
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bactericidal activity of ZnO nanoparticles–anti-TB drug combination towards the H37Rv strain and multidrug-resistant isolates of Mycobacterium tuberculosis via SufB splicing inhibition†
Deepak Kumar
Ojha
a,
Ashwaria
Mehra
a,
Sunil Swick
Rout
a,
Sidhartha
Giri
b and
Sasmita
Nayak
 *a
*a
aSchool of Biotechnology, Kalinga Institute of Industrial Technology Deemed to be University (KIIT-DU), Bhubaneswar, Odisha 751024, India. E-mail: sasmita.n@kiitbiotech.ac.in
bNational Reference Laboratory for Tuberculosis, ICMR-Regional Medical Research Centre (RMRC), Bhubaneswar, Odisha 751023, India
First published on 18th June 2025
Abstract
Tuberculosis (TB) remains a significant global health threat, claiming millions of lives annually. Despite advancements in treatment, the emergence of drug-resistant strains has hindered effective TB control. The current management of TB involves prolonged treatment duration with severe side effects, leading to poor patient compliance. Metal-based nanoparticles are shown to manage drug-sensitive TB when combined with anti-TB drugs. However, the mycobactericidal potential of nanoparticles towards drug-resistant TB is not confirmed yet. This work explores the bactericidal potential of zinc oxide nanoparticles (ZnONPs, 40 nm) in managing both drug-sensitive and drug-resistant TB in combination with anti-TB drugs. It was found that ZnONPs inhibit the generation of active SufB protein via splicing inhibition, an essential event for Mycobacterium tuberculosis (Mtb) survival. While TEM and UV-visible spectroscopy identified NPs–protein interaction, SEM visualised extensive membrane damage in H37Rv and multidrug-resistant (MDR) Mtb cells. Alamar blue assay and the spread plate method detected minimum inhibitory concentration and minimum bactericidal concentration of ZnONPs towards the H37Rv strain and MDR Mtb isolates. In vitro studies identified a combination with ZnONPs that reduced effective doses for anti-TB drugs towards H37Rv and MDR Mtb isolates. A correlation to splicing inhibition was made by performing Alamar blue assay in an SufB intein-less microbe, Mycobacterium smegmatis. A similar drug combination attenuated the mycobacterial load and inflammation in the spleen and lungs and protected against Mtb induced splenomegaly in infected mice. Thus, ZnONPs can be used as a potent additive in the anti-TB regimen to manage drug-susceptible and drug-resistant TB, addressing challenges such as prolonged therapy, drug toxicity and poor patient compliance.
1. Introduction
Tuberculosis (TB) is one of the leading causes of death worldwide, ranking among the top ten deadliest diseases globally, and is the single most lethal infectious disease, surpassing even HIV/AIDS.1,2 However, efforts to combat TB are often impeded by several challenges, including the absence of an effective vaccine, the complexities of treatment regimens, and the rapid emergence of drug-resistant Mycobacterium tuberculosis (Mtb) strains.3,4 Moreover, prolonged therapy can lead to the phenomenon of cross-resistance, where bacteria become resistant to multiple drugs by exploiting shared mechanisms of action.5 The standard treatment for drug-susceptible and drug-resistant TB spans approximately 6 months and over 18 months, respectively. The intricacy of these regimens, along with drug-induced side effects, often leads to poor patient compliance, worsening the current situation due to the emergence of new drug-resistant strains.6 Hence, there is an urgency to develop next-generation strategies to mitigate the limitations and enhance the accessibility of TB management.One promising approach to control TB could be targeting the intein splicing, which is a critical intracellular process that enables Mycobacterium tuberculosis (Mtb) survival and persistence by generating essential proteins.7,8 Inteins are intervening polypeptides co-translated within the host precursor proteins and self-excised through protein splicing to generate active protein.9 Given that several human pathogens harbour inteins within vital proteins and intein splicing is critical for their survival, the regulation of splicing can function as a promising target for developing anti-microbial drugs especially towards drug-resistant organisms. Mtb SufB, which is an Fe–S cluster assembly protein, represents a notable example among mycobacterial proteins, for its significance in disease pathogenesis.10,11 The SUF system as a whole is required for metalloprotein biogenesis and plays an important role in mycobacterial survival during iron limitation including oxidative and nitrosative stresses within infected macrophages.12,13
In recent years, the synergistic integration of nanotechnology and medicine has emerged as a popular and diverse approach, particularly for the development of anti-microbial compounds. The convergence of these disciplines has demonstrated significant potential in addressing various challenges associated with bacterial infections.14–16 Furthermore, the interaction between nanoparticles and proteins holds significant importance in various areas of contemporary biomedical research, particularly in the fields of nanomedicine and nano-diagnostics.17–21 When nanoparticles interact with proteins, they form a protein corona, which alters the biological activity of the bound protein. This phenomenon has profound implications for understanding the behaviour of nanoparticles within biological environments and their potential applications in drug delivery, diagnostics, and therapeutics.22–29
Thus, nanotechnology is gaining popularity as the cornerstone of modern research and therapeutics. This frequently involves metal-based NPs like ZnONPs that offer various uses in emerging applications such as antimicrobial, anti-inflammatory, anti-tumour, and wound healing.30–35 ZnONPs exhibit superior bioactivity, primarily due to their small size and increased surface area to volume ratio.36 ZnONPs are also preferred as efficient drug delivery vehicles for various diseases including cancer, because of their safety, stability, and low cost.36,37
Previous research has shown the bacteriostatic effects of ZnONPs towards both drug-sensitive and extensively drug-resistant (XDR) Mtb strains at concentrations equal to or higher than 1 μg ml−1.38 The same study has also determined 4 μg ml−1 as the inhibitory concentration of ZnONPs against the multidrug-resistant (MDR) strain of Mtb. Another work has reported the bactericidal role of ZnONPs–Se against Mtb BCG and H37Rv strains.39 The antimicrobial mechanisms of ZnONP effects are mostly attributed to a cumulative effect of cell membrane disruption, ROS production, release of toxic Zn2+ ions, and toxicity induced by their photocatalytic action.40 Although ZnONPs are known to exhibit potent antimicrobial and fungicidal activities towards different microbes,41 their bactericidal effect on drug-resistant Mtb strains is not shown to date. In a different context, the inhibitory effects of divalent metal ions such as Zn2+ and Cu2+ on the splicing of various intein containing precursor proteins were explored extensively, but studies showing intein splicing attenuation in the presence of metal-based NPs are missing to date.8,42–47
The current study addresses the following questions: (1) whether ZnONPs can regulate Mycobacterium tuberculosis (Mtb) SufB intein splicing and N-terminal cleavage reactions, (2) since intein splicing generates a functional SufB protein that is critical for mycobacterial survival, whether ZnONP mediated regulation of SufB splicing can affect the viability of whole cell Mtb, (3) importantly, whether the viability of both drug-sensitive and drug-resistant strains of Mtb is influenced by ZnONPs, (4) the minimum effective concentration of ZnONPs for the observed effect, (5) whether ZnONP combination can reduce the required dosage of existing anti-TB drugs to control both drug-sensitive and drug-resistant Mtb infections, (6) whether ZnONPs exhibit a bacteriostatic or bactericidal effect towards drug-sensitive and drug-resistant Mtb strains, and (7) whether ZnONPs protect against the effects of Mtb infection in an animal model.
In this work, ZnONP (40 nm) activity in the splicing and N-terminal cleavage reactions of the Mycobacterium tuberculosis (Mtb) SufB precursor protein was examined. Initial cytotoxicity evaluation via MTT assay indicated that ZnONPs are biocompatible towards HEK293T cells (human embryonic kidney cell lines), up to a concentration of 50 μg ml−1; consequently, the rest of the experiments were performed within this limit. UV-vis spectroscopy confirmed ZnONP–SufB interaction, further visualised by transmission electron microscopy (TEM), which identified protein “corona” formation around the ZnONPs. Additional experiments like Dynamic Light Scattering (DLS) and zeta potential analysis confirmed ZnONP–Mtb SufB interaction. Next, ZnONPs with a varied concentration range (0.5–50 μg ml−1) significantly inhibited the splicing and N-terminal cleavage reactions of Mtb SufB precursor protein as shown by in vitro protein refolding assay. SDS-PAGE and western blot analyses visualized and confirmed the splicing and cleavage products. Furthermore, Alamar blue assay and the spread plate method detected the anti-mycobacterial activity of ZnONPs with minimum inhibitory concentrations (MIC)/minimum bactericidal concentrations (MBC) of 5 μg ml−1/17 μg ml−1 and 14 μg ml−1/23 μg ml−1 towards the H37Rv strain (drug-sensitive) and multidrug-resistant (MDR) Mtb isolates respectively. In a comparative study, we also observed a reduction in the effective dose of anti-TB drugs INH/RIF and LFX/MXF in combination with ZnONPs that could efficiently kill the H37Rv strain and multidrug-resistant (MDR) Mtb isolates respectively. The observed activity was correlated to splicing inhibition by performing Alamar blue assay in the SufB intein-less microbe Mycobacterium smegmatis. Scanning electron microscopy (SEM) demonstrated the mycobactericidal activity of ZnONPs towards both drug-sensitive and drug-resistant Mtb (H37Rv) strains. Finally, the infected mouse model study revealed that ZnONPs in combination with anti-TB drugs can effectively reduce mycobacterial load in the lungs and spleen. The observed effects were noticed when ZnONPs were administered as a solo therapy or in combination with a reduced dose of RIF compared to a previously recommended amount of RIF. Likewise, the same combination of ZnONPs–RIF not only attenuated the inflammation in lungs and spleen, but also provided resistance to splenomegaly in the infected mice. Therefore, we suggest the use of ZnONPs as a promising adjunctive therapy along with standard anti-TB drugs to combat both drug-sensitive and multidrug-resistant Mtb infections, by inhibiting the splicing of an essential protein, Mtb SufB. This approach may not only mitigate the toxicity associated with conventional and contemporary anti-TB regimens but also potentially reduce the duration of therapy, thereby enhancing patient compliance.
2. Materials and methods
2.1 ZnO nanoparticles (ZnONPs)
ZnONPs (≥40 nm) were procured from Sigma Aldrich (product #721077).2.2 MTT assay48–50
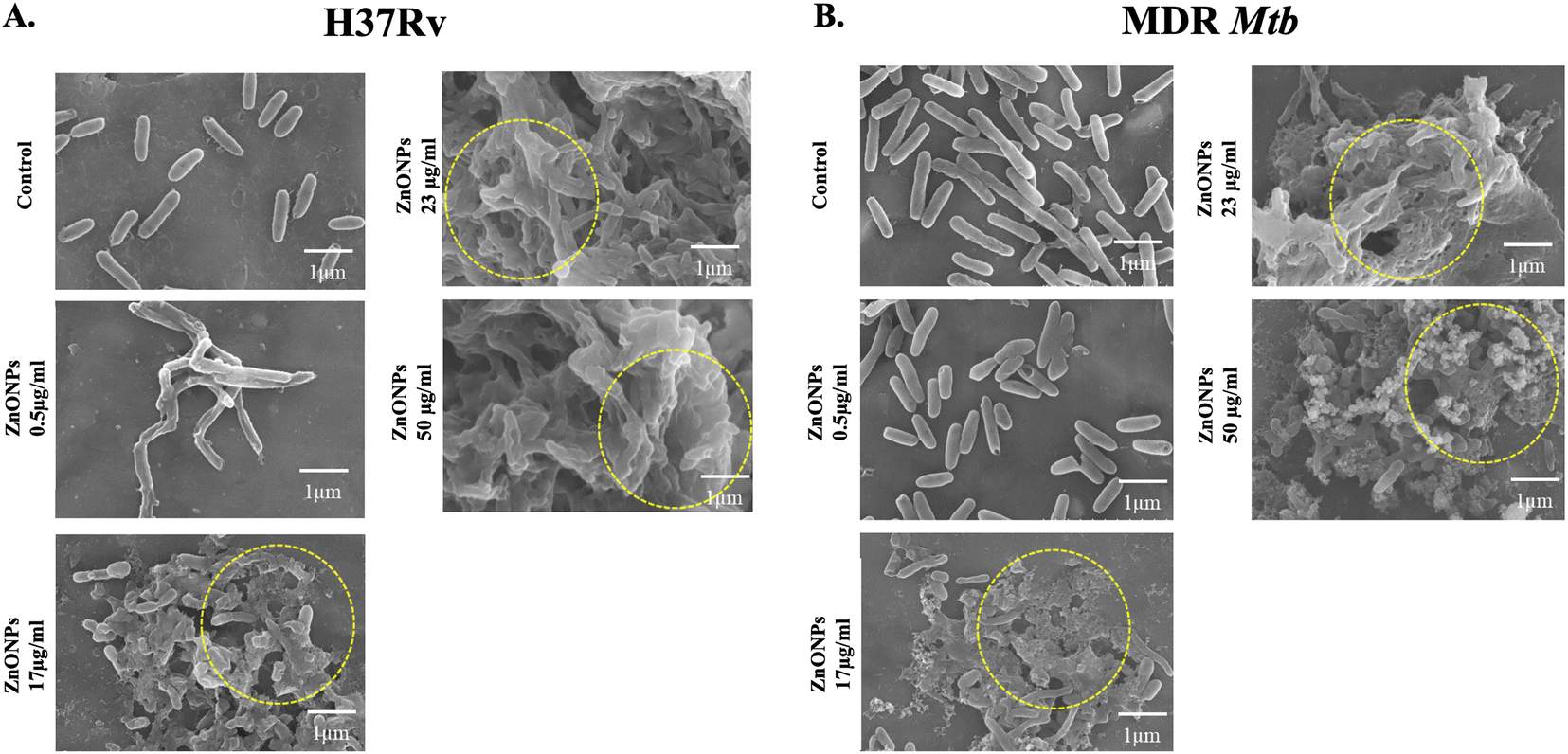
The cytotoxic effects of ZnONPs were evaluated using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay over a period of 72 h. HEK239T cells (104 cells per well) [Product # HEK239T (human embryonic kidney cell line), ATCC (CRL-3216)] were seeded in Dulbecco's Modified Eagle Medium (DMEM, Gibco, Invitrogen-2562497) in a 96-well plate (200 μl per well) and allowed to adhere for 24 hours at 37 °C in a 5% CO2 incubator. Subsequently, various concentrations of ZnONPs (5 μg ml−1, 14 μg ml−1, 17 μg ml−1, 23 μg ml−1 μg ml−1, 26 μg ml−1, 50 μg ml−1, 100 μg ml−1, and 150 μg ml−1) were added, followed by incubation at 37 °C in a 5% CO2 incubator for 72 hours. HEK239T cells (104 cells per well) incubated in the absence of nanoparticles were considered as the control for 100% viable cell growth. DMEM culture medium alone was used as blank control. 20 μl of MTT (EZcount™ MTT cell assay kit, Himedia-CCK003) solution (5 mg ml−1 in incomplete medium) was added to each well and incubated for 3 h to facilitate formation of purple formazan crystals under standard culture conditions. Next, MTT solution was discarded, and the crystals were solubilized by gently stirring on an orbital shaker for 30 minutes. After solubilization, the absorbance (570 nm) of each well was measured by using the ELISA plate reader (Bio Teck Epoch Microplate Spectrophotometer). The percentage of cell viability was calculated using the following formula.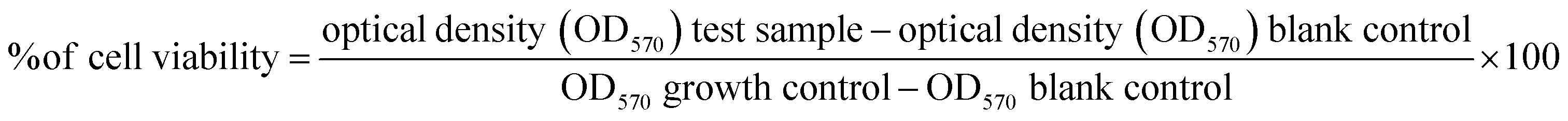
2.3 UV-visible spectroscopy51–53
Purified Mtb SufB precursor protein was refolded in the presence of different concentrations of ZnONPs (0.5 μg ml−1, 26 μg ml−1, and 50 μg ml−1) as explained in Section 2.7 for in vitro refolding assay. Then, the treated samples were analysed via UV-visible spectroscopy (UV-1800 Shimadzu) over the visible region of the spectrum (200–800 nm). To account for potential buffer–nanoparticle interactions, control samples included refolding buffer (20 mM sodium phosphate, 0.5 M NaCl, 0.5 M arginine, and 8 M urea) alone and the buffer with 50 μg ml−1 nanoparticles incubated under the same conditions. The nanoparticle-treated buffer served as a blank for baseline correction. The absorbance results were analysed by plotting absorbance versus wavelength using OriginPro 8.5 (Version 8.5, OriginLab Corporation, Northampton, MA, USA).2.4 Quantification of NP-bound proteins via Bradford assay54–56
The quantification of adsorbed protein was conducted using the Bradford assay. Mtb SufB precursor protein was renatured in vitro both in the presence and absence of various concentrations of ZnONPs (0.5 μg ml−1, 26 μg ml−1, and 50 μg ml−1) for 4 hours, as mentioned in Section 2.7. Next, the protein–NPs mixture was centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 minutes, at 4 °C, to separate the protein-bound nanoparticles. Then, the pellet containing the protein–NPs was resuspended in 100 μl of 1× PBS and analysed using the Bradford reagent to quantify the amount of adsorbed proteins. Control samples included nanoparticles in 1× PBS and test proteins resuspended in 1× PBS.
000 rpm for 30 minutes, at 4 °C, to separate the protein-bound nanoparticles. Then, the pellet containing the protein–NPs was resuspended in 100 μl of 1× PBS and analysed using the Bradford reagent to quantify the amount of adsorbed proteins. Control samples included nanoparticles in 1× PBS and test proteins resuspended in 1× PBS.
2.5 TEM analysis
Mtb SufB precursor protein–ZnONP interaction was analysed by transmission electron microscopy (Jeol JEM-1400) for visualising protein corona formation. Mtb SufB precursor protein was overexpressed, purified and refolded with 50 μg ml−1 ZnONPs as mentioned in Section 2.7. The protein–ZnONP complex was collected via centrifugation (13000 rpm for 30 minutes) at 4 °C and washed twice with phosphate buffered saline (1× PBS), and the supernatant was discarded. Then the pellet (protein–ZnONP complex) was resuspended and fixed with 100 μl of 2.5% glutaraldehyde in 1× PBS for 1 h. Finally, 5 μl of ZnONP-complex solution was placed onto the carbon-coated copper grid and imaged under TEM with an acceleration voltage of 120 kV.57,58
Additionally, to visualise the protein corona clearly, TEM analysis was performed using a negative staining technique. 5 μl of the protein–ZnONP complex was drop-cast onto a carbon-coated copper grid and incubated for 5 minutes. Excess complex was carefully removed using filter paper. Subsequently, 2% uranyl acetate was applied for negative staining and incubated for 2–3 min. The excess stain was removed, and the grid was gently rinsed with nuclease-free water. The sample was air dried at room temperature overnight. Imaging was performed using a Jeol JEM-2100 Plus Electron Microscope operating at an accelerating voltage of 200 kV.59,60
2.6 Purification of overexpressed proteins
The details of the plasmids used in this study are provided elsewhere.7 The Mtb SufB wild type precursor and Mtb SufB splicing inactive double mutant (C1A/N359A) (splicing negative control), all carrying an N-terminal 6× His tag, were overexpressed in BL21 (DE3) E. coli cells via IPTG (500 μM, Sigma 367-93-1) induction. The cells were resuspended in lysis buffer (20 mM sodium phosphate, 0.5 M NaCl, pH 7.4) and lysed using a tip sonicator (Sonics vibra cell VCX-130).61 The cell lysate was centrifuged at 16500 × g for 20 minutes at 4 °C and the supernatant was discarded. The collected IB materials were washed thrice via centrifugation in lysis buffer, solubilized in 8 M urea (Merck, 1084870500) buffer (lysis buffer, 8 M urea, 10 mM imidazole) (MP–biochemicals-288-32-4), and centrifuged at 16500 × g for 20 minutes at 4 °C to collect the supernatant. The solubilized proteins were purified using a Ni-NTA affinity column (Ni-NTA His trap, HP GE healthcare life sciences-17524802).62–66 Before sample application, the columns were equilibrated with binding buffer (20 mM sodium phosphate, 0.5 M NaCl, 8 M urea, 10 mM imidazole), and after loading the samples, the columns were washed several times (15 column volumes) with the wash buffer (20 mM sodium phosphate, 0.5 M NaCl, 8 M urea, 40 mM imidazole). Finally, the test proteins were eluted in elution buffer (20 mM sodium phosphate, 0.5 M NaCl, 500 mM imidazole, 8 M urea, pH 7.4) and quantified using Bradford's assay.
2.7 In vitro protein refolding and splicing analysis via SDS gradient PAGE
Denatured test proteins (2.5 μM) in 8 M urea buffer were renatured in a refolding buffer containing 20 mM sodium phosphate, 0.5 M NaCl, 0.5 M L-arginine (A5006 Sigma Aldrich), and 2 mM TCEP-HCl (Sigma-51805-45-9). Protein refolding was performed for 4 hours at 25 °C61–66 in the presence and absence of ZnO nanoparticles (ZnONPs) over a varied concentration range (0.5 μg ml−1 to 50 μg ml−1). For the 0-hour sample, the protein sample was collected before initiation of the refolding, followed by immediate addition of the loading dye (0.1% bromophenol blue, 50% glycerol, β-mercaptoethanol, 10% SDS, tris, pH 6.8) and rapid freezing at −20 °C. After 4 h of protein renaturation, the rest of the samples were collected, loading dye was added, and samples were boiled at 95 °C for 5 minutes. Then protein products were resolved through a 5–10% gradient SDS-PAGE gel. The protein bands were visualized by staining with Coomassie brilliant blue R-250 (catalogue no. #112553, Sigma-Aldrich), and densitometric analysis was performed using the GelQuant.Net biochemical solutions software. The splicing and cleavage efficiencies were calculated as the percentage ratio of the total splicing product (LE and I) over the total proteins (LE + I + P) and the total N-terminal cleavage product (NE + NC) over the total proteins (NE + NC + P), respectively. 0-Hour splicing and cleavage product values were subtracted from respective sample values for baseline corrections. The results were analysed using one-way ANOVA and plotted using GraphPad Prism version 5.01 for Windows, GraphPad Software, San Diego, CA, USA, www.graphpad.com.2.8 Western blot analysis8
For western blot analysis, an anti-His antibody was utilized to confirm the presence of splicing and N-terminal cleavage products. The test proteins were transferred from the 5–10% SDS-PAGE to the PVDF membrane (Millipore, IPVH 00010) at a voltage of 10 V, overnight at 4 °C. Subsequently, the blot was blocked with 5% skim milk and incubated overnight at 4 °C. Then, the membrane was washed once with 1× TBST (Tris-buffered saline with Tween-20) followed by incubation with a primary anti-His antibody (abgenex-32-6116) at 1:5000 dilution overnight at 4 °C. In the next step, the membrane was washed thrice for 5 minutes with 1× TBST and incubated with a secondary antibody (anti-mouse, Abgenex 11-301) at a dilution of 1:6000, for 6 hours at 25 °C. To enhance the detection of the N-extein band, higher dilutions of the primary antibody (1:2500) and secondary antibody (1:4000) were used. Finally, the blots were washed with 1× TBST and developed using an enhanced chemiluminescence substrate (ECL) (Abcam, ab65628) to visualize the protein bands and images were captured using an in-house ImageQuant™ LAS 500 facility (GE Healthcare).
2.9 Anti-mycobacterial study

A graphical representation of the same is provided in the ESI.†
:1000 dilution) from each well were evenly spread onto respective 7H10 agar (HiMedia-M199) plates using a sterile spreader. The plates were incubated at 37 °C for 21 days to grow mycobacterial colonies. Subsequently, colonies were counted by a colony counting method and the MBC of ZnONPs was determined as the lowest concentration at which no colony growth was visualised on the respective agar plates. Control plates included mycobacterial cells without NP treatment.
2.10 Statistical analysis
For each method, numerical data from three to six independent sets of experiments were analysed using GraphPad Prism 6.0 (La Jolla, CA, United States) and were presented as mean ± 1 standard deviation or standard error of the mean. Statistical significance between different groups was assessed using one-way ANOVA, with significance denoted by p < 0.05. Specific p-values of <0.05, <0.01, <0.001, and <0.0001 were indicated as *, **, ***, and ****, respectively.3. Results and discussion
3.1 ZnONPs interact with Mtb SufB precursor protein
Previous studies have reported interaction of nanoparticles with biological molecules such as proteins and DNA.75 Before examining the role of ZnONPs in the splicing activity, we evaluated possible interaction between ZnONPs and Mtb SufB precursor protein. Through a combination of analytical techniques such as UV-visible spectroscopy, Bradford analysis, and transmission electron microscopy (TEM), we have evaluated ZnONPs–protein binding activity (Fig. 1). | ||
| Fig. 1 Evaluation of ZnONP interaction with Mtb SufB. (A) UV-visible spectroscopy: Mtb SufB precursor protein was treated with ZnONPs (0.5 μg ml−1, 26 μg ml−1, and 50 μg ml−1), and sample absorbance was measured by scanning over a 200–500 nm wavelength. (B) Bradford assay: denatured test proteins were renatured in vitro in the presence of various ZnONP concentrations for a period of 4 h at 25 °C. Then, test samples were processed, and the concentration of adsorbed protein was determined using the Bradford reagent. (C) TEM analysis using the negative staining technique: images showing (i) ZnONP aggregates, (ii) protein corona formation on the surface of ZnONP aggregates after interaction with Mtb SufB (white arrows). | ||
Initially, cytotoxicity of the ZnONPs was assessed via MTT assay using a human embryonic kidney cell line, HEK239T cells, since potentially harmful NPs are mostly excreted by the kidneys through urine.76–78 It was demonstrated that up to a concentration of 50 μg ml−1, ZnONPs exhibited no cytotoxicity against HEK239T cells (Fig. S1, ESI†). This is further supported by a prior research study, which has shown similar results.79 Consequently, we decided to explore SufB interaction with various ZnONP concentrations up to this limit (0.5, 26, and 50 μg ml−1). UV-visible spectra of both the Mtb SufB precursor protein and ZnONPs–SufB complexes were recorded by scanning over the visible range (200 to 800 nm), as depicted in Fig. 1(A). Buffer alone and buffer along with nanoparticles (50 μg ml−1) were used for baseline correction and as the reference blank respectively. An observable broadening and blue shift were differentiated in the absorbance spectra of the Mtb SufB protein following interaction with increasing concentrations of ZnONPs, when compared to the spectra of protein solubilised in buffer alone. This observation suggests the binding of ZnONPs with the Mtb SufB precursor protein over the test concentration range.
To further validate such interaction, we utilized Bradford assay, where Mtb SufB precursor protein was refolded along with various concentrations of ZnONPs (0.5, 26, and 50 μg ml−1), causing adsorption of proteins onto the NP surface. After isolating ZnONPs–protein complexes, quantification of bound proteins was done using Bradford assay. As illustrated in Fig. 1(B), an increase in nanoparticle concentration correlated with a proportional increase in the bound proteins. This result further supported the interaction between ZnONPs and Mtb SufB protein.
The formation of a protein corona on NP surfaces was investigated by TEM, with and without a negative staining technique, as shown in Fig. 1(C) and Fig. S2 (ESI†) respectively. While standalone ZnONPs were seen as aggregates [Fig. 1(C)-(i), and Fig. S2A-(i), ESI†] or discrete spheres [Fig. S2B-(i), ESI†], the NP (50 μg ml−1)-treated protein samples displayed a distinct feature. A thin, transparent layer encasing the NPs was evident in the treated sample (white arrows) as seen in Fig. 1(C)-(ii) and Fig. S2 (yellow arrows), ESI.† This is indicative of the protein corona formed on the surface of the ZnONPs upon interaction with the Mtb SufB protein.59
DLS measurements demonstrated a significant increase in hydrodynamic diameter following interaction of ZnONPs (26 μg ml−1 and 50 μg ml−1) with Mtb SufB compared to controls (buffer; buffer with Mtb SufB; buffer with ZnONPs) (Fig. S3A and Table S1, ESI†).80 Concomitant zeta potential analysis revealed a charge inversion from +5.1 mV (bare ZnONPs) to −4 mV for the ZnONPs–SufB complex (Fig. S3B and Table S1, ESI†).80,81 This transition from a net positive to a net negative surface charge suggests a protein–nanoparticle binding event. Therefore, the observed changes in both the hydrodynamic size and the surface charge distribution shows evidence for ZnONPs–SufB interaction.
The comprehensive assessment involving UV-visible spectroscopy, Bradford analysis, DLS, zeta potential analysis and TEM successfully indicated the binding activity and protein corona formation following ZnONP interaction with Mtb SufB.
3.2 ZnONPs inhibit the generation of functional SufB protein
The SUF system provides a unique pathway for [Fe–S] cluster biosynthesis and Mtb survival during intracellular stress like iron deprivation.82 In such environments, the functionality of the SUF complex relies exclusively on the splicing of the Mtb SufB precursor to generate active SufB protein.13 Hence, splicing regulation under such physiological stress can lead to potential anti-TB drug development strategies by influencing mycobacterial cell viability. Although metals are known inhibitors of intein splicing,42,45,46 NP mediated splicing inhibition has not been reported yet. Furthermore, higher reactivity of nanoparticles in a biological system has prompted us to evaluate ZnONPs and Mtb SufB interactions with possible effects on SufB precursor splicing.In vitro splicing assay was conducted both in the presence and absence of ZnONPs, as detailed in the Materials and methods section. Since MTT assay demonstrated that HEK239T cells (human embryonic kidney cell lines) were tolerant to ZnONPs up to a concentration of 50 μg ml−1 (Fig, S1, ESI†), splicing efficiency was evaluated using NP concentrations up to this limit. Fig. 2(A) illustrates different structural domains of Mtb SufB precursor protein; N-extein (29.9 kDa), intein (40.2 kDa), and C-extein (25.8 kDa). The details of Mtb SufB splicing and N-terminal cleavage reactions are published elsewhere.7 The SufB precursor undergoes splicing reactions to generate ligated exteins (LE) and intein (I), whereas N-terminal cleavage reactions yield an N-terminal cleavage product (NC, 65.2 kDa) and N-extein (NE) (Fig. 2(A)). Fig. 2(B)-(i) and B-(ii) depict the concentration-dependent inhibition of these reactions by ZnONPs, providing crucial insights into their biological role.
 | ||
| Fig. 2 Effect of ZnONPs on Mtb SufB precursor splicing and N-terminal cleavage reactions. (A) Schematic diagram representing different structural domains of Mtb SufB precursor protein and splicing & N-terminal cleavage reactions of the precursor protein generating different protein products. (B) Products from the in vitro protein refolding experiment were resolved through 5–10% gradient SDS-PAGE. (T0): splicing and cleavage reactions at 0 h, (UT): untreated protein sample; and lanes 4–13 show protein products induced by various concentrations of ZnONPs: (i) (0.5–26 μg ml−1), (ii) (29 μg ml−1 to 50 μg ml−1), lane 14 (SI): splicing inactive SufB double mutant (Cys1Ala/Asn359Ala) is used as a negative control for splicing, lane 15: ZnCl2 (2 mM) was used as a positive control, since it is a known inhibitor of Mtb SufB splicing reactions.8 (C) Line plots showing the quantitative results of splicing and N-terminal cleavage reaction inhibition. Values were extracted from Fig. 2(B) and plotted after densitometric analysis using GelQuant.Net biochemical solutions. 0 h splicing and cleavage values were subtracted from each sample as baseline correction. The red arrow and blue arrow denote complete inhibition of SufB splicing and N-terminal cleavage reactions by ZnONPs at concentrations of 47 μg ml−1 and 26 μg ml−1, respectively. Error bars represent (±1) SEM from (n = 3) three independent sets of experiments (p < 0.0001). P: precursor, NC: N-terminal cleavage product, LE: ligated extein, I: intein, and NE: N-extein. | ||
The samples were analysed through gradient SDS-PAGE, and the bands were quantified using the GelQuant.NET software. Our findings indicated a substantial inhibition of Mtb SufB precursor splicing (p < 0.0001) over all the test concentrations, with discernible effects commencing at a ZnONP concentration of 0.5 μg ml−1. Remarkably, a complete inhibition of splicing was observed at a concentration of 47 μg ml−1 [Fig. 2(B)-(ii) and (C)]. These findings suggest that ZnONPs exert a dose-dependent inhibition of splicing in Mtb SufB precursor protein, which could lead to critical consequences, such as loss of functionality of this protein in a cellular environment.
Similarly, the N-terminal cleavage reaction of the Mtb SufB precursor was notably inhibited (p < 0.0001) in the presence of ZnONPs. A reduction in the cleavage activity was observed at the minimal tested concentration of 0.5 μg ml−1, culminating in a complete inhibition at a concentration of 26 μg ml−1 [Fig. 2(B)-(i) and (C)]. Thus, ZnONPs exert a potent inhibitory effect on the Mtb SufB splicing and N-terminal cleavage reactions, underscoring their potential interference with SufB-associated cellular processes. These findings also highlight the significant biological implications of ZnONPs on Mtb survival within host cells. To substantiate these findings, we performed western blot analysis to validate identity of protein products (Fig. S4, ESI†), which further confirmed the inhibitory role of ZnONPs in the splicing and N-terminal cleavage reactions of SufB. Additional intermediate concentrations of ZnONPs were used to check the robustness of such effects on the splicing and cleavage activity of the precursor protein (Fig. S5, ESI†).
Current research presents evidence that ZnONPs interact with the Mtb SufB precursor protein, leading to a concentration-dependent inhibition of both splicing and N-terminal cleavage reactions. These findings also emphasize the notable impact ZnONPs could have on the essential cellular processes linked to Mtb SufB protein and their potential implications in biological systems. Specifically, they call for further exploration into the influence of ZnONPs on SufB splicing, correlating this process to the growth and viability of Mtb.
3.3 ZnONPs inhibit the growth and viability of mycobacterial cells
Since ZnONPs were shown to interact with the SufB precursor protein and caused the inhibition of protein splicing, it was imperative that this may retard the growth and viability of Mtb by blocking the generation of an essential protein SufB. Both H37Rv cells and MDR Mtb isolates were cultured in a nutrient medium along with various concentrations (0.5–50 μg ml−1) of ZnONPs, as discussed in the Materials and methods section. The test concentration range for the ZnONPs was chosen based on their activity range as outlined in Fig. 2(B) and (C). Furthermore, up to a concentration of 50 μg ml−1, ZnONPs exhibited no cytotoxicity against HEK239T cells (Fig. S1, ESI†). Besides, ZnONPs at a concentration of ≥50 μg ml−1 were also found to be toxic for human cells, elucidated by earlier research.83We performed Alamar blue assay, a well-established metric for cell viability, which yielded data on the inhibitory roles of ZnONPs in the growth and survival of H37Rv and MDR Mtb cells. The Alamar blue dye, initially blue, undergoes reduction in viable cells, resulting in a colour change to pink, whereas persistence of the blue colour indicates lack of viable cells.84 Spectral quantification for the same was performed by measuring the absorbance at 570 nm (OD570), and the % loss of cell viability was measured using a standard formula. In the Alamar blue assay, metabolically active cells reduce resazurin to pink fluorescent resorufin, with a specific absorbance measured at 570 nm. The intensity of this colour change is directly proportional to the number of viable cells present. ZnONPs at 0.5 μg ml−1 and 2 μg ml−1 did not have any effect on H37Rv cells. 5 μg ml−1 was identified as the minimum inhibitory concentration (MIC) of ZnONPs at which H37Rv cells turned purple due to partial loss of cell viability (Fig. 3(A) and Fig. S6A, ESI†). A complete loss of cell viability and retention of blue colour was noted at a concentration of 17 μg ml−1 of ZnONPs.
 | ||
| Fig. 3 Alamar blue assay to determine the minimum inhibitory concentrations (MIC) of ZnONPs towards H37Rv Mtb cells. Mycobacterial cells (106 CFU) were incubated in a 96 well plate, in the presence of various treatment conditions for 14 days, followed by addition of Alamar blue reagent. Further incubation at 37 °C for 24 hours led to a colour change in the wells. (A) Cells were cultured in the presence of various concentrations (0.5 to 50 μg ml−1) of ZnONPs. The MIC of ZnONPs was identified as 5 μg ml−1, at which transition of blue to purple colour occurred, suggesting partial loss of cell viability among the cells. Persistence of blue colour correlating to complete loss of viable cell population was seen at 17 μg ml−1 ZnONPs. Pink cells refer to healthy viable cells, resisting the inhibitory effect of ZnONPs. (B) Comparison of the activity of first line anti-TB drugs; INH & RIF and various concentration of ZnONPs. All the cells were rendered non-viable (blue) at a concentration of 17 μg ml−1 for the NPs, 2 μg ml−1 for RIF, 0.5 μg ml−1 for INH, and at respective higher concentrations. The MIC of RIF & INH as shown in this study coincided with the known values of 1 and 0.1 μg ml−1 respectively.85 Synergistic activity of ZnONPs and anti-TB drugs; (C) RIF and (D) INH. A combination of RIF (0.5 μg ml−1) + ZnONPs (11 μg ml−1) and RIF (1 μg ml−1) + ZnONPs (5 μg ml−1) successfully yielded non-viable cells (blue). Likewise, INH (0.05 μg ml−1) + ZnONPs (11 μg ml−1) & INH (0.1 μg ml−1) + ZnONPs (5 μg ml−1) combinations resulted in non-proliferating cells (blue). Red stars – complete loss of viability and yellow stars – partial loss of viable cells (purple) at a lower concentration of NPs. DS: drug sensitive H37Rv Mtb, RIF: rifampicin, INH: isoniazid. | ||
Next, a comparative study was conducted to assess the efficacy of ZnONPs in tandem with first-line anti-TB drugs such as rifampicin (RIF) and isoniazid (INH) for which the Minimum Inhibitory Concentrations (MICs) and Minimum bactericidal Concentrations (MBC) are well-documented.85,86 RIF and INH, at known MICs of 1 μg ml−1 and 0.1 μg ml−1 respectively, resulted in partial loss of mycobacterial cell viability (purple colour) in the respective wells. There was a complete loss of viable cell population (blue colour) at 2 μg ml−1 RIF and 0.5 μg ml−1 INH, comparable to the activity of ZnONPs at 17 μg ml−1 (Fig. 3(B) and Fig. S6B, ESI†). All the cells were rendered non-viable at concentrations higher than these values.
Fig. 3(C), (D) and Fig. S6C, D, ESI† depict the synergistic activity of ZnONPs and first line anti-TB drugs, RIF and INH. A combination of RIF (0.5 μg ml−1) + ZnONPs (11 μg ml−1) and RIF (1 μg ml−1) + ZnONPs (5 μg ml−1) successfully yielded non-viable cells (blue). Likewise, INH (0.05 μg ml−1) + ZnONPs (11 μg ml−1) and INH (0.1 μg ml−1) + ZnONPs (5 μg ml−1) combinations resulted in non-proliferating cells (blue). Thus, both RIF and INH at their respective MIC and half reduced MIC values could completely abolish mycobacterial cell viability when added together with ZnONPs. These results emphasize the diminution in the effective concentration of the conventional anti-TB drugs (INH and RIF) in combination with ZnONPs.
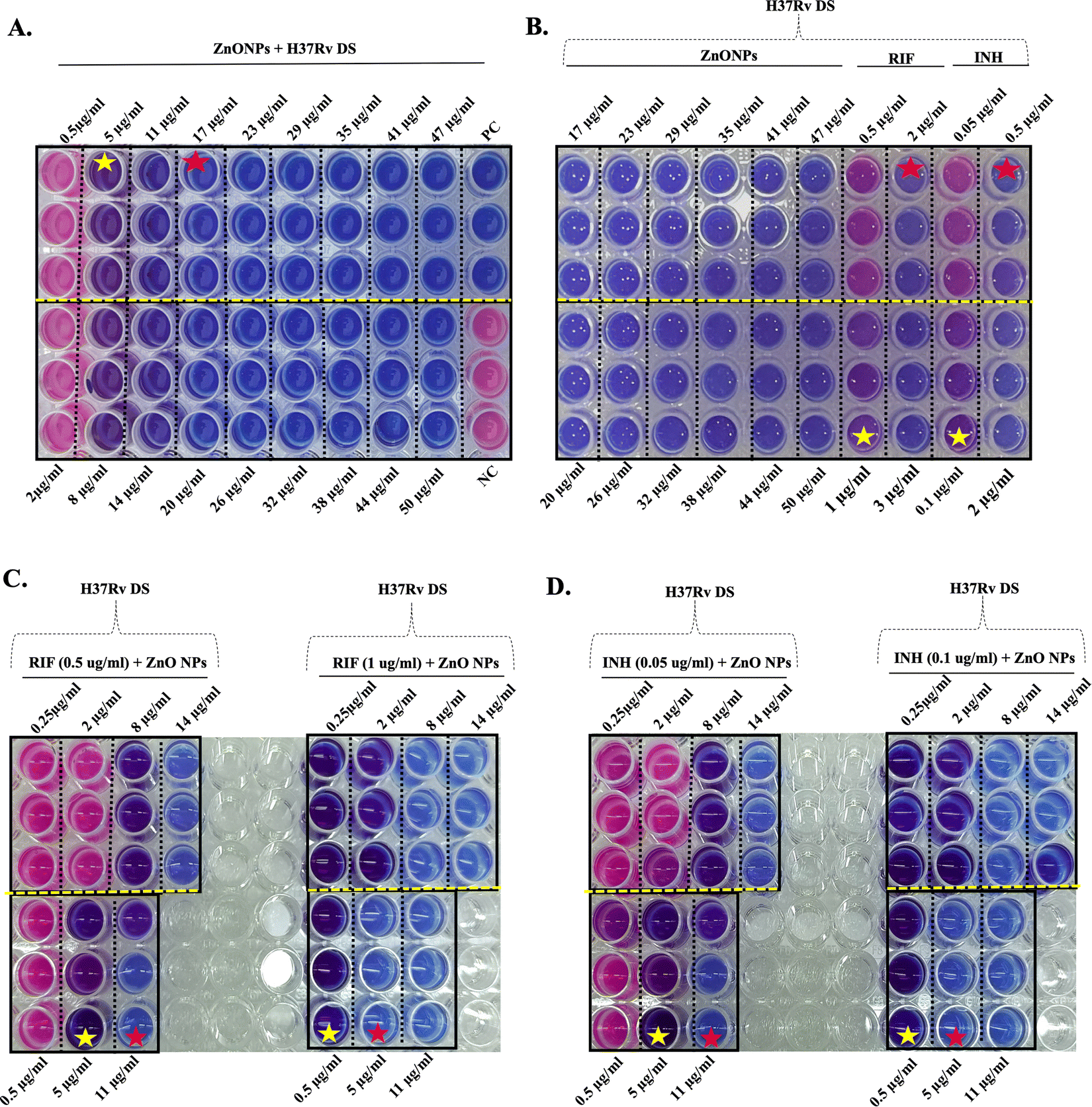
MIC values of ZnONPs towards multidrug-resistant (MDR) Mtb isolates were found to be higher at 14 μg ml−1 (Fig. 4(A) and Fig. S7A, ESI†). The MBC values for the ZnONPs [17 μg ml−1 for drug-sensitive H37Rv and 23 μg ml−1 for multidrug-resistant (MDR) Mtb isolates] were further confirmed through the agar spread plate method as shown in the ESI† (Fig. S8). Although MDR cells were rendered nonviable at 20 μg ml−1 ZnONPs (Fig. 4(A)), based on the data obtained from the agar spread plate method (Fig. S8, ESI†), for the remaining experiments 23 μg ml−1 was considered as the MBC for ZnONPs towards MDR Mtb cells.
 | ||
| Fig. 4 Alamar blue assay to determine the MIC of ZnONPs towards MDR Mtb cells. Mycobacterial cells (106 CFU) were incubated in a 96 well plate, in the presence of varied treatment conditions for 14 days, followed by addition of the Alamar blue reagent. Further incubation at 37 °C for 24 hours led to a colour change in the wells. (A) Cells were cultured in the presence of varied concentrations (0.5 to 50 μg ml−1) of ZnONPs. MIC of ZnONPs was identified as 14 μg ml−1 where transition of blue to purple colour occurred, suggesting partial loss of cell viability among the cells. In contrast, persistence of a blue colour due to complete loss of viable cell population was seen at 23 μg ml−1 ZnONPs. Pink cells refer to healthy viable cells resisting the inhibitory effect of ZnONPs. (B) Comparison of the activity of anti-TB drugs; levofloxacin (LFX) & moxifloxacin (MXF) and various concentration of ZnONPs. All the cells were rendered non-viable (blue) at a concentration of 23 μg ml−1 for the ZnONPs and in the presence of LFX (0.25 μg ml−1) and MXF (0.125 μg ml−1) and at higher concentration. The MIC of LFX and MXF as shown in this study coincided with the known values of 0.125 and 0.0625 μg ml−1 respectively.87,88 Synergistic activity of ZnONPs and anti-TB drugs; (C) LFX and (D) MXF. A combination of LFX (0.0625 μg ml−1) + ZnONPs (17 μg ml−1) and LFX (0.125 μg ml−1) + ZnONPs (11 μg ml−1) successfully yielded non-viable cells (blue). Likewise, MXF (0.031 μg ml−1) + ZnONPs (17 μg ml−1) and MXF (0.0625 μg ml−1) + ZnONPs (11 μg ml−1) combinations resulted in non-proliferating cells (blue). Red stars -complete loss of viability and yellow stars - partial loss of viable cells (purple) at a lower concentration of NPs. MDR: multidrug-resistant, LFX: levofloxacin, MXF: moxifloxacin. | ||
In the case of MDR isolates, the anti-TB drugs moxifloxacin (MXF) and levofloxacin (LFX), with known MICs of 0.0625 μg ml−1 and 0.125 μg ml−1 respectively,87,88 were compared with the activity of the ZnONPs (Fig. 4(B) and Fig. S7B, ESI†). There was a complete loss of viable cell population (blue colour) at LFX (0.25 μg ml−1)/MXF (0.125 μg ml−1) and their activity was comparable to the effect of ZnONPs at 23 μg ml−1 (MBC) (Fig. 4(B) and Fig. S7B, ESI†). All the cells were rendered non-viable (blue) at concentrations higher than these values for the test compounds. Thus, the antimycobacterial activity of ZnONPs towards MDR Mtb isolates was comparable to that of anti-TB drugs MXF and LFX at their respective MIC and higher values. This underscores the potential of ZnONPs as a potent alternative for managing Mtb infections.
Next, the synergistic activity of ZnONPs along with LFX and MXF was evaluated towards MDR Mtb isolates (Fig. 4(C), (D) and Fig. S7C, D, ESI†). A combination of LFX (0.0625 μg ml−1) + ZnONPs (17 μg ml−1) and LFX (0.125 μg ml−1) + ZnONPs (11 μg ml−1) resulted in non-viable cells (blue). A similar observation was made for, MXF (0.031 μg ml−1) + ZnONPs (17 μg ml−1) and MXF (0.0625 μg ml−1) + ZnONPs (11 μg ml−1) combinations, towards the viability of MDR cells.
Thus, both LXF and MXF at their respective MIC and half reduced MIC values could completely abolish mycobacterial cell viability when added together with ZnONPs. In toto, these results suggest that ZnONPs exhibit anti-mycobacterial properties against both H37Rv and MDR Mtb cells, indicating their potential utility as a supportive therapy in the fight against TB.
On the whole, from the results of in vitro splicing and Alamar blue assays, ZnONP-mediated inhibition of mycobacterial growth could be attributed to splicing attenuation induced by interactions between ZnONPs and Mtb SufB precursor protein. By targeting essential proteins like the splicing of SufB, ZnONPs are likely to disrupt vital cellular processes in the mycobacteria, leading to growth inhibition and loss of cell viability. To further correlate these events, Alamar blue assay was performed to examine the effect of ZnONPs (0.5 μg ml−1 to 50 μg ml−1) on the growth and viability of Mycobacterium smegmatis (M. sm), in which the SufB polypeptide chain lacks an intein sequence (Fig. S9, ESI†).89 Live viable cells (pink) (Fig. S9, ESI†) indicated no effect of ZnONPs (0.5 μg ml−1 to 50 μg ml−1) on M. sm viability, possibly due to lack of regulatory influence on SufB splicing and cleavage reactions. This provided further evidence to support loss of Mtb cell viability (Alamar blue assay) as a result of ZnONP mediated inhibition of Mtb SufB splicing and cleavage reactions, although additional mechanisms such as NP-mediated membrane damage cannot be ruled out.
Furthermore, our investigation into combinatorial therapy, integrating lower effective concentrations of known anti-TB drugs alongside ZnONPs, revealed a notable inhibitory effect on the growth and viability of Mycobacterium tuberculosis (Mtb) cells. Particularly noteworthy was the complete loss of cell viability observed at concentrations half of the MIC values for the individual drugs along with a reduced effective concentration for the NPs (Fig. 3(C), (D), 4(C), (D), and Fig. S6C–S7D, ESI†). As shown in Fig. 3(C) and Fig. S6C, ESI† when the concentration of RIF was reduced to 0.5 μg ml−1 (half of MIC), along with 11 μg ml−1 ZnONPs, this led to no observable viable growth of H37Rv Mtb. Similarly, INH at a concentration of 0.05 μg ml−1 (half of MIC) completely blocked the viability of mycobacterial cells in the presence of lower dose of ZnONPs (11 μg ml−1) (Fig. 3(D) and Fig. S6D, ESI†). In another approach, when RIF and INH were added at their respective MIC (1 μg ml−1 and 0.1 μg ml−1 respectively), in the presence of MIC of ZnONPs (5 μg ml−1), no viable cells were observed (Fig. 3(C), (D) and Fig. S6C, D, ESI†). Similarly, in the case of MDR cells, when LFX and MXF were added at their MIC (0.125 and 0.0625 μg ml−1 respectively),87,88 a reduction in ZnONPs' MIC occurred from 14 μg ml−1 to 0.5 μg ml−1, with no growth observed at 11 μg ml−1 (Fig. 4(C), (D) and Fig. S7C, D, ESI†).
This strategy demonstrates the potential to achieve significant inhibition of H37Rv as well MDR Mtb cells at markedly lower concentrations of conventional anti-TB drugs, with the potential to mitigate the cytotoxic effects associated with prolonged drug administration. The observed synergy between lower doses of existing drugs and ZnONPs presents a promising avenue for enhancing the efficacy of TB management while minimizing potential adverse effects.
3.4 ZnONPs exhibit bactericidal effects on drug-sensitive and MDR Mtb cells
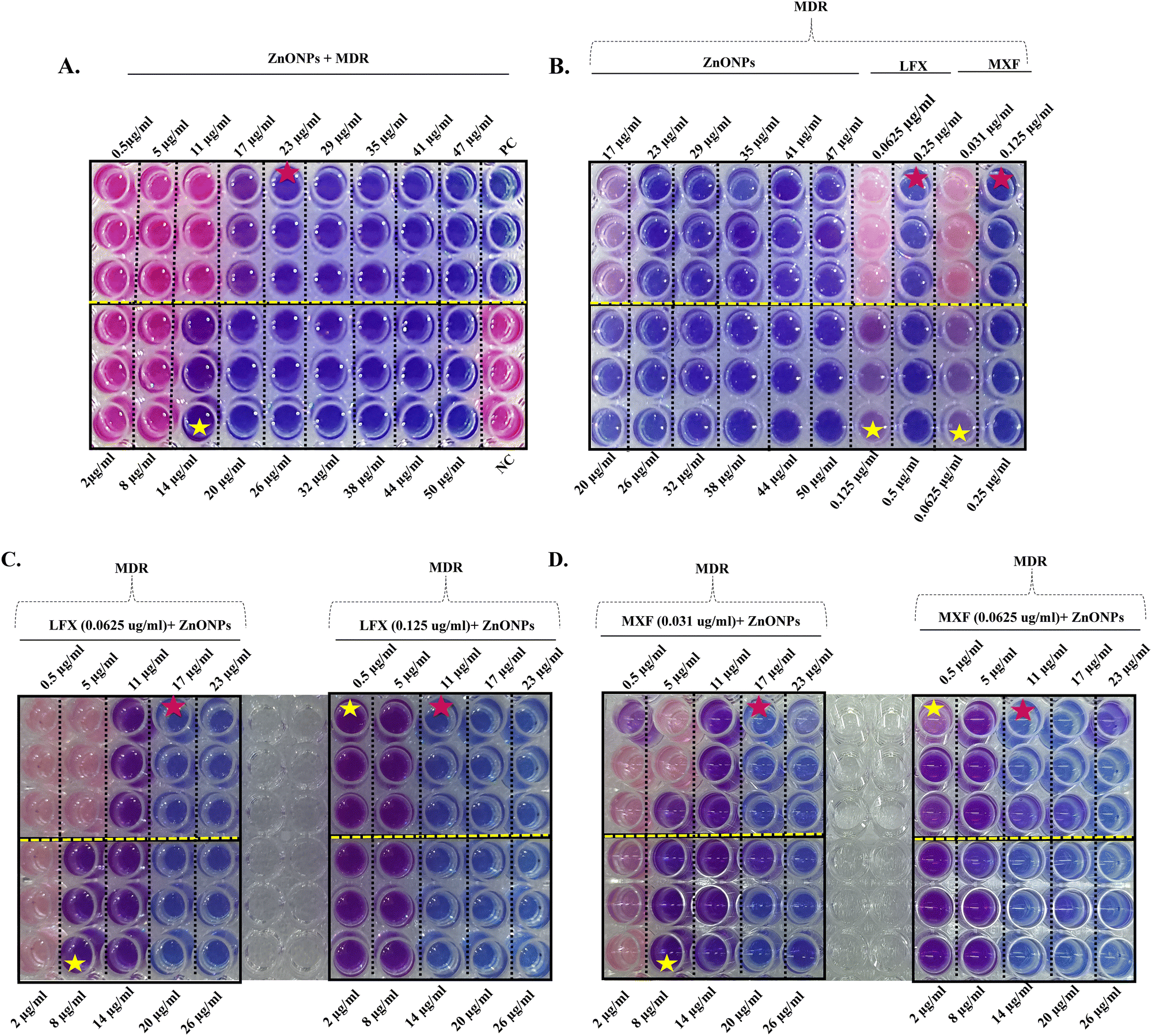
Current work validated the interaction between ZnONPs and Mtb SufB, which influenced cell viability, and this can be attributed to the production of a non-functional SufB protein via splicing inhibition, a vital component for Mtb survival.11,13 The observed decrease in viability could also be associated with a cumulative effect of mycobacterial membrane damage activity induced by ZnONPs.40 To substantiate this hypothesis, we performed a scanning electron microscopy (SEM) analysis, employing various ZnONP concentrations; a lower concentration of 0.5 μg ml−1, two effective concentrations of 17 and 23 μg ml−1 (MBC for ZnONPs towards H37Rv and MDR Mtb cells), and 50 μg ml−1, the upper limit for toxicity. These concentrations were selected to cover the effective range of ZnONPs for in vitro splicing studies, and MBC values were identified by Alamar blue assay and the spread plate method.Treatment with ZnONPs at a concentration of 0.5 μg ml−1 induced minor alterations in the bacterial morphology. However, exposure to concentrations of 17 μg ml−1, 23 μg ml−1, and 50 μg ml−1 led to extensive membrane disruption, cell-fusion, and cytoplasmic leakage accompanied by a complete loss of cellular morphology (Fig. 5(A) and (B)). Thus, SEM analysis offers substantial evidence of the membrane-damaging activity of ZnONPs towards H37Rv and MDR Mtb isolates. This uniform activity of ZnONPs against both drug-sensitive and MDR Mtb cells validates our previous observations on antimycobacterial effectiveness detected via Alamar blue assay. This result is particularly significant in the context of MDR-TB, where conventional antibiotic treatments encounter substantial obstacles.
 | ||
| Fig. 5 SEM analysis displaying the bactericidal effect of ZnONPs on Mycobacterium tuberculosis (Mtb) cells: (A) H37Rv and (B) MDR Mtb isolates. Control (untreated) mycobacterial cells were uniform looking, without any changes in the cell morphology or any evidence of membrane damage. Mild alterations in cellular morphology were noted for both H37Rv and MDR Mtb cells in the presence of 0.5 μg ml−1 ZnONPs. Dose dependent increase in the severity of cell membrane damage, cell fusion, and loss of cellular architecture were identified among the (A) H37Rv and (B) MDR Mtb isolates, across the rest of the ZnONP test concentration ranges (17 μg ml−1, 23 μg ml−1, and 50 μg ml−1) (outlined by yellow circles). MDR: multidrug-resistant Mtb. | ||
3.5 ZnONPs rescue Mtb induced changes in the infected mouse model
So far, in vitro data obtained from this work suggested the potent mycobactericidal activity of ZnONPs against H37Rv and MDR Mtb isolates, possibly by inhibiting the splicing of essential protein Mtb SufB, leading to loss of viability and membrane damage. To further assess this activity in an in vivo setting, we conducted experiments in a mouse model.Mtb H37Ra (H37Rv attenuated strain)-infected mice serve as an established in vivo model to evaluate TB infection.90 Both H37Ra and H37Rv Mtb strains are inhibited by similar concentrations of anti-TB drugs as suggested by an earlier work.91 Hence, the efficacy of ZnONPs–drug combination and recovery from the infection were further tested in H37Ra-infected mice.
BALB/6 mice were infected with Mtb H37Ra; 1× PBS (placebo), ZnONPs, and drug (RIF) administration commenced four days post-infection and continued for a total of seven days, with mice sacrificed on day 18 post-infection for subsequent histopathological and bacterial burden analysis (experimental design elucidated in Fig. 6(A)). Administration of RIF dosage is based on earlier works published elsewhere.92–94 Our findings revealed a strong anti-Mtb effect of ZnONPs, as evidenced by the analysis of the gross morphology of spleen, histopathological analysis and bacterial burden assay.
 | ||
| Fig. 6 In vivo antimycobacterial effects of ZnONPs on the H37Ra infected mouse model. (A) Schematic representation of the experimental design in BALB/c mice (n = 6 in each group). Mice were infected with H37Ra; 1× PBS (placebo), NPs and drug administration commenced four days post-infection and continued for a total of seven days, with mice sacrificed on day 18 post-infection. (B) Significant splenomegaly was noticed in the infected mouse, which had received 1× PBS only. This was not observed in the infected mice, who were given RIF (15 mg kg−1) alone, and combinations of RIF (10 mg kg−1) + ZnONPs (26 μg ml−1), and RIF (10 mg kg−1) + ZnONPs (50 μg ml−1), where the spleen size was comparable to the normal sized spleen in the healthy cage control mouse. The splenomegaly was partially rescued in mice who had received ZnONPs (26 or 50 μg ml−1) alone. Bacterial burden in the (C) lungs and (D) spleen of the infected mice was determined by plating the organ lysate on the 7H10 plates after treatment with ZnONPs or 1× PBS for 7 days. Significant reduction in mycobacterial loads was observed in mouse groups treated with a combination of RIF (10 mg kg−1) + ZnONPs (26 μg ml−1), and RIF (10 mg kg−1) + ZnONPs (50 μg ml−1), emphasizing the superior anti-TB activity of the ZnONPs–RIF combination as compared to RIF alone. Data are presented as mean ± 1 SD (p > 0.0001) based on values obtained for (n = 6) mice. | ||
A significant splenomegaly was observed in the infected mouse, which had received 1× PBS as a placebo following infection with the H37Ra strain.95–99 Resistance to this morphological effect was observed in infected mice, receiving RIF (15 mg kg−1) alone, ZnONPs (26 and 50 μg ml−1), RIF (10 mg kg−1) + ZnONPs (26 μg ml−1), and RIF (10 mg kg−1) + ZnONPs (50 μg ml−1). The splenomegaly following H37Ra infection in the aforementioned experimental groups was rescued significantly when ZnONPs were given as a solo treatment at concentrations of 26 μg ml−1 and 50 μg ml−1. Furthermore, the efficacy of RIF at 15 mg kg−1 was similar to a combination containing a reduced dosage of RIF at 10 mg kg−1 along with ZnONPs (26 μg ml−1, and 50 μg ml−1), where a complete rescue from splenomegaly was seen comparable to the normal sized spleen in the healthy cage control mouse (Fig. 6(B)). Likewise, a substantial reduction in mycobacterial load in lungs and spleen was observed in similarly treated mice groups, emphasizing the superior anti-TB activity of the ZnONPs/RIF combination (Fig. 6(C) and (D)). Bacterial loads were equivalently attenuated when RIF was administered solo at 15 mg kg−1 or in combination (RIF 10 mg kg−1 + ZnONPs 26 μg ml−1 or RIF 10 mg kg−1 + ZnONPs 50 μg ml−1) at a lower concentration. This highlights the synergistic activity of these anti-Mtb compounds displaying a higher potential to manage mycobacterial infection.
Next, histopathological examination of infected lungs and spleen tissues provided additional evidence for the anti-mycobacterial efficacy of ZnONPs. Lung sections from cage control healthy mice displayed clear lung fields without any evidence of inflammation [Fig. 7(A)-(a)]. The infected mouse group administered with 1× PBS displayed distinct pathological changes in the lungs with noticeable inflammatory infiltrates, indicative of infection induced immune response [Fig. 7(A)-(b)].100 When administered individually, ZnONPs (26 μg ml−1), ZnONPs (50 μg ml−1), and RIF (15 mg kg−1) caused a partial reduction in inflammatory cells within the lungs [Fig. 7(A)(c)–(e)]. In contrast, mice treated with a combination of ZnONPs (26 μg ml−1) + RIF (10 mg kg−1) and ZnONPs (50 μg ml−1) + RIF (10 mg kg−1), respectively, demonstrated no inflammatory infiltrations, suggesting their superior anti-mycobacterial action (Fig. 7(A)-(f) and (g)).
 | ||
| Fig. 7 Histopathology of lungs and spleen from H37Ra infected mice (H37Rv attenuated strain), comparing the efficacy of ZnONPs, RIF, and ZnONPs–RIF combinations. (A) Lungs sections are stained by H&E staining and processed for visualization by microscopy. (a) Histology of uninfected lungs in healthy control mice. (b)–(e) represent tissue sections from infected mice who have received different solo treatments, (b) 1× PBS exhibiting prominent aggregates of inflammatory cells; (c) 26 μg ml−1 ZnONPs, (d) 50 μg ml−1 ZnONPs, and (e) RIF 15 mg kg−1 displaying partial reduction in immune cell infiltrates; (f) RIF (10 mg kg−1) + ZnONPs (26 μg ml−1), and (g) RIF (10 mg kg−1) + ZnONPs (50 μg ml−1) represent no inflammatory changes and restoration of normal lung histology. (B) Tissue sections from spleen are stained by H&E staining and processed for visualization by microscopy. (a) Histology of spleen in uninfected healthy control mice showing well differentiated white and red pulp structures. (b)–(e) represent tissue sections from infected mice who have received different solo treatments; (b) 1× PBS exhibiting heavy immune cell aggregates obliterating white and red pulps; (c) 26 μg ml−1 ZnONPs and (d) 50 μg ml−1 ZnONPs display partial rescue from inflammatory infiltrates; (e) RIF 15 mg kg−1, (f) RIF (10 mg kg−1) + ZnONPs (26 μg ml−1), and (g) RIF (10 mg kg−1) + ZnONPs (50 μg ml−1), either of these treatments when given to infected mice, restored normal spleen histology with no evidence of inflammation. Images represent different experimental groups of mice visualized under 10× magnification. H&E – hematoxylin and eosin staining; RIF – Rifampicin. | ||
Similarly, inflammatory changes were noted within the white pulp of the infected splenic tissue, which is a critical component of the organ's immune function.101 Healthy mice from the cage control group exhibited normal spleen histology with clear differentiation into white and red pulps [Fig. 7(B)-(a)],102 whereas infected mice treated with 1× PBS demonstrated rich inflammatory infiltrates obscuring normal spleen histology [Fig. 7(B)-(b)].103 A partial reduction in inflammation was observed, along with restoration of the white and red pulp structures in the spleen of the mice treated with ZnONPs (26 μg ml−1) and ZnONPs (50 μg ml−1), respectively [Fig. 7(B)-(c), and (d)]. Notably, in the mice group that received RIF (15 mg kg−1) alone or combination therapy [ZnONPs (26 μg ml−1) + RIF (10 mg kg−1) and ZnONPs (50 μg ml−1) + RIF (10 mg kg−1)], there were no inflammatory infiltrations, and distinct white and red pulp structures were evident (Fig. 7(B)-(e)–(g)). Hence, the efficacy of the combinatorial approach with a reduced dose of RIF (10 mg kg−1) was similar to RIF solo therapy at a higher concentration (15 mg kg−1).
These observations suggest a potential therapeutic effect of the ZnONPs against Mycobacterium tuberculosis (Mtb). A concurrent mode of action combining SufB splicing inhibition by ZnONPs along with inhibition of bacterial RNA polymerase due to RIF and ZnONP mediated membrane damage possibly facilitates complete resolution of inflammation restoring normal spleen and lung histology.104 Taken together, these findings provide a strong foundation for further investigation into the mechanisms underlying the in vivo anti-mycobacterial activity of ZnONPs towards drug-sensitive as well as MDR TB. Additionally, long-term studies evaluating the safety and sustained efficacy of ZnONPs will be crucial for assessing their potential as a viable therapeutic option.
4. Conclusion
One of the major milestones of the United Nations Sustainable Development Goals (SDGs) is to end TB by 2030.105 Besides aiming for accessible patient care with the attenuated hospitalization rate, WHO also calls for “adoption to innovations” in TB management.2 The escalating challenge of drug-resistant TB necessitates the exploration of novel therapeutic strategies such as nanomedicine. Current work investigates the mycobactericidal effect of the ZnONPs against both the H37Rv Mtb strain and MDR Mtb isolates, by blocking the generation of crucial SufB protein via intein splicing inhibition.A series of in vitro experiments, encompassing Bradford assays, UV-visible spectroscopy, DLS, zeta potential analysis, and transmission electron microscopy (TEM), have collectively substantiated the interaction between the ZnONPs and the SufB protein. The Alamar blue assay and scanning electron microscopy (SEM) analysis further elucidated the bactericidal effects of the ZnONPs on H37Rv and MDR Mtb cells, which was correlated to the inhibitory effects of the ZnONPs on splicing and cleavage reactions of SufB precursor protein. Thus ZnONPs and the SufB protein interaction induced a significant alteration in the Mtb viability possibly due to loss of protein's biological activity. While SEM confirmed extensive mycobacterial membrane damage, the Alamar blue assay highlighted the superior efficacy of combination therapy (ZnONPs–INH/RIF and ZnONPs-LFX/MXF), where the effective dosage of the anti-TB drugs was reduced by half towards both the drug-sensitive strain and MDR Mtb isolates.85–88 Next, in vivo experiments in the infected mouse model validated the substantial anti-mycobacterial activity of ZnONPs–RIF combination against the Mtb H37Ra strain.
In the current study, histopathological examination of infected lungs and spleen tissues (H&E staining) has provided evidence for the anti-inflammatory efficacy of ZnONPs (Fig. 7). These results provided insight into the extent of inflammation and immune cell recruitment, which are the key indicators of the host immune response during Mtb infection and management.106–108 When administered individually, ZnONPs (26 μg ml−1), ZnONPs (50 μg ml−1), and RIF (15 mg kg−1) caused a partial reduction in inflammatory cells within the infected lungs and spleen. In contrast, mice treated with a combination of ZnONPs (26 μg ml−1) + RIF (10 mg kg−1) and ZnONPs (50 μg ml−1) + RIF (10 mg kg−1), demonstrated no inflammatory cell infiltrations, suggesting their superior anti-inflammatory action (Fig. 7). The observed reduction in inflammatory cells in the ZnONPs + RIF treated mouse group suggests a favourable anti-inflammatory and anti-mycobacterial effect.109,110
These collective findings underscore the potential of ZnONPs as a promising additive along with existing anti-TB drugs in the fight against tuberculosis. Herein, we propose a dual mode of action for this ZnONP-based combinatorial approach: ZnONPs can significantly influence mycobacterial viability by complete blockage of splicing and cleavage reactions to inhibit the generation of essential protein SufB, further supported by the potent antimycobacterial action of anti-TB drugs (Fig. 8). Since eukaryotes such as humans lack intein sequences and microbial intein sequences resist mutations, targeted drug development strategies aiming splicing regulation of critical proteins have been proposed as novel mechanisms to manage drug-resistant infections.111 The current study also highlights the reduction in the effective dosage of the anti-TB drugs in combination with ZnONPs, which will address caveats such as drug toxicity. However, further research and clinical trials will be essential to validate the safety and efficacy of ZnONPs as a potential supportive treatment option for the infected patients.
 | ||
| Fig. 8 Schematic diagram illustrating the proposed mechanism for higher efficacy of combination therapy in managing drug-susceptible and MDR TB, contrasting with conventional drug therapy. ZnONPs and RIF/INH or MXF/LFX combination exhibit superior therapeutic potential possibly via a dual approach: targeting the mycobacterial viability by splicing regulation to inhibit the generation of essential protein SufB, further supported by the potent antimycobacterial action of anti-TB drugs. Multidrug-resistant (MDR), rifampicin (RIF), isoniazid (INH), moxifloxacin (MXF), levofloxacin (LFX). | ||
This study's uniqueness lies in its innovative approach that demonstrated bactericidal efficacy against both drug-sensitive and MDR Mtb isolates as a significant improvement over earlier research. Furthermore, the inhibition of SufB splicing by ZnONPs provides a new mechanism for managing TB, distinct from existing treatments. Additionally, the reduced dose of RIF/INH and MXF/LFX when combined with ZnONPs may lead to fewer adverse effects and improved patient compliance, especially addressing concerns during extended treatment periods (as in the case of disseminated TB). Moreover, the potential toxicity of ZnONPs can be further eliminated by obtaining NPs via green synthesis routes.112,113 Howbeit, future research should focus on clinical trials to validate these findings and explore the long-term benefits and safety of this combinatorial approach. The current study addresses the growing issue of drug-resistant TB by exploring a new therapeutic approach that can have a significant contribution to the field of TB research and management. Higher efficacy along with lower drug dosage and probable shorter treatment duration may have a positive impact on the overall health cost, facilitating global effort to fight against TB.
Ethics statement
The animal study was reviewed and approved/recommended by the IAEC of Kalinga Institute of Industrial technology Deemed to be University (KIIT-DU), Bhubaneswar, Odisha (approval no. KSBT/IAEC/2023/2/14.2).Author contributions
Deepak Kumar Ojha: conceptualization, resources, data curation, formal analysis, validation, visualization, methodology, writing – original draft, writing – reviewing and editing; Ashwaria Mehra: resources, data curation, funding acquisition, validation, methodology, writing – reviewing and editing; Sunil Swick Rout: resources, data curation, software, validation, visualization; Sidhartha Giri: resources, validation, reviewing and editing; and Sasmita Nayak: conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing – original draft, project administration, writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Abbreviations
| Ab | Antibody |
| ANOVA | Analysis of variance |
| CC | C-terminal cleavage product |
| IB | Inclusion bodies |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| I | Intein |
| CC | C-terminal cleavage |
| P | Precursor |
| LE | Ligated extein |
| DS | Drug sensitive |
| DR | Dug resistance |
| Mtb | Mycobacterium tuberculosis |
| NC | N-terminal cleavage product |
| NE | N-extein |
| Ni-NTA | Nickel-nitrilotriacetic acid |
| PAGE | Polyacrylamide gel electrophoresis |
| PVDF | Polyvinylidene fluoride |
| SI | Splicing inactive |
| SDS | Sodium dodecyl sulfate |
| SEM | Scanning electron microscope |
| SUF | Mobilization of sulphur |
| TB | Tuberculosis |
| TCEP.HCl | Tris 2-carboxyl ethyl phosphine hydrochloric acid |
| TNB | 2-Nitro-5-thiobenzoic acid |
| UV-vis | UV-visible spectroscopy |
| WT | Wild-type |
| NP | Nanoparticle |
| ZnO | Zinc oxide |
| H&E | Hematoxylin and eosin |
| MDR | Multidrug resistant |
| RIF | Rifampicin |
| INH | Isoniazid |
| MXF | Moxifloxacin |
| LFX | Levofloxacin |
Data availability
The data underlying the present article are available in the article and in its online ESI.†Acknowledgements
This work is supported by the Indian Council of Medical Research (ICMR), India (ICMR Project ID: IIRP-2023-7834/F1). A. M. is supported by the INSPIRE fellowship (DST/INSPIRE/03/2021/002800/IF 190921), INSPIRE Division, DST, Government of India. The plasmids used in the present study are borrowed from Prof. Marlene Belfort's Lab, SUNY, Albany, NY, USA Our sincere gratitude to the Central Instrumentation facility (CIF) at OUAT (Odisha University of Agriculture & Technology), Bhubaneswar and Dr S. K. Dash for his help in conducting SEM analysis at OUAT Bhubaneswar. We thank Dr Sravan Kumar for his help in conducting TEM analysis at the Central Institute of Petrochemicals Engineering and Technology (CIPET), Bhubaneswar. We extend our gratitude to Dr Sidhartha Giri for providing access to the Biosafety Level 3 (BSL3) laboratory facility at ICMR-RMRC, Bhubaneswar, for smooth conductance of mycobacterial experiments.References
- D. R. Silva, F. C. de Queiroz Mello and G. B. Migliori, J. Bras. Pneumol., 2020, 46, e20200027 CrossRef PubMed.
- Geneva: World Health Organization, Global tuberculosis report 2023, Global tuberculosis report 2023, https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2023, (accessed 2 July 2024).
- K. J. Seung, S. Keshavjee and M. L. Rich, Cold Spring Harbor Perspect. Med., 2015, 5(9), a017863, DOI:10.1101/cshperspect.a017863.
- A. Prasanna and V. Niranjan, Bioinformation, 2019, 15, 261–268 CrossRef PubMed.
- S. N. Goossens, S. L. Sampson and A. Van Rie, Clin. Microbiol. Rev., 2020, 34(1), 10–1128, DOI:10.1128/CMR.00141-20.
- K. H. Alzahabi, O. Usmani, T. K. Georgiou, M. P. Ryan, B. D. Robertson, T. D. Tetley and A. E. Porter, Emerging Top. Life Sci., 2020, 4(6), 581–600, DOI:10.1042/ETLS20190154.
- S. Panda, A. Nanda, N. Sahu, D. K. Ojha, B. Pradhan, A. Rai, A. R. Suryawanshi, N. Banavali and S. Nayak, Biosci. Rep., 2022, 42(3), BSR20212207, DOI:10.1042/BSR20212207.
- S. Panda, A. Nanda, S. S. Nasker, A. Mehra, D. K. Ojha, P. S. Mohanty and S. Nayak, Clin. Sci., 2023, 137(14), 1027–1048, DOI:10.1042/CS20230307.
- O. Novikova, P. Jayachandran, D. S. Kelley, Z. Morton, S. Merwin, N. I. Topilina and M. Belfort, Mol. Biol. Evol., 2016, 33(3), 783–799, DOI:10.1093/molbev/msv271.
- F. Wayne Outten, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853(6), 1464–1469, DOI:10.1016/j.bbamcr.2014.11.001.
- G. Huet, M. Daffé and I. Saves, J. Bacteriol., 2005, 187, 6137–6146 CrossRef CAS PubMed.
- H. K. Chahal, Y. Dai, A. Saini, C. Ayala-Castro and F. W. Outten, Biochemistry, 2009, 48(44), 10644–10653, DOI:10.1021/bi901518y.
- G. Huet, J. P. Castaing, D. Fournier, M. Daffé and I. Saves, J. Bacteriol., 2006, 188, 3412–3414 CrossRef CAS PubMed.
- K. W. Jayawardana, H. S. N. Jayawardena, S. A. Wijesundera, T. De Zoysa, M. Sundhoro and M. Yan, Chem. Commun., 2015, 51(60), 12028–12031, 10.1039/c5cc04251h.
- P. V. Mohanan, V. G. Reshma, S. Syama, S. Sruthi, S. C. Reshma and N. S. Remya, Curr. Drug Metab., 2017, 18(11), 1040–1054, DOI:10.2174/1389200218666170925122201.
- M. Vallet-Regí, B. González and I. Izquierdo-Barba, Int. J. Mol. Sci., 2019, 20(15), 3806, DOI:10.3390/ijms20153806.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2016, 1(1), 10–29, DOI:10.1002/btm2.10003.
- G. T. Tietjen and W. M. Saltzman, Sci. Transl. Med., 2015, 7(314), 314fs47, DOI:10.1126/scitranslmed.aad6645.
- A. Gallud, H. Líbalová and B. Fadeel, Nanomedicine, 2015, 10(12), 1859–1861, DOI:10.2217/nnm.15.79.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Nanomater. Neoplasms, 2021, 31–142, DOI:10.1002/anie.201403036.
- J. N. Barry and A. A. Vertegel, Nano LIFE, 2013, 3(04), 1343001, DOI:10.1142/s1793984413430010.
- S. J. Park, Int. J. Nanomed., 2020, 15(2020), 5783–5802, DOI:10.2147/IJN.S254808.
- S. R. Saptarshi, A. Duschl and A. L. Lopata, J. Nanobiotechnol., 2013, 11, 1–12, DOI:10.1186/1477-3155-11-26.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nano-Enabled Med. Appl., 2020, 61–91, DOI:10.1038/nnano.2007.387.
- K. K. Jain, Clin. Chim. Acta, 2005, 358(1–2), 37–54, DOI:10.1016/j.cccn.2005.03.014.
- Y. Barenholz, J. Controlled Release, 2012, 160(2), 117–134, DOI:10.1016/j.jconrel.2012.03.020.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6(12), 1078–1094, DOI:10.1038/s41578-021-00358-0.
- J. Theron, J. A. Walker and T. E. Cloete, Crit. Rev. Microbiol., 2008, 34(1), 43–69, DOI:10.1080/10408410701710442.
- A. Gogos, K. Knauer and T. D. Bucheli, J. Agric. Food Chem., 2012, 60(39), 9781–9792, DOI:10.1021/jf302154y.
- J. Mou, Z. Liu, J. Liu, J. Lu, W. Zhu and D. Pei, Drug Delivery, 2019, 26(1), 179–187, DOI:10.1080/10717544.2019.1571121.
- C. Pushpalatha, J. Suresh, V. S. Gayathri, S. V. Sowmya, D. Augustine, A. Alamoudi, B. Zidane, N. H. Mohammad Albar and S. Patil, Front. Bioeng. Biotechnol., 2022, 10, 917990, DOI:10.3389/fbioe.2022.917990.
- X. Li, C. Yuan, Q. Chen, Q. Xue, J. Mou and P. Wang, Med. Oral Patol. Oral Cir. Bucal, 2023, 28, e487–e495 CrossRef CAS PubMed.
- Y. Wang, H. Hua, W. Li, R. Wang, X. Jiang and M. Zhu, J. Dent., 2019, 80, 23–29 Search PubMed.
- C. Petchthanasombat, T. Tiensing and P. Sunintaboon, J. Colloid Interface Sci., 2012, 369, 52–57 CrossRef CAS PubMed.
- E. M. El-Diasty, M. A. Ahmed, N. Okasha, S. F. Mansour, S. I. El-Dek, H. M. A. El-Khalek and M. H. Youssif, Rom. J. Biophys., 2013, 23, 191–202 CAS.
- S. Raha and M. Ahmaruzzaman, Nanoscale Adv., 2022, 4(8), 1868–1925, 10.1039/d1na00880c.
- S. Jha, R. Rani and S. Singh, J. Inorg. Organomet. Polym. Mater., 2023, 33(6), 1437–1452, DOI:10.1007/s10904-023-02550-x.
- M. Heidary, S. Z. Bostanabad, S. M. Amini, A. Jafari, M. G. Nobar, A. Ghodousi, M. Kamalzadeh and D. Darban-Sarokhalil, Infect Drug Resist, 2019, 12(2019), 3425–3435, DOI:10.2147/IDR.S221408.
- W. Lin, S. Fan, K. Liao, Y. Huang, Y. Cong, J. Zhang, H. Jin, Y. Zhao, Y. Ruan, H. Lu, F. Yang, C. Wu, D. Zhao, Z. Fu, B. Zheng, J. F. Xu and J. Pi, Front. Cell. Infect. Microbiol., 2023, 12, 1074533, DOI:10.3389/fcimb.2022.1074533.
- A. J. Caron, I. J. Ali, M. J. Delgado, D. Johnson, J. M. Reeks, Y. M. Strzhemechny and S. M. McGillivray, Front. Microbiol., 2024, 15, 1394078 Search PubMed.
- S. V. Gudkov, D. E. Burmistrov, D. A. Serov, M. B. Rebezov, A. A. Semenova and A. B. Lisitsyn, Front. Phys., 2021, 9, 641481, DOI:10.3389/fphy.2021.641481.
- P. Sun, S. Ye, S. Ferrandon, T. C. Evans, M. Q. Xu and Z. Rao, J. Mol. Biol., 2005, 353(5), 1093–1105, DOI:10.1016/j.jmb.2005.09.039.
- B. W. Poland, M. Q. Xu and F. A. Quiocho, J. Biol. Chem., 2000, 275(22), 16408–16413, DOI:10.1074/jbc.275.22.16408.
- L. Zhang, Y. Zheng, Z. Xi, Z. Luo, X. Xu, C. Wang and Y. Liu, Mol. BioSyst., 2009, 5(6), 644–650, 10.1039/b903144h.
- P. Van Roey, B. Pereira, Z. Li, K. Hiraga, M. Belfort and V. Derbyshire, J. Mol. Biol., 2007, 367(1), 162–173, DOI:10.1016/j.jmb.2006.12.050.
- S. Panda, A. Nanda, S. S. Nasker, D. Sen, A. Mehra and S. Nayak, Biochimie, 2021, 185, 53–67, DOI:10.1016/j.biochi.2021.03.006.
- D. Woods, S. Vangaveti, I. Egbanum, A. M. Sweeney, Z. Li, V. Bacot-Davis, D. S. Lesassier, M. Stanger, G. E. Hardison, H. Li, M. Belfort and C. W. Lennon, mBio, 2020, 11(4), e01403-20, DOI:10.1128/mBio.01403-20.
- R. Afarin, F. Ahmadpour, M. Hatami, S. Monjezi and S. Igder, Heliyon, 2024, 10, e31925 Search PubMed.
- Q. Lin, Y. Jing, C. Yan, X. Chen, Q. Zhang, X. Lin, Y. Xu and B. Chen, Int. J. Nanomed., 2024, 19, 5381–5395 CrossRef PubMed.
- B. Naiel, M. Fawzy, A. E. D. Mahmoud and M. W. A. Halmy, Sci. Rep., 2024, 14, 13459 Search PubMed.
- A. Nanda, S. S. Nasker, A. K. Kushwaha, D. K. Ojha, A. K. Dearden, S. K. Nayak and S. Nayak, Front. Bioeng. Biotechnol., 2021, 9, 773303, DOI:10.3389/fbioe.2021.773303.
- E. Suhartono, I. Thalib, I. Aflanie, Z. Noor and R. Idroes, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 350, 012008, DOI:10.1088/1757-899X/350/1/012008.
- P. K. Grzyska, R. P. Hausinger and D. A. Proshlyakov, Anal. Biochem., 2010, 399, 64–71 Search PubMed.
- J. Saikia, M. Yazdimamaghani, S. P. Hadipour Moghaddam and H. Ghandehari, ACS Appl. Mater. Interfaces, 2016, 8(50), 34820–34832, DOI:10.1021/acsami.6b09950.
- G. Su, H. Jiang, B. Xu, Y. Yu and X. Chen, Mol Pharm, 2018, 15(11), 5019–5030, DOI:10.1021/acs.molpharmaceut.8b00612.
- A. M. Clemments, C. Muniesa, C. C. Landry and P. Botella, RSC Adv., 2014, 4, 29134–29138 Search PubMed.
- A. Nanda, S. S. Nasker, A. K. Kushwaha, D. K. Ojha, A. K. Dearden, S. K. Nayak and S. Nayak, Front. Bioeng. Biotechnol., 2021, 9, 773303 Search PubMed.
- A. Sasidharan, J. E. Riviere and N. A. Monteiro-Riviere, J. Mater. Chem. B, 2015, 3, 2075–2082 Search PubMed.
- M. Kokkinopoulou, J. Simon, K. Landfester, V. Mailänder and I. Lieberwirth, Nanoscale, 2017, 9, 8858–8870 Search PubMed.
- M. G. Soliman, A. Martinez-Serra, M. Dobricic, D. N. Trinh, J. Cheeseman, D. I. R. Spencer and M. P. Monopoli, Front. Toxicol., 2024, 6, 1393330, DOI:10.3389/ftox.2024.1393330.
- K. Friedel, M. A. Popp, J. C. J. Matern, E. M. Gazdag, I. V. Thiel, G. Volkmann, W. Blankenfeldt and H. D. Mootz, Chem. Sci., 2019, 10(1), 239–251, 10.1039/C8SC01074A.
- L. Zhang, Y. Zheng, B. Callahan, M. Belfort and Y. Liu, J. Biol. Chem., 2011, 286(2), 1277–1282, DOI:10.1074/jbc.M110.171124.
- K. V. Mills, B. M. Lew, S. Q. Jiang and H. Paulus, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(7), 3543–3548, DOI:10.1073/pnas.95.7.3543.
- K. V. Mills and H. Paulus, J. Biol. Chem., 2001, 276(14), 10832–10838, DOI:10.1074/jbc.M011149200.
- J. P. Gangopadhyay, S. Qin Jiang and H. Paulus, Anal. Chem., 2003, 75(10), 2456–2462, DOI:10.1021/ac020756b.
- H. Chan, C. S. Pearson, C. M. Green, Z. Li, J. Zhang, G. Belfort, A. Shekhtman, H. Li and M. Belfort, J. Biol. Chem., 2016, 291(43), 22661–22670, DOI:10.1074/jbc.M116.747824.
- K. Punjabi, S. Mehta, R. Chavan, V. Chitalia, D. Deogharkar and S. Deshpande, Front. Microbiol., 2018, 9, 2207, DOI:10.3389/fmicb.2018.02207.
- W. Zhou, B. Yang, Y. Zou, K. Rahman, X. Cao, Y. Lei, R. Lai, Z. F. Fu, X. Chen and G. Cao, Front. Microbiol., 2021, 12, 658637, DOI:10.3389/fmicb.2021.658637.
- E. M. Longhin, N. El Yamani, E. Rundén-Pran and M. Dusinska, Front. Toxicol., 2022, 4, 981701, DOI:10.3389/ftox.2022.981701.
- C. Mahendra, M. N. Chandra, M. Murali, M. R. Abhilash, S. B. Singh, S. Satish and M. S. Sudarshana, Process Biochem., 2020, 89, 220–226, DOI:10.1016/j.procbio.2019.10.020.
- G. Biosciences, Alamar blue Cell Viability Assay, https://cdn.gbiosciences.com/pdfs/protocol/786-921_protocol.pdf, (accessed 18 June 2025).
- E. M. Longhin, N. El Yamani, E. Rundén-Pran and M. Dusinska, Front. Toxicol., 2022, 4, 981701, DOI:10.3389/ftox.2022.981701.
- S. Sharma, E. Gelman, C. Narayan, D. Bhattacharjee, V. Achar, V. Humnabadkar, V. Balasubramanian, V. Ramachandran, N. Dhar and N. Dinesh, Antimicrob. Agents Chemother., 2014, 58(10), 5801–5808, DOI:10.1128/AAC.03205-14.
- S. Panda, T. K. Rout, A. D. Prusty, P. M. Ajayan and S. Nayak, Adv. Mater., 2018, 30(7), 1702149, DOI:10.1002/adma.201702149.
- L. Abarca-Cabrera, P. Fraga-García and S. Berensmeier, Biomater. Res., 2021, 25(1), 12, DOI:10.1186/s40824-021-00212-y.
- C. Eisner, H. Ow, T. Yang, Z. Jia, E. Dimitriadis, L. Li, K. Wang, J. Briggs, M. Levine, J. Schnermann and M. G. Espey, J. Appl. Physiol., 2012, 112(4), 681–687, DOI:10.1152/japplphysiol.01068.2011.
- G. Yan, Y. Huang, Q. Bu, L. Lv, P. Deng, J. Zhou, Y. Wang, Y. Yang, Q. Liu, X. Cen and Y. Zhao, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2012, 47(4), 577–588, DOI:10.1080/10934529.2012.650576.
- A. M. Florea, F. Splettstoesser and D. Büsselberg, Toxicol. Appl. Pharmacol., 2007, 220(3), 292–301, DOI:10.1016/j.taap.2007.01.022.
- V. G. Reshma and P. V. Mohanan, Colloids Surf., B, 2017, 157, 182–190, DOI:10.1016/j.colsurfb.2017.05.069.
- E. L. Han, S. Tang, D. Kim, A. M. Murray, K. L. Swingle, A. G. Hamilton, K. Mrksich, M. S. Padilla, R. Palanki, J. J. Li and M. J. Mitchell, Nano Lett., 2024, 25(2), 800–810, DOI:10.1021/acs.nanolett.4c05186.
- B. Baral, B. Panigrahi, A. Kar, K. D. Tulsiyan, U. Suryakant, D. Mandal and U. Subudhi, Mol. Ther.–Nucleic Acids, 2023, 33, 493–510, DOI:10.1016/j.omtn.2023.07.017.
- G. Huet, M. Daffé and I. Saves, J. Bacteriol., 2005, 187(17), 6137–6146, DOI:10.1128/JB.187.17.6137-6146.2005.
- V. Valdiglesias, A. Alba-González, N. Fernández-Bertólez, A. Touzani, L. Ramos-Pan, A. T. Reis, J. Moreda-Piñeiro, J. Yáñez, B. Laffon and M. Folgueira, Int. J. Mol. Sci., 2023, 24(15), 12297, DOI:10.3390/ijms241512297.
- B. N. Patil and T. C. Taranath, Int. J. Mycobact., 2016, 5(2), 197–204, DOI:10.1016/j.ijmyco.2016.03.004.
- S. S. Hakkimane, V. P. Shenoy, S. L. Gaonkar, I. Bairy and B. R. Guru, Int. J. Nanomed., 2018, 13, 4303–4318, DOI:10.2147/IJN.S163925.
- M. Getahun, H. M. Blumberg, G. Ameni, D. Beyene and R. R. Kempker, PLoS One, 2022, 17, e0274426, DOI:10.1371/journal.pone.0274426.
- H. Zheng, W. He, W. Jiao, H. Xia, L. Sun, S. Wang, J. Xiao, X. Ou, Y. Zhao and A. Shen, BMC Infect. Dis., 2021, 21, 1–6, DOI:10.1186/s12879-021-06024-8.
- Q. Sun, K. Cheng, X. Liao, W. Zhao, C. Wang, C. Wang, J. Yan, L. Dong, F. Wang, G. Jiang, H. Huang, Z. Guo and G. Wang, J. Med. Microbiol., 2024, 73, 001825, DOI:10.1099/jmm.0.001825.
- F. B. Perler, Nucleic Acids Res., 2002, 30(1), 383–384, DOI:10.1093/nar/30.1.383.
- N. Kumari, R. Sharma, J. Ali, G. Chandra, S. Singh and M. Y. Krishnan, Tuberculosis, 2024, 145, 102479, DOI:10.1016/j.tube.2024.102479.
- M. T. Heinrichs, R. J. May, F. Heider, T. Reimers, S. K. B. Sy, C. A. Peloquin and H. Derendorf, Int. J. Mycobact., 2018, 7(2), 156–161, DOI:10.4103/ijmy.ijmy_33_18.
- J. E. M. De Steenwinkel, R. E. Aarnoutse, G. J. De Knegt, M. T. Ten Kate, M. Teulen, H. A. Verbrugh, M. J. Boeree, D. Van Soolingen and I. A. J. M. Bakker-Woudenberg, Am. J. Respir. Crit. Care Med., 2013, 187(10), 1127–1134, DOI:10.1164/rccm.201207-1210OC.
- I. M. Rosenthal, R. Tasneen, C. A. Peloquin, M. Zhang, D. Almeida, K. E. Mdluli, P. C. Karakousis, J. H. Grosset and E. L. Nuermberger, Antimicrob. Agents Chemother., 2012, 56(8), 4331–4340, DOI:10.1128/AAC.00912-12.
- H. M. Blumberg, W. J. Burman, R. E. Chaisson, C. L. Daley, S. C. Etkind, L. N. Friedman, P. Fujiwara, M. Grzemska, P. C. Hopewell, M. D. Iseman, R. M. Jasmer, V. Koppaka, R. I. Menzies, R. J. O’Brien, R. R. Reves, L. B. Reichman, P. M. Simone, J. R. Starke and A. A. Vernon, Am. J. Respir. Crit. Care Med., 2003, 167, 603–662 Search PubMed.
- B. Pottakkat, A. Kumar, A. Rastogi, N. Krishnani, V. K. Kapoor and R. Saxena, Gut Liver, 2010, 4(1), 94, DOI:10.5009/gnl.2010.4.1.94.
- D. A. Lestari, N. Rahadiani and R. A. Syaiful, Int. J. Surg. Case Rep., 2021, 83, 105966, DOI:10.1016/j.ijscr.2021.105966.
- Y. Tojo, S. Yanagisawa, E. Miyauchi and M. Ichinose, Int. J. Infect. Dis., 2018, 67, 41–42, DOI:10.1016/j.ijid.2017.12.009.
- L. Kremer, J. Estaquier, I. Wolowczuk, F. Biet, J. C. Ameisen and C. Locht, Infect. Immun., 2000, 68(7), 4264–4273, DOI:10.1128/IAI.68.7.4264-4273.2000.
- A. Masumi, K. Mochida, K. Takizawa, T. Mizukami, M. Kuramitsu, M. Tsuruhara, S. Mori, K. Shibayama, K. Yamaguchi and I. Hamaguchi, Inflammation Regener., 2016, 36, 1–8, DOI:10.1186/s41232-016-0024-3.
- A. Nava, A. C. Hahn, T. H. Wu and T. F. Byrd, Front. Immunol., 2022, 13, 1017540, DOI:10.3389/fimmu.2022.1017540.
- S. M. Lewis, A. Williams and S. C. Eisenbarth, Sci. Immunol., 2019, 4(33), eaau6085, DOI:10.1126/sciimmunol.aau6085.
- B. S. Steiniger, Immunology, 2015, 145(3), 334–346, DOI:10.1111/imm.12469.
- S. M. Szczepanek, J. T. McNamara, E. R. Secor, P. Natarajan, L. A. Guernsey, L. A. Miller, E. Ballesteros, E. Jellison, R. S. Thrall and B. Andemariam, Am. J. Pathol., 2012, 181(5), 1725–1734, DOI:10.1016/j.ajpath.2012.07.034.
- W. Wehrli, Rev. Infect. Dis., 1983, 5, s407–s411, DOI:10.1093/clinids/5.Supplement_3.S407.
- United Nations, The Sustainable Development Goals Report 2023, https://unstats.un.org/sdgs/report/2023/, (accessed 2 July 2024).
- B. Jee, Cent. Eur. J. Immunol., 2020, 45, 99–103 Search PubMed.
- F. Rahman, Front. Immunol., 2024, 15, 1437901 Search PubMed.
- B. Singh, I. Pahuja, P. Yadav, A. Shaji, S. Chaturvedi, A. Ranganathan, V. P. Dwivedi and G. Das, J. Infect. Dis., 2024, 229, 1509–1518 Search PubMed.
- F. E. A. Hayford, R. C. Dolman, R. Blaauw, A. Nienaber, C. M. Smuts, L. Malan and C. Ricci, Respir. Res., 2020, 21, 1–14 Search PubMed.
- V. M. Kroesen, M. I. Gröschel, N. Martinson, A. Zumla, M. Maeurer, T. S. van der Werf and C. Vilaplana, Front. Immunol., 2017, 8, 280326 Search PubMed.
- H. Paulus, Front. Biosci.-Landmark, 2003, 8, 1157–1165 Search PubMed.
- R. Álvarez-Chimal and J. Ángel Arenas-Alatorre, Green Chemistry for Environmental Sustainability – Prevention-Assurance-Sustainability (P–A–S) Approach, 2023 Search PubMed.
- J. Iqbal, B. A. Abbasi, T. Yaseen, S. A. Zahra, A. Shahbaz, S. A. Shah, S. Uddin, X. Ma, B. Raouf, S. Kanwal, W. Amin, T. Mahmood, H. A. El-Serehy and P. Ahmad, Sci. Rep., 2021, 11(1), 20988, DOI:10.1038/s41598-021-99839-z.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01224k |
| This journal is © The Royal Society of Chemistry 2025 |