacuum at 50 °C for 24 h.
:lignin mass ratio of 3:1 in a customized version of a Model 4541, 2-L Parr reactor. The reactor and contents were heated to 60 °C with stirring, with these conditions resulting in the formation of two liquid phases, αLR and αLP (see Fig. 1).35 To ensure that the solid impurities did not inadvertently contaminate αLP, the contents of the vessel were vacuum-filtered (Grade 4 filter paper, Whatman), resulting in retentate (i.e., αLR) and filtrate (αLP) streams. Both streams were dried (to remove the solvent) under vacuum at 50 °C for 12 h and then atmospheric pressure and 120 °C for 2 h. ALPHA Stage 1 was performed on three different batches of BLF having different impurities (i.e., polysaccharide and mineral/ash) levels to obtain αLR-a, αLR-b, αLR-c. For all three of these batches, biopolymer and impurities compositions in the αLP phase were essentially the same, so all three were combined to create a single sample.
:1 liquor-to-wood ratio by weight. The reactor vessel was heated to 150 °C and held for 3 h, after which the vessel was cooled to <100 °C prior to opening. The resulting alkaline black liquor was decanted from the residual solids. The pH of the liquor was adjusted to 2.0 through the addition of 96.6 wt% sulfuric acid, and the bulk lignin feed (BLF; ∼95 wt% lignin) solids precipitated from the black liquor. The resulting slurry was centrifuged for 2 h at 8000 rpm, the supernatant was decanted, and DI water was added (lignin:water ratio of 1:5 by mass). The process of centrifuging and washing was repeated, and the BLF solids were dried under vacuum at 50 °C for 24 h.
:lignin mass ratio of 3:1 in a customized version of a Model 4541, 2-L Parr reactor. The reactor and contents were heated to 60 °C with stirring, with these conditions resulting in the formation of two liquid phases, αLR and αLP (see Fig. 1).35 To ensure that the solid impurities did not inadvertently contaminate αLP, the contents of the vessel were vacuum-filtered (Grade 4 filter paper, Whatman), resulting in retentate (i.e., αLR) and filtrate (αLP) streams. Both streams were dried (to remove the solvent) under vacuum at 50 °C for 12 h and then atmospheric pressure and 120 °C for 2 h. ALPHA Stage 1 was performed on three different batches of BLF having different impurities (i.e., polysaccharide and mineral/ash) levels to obtain αLR-a, αLR-b, αLR-c. For all three of these batches, biopolymer and impurities compositions in the αLP phase were essentially the same, so all three were combined to create a single sample.
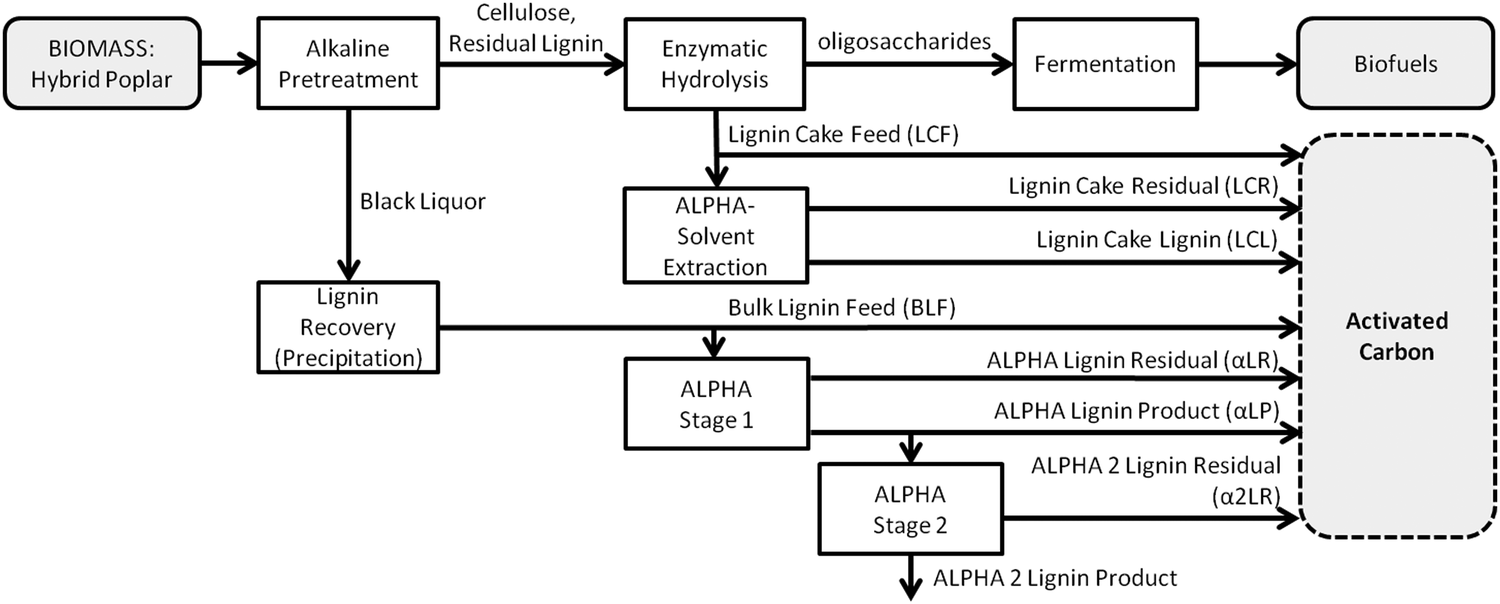 | ||
| Fig. 1 Flowsheet depicting the production of activated carbons from two, lignin-rich byproduct streams of a lignocellulosic biorefinery: bulk lignin feed (BLF) and lignin cake feed (LCF). Seven additional biomass streams for AC production were generated by applying ALPHA-based fractionation to BLF and LCF. | ||
:lignin mass ratio of 3:1.32,36 After heating to 60 °C with stirring, lignin-rich (α2LR) and solvent-rich (α2LP) phases were formed as described above, see Fig. 1. However, in this case no discernible insoluble impurities were present in the lignin-rich phase, obviating the need for a filtration step; thus, the solvent-rich phase was simply decanted off. A2LR was dried under vacuum at 50 °C and then under atmosphere at 120 °C. α2LP was not further used in this study.
:LCF39 mass ratio of 9:1, with 5 g of sulfuric acid/100 g of LCF added as a catalyst. A 50-ml reactor (model 18553, Parr) were used to carry out the experiments. After heating to 200 °C and stirring for 1 h, the reactor was allowed to cool to 65 °C before the contents were separated via vacuum filtration (Grade 4 filter paper, Whatman). The retentate and filtrate were dried under vacuum at 50 °C for 12 h and then under atmosphere at 120 °C for 2 h. The dried retentate is referred to as lignin cake residue (LCR) and the dried filtrate is referred to as lignin cake lignin (LCL).
Biomass sample characterization.
Polysaccharide (as sugars) analysis was performed using an excerpt from NREL/TP-510-42618.40 Samples of nominally 100 mg were added to 35-mL, pressure-rated vials with 72% sulfuric acid. The vials were then held at 30 °C for 1 h with stirring every 10 min. Next, 28 mL of DI water were added to the vials, which were then capped and placed in an autoclave set to 121 °C for 1 h. After cooling, a portion of the (homogeneous) supernatant was filtered through a 0.22 μm PTFE syringe filter. This digested filtrate was then run through a high-performance liquid chromatography (HPLC) system with an Aminex HPX-87H stationary phase maintained at 65 °C and a mobile phase of 5 mM sulfuric acid in DI water at 0.6 mL min−1. Detection of sugars was performed using a Waters 2414 Refractive Index Detector, with external calibration performed using standard solutions of xylose, glucose, and arabinose.Ash content in a given biomass sample (as an indirect measurement of mineral/metals content) was determined via thermogravimetric analysis (TGA). ∼10 mg of sample were placed in a platinum holder of an automatic sample processer (Q5000, TA Instruments), heated to 100 °C, held for 15 min, and then heated to 800 °C. The purge gas was air and the heating ramp was 10 °C min−1.
Metal contents of the lignin and lignin fractions were measured via Inductively coupled plasma atomic emission spectroscopy (ICP-AES, model ACROS, spectro analytical instruments). Before measurement, a weight of 100 mg dried lignin was digested in 5 mL concentrated nitric acid at 25 °C for 30 min and then further digested by heating to 125 °C for 90 min, followed by adding 3 mL hydrogen peroxide (H2O2) and heating at 125 °C for 60 min. Afterward, 3 mL additional H2O2 was added, and the sample was kept heated at 125 °C for 60 min. Finally, the sample was air dry at 200 °C for 1 h and the dried sample was diluted in 10 mL 1.6 M nitric acid and another 50 mL deionized water after cooling. The resulting liquid was transferred to the ICP tube for detection.
The molecular weight (MW) of lignin-rich samples was determined using SEC-MALS (size exclusion chromatography, followed by multi-angle light scattering). Lignin samples containing <2 wt% sugars were first dissolved at a nominal concentration of 1.5 mg mL−1 in a solution of 0.05 M LiBr in dimethyl formamide (DMF). Samples were sonicated for approximately 1 h and filtered through 0.22 μm PTFE syringe filters. The samples were then injected into an Agilent 1200 series HPLC system, with a 0.05 M LiBr in DMF mobile phase, flowing at 0.6 mL min−1. A stationary phase of one HT5 Styragel (WAT045945, Waters) followed by one Polargel-L (PL1117-6830, Agilent) was used for separation, in conjunction with an Optilab-WREX-08 differential refractometer and a Wyatt Technology DAWN MALS instrument (with filtered detectors) used for detection.
Activated carbon synthesis
1.8 g of a given biomass sample was dried in a vacuum oven at 60 °C for 12 h, and then sieved through a 60-mesh sieve. The biomass sample was mixed with a ZnCl2 solution (made by combining ZnCl2 (anhydrous, 98 + %, Alfa Aesar)) at a ratio of 2:1 by weight (ZnCl2:biomass) and enough DI water to achieve a ratio of 2.0 mL water per gram of total solids. The mixture was stirred at 350 rpm for 24 h. The ZnCl2/biomass solution was then dried by rotary evaporation and placed under vacuum at 110 °C for 24 h. Next, the ZnCl2-impregnated biomass was packed in graphite foil and placed in the center of a horizontal quartz tube in an electric furnace (Lindberg/Blue M, Thermo Scientific). The tube was purged with high-purity (+99.99%) nitrogen (N2) at a flow rate of 1000 cm3 min−1 for 30 min. For carbonization, the N2 flow was adjusted to 300 cm3 min−1, and the sample was heated at 10 °C min−1 to 500 °C and held for 1 h. After cooling to ambient, the AC was washed with 150 mL 3 M hydrochloric acid (HCl) for 1 h while stirring at 350 rpm, filtered (0.45 μm, Nylon filter, Sigma Aldrich), and then repeatedly rinsed with 60 °C DI water and filtered until the conductivity of the washing water was near the conductivity of the DI water (∼0.6 μS cm−1). The sample was then dried in vacuum at 110 °C for 24 h.
Activated carbon characterization: N2 sorption analysis
N2 adsorption and desorption isotherms of AC were measured at 77 K by using an automated gas sorption analyzer (Autosorb iQ, Quantachrome Instruments), after samples of approximately 150 mg were degassed at 250 °C under vacuum. Using Brunauer–Emmett–Teller (BET) theory, the gas sorption behavior was connected to the porosity of the AC. Total pore volume was determined by the amount of N2 adsorption expressed in liquid form at 77 K and a relative pressure (P/P0) of approximately 0.95. The average pore diameter was calculated based upon the specific surface area data and the total pore volume by the Gurwitch rule. Pore size distribution data were obtained by analyzing N2 desorption data using density functional theory (DFT).Carbon fractional conversion
Carbon fractional conversion is defined as the mass of carbon in AC divided by the mass of carbon in the starting biomass sample. These conversions in biomass and AC samples were determined by elemental analysis of combustion products using automatic analyzers in Atlantic Microlab, Inc (Atlanta, Georgia, USA). Overall yield is defined as the mass of AC divided by mass of biomass sample. In preparation for the above analyses, biomass samples were vacuum-dried (0.015 mm Hg) at 120 °C for 2 h, and AC samples were vacuum-dried (0.015 mm Hg) at 250 °C for 4 h.Results and discussion
Precursors for activated carbon
The overall flowsheet for producing biomass samples of varying lignin, sugars, and ash content as activated carbon (AC) precursors is depicted in Fig. 1. The generation of biomass samples encompassing a wide range of biopolymer and metals/ash content was made possible by two factors: (1) the multi-step process for generating bulk lignin feed (BLF) from the starting HP wood chips resulted in significant batch-to-batch variation in BLF composition; (2) by using ALPHA separations technology to induce a liquid–liquid phase split, the sugars and impurities content of the starting BLF could be enhanced up to 10× in a given generated sample of αLR. In this work three different BLF batches were used to generate αLR-a, αLR-b, and αLR-c. Incidentally, for all three BLF batches the 2nd liquid phase of the phase split, namely αLP, was always comprised of >99% lignin (dry basis) – and so was of lesser interest in this work. However, such high-purity lignins have served as precursors for carbon fibers, as described elsewhere.35Extracting the lignin cake feed LCF with an ALPHA-based solvent produced a 97% pure lignin cake lignin LCL,39 which was also of lesser interest for this work. However, the solid residue LCR remaining after this extraction, being rich in both sugars and lignin, served as a useful precursor for AC.
To produce a biomass sample with very low ash and sugar composition, a second stage of ALPHA was carried out on biomass sample αLP, generating α2LR. The lignin present in α2LP was not used. In summary, then, a total of nine samples with varying sugars and ash content, which included the feed streams BLF and LCF, were used to synthesize AC.
Biomass sample characterization
Fig. 2(a) shows the sugar composition of the biomass samples via high-performance liquid chromatography (HPLC), along with the corresponding metals/ash content by thermal gravimetric analysis (TGA). Here we see how access to the aforementioned lignin byproduct streams from the conversion of lignocellulosic biomass to ethanol provides a set of biomass samples with a wide range of sugar compositions. The lignin cake feed (LCF) stream has a much higher sugar composition (26.9 wt%) than the bulk lignin feed (BLF, 2 wt%) because it contains residual sugars from the enzymatic digestion process. However, by applying ALPHA to BLF, we see that the three αLR fractions a–c have 10.2, 15.9, and 28.8% sugars content, thus supplying us with a wide range useful for evaluating the effect of sugars on AC performance. On the other hand, the αLP and α2LR fractions from ALPHA, with their very low sugars and ash content, are useful in this work as a reference point, that is, to assess the ability of ultrapure lignins (99 + % pure, <0.05% sugars and <0.1% ash) to serve as precursors for AC. | ||
| Fig. 2 (a) Thermogravimetric analysis curves of biomass samples with different sugar compositions. (b) Thermogravimetric analysis curves of biomass samples with different ash contents. (c) Sugar compositions and ash contents (inset) of biomass samples. (d) Overall yield (mass of AC/mass of biomass) and carbon fractional conversion (mass of C in AC/mass of C in biomass) as a function of sugar composition in the biomass sample. BL and LC refer to the biomass samples from the black liquor and lignin cake streams, respectively. | ||
With respect to the ALPHA-solvent extraction of LCF, the lignin-rich (97%) extract LCL contained only 1.7% sugars. Thus, virtually all of the sugars in LCF were concentrated into the unextracted residual solids LCR. As a result, LCR had the highest sugars content (55.6%) of all biomass samples. Finally, it is noteworthy that 90% of the sugars in samples derived from BLF were hemicellulose (xylan), while >85% of the sugars in samples derived from LCF were cellulose (glucan). Table S1 in ESI† presents additional information about the types of sugars in each biomass sample.
The metals/ash content of each biomass sample is shown in the Fig. 2(a) inset. Note that, like the sugars, the metals/ash were highly concentrated (to over 7%) in the residual (i.e., αLR-x and LCR) phases by ALPHA, even though the starting lignins, BLF (0.3%) and LCF (1.3% ash), contained only moderate levels of ash. With the 2-stage ALPHA stream (α2LR) containing ∼0.01% ash, we thus had a wide range of metals/ash levels to evaluate how these impurities affect AC properties. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) was used to determined the primary source of metals in the biomass samples, along with the sulfur content (ESI,† Fig. S#). Samples had less than 0.1 wt% S and the primary metal in the black liquor was sodium (∼0.1 wt% in BLF) and the main metal in the lignin cake was calcium (∼0.5 wt% in the LCF). In summary and most importantly, we had a set of biomass samples in which sugars and ash contents varied independently of each other, that is, (1) biomass samples encompassing a wide range of sugars content and (2) low-sugar samples with varying ash content – and with all samples from the same HP source material. Such a sample set was critical to separately isolating the effect of sugars and ash content on AC properties.
Activated carbon overall yield and carbon fractional conversion
AC was synthesized from biomass samples using a constant ZnCl2 activation method with low-temperature carbonization. ZnCl2 activation, compared to other common methods such as physical activation and KOH activation, requires a much lower processing temperature (500 °C) to optimize surface area and pore structure.41 Unlike phosphoric acid activation, ZnCl2 activation does not introduce additional functional groups on the surface of the AC. Similar studies suggest that an impregnation ratio of 2 is optimal for achieving high performance AC from biomass and lignin; therefore, the same ratio was used in this study.21,42Fig. 2(b) shows the carbon fractional conversion and AC overall yield plotted as a function of sugar composition. Raw data for the wt% carbon in all biomass and AC samples can be found in the ESI,† Fig. S1. Because the sugar components (hemicellulose and cellulose) in the biomass precursor have a higher percentage of oxygen and hydrogen relative to lignin, we expected (1) a higher overall AC yield from samples enriched with lignin and (2) a higher carbon fractional conversion because lignin is more thermally stable and recalcitrant.43,44 Although the former expectation is obvious due to the chemical functionality (elemental composition) of the components, the latter is less clear because of inconsistent conclusions in the literature.
As expected, the overall yield decreased monotonically with increasing sugar composition because the sugar has more non-carbon species that do not remain in the final AC product. From elemental analysis of the combustion products, we observed a fairly consistent composition of carbon in all ACs, ranging between 87–89%, which did not correlate with the starting carbon composition of the biomass sample. This further confirms that non-carbon species are released as volatiles and do not remain in the final AC product. Because the elemental analysis method only measures carbon and hydrogen content – and the hydrogen composition in all AC samples is approximately 2%. – it is assumed that the remaining content is primarily oxygen (consistent with 500 °C activation/carbonization).
The maximum carbon fractional conversion achieved was nearly 81%, and it's likely this value would be higher (closer to 85%) if we corrected for the adsorbed water in the final AC product. Importantly, all carbon conversions observed were higher than 65%, because using ZnCl2 as the activating agent inhibits both gasification of the carbonaceous material and tar formation in the carbonized material while also allowing for the use of lower temperatures for carbonization.45–47 As shown in Fig. 2(b), we found that the carbon fractional conversion is low for samples with less than 2 wt% sugar and significantly increases when the sugar composition increases up to 10 wt% after which the conversion remains fairly constant. This was a surprising result, as conventional wisdom suggests that lignin is the primary char formed in the final AC.21 When activated carbon was previous synthesized using low-temperature carbonization, the authors found that a lower amount of carbon was burned off when the samples were rich in sugar compared to those that were nearly pure lignin.48–51
When sugars, such as hemicellulose and cellulose, are pyrolyzed at low temperatures (500 °C), deoxygenation reactions result in the formation and release of more oxygen-containing volatiles such as H2O, CO2, and CO from the cracking of hydroxyl, carboxyl, and carbonyl groups.48–51 During these reactions, only very few short-chain hydrocarbons are released during pore formation and growth, resulting in more carbon remaining in the final product. Pyrolysis of lignin, on the other hand, results in a significantly higher release of H2, CH4, or other short-chain hydrocarbons from the cracking of C–C, C–H, and alkoxy groups during pore formation and growth.48–51 This means that more carbon bonds are broken during carbonization of lignin; thus, a lower carbon fractional conversion is obtained for biomass samples enriched in lignin. When the sugar composition was greater than 30 wt%, the carbon fractional conversion was fairly constant because enough sugars are present to promote pore formation. The influence of sugar composition on pore formation and growth will be discussed in more detail in the next section.
Pore structure of activated carbon
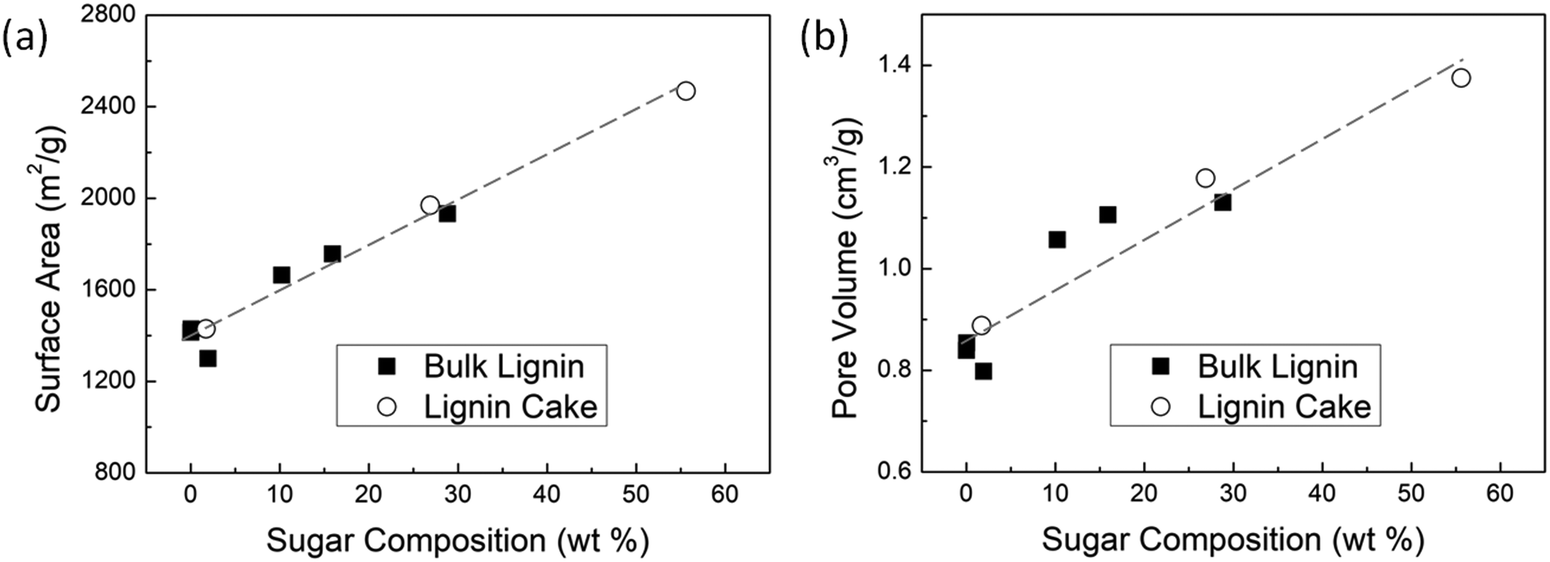 | ||
| Fig. 3 (a) Specific surface area and (b) total pore volume of activated carbon as a function of sugar composition in the biomass sample. | ||
The correlation between AC surface area and sugar composition of the biomass sample is highly linear, regardless of whether the lignin precursor sample came from the bulk lignin feed or the lignin cake remaining after enzymatic digestion. Because the samples from the black liquor (bulk lignin) contain primarily hemicellulose, and those from the lignin cake contain primarily cellulose, the type of sugar appears to be insignificant in comparison to the amount of sugar in the sample. We found that the surface area varied from 1500 to 2500 m2 g−1 for biomass samples ranging from 0.02% (i.e., essentially pure lignin) up to 56% sugar, which is consistent with literature observations27,52 that the surface area of AC from pure lignin is consistently lower than that of lignocellulosic biomass.
A similar linear correlation was found between AC pore volume and sugar composition. Pores are more readily formed during the carbonization of biomass samples rich in sugars because of the differences in structure and thermal reactivity of cellulose/hemicellulose relative to lignin. The sugars are comprised of aliphatic groups with an abundance of oxygen which, during pyrolysis, are readily converted to various oxygen-containing volatiles and released in the off-gas, thus leaving behind many small pores. Lignin, on the other hand, has a highly aromatic structure with less oxygen, and because it is less reactive than aliphatic sugar molecules, pore development tends to favor pore enlargement over pore formation. Once a pore has formed and disrupted the aromatic structure, reactions on the disrupted carbon system are more favorable than creating new pores, resulting in pore enlargement rather than pore formation, and thus generally leading to relatively lower surface area. In terms of how the sugars affect the resulting AC, it doesn’t matter whether they are cellulose or hemicellulose. For example, AC synthesized from αLR-c (28.8%, mostly hemicellulose) and LCF (26.9%, mostly cellulose) have a similar surface area and pore volume. Even when using a different activation agent (KHCO3), Deng et al.31 also reported that AC synthesized from cellulose or hemicellulose under low-temperature carbonization at 400 °C display similar pore morphologies.
Fig. 4 shows the average pore width of the ACs as a function of sugar composition (panel a) and ash content (panel b) of the biomass sample. Over the full range of sugar composition, the average pore width generally decreases as the sugar composition increases, particularly when the sugar composition is above 10%. This observation indicates that the presence of sugars result in the development of smaller pores (micropores) during carbonization, contradicting various reports27,29 which suggest cellulose is the key component for forming mesopores, while lignin is the main contributor to micropore formation when using ZnCl2 or H3PO4 activation. As discussed above, pore formation readily occurs in biomass samples containing sugars, but pore enlargement is more favorable in lignin because fewer oxygen-containing groups are present, and it is easier to break a disrupted carbon bond in an existing pore than to form a new pore by breaking an unperturbed bond.
 | ||
| Fig. 4 AC average pore width as a function of (a) sugar composition and (b) ash content of the biomass sample. | ||
However, as seen in Fig. 4(a), the average pore width appears to increase with increasing sugar composition when the sugar composition is below 10%. This is because ash content also affects the pore width, but up to now literature results have been mixed. In this study, we used not only the same biomass source (HP) but even the same cultivar; furthermore, we had available the ALPHA process to control sugars and ash content in a well-behaved manner. Thus, as shown below, the effect of each biomass component is clear.
Fig. 4(b) shows the average pore width as a function of ash content. Except for the samples of sugar compositions in excess of 20%, the average pore width of AC increases with increasing ash content. We have already demonstrated in Fig. 4(a) that the pore width decreases with increasing sugar content over a wide range of sugar composition. Furthermore, using ALPHA we were able to isolate lignin samples with both very low sugar composition and varying ash impurities. Pores are more easily formed and enlarged in higher-ash samples because the minerals weaken the intermolecular forces between the biopolymers. These observations are consistent with previous literature,42 which demonstrates that the presence of ash blocks the chemical-activating agents from penetrating into the particles. Therefore, the pores formed on the exterior of the biomass particles undergo pore enlargement in the presence of the excess ZnCl2. Furthermore, metals such as Na (or Ca) can act as catalysts during the carbonization process to accelerate thermal decomposition of carbohydrates and thus enlarge existing pores.48,53
In summary, the average pore width in the AC increases as the mineral/ash content in the biomass precursor increases. The pore width decreases with increasing sugar composition because pore formation readily occurs due to the aliphatic groups with an abundance of oxygen. Only our ability to decouple sugars and ash content, using our unique set of biomass samples, has made it possible for us to arrive at the above conclusion.
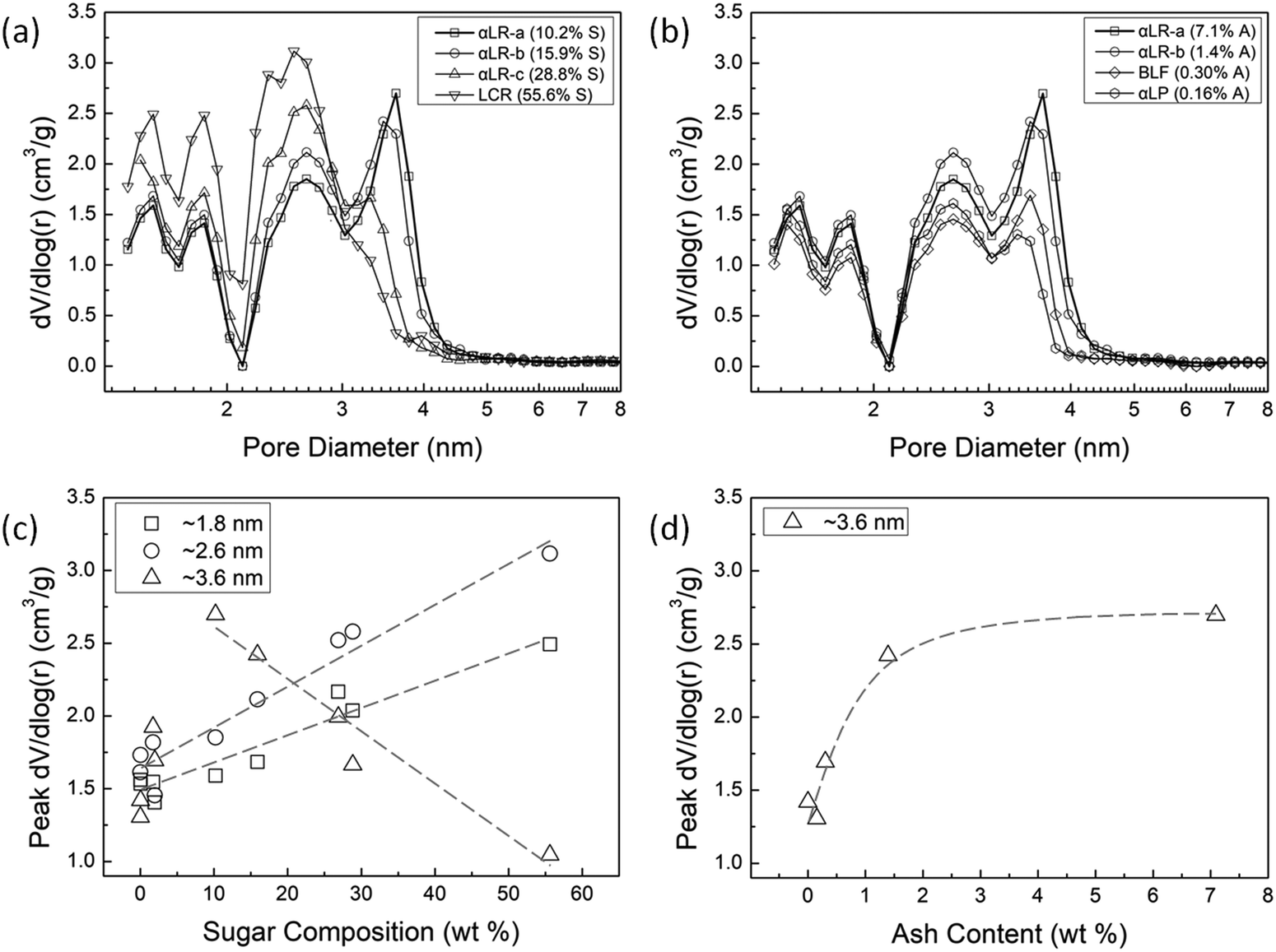 | ||
| Fig. 5 Pore size distributions in AC from biomass samples (determined from the differential of pore volume with respect to log(radius)) with (a) varying sugar composition and (b) varying ash content. Peak values of differential pore volume as a function of (c) sugar composition at pore diameters of ∼1.8 nm, ∼2.6 nm, and ∼3.6 nm and (d) ash content at a pore diameter of ∼3.6 nm. | ||
Fig. 5(a) shows the pore size distributions for selected ACs synthesized from samples with increasing sugar composition. Four peaks in pore volume (i.e., ∼1.5 nm, ∼1.8 nm, ∼2.6 nm, and ∼3.6 nm) were observed, indicating the primary pore diameters within the AC. In other words, a peak value in “the rate of change in pore volume” around a specific diameter indicates that the AC has many pores of this size. Therefore, we can equate the relative size of the peak in dV/dlog(r) to the number of pores with the corresponding pore diameter on the abscissa. For example, AC synthesized from LCR, the biomass with the highest sugar (55.6%) composition, has a high number of pores with a diameter of ∼1.5 nm, ∼1.8 nm, and ∼2.6 nm, but few pores with a diameter of ∼3.6 nm. As the sugar composition in the biomass precursor decreases (and the lignin increases), we observe the following trends in pore diameter: the number of micropores (<2.0 nm) and small mesopores (∼2.6 nm) decreases, while the number of mesopores (∼3.6 nm) increases. Because we observe more micropores and small mesopores with increasing sugar content, we can conclude that the mechanism for pore development is governed by pore formation rather than pore enlargement. In fact, for AC made from samples with the highest sugar compositions, we see little contribution from the larger mesopores to the total pore volume, see Fig. 5(a). However, as the composition of lignin increases (and that of sugar decreases), the peak in dV/dlog(r) around the larger mesopore diameter of ∼3.6 nm emerges, and even becomes a maximum for biomass samples in which lignin dominates. As discussed above, pore formation is not favorable in lignin, so excess ZnCl2 is available and promotes pore enlargement around the existing pores. Furthermore, as shown in Fig. 5(b), when sugars content drops below 20%, mineral/ash content in the biomass sample becomes important. Specifically, as ash content increases, we observe a significant increase in the peak for larger mesopores (∼3.6 nm). This behavior occurs because the activating agent ZnCl2 accumulates on the surface of lignin-rich/sugars-poor particles, rather than penetrating inside. As no change in the number of micropores and small mesopores with increasing ash content is observed, we conclude that primarily pore enlargement via the mechanism proposed above is occurring.
Peak values in differential pore volume are plotted vs. sugars composition in Fig. 5(c) and vs. ash content in Fig. 5(d) for various pore diameters in order to show the trends in pore size distribution. Because the peaks at ∼1.5 nm and ∼1.8 nm are essentially identical, only the peak values at ∼1.8 nm for clarity. Fig. 5(c) indicates that there is a linear increase in the number of micropores (<2.0 nm) and small mesopores (∼2.6 nm) with increasing sugar composition, and a corresponding decrease in large mesopores (∼3.6 nm) as lignin levels decrease. The trend in the number of larger mesopores (Fig. 5(d)) is similar to the trend observed in Fig. 4(b) for average pore diameter with increasing ash content, when we factor out those biomass samples containing >20% sugars. However, the ability of ash content to effect pore enlargement is essentially ameliorated as the sugars content of a biomass sample exceeds 20% sugars. Fig. S2 in the ESI† shows the peak in dV/dlog(r), an indicator of the number of pores, for AC synthesized from biomass samples with nearly 30% sugar and relatively high ash content. Samples with marginally higher sugar (28.8 vs. 26.9%) have a fewer number of large mesopores (3.6 nm) even though they have over double the ash content (2.7 vs. 1.3%). If ash content was the primary driver of pore formation, the trend observed would be reversed.
The effect of lignin molecular weight on pore properties of activated carbon
Multiple, high-purity (96.5–99.9% pure) lignin samples with a sugar composition below 2 wt% were generated and used as AC precursors. These samples were comprised of lignin with narrow MW distributions and having number average MWs ranging from 11800 Da for Lignin Cake Lignin (LCL) to 18200 Da for 1st-stage ALPHA Lignin Product (αLP) and 22,740 Da for 2nd-stage ALPHA Lignin Residue (α2LR). Even with this variation, the surface area, pore volume, and pore diameter of AC synthesized from these samples were quite similar, exhibiting overlapping data points in Fig. 3. Therefore, we can conclude that lignin MW has little influence on pore formation and development in AC relative to the sugars and ash content. However, significantly lower (2000 Da) and higher (52000 Da) MW materials have been isolated via ALPHA technology,32,35 and these may be worth investigating.
Conclusions
Using chemical activation with ZnCl2 and low-temperature carbonization, AC was synthesized from a hybrid poplar cultivar, with the sugars and mineral/ash content of said biomass being controlled by the ALPHA process. We showed that sugars content of a given biomass sample is the primary driver that governs the surface area and pore structure of AC. Positive linear correlations were found between both the surface area and total pore volume vs. sugar composition of the biomass precursor for AC, as pore formation governs the pore-development mechanism of the sugars. For example, an AC with a surface area of 2500 m2 g−1 was achieved when the biomass precursor with the highest sugar composition (55.6%) was used. For precursors comprising mostly lignin, the surface area does not vary significantly; however, higher ash content promotes pore enlargement. In addition, we showed that, somewhat unexpectedly, carbon fractional conversion increases when precursors with higher sugars composition are used as the AC starting material. As a result of this work, AC surface area and pore sizes (and distribution) can now be predicted based on the sugars and mineral/ash composition of a given biomass. Furthermore, these correlations may be used to guide the selection of AC precursors for target applications, assuming a given constant activation/carbonization process. This newly achieved ability to tailor surface area, and pore size and distribution, is important for creating AC for various applications, such as gas or liquid purification.Data availability
The data supporting this article have been included in the article and as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based on work supported by the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) (under Bioenergy Technology Office award no. DE-EE0008502). Graham Tindall would also like to acknowledge support by the Department of Education under the Graduate Assistance in Areas of National Needs Fellowship (GAANN) Fellowship (award no. P200A180076). The authors would like to thank Prof. David Hodge's group (including Villo Becsy-Jakab and Ryan Stoklosa) at Chemical and Biological Engineering, Montana State University, for generating the bulk lignin feed recovered via alkaline pretreatment and the lignin cake byproduct from enzymatic hydrolysis. At Clemson University, Zachariah Pittman is acknowledged for his MW work via SEC-MALS, and Bronson Lynn and Oreoluwa Agede for their efforts in sugar analysis and HPLC.References
- J. M. Dias, M. C. Alvim-Ferraz, M. F. Almeida, J. Rivera-Utrilla and M. Sánchez-Polo, Waste Materials for Activated Carbon Preparation and Its Use in Aqueous-Phase Treatment: A Review, J. Environ. Manage., 2007, 85(4), 833–846 CrossRef.
- A. Bhatnagar, W. Hogland, M. Marques and M. Sillanpää, An Overview of the Modification Methods of Activated Carbon for Its Water Treatment Applications, Chem. Eng. J., 2013, 219, 499–511 CrossRef.
- N. M. Nor, L. C. Lau, K. T. Lee and A. R. Mohamed, Synthesis of Activated Carbon from Lignocellulosic Biomass and Its Applications in Air Pollution Control—a Review. Journal of Environmental, Chem. Eng., 2013, 1(4), 658–666 Search PubMed.
- P. Serp and J. L. Figueiredo, Carbon Materials for Catalysis, John Wiley & Sons, 2008 Search PubMed.
- E. Frackowiak, Carbon Materials for Supercapacitor Application, Phys. Chem. Chem. Phys., 2007, 9(15), 1774–1785 RSC.
- M. Sevilla and R. Mokaya, Energy Storage Applications of Activated Carbons: Supercapacitors and Hydrogen Storage, Energy Environ. Sci., 2014, 7(4), 1250–1280 RSC.
- A. M. Abioye and F. N. Ani, Recent Development in the Production of Activated Carbon Electrodes from Agricultural Waste Biomass for Supercapacitors: A Review, Renewable Sustainable Energy Rev., 2015, 52, 1282–1293 CrossRef.
- A. Ahmadpour and D. D. Do, The Preparation of Active Carbons from Coal by Chemical and Physical Activation, Carbon, 1996, 34(4), 471–479 CrossRef.
- O. Ioannidou and A. Zabaniotou, Agricultural Residues as Precursors for Activated Carbon Production—A Review, Renewable Sustainable Energy Rev., 2007, 11(9), 1966–2005 CrossRef.
- A. Jain, R. Balasubramanian and M. P. Srinivasan, Hydrothermal Conversion of Biomass Waste to Activated Carbon with High Porosity: A Review, Chem. Eng. J., 2016, 283, 789–805 CrossRef.
- M. A. Yahya, Z. Al-Qodah and C. Z. Ngah, Agricultural Bio-Waste Materials as Potential Sustainable Precursors Used for Activated Carbon Production: A Review, Renewable Sustainable Energy Rev., 2015, 46, 218–235 CrossRef.
- M. Danish and T. Ahmad, A Review on Utilization of Wood Biomass as a Sustainable Precursor for Activated Carbon Production and Application, Renewable Sustainable Energy Rev., 2018, 87, 1–21 CrossRef.
- P. Hadi, M. Xu, C. Ning, C. S. K. Lin and G. McKay, A Critical Review on Preparation, Characterization and Utilization of Sludge-Derived Activated Carbons for Wastewater Treatment, Chem. Eng. J., 2015, 260, 895–906 CrossRef.
- H. L. Tekinalp, E. G. Cervo, B. Fathollahi and M. C. Thies, The Effect of Molecular Composition and Structure on the Development of Porosity in Pitch-Based Activated Carbon Fibers, Carbon, 2013, 52, 267–277 CrossRef.
- Z. Heidarinejad, M. H. Dehghani, M. Heidari, G. Javedan, I. Ali and M. Sillanpää, Methods for Preparation and Activation of Activated Carbon: A Review, Environ. Chem. Lett., 2020, 18, 393–415 CrossRef.
- Y. Gao, Q. Yue, B. Gao and A. Li, Insight into Activated Carbon from Different Kinds of Chemical Activating Agents: A Review, Sci. Total Environ., 2020, 746, 141094 CrossRef PubMed.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Features of Promising Technologies for Pretreatment of Lignocellulosic Biomass, Bioresour. Technol., 2005, 96(6), 673–686 CrossRef.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Bimetallic Catalysts for Upgrading of Biomass to Fuels and Chemicals, Chem. Soc. Rev., 2012, 41(24), 8075–8098 Search PubMed.
- C. E. Wyman, B. E. Dale, R. T. Elander, M. Holtzapple, M. R. Ladisch and Y. Y. Lee, Coordinated Development of Leading Biomass Pretreatment Technologies, Bioresour. Technol., 2005, 96(18), 1959–1966 CrossRef CAS PubMed.
- E. Sjöström and R. Alén, Analytical Methods in Wood Chemistry, Pulping, and Papermaking, Springer Science & Business Media, 1998 Search PubMed.
- P. J. M. Carrott and M. R. Carrott, Lignin–from Natural Adsorbent to Activated Carbon: A Review, Bioresour. Technol., 2007, 98(12), 2301–2312 CrossRef.
- R. Davis and A. Bartling, Biochemical Conversion of Lignocellulosic Biomass to Hydrocarbon Fuels and Products: 2021 State of Technology and Future Research, NREL/TP-5100-76567; National Renewable Energy Lab (NREL), Golden, CO (United States), 2022 Search PubMed.
- M. H. Langholtz; B. J. Stokes and L. M. Eaton, 2016 Billion-Ton Report: Advancing Domestic Resources for a Thriving Bioeconomy, EERE Publication and Product Library, Washington, DC (United States), 2016 Search PubMed.
- US Department of Energy Bioenergy Technologies Office. BETO 2020 R&D State of Technology; EERE Publication and Product Library: Washington, DC (United States), 2020.
- E. Menya, P. W. Olupot, H. Storz, M. Lubwama and Y. Kiros, Production and Performance of Activated Carbon from Rice Husks for Removal of Natural Organic Matter from Water: A Review, Chem. Eng. Res. Des., 2018, 129, 271–296 CrossRef CAS.
- Y. Guo, S. Yang, K. Yu, J. Zhao, Z. Wang and H. Xu, The Preparation and Mechanism Studies of Rice Husk Based Porous Carbon, Mater. Chem. Phys., 2002, 74(3), 320–323 CrossRef CAS.
- Y. Xue, C. Du, Z. Wu and L. Zhang, Relationship of Cellulose and Lignin Contents in Biomass to the Structure and RB-19 Adsorption Behavior of Activated Carbon, New J. Chem., 2018, 42(20), 16493–16502 RSC.
- C. R. Correa, T. Otto and A. Kruse, Influence of the Biomass Components on the Pore Formation of Activated Carbon, Biomass Bioenergy, 2017, 97, 53–64 CrossRef.
- B. Tiryaki, E. Yagmur, A. Banford and Z. Aktas, Comparison of Activated Carbon Produced from Natural Biomass and Equivalent Chemical Compositions, J. Anal. Appl. Pyrolysis, 2014, 105, 276–283 CrossRef CAS.
- B. Cagnon, X. Py, A. Guillot, F. Stoeckli and G. Chambat, Contributions of Hemicellulose, Cellulose and Lignin to the Mass and the Porous Properties of Chars and Steam Activated Carbons from Various Lignocellulosic Precursors, Bioresour. Technol., 2009, 100(1), 292–298 CrossRef CAS.
- J. Deng, T. Xiong, H. Wang, A. Zheng and Y. Wang, Effects of Cellulose, Hemicellulose, and Lignin on the Structure and Morphology of Porous Carbons, ACS Sustainable Chem. Eng., 2016, 4(7), 3750–3756 CrossRef CAS.
- G. Tindall, B. Lynn, C. Fitzgerald, L. Valladares, Z. Pittman, V. Bécsy-Jakab, D. Hodge and M. Thies, Ultraclean Hybrid Poplar Lignins via Liquid–Liquid Fractionation Using Ethanol–Water Solutions, MRS Commun., 2021, 11, 692–698 CrossRef CAS.
- M. C. Thies, A. S. Klett and D. A. Bruce, Solvent and Recovery Process for Lignin, US Pat., 10053482, 2018 Search PubMed.
- A. S. Klett, P. V. Chappell and M. C. Thies, Recovering Ultraclean Lignins of Controlled Molecular Weight from Kraft Black-Liquor Lignins, Chem. Commun., 2015, 51(64), 12855–12858 RSC.
- S. V. Kanhere, G. W. Tindall, A. A. Ogale and M. C. Thies, Carbon Fibers Derived from Liquefied and Fractionated Poplar Lignins: The Effect of Molecular Weight, iScience, 2022, 25(12), 105449 CrossRef CAS PubMed.
- G. W. Tindall, J. Chong, E. Miyasato and M. C. Thies, Fractionating and Purifying Softwood Kraft Lignin with Aqueous Renewable Solvents: Liquid–Liquid Equilibrium for the Lignin–Ethanol–Water System, ChemSusChem, 2020, 13(17), 4587–4594 CrossRef CAS.
- G. W. Tindall, S. C. Temples, M. Cooper, V. E. Bécsy-Jakab, D. B. Hodge, M. Nejad and M. C. Thies, Liquefying Lignins: Determining Phase-Transition Temperatures in the Presence of Aqueous Organic Solvents, Ind. Eng. Chem. Res., 2021, 60(47), 17278–17282 CrossRef.
- R. J. Stoklosa and D. B. Hodge, Fractionation and Improved Enzymatic Deconstruction of Hardwoods with Alkaline Delignification, BioEnergy Res., 2015, 8, 1224–1234 CrossRef.
- C. Fitzgerald, via, PhD dissertation, Clemson University, 2023.
- A. Sluiter; B. Hames; R. Ruiz; C. Scarlata; J. Sluiter; D. Templeton and D. Crocker, Determination of Structural Carbohydrates and Lignin in Biomass, NREL/TP-510-42618; National Renewable Energy Lab (NREL), Golden, CO (United States), 2008, pp. 1–16 Search PubMed.
- K. Mohanty, M. Jha, B. Meikap and M. Biswas, Preparation and Characterization of Activated Carbons from Terminalia Arjuna Nut with Zinc Chloride Activation for the Removal of Phenol from Wastewater, Ind. Eng. Chem. Res., 2005, 44(11), 4128–4138 CrossRef.
- T.-H. Liou, Development of Mesoporous Structure and High Adsorption Capacity of Biomass-Based Activated Carbon by Phosphoric Acid and Zinc Chloride Activation, Chem. Eng. J., 2010, 158(2), 129–142 CrossRef.
- M. Li, Y. Pu and A. J. Ragauskas, Current Understanding of the Correlation of Lignin Structure with Biomass Recalcitrance, Front. Chem., 2016, 4, 45 CrossRef PubMed.
- M. E. Himmel, S.-Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Biomass Recalcitrance: Engineering Plants and Enzymes for Biofuels Production, Science, 2007, 315(5813), 804–807 CrossRef PubMed.
- C. D. Blasi, C. Branca and A. Galgano, Products and Global Weight Loss Rates of Wood Decomposition Catalyzed by Zinc Chloride, Energy Fuels, 2008, 22(1), 663–670 CrossRef.
- C. Branca, C. Di Blasi and A. Galgano, Pyrolysis of Corncobs Catalyzed by Zinc Chloride for Furfural Production, Ind. Eng. Chem. Res., 2010, 49(20), 9743–9752 CrossRef.
- C. Di Blasi, C. Branca, A. Galgano and F. Zenone, Modifications in the Thermicity of the Pyrolysis Reactions of ZnCl2-Loaded Wood, Ind. Eng. Chem. Res., 2015, 54(51), 12741–12749 CrossRef.
- S. Wang, G. Dai, H. Yang and Z. Luo, Lignocellulosic Biomass Pyrolysis Mechanism: A State-of-the-Art Review, Prog. Energy Combust. Sci., 2017, 62, 33–86 CrossRef.
- H. Yang, R. Yan, H. Chen, D. H. Lee and C. Zheng, Characteristics of Hemicellulose, Cellulose and Lignin Pyrolysis, Fuel, 2007, 86(12–13), 1781–1788 CrossRef.
- H. Yang, R. Yan, H. Chen, C. Zheng, D. H. Lee and D. T. Liang, In-Depth Investigation of Biomass Pyrolysis Based on Three Major Components: Hemicellulose, Cellulose and Lignin, Energy Fuels, 2006, 20(1), 388–393 Search PubMed.
- T. Siengchum, M. Isenberg and S. S. Chuang, Fast Pyrolysis of Coconut Biomass–An FTIR Study, Fuel, 2013, 105, 559–565 CrossRef CAS.
- H. M. Boundzanga, B. Cagnon, M. Roulet, S. de Persis, C. Vautrin-Ul and S. Bonnamy, Contributions of Hemicellulose, Cellulose, and Lignin to the Mass and the Porous Characteristics of Activated Carbons Produced from Biomass Residues by Phosphoric Acid Activation, Biomass Convers. Biorefin., 2020, 1–16 Search PubMed.
- N. R. Khalili, M. Campbell, G. Sandi and J. Golaś, Production of Micro-and Mesoporous Activated Carbon from Paper Mill Sludge: I. Effect of Zinc Chloride Activation, Carbon, 2000, 38(14), 1905–1915 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01234h |
| This journal is © The Royal Society of Chemistry 2025 |