
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA hypervalent metal MOF catalyst as an avenue to go beyond heterogeneous Fenton-like processes for organic contaminant removal in water†
Lucas
Santos-Juanes
a,
Noelia
Rodriguez-Sanchez
 bc,
Salvador R. G.
Balestra
cde,
Nuria O.
Núñez
f,
Antonio
Arques
a,
A. Rabdel
Ruiz-Salvador
*cd and
Menta
Ballesteros
*cg
bc,
Salvador R. G.
Balestra
cde,
Nuria O.
Núñez
f,
Antonio
Arques
a,
A. Rabdel
Ruiz-Salvador
*cd and
Menta
Ballesteros
*cg
aGrupo de Procesos de Oxidación Avanzada, Departamento de Ingeniería Textil y Papelera, Universitat Politècnica de València, Campus de Alcoy, Alcoy, Spain
bBAM Federal Institute for Materials Research and Testing, Richard-Willstatter-Str, Berlin, 11 12489, Germany
cCentro de Nanociencia y Tecnologías Sostenibles (CNATS), Universidad Pablo de Olavide, Carretera de Utrera km. 1, Sevilla, Spain. E-mail: rruisal@upo.es; mmbalmar@upo.es
dDepartamento de Sistemas Físicos, Químicos y Naturales, Universidad Pablo de Olavide, Ctra. Utrera, km. 1, Sevilla, Spain
eDepartamento de Física Atómica, Molecular y Nuclear, Área de Física Teórica, Universidad de Sevilla, Avenida de Reina Mercedes s/n, Sevilla, 41012, Spain
fInstituto de Ciencia de Materiales de Sevilla, ICMS (CSIC-US), c/Américo Vespucio, 49, Sevilla, Spain
gDepartamento de Biología Molecular e Ingeniería Bioquímica, Universidad Pablo de Olavide, Ctra. Utrera km. 1, Sevilla, Spain
First published on 1st May 2025
Abstract
Metal–organic frameworks (MOFs) have recently been proposed as a plausible solution to the pressing issue of water scarcity and as a means of remediating contaminated water bodies. In light-assisted water treatment, they have so far only been exploited via the hydroxyl radical route, through Fenton-like processes. A new avenue is introduced here by the biomimetic conceptual design of MOF bearing hypervalent metal atoms for photocatalytic water treatment. We report a zeolitic imidazole framework (ZIF) material doped with iron (Fe-ZIF-7-III; UPO-4) synthesized via a novel mild treatment to stabilize photoactive hypervalent ferryl ions for the first time in a MOF for water treatment. The successful synthesis of the 2D material and the adequate incorporation of iron into the structure were demonstrated using X-ray diffraction (XRD), Fourier-transform infrared (FTIR) spectroscopy, and scanning electron microscopy (SEM). A simulation study analyzed the structure and stability of the Fe-ZIF-7-III material as well as the involvement of ferryl ions in the photo-Fenton-type process. Furthermore, the calculated band gap of this material shows its viability for use in photocatalysis using sunlight. This was confirmed by evaluating the photodegradation of caffeine, a model pollutant in water, without the assistance of hydroxyl radicals as indicated by a scavenger test. The recyclability test revealed that Fe-ZIF-7-III could be used continuously with effective catalytic activity, thus opening the door to the field of studying hypervalent metal MOFs not yet explored in water treatment.
Introduction
Metal–organic frameworks have been in focus for the past 25 years, initially motivated by their spectacular crystalline porous structures1,2 and further fuelled by the amenable design following the principles of reticular chemistry.3,4 These materials have exhibited significant performance in high-profile adsorption separation applications, such as CO2 adsorption,5 and small molecules6 and hydrocarbon separations.7 Magnetism and sensing have also been the subject of relevant research,8,9 along with increasing interest in catalytic properties.10 Progress in the design of new catalytic MOFs has been achieved by, among the most successful routes, using biomimetic approaches that have exploited the versatility of the materials to be easily modified at both metal centres and ligands to reproduce high yield reactions that are observed in living species, such as is the conversion of methane or ethane to methanol or ethanol.11,12 This has opened an avenue for the production of building unit molecules from natural renewable sources, such as biogas, for the next generation biorefinery industry. The structural motif behind this application is the presence of hypervalent iron, which is not common but can be encountered in specific chemical environments, and can account for the reactivity appearing as relevant intermediates, according to DFT calculations.11–16 In a recent study, Hou et al.17 successfully stabilized hypervalent ferryl (Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) ions in a MOF that demonstrated effective catalytic activity in the oxygenation of cyclohexane and the conversion of ethane to ethanol. This interesting result may stimulate applications where chemical oxidation would be the key point, as can be the oxidative water treatment, a field that is receiving increasing attention due to the scarcity of access to water resources. MOFs supporting high-valent iron metal species offer potential advantages over traditional photo-Fenton reactions by providing a higher concentration and stability of oxidant species less susceptible to matrix inhibition,18 the possibility of greater selectivity in the degradation of some specific contaminants poorly reactive with hydroxyl radicals (˙OH),19 the stabilization and activation of these species by the MOF structure16 and the promotion of alternative reaction mechanisms and the ability to design tailored catalysts for specific organic pollutant degradation applications.19 However, a comprehensive search of the most widely used bibliographic databases for academic research and literature analysis, such as Web of Science or Scopus (as of 20 November 2024), does not yield any results on water treatment when the words “MOF” and “hypervalent” or “ferryl” are used as search terms. This suggests that there is an opportunity for the scientific community to investigate the potential of utilizing MOFs in which hypervalent metal ions are involved, representing a novel and promising area for further study and development, including water treatment.
O) ions in a MOF that demonstrated effective catalytic activity in the oxygenation of cyclohexane and the conversion of ethane to ethanol. This interesting result may stimulate applications where chemical oxidation would be the key point, as can be the oxidative water treatment, a field that is receiving increasing attention due to the scarcity of access to water resources. MOFs supporting high-valent iron metal species offer potential advantages over traditional photo-Fenton reactions by providing a higher concentration and stability of oxidant species less susceptible to matrix inhibition,18 the possibility of greater selectivity in the degradation of some specific contaminants poorly reactive with hydroxyl radicals (˙OH),19 the stabilization and activation of these species by the MOF structure16 and the promotion of alternative reaction mechanisms and the ability to design tailored catalysts for specific organic pollutant degradation applications.19 However, a comprehensive search of the most widely used bibliographic databases for academic research and literature analysis, such as Web of Science or Scopus (as of 20 November 2024), does not yield any results on water treatment when the words “MOF” and “hypervalent” or “ferryl” are used as search terms. This suggests that there is an opportunity for the scientific community to investigate the potential of utilizing MOFs in which hypervalent metal ions are involved, representing a novel and promising area for further study and development, including water treatment.
Advanced oxidation processes (AOPs) play a central role in water treatment for the removal of hazardous contaminants. Among them, those using environmentally friendly energy sources are being extensively investigated, especially those using solar energy, which have gained attention for their ability to degrade persistent pollutants, often using photo-assisted reactions such as the photo-Fenton process or semiconductor-mediated photocatalysis.20,21 In this context, heterogeneous catalysis is attracting much attention in the field of AOPs, stimulated by its well-known advantages in implementation and reuse.22–25 The versatility and amenable design of MOFs for catalytic applications22,26–28 lead these materials to be used as heterogeneous catalysts in AOPs for water treatment. The work reported to date using MOFs in this area uses Fenton and Fenton-like reactions, which are based on radicals derived from hydrogen peroxide, sulphate, or other oxidants as reactive species. Since AOPs can also be achieved by routes that do not involve these radicals, but rather hypervalent metal ions,23 it is interesting to investigate whether such an approach could be implemented with MOFs.
We have noted that the use of ferrihydrite iron oxydroxide with hypervalent ferryl ions as a key oxidant in organic matter degradation reactions has been described.29 Similar to Long et al., we have chosen a biomimetic molecular environment to design our material with stable isolated hypervalent metal sites for water treatment. Learning from nature, it is known that iron coordinated with multiple nitrogen atoms, as in porphyrinoid systems, can form ferryl ions with significant catalytic activity.30,31 Such an N-rich molecular environment could be replicated in Fe-bearing MOFs, making Fe-ZIFs (ZIF stands for zeolitic imidazolate framework) a natural choice for exploration. ZIFs, with topologies similar to those of zeolites, are a subclass of MOFs composed of tetrahedrally coordinated transition metal ions and organic imidazole ligands.32 They have excellent thermal and hydrolytic stability, high surface area, and abundant catalytic sites.33,34 Previous studies have primarily used specific ZIFs, such as ZIF-9 and ZIF-67, to activate ˙OH or other reactive species for the degradation of environmental contaminants. For example, ZIF-9 and ZIF-12 have been used to activate peroxymonosulfate (PMS) for the degradation of contaminants such as rhodamine B and tetracycline.35,36 Cu-doped ZIF-67 and CuCo-ZIFs have shown promise as visible light-driven photocatalysts for dye degradation via Fenton-related reactions and α-Fe2O3/ZIF-67 was able to completely degrade ciprofloxacin in 30 min via SO4˙− and ˙OH radicals.37
In designing the targeted hypervalent ferryl ion-containing MOF for water remediation, we would need a very stable material as a proof of concept. According to Amombo Noa et al.,38 the order of stability increases from dot, rod to sheet or 2-D MOFs. In the versatile ZIF-7 family, the 2-D ZIF-7-III member emerges as the most stable phase.39 It should be noted that the 2-D ZIF-L solid, derived from ZIF-8, is also the most stable phase of this system.40 The choice of ZIF-7-III over ZIF-L is due to the presence of benzene chromophores, which enhance its stability and light absorption properties, making it effective for photocatalysis.39,41 The incorporation of benzimidazole lowers the conduction band energy compared to pure imidazole and methylimidazole, further improving the photocatalytic efficiency.42 In addition, the incorporation of transition metals can optimize light absorption by enhancing the valence band.42
Building on these insights, our study aims to explore the potential of Fe-doped ZIF-7-III (UPO-4) as a heterogeneous catalyst for photocatalytic AOPs at circumneutral pH under simulated solar irradiation. Caffeine was selected as the target pollutant due to its stability and inability to complex with iron, which ensures that it does not interfere with the Fenton process.43,44 We further investigated the nature of photoreactivity using radical scavengers and conducted DFT-based simulations to better understand the structural and electronic properties of Fe-ZIF-7-III in relation to its photocatalytic performance.
Materials and methods
Materials
All chemicals and reagents used in this study were of analytical grade and were used without further purification. Toluene (≥99.5%, ACS Reagent), benzimidazole (≥98%, Sigma Aldrich), ethanol (≥99.8%, Honeywell), ammonium hydroxide (30%, ITW Reagents), zinc acetate dihydrate (≥98%, VWR Chemicals), caffeine (>99%, Fluka Analytical), acetone (≥99.8%, ACS Reagent), FeCl3 (97%, Sigma Aldrich), H2O2 (30%, PanReac), dimethyl sulfoxide (≥99.9%, Sigma Aldrich) and tert-butyl alcohol (>99.5%, Carlo Erba reagents) were used. Deionized water was used to prepare the aqueous solutions.Characterization
A Bruker D8 Discover diffractometer equipped with a high-resolution image plate detector was used to obtain powder X-ray diffraction patterns. Patterns were collected using Cu Kα radiation (λ = 1.5406 Å) in the 2θ range of 5–50° with a step size of 0.02° and a counting time of 3 s per step. The morphology and chemical composition of the samples were analysed using an SEM microscope (FEGSEM, Hitachi S4800, 20 kV), and to determine the Fe content in the sample, the inductively coupled plasma atomic emission spectroscopy technique was used (ICP-AES, Horiba Jobin Yvon, Ultima 2). To perform the FTIR experiments, the samples were placed 48 h in a desiccator to remove moisture. Infrared spectra were recorded using a JASCO FT/IR-480 plus spectrometer at room temperature using disks prepared with 1% of the samples in KBr. Spectra were recorded by adding 24 scans at a resolution of 4 cm−1 in the 4000–400 cm−1 region. The optical characteristics of the samples were determined by UV-vis diffuse reflectance spectroscopy with a Varian Cary 300 UV-vis spectrophotometer equipped with an integrating sphere and using BaSO4 as a blank reference. Analyses were carried out on powder samples. X-ray photoelectron spectroscopy (XPS) analysis was performed using a SPECS Phoibos 150 1D-DLD spectrometer equipped with a monochromated Mg Kα radiation X-ray source at 13 kV and 15.4 mA. The binding energies were normalised to the C 1s at 284.6 eV. Spectra were analysed using CasaXPS processing software.Fe-ZIF-7-III (UPO-4) synthesis
Pure-phase Fe-ZIF-7-III was synthesized using a novel method, by soaking ZIF-11 with an iron(III) chloride solution containing a small amount of acetone. ZIF-11 is a metastable MOF that undergoes a transition to the more stable phase ZIF-7-III.41 ZIF-11 was prepared following the procedure of He et al.45 with a small modification in the content of ammonium hydroxide, dissolving 0.12 g of benzimidazole (1 mmol) in 6.8 g of ethanol, followed by the addition of 4.6 g of toluene (50 mmol) and 0.20 g of ammonium hydroxide (1 mmol NH3) under stirring at room temperature. Subsequently, 0.11 g of zinc acetate dihydrate (0.5 mmol) were added and stirred for 3 h at room temperature (∼25 °C) to complete crystallization.45 Then, 70 mg of ZIF-11 were mixed with 100 μL of acetone and 8 mL of FeCl3 solution (1.8 g L−1) and stirred for 24 h at room temperature. After that, the solid was washed three times with distilled water, to separate the formed brown gelatin from the yellow precipitate, and dried at room temperature. ZIF-7-III was also obtained following the same steps but using distilled water instead of the FeCl3 solution and adjusting the pH to 2.6 with HCl.Photocatalytic assays
Solutions containing 4 mg L−1 of caffeine and 200 mg L−1 of Fe-ZIF-7-III (UPO-4) were prepared in Milli-Q grade water without adjustment of pH (6.4) and placed in 100 mL cylindrical open glass reactors. For (photo)-Fenton-related experiments, 20 mg L−1 of hydrogen peroxide were added, which represents twice the stoichiometric amount required to reach complete mineralization of the pollutant.The irradiation was performed with a solar simulator (Oriel Instruments, Model 81160 equipped with a 300W xenon lamp). Glass filters were used to cut off the transmission of wavelengths λ < 300 nm. UV-A irradiance in the 315–380 nm region was 32 W m−2. The irradiation started simultaneously with the addition of hydrogen peroxide and was kept for up to 180 min. Samples were periodically taken from the solution for analysis; those to be analysed by HPLC were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 with methanol to quench the excess peroxide. For the reuse of the catalyst test, three cycles of oxidation were performed with the same catalyst, adding the corresponding amount of caffeine to achieve the same initial concentration. In a complementary experiment, tert-butyl alcohol (100 μM) was added to study if ˙OH radicals contribute to caffeine degradation, as expected in Fenton-like processes. To check the possible involvement of hypervalent iron ions, tests were performed in the presence of dimethyl sulfoxide (DMSO) at a concentration of 100 μM.
:2 with methanol to quench the excess peroxide. For the reuse of the catalyst test, three cycles of oxidation were performed with the same catalyst, adding the corresponding amount of caffeine to achieve the same initial concentration. In a complementary experiment, tert-butyl alcohol (100 μM) was added to study if ˙OH radicals contribute to caffeine degradation, as expected in Fenton-like processes. To check the possible involvement of hypervalent iron ions, tests were performed in the presence of dimethyl sulfoxide (DMSO) at a concentration of 100 μM.
Dark controls were performed with the catalyst and with and without hydrogen peroxide and irradiation of the caffeine solution with H2O2 and in the absence of the catalyst was also performed.
All experiments were performed in duplicate.
Analytical measurements
The concentration of caffeine was determined using HPLC (Hitachi Chromaster chromatograph; VWR) equipped with a UV-vis detector. A Prevail Hichrom column (C18-Select; 250 × 4.6 mm; 5 μm) was employed as the stationary phase. The mobile phase (flow rate of 1 mL min−1) consisted of a binary mixture of solvents A (acetonitrile) and B (10 mM aqueous solution of formic acid). The wavelength used for the quantification was 275 nm. The concentration of iron species in solution was measured according to the standardized 1,10-phenanthroline spectrometric procedure (ISO 6332:1988) using a UH5300-Hitachi spectrophotometer.Computational modelling
The calculations were based on density functional theory (DFT), with the CP2K code, which uses mixed Gaussian-plane wave (GPW) basis sets.46 All calculations were conducted using periodic cells, while for visualization, the structures are shown as discrete slabs. The meta-GGA r2SCAN exchange correlation functional of Furness et al.,47 and the rVV10 nonlocal dispersion model of Sabatini et al.48 were used to accurately describe the bonding and van der Waals interactions.49 According to the SCAN family of functionals, a tailor-made triple-ζ Gaussian atomic orbital basis set (TZVP-MOLOPT-SCAN-GTH) was used. Geometry optimization was conducted by energy minimization using a convergence tolerance of 0.534 kcal mol−1 Å−1 (4.5 × 10−4 Ha Bohr−1). Spin polarization was conducted for iron-containing systems. We placed two Fe atoms per unit cell, with the remaining 10 tetrahedral nodes occupied by Zn atoms. The initial crystal structure of ZIF-7-III was obtained from the isostructural ZIF-9-III.50 The relatively flat energy surfaces associated with the 2D structure of these solids, even for undoped ZIF-7-III, make the calculations computationally expensive in terms of structure optimization. We have used the nucleophilic, electrophilic, and radical attack susceptibility Fukui functions (f+(r), f−(r), and f0(r), respectively) as defined by Parr and Yang51 and the dual descriptor function, Δf(r), as defined by Morell et al.52,53 to study the local reactivity by analysing the electron density using DFT calculations at the r2SCAN level. The hybrid HSE06 functional was also used to calculate the band gap,54,55 as it has been shown to be efficient for such a purpose in many types of materials, including MOFs.42,56,57 Since it is also possible that ferryl ions are formed by hydrogen peroxide and would play a relevant role in catalytic properties, we also modelled the structure of the material having the ferryl ion, as well as the relative stability with respect to Fe2+ bearing ZIF. To do this, we studied the initial system in the presence of H2O2 and H2O and the reacted one with the FeO ferryl ion formed and two H2O molecules. To show the electron redistribution upon the formation of the ferryl ion, the REPEAT charges were computed58 as they are known to be a good descriptor in MOFs.59 The optimized structures are given as Crystallographic Information Files (CIFs) in the ESI.†
Results and discussion
XRD was analysed to confirm the successful synthesis of ZIF-7-III from ZIF-11 through interaction with a water–acetone solution. Both Fe-incorporated and bare ZIF-7-III showed diffraction patterns analogous to those simulated from the reported ZIF-7-III,60,61 (Fig. 1) since there is a concordance in the position of the peaks with those appearing in the simulated pattern. Park et al.60 recently synthesized a mixed phase and calculated the percentages of each ZIF-7 phase by integrating the intensive XRD peaks of (101) and (110) for ZIF-7-I and (002) for ZIF-7-III, respectively. No typical peak corresponding to ZIF-7-I (or ZIF-7-II) was observed. ZIF-7-II is achieved by phase transition from ZIF-7-I during complete elimination of the occupied solvent in the synthesized nanocrystals,62 which clearly does not occur in our synthesis. In contrast to bare ZIF-7-III, Fe-ZIF-7-III exhibited some very wide diffraction peaks at 2θ between 16 and 24°. This suggests a partial loss of crystallinity of the layers on the basal plane. These layers are stabilized by van der Waals interactions and the presence of iron might enhance the occurrence of small displacements and rotations of the layers. Note that the main peak related to the basal plane 002 in Fe-ZIF-7-III at 2θ = 9.6° is as narrow as the one appearing in the bare ZIF-7-III, indicating that the 2D layer structure is kept. In a previous study, wide peaks or peaks with much less intensity than those of simulated patterns have been reported.50,61 In particular, the ball milling route for preparing ZIF-7-III gives a diffractogram very similar to our Fe-ZIF-7-III one, which supports our view.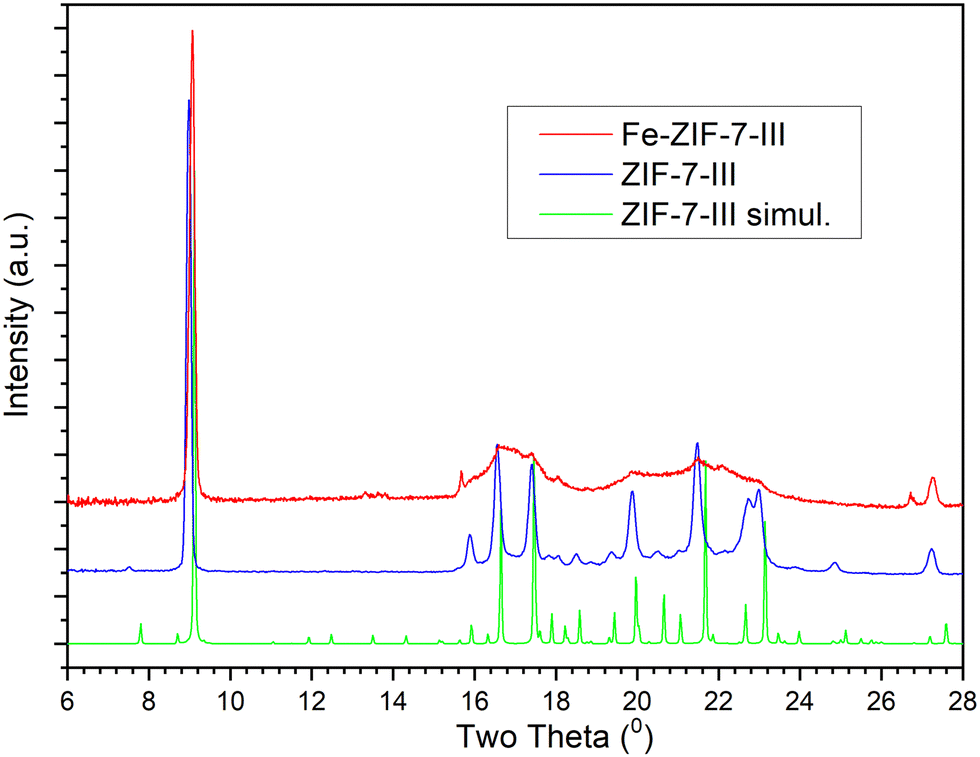 | ||
| Fig. 1 XRD patterns of experimental Fe-ZIF-7-III, experimental ZIF-7-III, and simulated ZIF-7-III. | ||
The DFT-simulated structures of ZIF-7-III with and without Fe atoms were very similar (Fig. 2). This suggests that the layers are sufficiently flexible to host Fe atoms without a large overall deformation. Note that both Fe2+ and Fe3+ were considered, as they are expected to appear in the prepared materials. A closer look at the structures shows that the local environment is modified by the presence of Fe, shown in Fig. 2, which induces orientation of the benzimidazole ligands, along with small displacement and together lead to reduce the atomic overlap. This view helps rationalize the partial loss of crystallinity observed in X-ray diffraction (Fig. 1, ∼ 16–24°). Due to the high cost of DFT calculations, our simulated systems contained only 466 atoms.
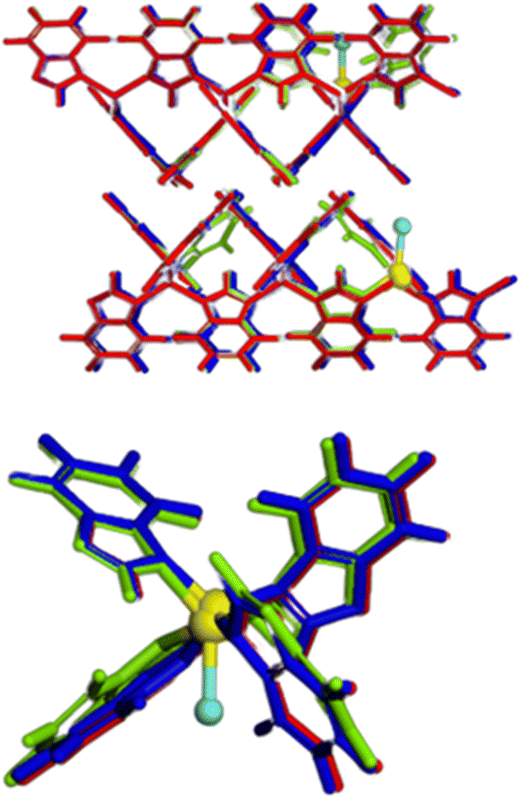 | ||
| Fig. 2 Top: overlapped view of ZIF-7-III (red) Fe3+-Cl−-ZIF-7-III (green) and Fe2+-ZIF-7-III (blue). Fe atoms are highlighted in yellow, and Cl atoms are shown in light blue. Bottom: zoom around Fe atoms to illustrate the local structural deformation. | ||
In real systems, many different configurations are likely to appear, giving rise to a variety of local deformations, which explains the partial loss of crystallinity. It is also noted that the main periodicity perpendicular to the basal plane, retrieved from the diffraction peak at 9.6° in Fig. 1, is apparent from the left panel of Fig. 2, as the layers are nicely parallel. Using infrared spectroscopy, as a local probe, the structural similarity of ZIF-7-III with and without Fe was confirmed (Fig. 3). The bands observed in the FTIR spectra agree with those found by other authors for ZIF-7-III,63 where most of the observed peaks were obtained below 2000 cm−1 due to the vibration energy of the different types of bonds in the structure. Therefore, the characteristic peaks from 550 to 900 cm−1 are due to aromatic sp2 C–H bending, those from 1000 to 1300 cm−1 are related to in-plane bending of the ring, and those at 1600 and 1800 cm−1 correspond to CN and CO stretching, respectively. The incorporation of Fe atoms was confirmed by the large peak appearing at 615 cm−1, which agrees with that measured in Fe-doped ZIF-8.64
 | ||
| Fig. 3 FTIR spectra of ZIF-7-III and Fe-ZIF-7-III prepared in this study. | ||
Although diffraction and FTIR did not show large differences in ZIF-7-III before and after Fe doping, SEM images did (Fig. 4). Therefore, iron affects the crystallization of the synthesized solids. The Fe-ZIF-7-III powder exhibited an almost 2D dense stacked layer structure, with several micrometres in size (around 2.0–2.5 μm) and a micrometre size planar shape and a smooth surface (Fig. 4), which is consistent with the previously reported ZIF-7-III.61,63 Under our synthesis conditions, the ZIF-7-III powder exhibited a whisker morphology with the presence of a minor amount of lamellar particles.
 | ||
| Fig. 4 SEM images of the samples ZIF-7-III (a) and Fe-ZIF-7-III (b) prepared in this work. | ||
While iron strongly modifies the morphology and generates disorder in the layer stacking, it contrasts with its low incorporation into the material. ICP revealed that the Fe concentration was only 0.263 ± 0.007 ppm (0.16 wt%). Nevertheless, this is sufficient to provide measurable visible-light absorption, as detected by diffuse-reflectance UV-vis spectroscopy (Fig. 5). To understand the electronic and optical properties of MOFs, it is necessary to consider that electron transitions can occur within MOFs involving the movement of electrons from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO).65 To achieve optimal excitation of the MOFs, incident light (Elight = 1240/λ > EHOMO–LUMO) was limited to the UV region, characterized by shorter wavelengths (λ < 365 nm). It is therefore evident that MOFs capable of responsively absorbing visible light (λ > 400 nm, or Elight < 3.1 eV) are to be preferred for the effective utilization of solar energy, as is commonly the case with Fe-MOFs.65,66
 | ||
| Fig. 5 DR-UV-vis spectra of ZIF-7-III and Fe-ZIF-7-III prepared in this study. | ||
Grau-Crespo et al.42 showed that Zn-ZIFs have band gaps that are determined by the HOMO–LUMO of the ligands. In the case of ZIF-7, the calculated band gap was 4.50 eV due to the benzimidazole ligand. This explains the presence of a high band in the experimental UV-vis spectrum of ZIF-7-III at 280 nm, since this MOF is formed by the same ligand (Fig. 5). We turn to the first-principles computed bandgap of ZIF-7-III, obtaining a value of 4.39 eV (282 nm), which confirms that indeed this band arises from tetrahedral zinc and benzimidazole. When iron was incorporated into ZIF-7-III the experimental spectrum showed an additional band at 375 nm (3.30 eV), and a shoulder at 550 nm (2.25 eV). To confirm that iron is related to observed differences in UV-vis spectra, the band gaps of Fe2+-ZIF-7-III and Fe3+Cl-ZIF-7-III were calculated. The calculated band gaps shift from 4.39 eV to 3.60 eV (344 nm) and 2.45 eV (505 nm) for Fe2+-ZIF-7-III and Fe3+Cl−-ZIF-7-III, respectively, which is in reasonable agreement with the experimental light absorption bands. Other authors have also found that the incorporation of iron in ZIF-8-NH2 reduces the band gap from 5.11 eV to 2.17 eV.67 We therefore expect photochemical activity in Fe-ZIF-7-III that could be exploited directly for solar heterogeneous photo-Fenton catalysis or for the photo-activation of hydrogen peroxide to form ferryl ions.
Considering that the presence of iron modifies the electronic structure of ZIF-7-III materials, it is expected that the reactivity would also be altered. To predict the reactivity potential of these materials, Fukui functions were analysed because they relate the change in chemical potential as a function of the variation in the number of electrons.68 A high value of f+(r) indicates that an atom is more likely to be attacked in a nucleophilic reaction, or that these regions will readily donate charge to the electron acceptor. In contrast, a high value of f−(r) indicates that the atom is more likely to undergo an electrophilic reaction or that these regions will stabilize the uptake of charge from electron donors.65,69 Therefore, Fig. 6 illustrates the clear differences among the three materials. In Fig. 6a, we demonstrate how ZIF-7-III (without Fe atoms) exhibits low reactivity, except for the metallic centers (Zn atoms), which are difficult to access due to steric hindrance. The presence of Fe2+ atoms (Fig. 6b) enhances the reactivity of the metallic center. Calculations revealed that within the three-dimensional lattice, ferrous ions can act as electrophilic agents, decomposing H2O2, which can result in two different scenarios (˙OH generation via a Fenton-like reaction or the formation of hypervalent ferryl ions). The presence of Cl atoms (Fig. 6c) significantly increased the reactivity of the material. Most surface aromatic groups become susceptible to electrophilic and nucleophilic attacks, such as those that initiate the Criegee mechanism.
 | ||
| Fig. 6 Dual Fukui descriptor functions for Δf(r) > 0.05 Ha (in blue, for electrophilic attack susceptibility) and Δf(r) < 0.05 Ha (in red, for nucleophilic attack susceptibility), of (a) ZIF-7-III (left) and (b) Fe2+-ZIF-7-III (middle) and (c) Fe3+Cl−-ZIF-7-III (right), indicative of local reactivities of the atoms. The atom code colours are silver, cyan, green, blue, grey, and white for Zn, Fe, Cl, N, C, and H atoms, respectively. In the red circles, we marked other highly reactive regions (mainly, aromatic carbons on the surface close to Cl atoms). | ||
Once the synthesized material was characterized and the presence of iron in the material was assessed, different experiments were carried out to check its activity in photo-degradation processes. First, caffeine irradiation alone, with H2O2 or with Fe-ZIF-7-III (UPO-4) in parallel experiments, resulted in negligible degradation of the pollutant after 90 min, which allowed us to rule out direct photolysis, oxidation by H2O2, or photocatalytic activity of Fe-ZIF-7-III. The caffeine concentration also remained constant in dark controls consisting of caffeine with Fe-ZIF-7-III, indicating that the adsorption of the pollutant on the catalyst surface was negligible.
Next, the ability of the material to drive a (photo)-catalytic decontamination process at pH = 6.4 was tested. For this purpose, experiments were carried out in the presence of Fe-ZIF-7-III (200 mg L−1) and hydrogen peroxide (twice the stoichiometric amount) to oxidize 4 mg L−1 caffeine in the dark and under solar simulated irradiation. Approximately 80% caffeine removal was reached after 90 min of irradiation and more than 90% abatement after 180 min (Fig. 7) and the dark experiment resulted in negligible pollutant degradation (data not shown). These results agree with the improved photoreduction of Fe(III) present in the solid material, which cannot be reached in the dark. Thus, light irradiation is an essential component in the catalytic activity of Fe-ZIF-7-III, at near neutral pH, at least for the removal of very stable molecules such as caffeine.
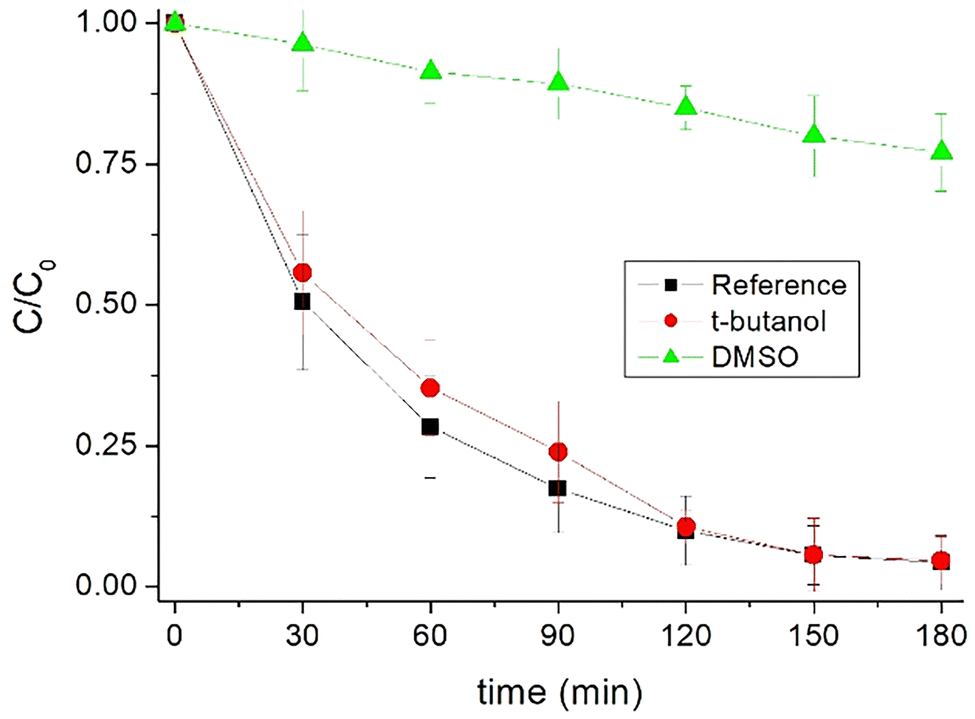 | ||
| Fig. 7 Evolution of caffeine degradation in the presence and absence of t-butanol and dimethyl sulfoxide (100 μM) under simulated solar light. Conditions: pH 6.4, 4 mg L−1 caffeine, 200 mg L−1 Fe-ZIF-7-III and 20 mg L−1 hydrogen peroxide. | ||
We have noted that there are no studies reporting caffeine degradation by photo-Fenton-like processes using ZIF catalysts. To provide a comparative view of the performance of our material, we show here some prominent examples of photocatalytic caffeine degradation. Refluxed MIL-125 over NTU-9 shows 85% of caffeine degradation under 105 min of visible light.70 A nano-TiO2 photocatalyst was used to remove 94% of caffeine within 220 min at pH 3 and pH 1, while 88% and 80% of caffeine were removed within 220 min at pH 6 and pH 9, respectively, under UV light.71 Additionally, the application of a three-layer perovskite Dion-Jacobson phase CsBa2M3O10 (M = Ta and Nb) and oxynitride crystals was applied for the removal of caffeine by photocatalysis under UVA and visible light irradiation, achieving 89% degradation under UVA light after 60 min.72 Also, ZnO nanoparticles achieved 98.6% degradation of a 1 ppm caffeine solution under sunlight in 120–140 min.73 Iron oxide magnetic nanoparticles (MH0.2) combined with H2O2 achieved significant caffeine removal (5 mg L−1) after 180 min.74 Similar to this photocatalyst, degradation of caffeine was achieved under UV-vis radiation using magnetic nanoparticles (NPs) based on iron oxides after 60 min.75 Fe-ZIF-7-III (UPO-4) exhibits significantly better performance under simulated sunlight, achieving higher degradation rates and superior catalytic activity while maintaining structural stability without detectable iron leaching, thus ensuring a truly heterogeneous process, unlike other systems that often suffer from stability issues and iron leaching, leading to a homogeneous-like mechanism.
It should be noted that in some cases solids have been reported to act as simple iron reservoirs, which are leached into the solution, thus leading to a predominating homogeneous photo-Fenton process.76 To determine the extent of this mechanism, dissolved iron was measured according to the o-phenanthroline colorimetric method and ICP. In none of the measurements, dissolved iron was detected, which indicates that the role, if any, of leached iron cations in the observed caffeine removal was very marginal. It was confirmed by EDX before and after the photocatalytic reactions, as the atomic percentage of Fe (Fig. S1, ESI†) showed only a slight decrease from 3.06% to 2.82%. The X-ray diffraction pattern and SEM also reveal the robustness of the material after photocatalysis (Fig. S2, ESI†). XPS analysis (Fig. S3, ESI†) further supports the stability of the material, showing that prior to the reaction, Fe was predominantly in the Fe3+ oxidation state with a small contribution of Fe2+. Before the reaction, a main peak was observed in the Fe 2p3/2 region, with a predominant contribution from Fe3+ (712.5 eV) and a smaller fraction of Fe2+ (710.9 eV). However, after photocatalysis, no clear Fe2+ or Fe3+ signals were detected, suggesting that the remaining iron has transitioned to a higher oxidation state, such as a ferryl species, which is not readily identified by XPS. This interpretation is consistent with previous reports indicating that XPS is unable to directly confirm the presence of high valence iron species.77
To determine whether the process was indeed controlled by the targeted ferryl ion or mediated by ˙OH radicals, as in photo-Fenton processes, photocatalytic experiments were repeated in the presence of t-butanol, a known scavenger of hydroxyl radicals. Fig. 7 shows that caffeine removal coincided with and without t-butanol. Based on these results, it seems that the role of ˙OH is not predominant in this case, in contrast to most information reported on homogeneous and heterogeneous photo-Fenton reactions.44,66 Therefore, these results support the hypothesis of the presence of ferryl ions in Fe-ZIF-7-III.
To determine the participation of hypervalent iron ions, that is, the ferryl ions, experiments were carried out in the presence of a compound that deactivates this transient state of iron. This compound was dimethyl sulfoxide (DMSO). In Fig. 7, the experiment in the presence of DMSO is presented and an important inhibition of caffeine removal can be observed. These results would indicate an important role of the ferryl ion in the degradation of the model pollutant. The reactions involved could be represented, in a simplified way, as follows:
| Fe(II) + H2O2 → [FeIVO]2+ + H2O |
| [FeIVO]2+ + pollutant → Fe (II) + pollutantox |
Fe in N-rich environments has been reported to be easily activated to adopt hypervalent states, such as ferryl ions.30,31 One distinctive feature from a structural point of view is the short Fe–O distance of ca. 1.63 Å.30,31 On the basis of this background and the above catalytic experiments, to confirm our hypothesis, we performed DFT calculations to elucidate whether hydrogen peroxide could lead to the formation of stable ferryl groups in Fe-ZIF-7-III. Fig. 8 shows the energy minimized structure of a hydrogen peroxide molecule and a water molecule adsorbed near the Fe atom in Fe-ZIF-7-III and a ferryl group with two adsorbed water molecules in Fe-ZIF-7-III. The FeO distance in the ferryl group is 1.646 Å, which is in good agreement with the experimental distances measured by EXAFS in N-rich environments. The formation of the FeO bond leads to an increase in the Fe atomic charge from 0.875e to 1.102e, while the Zn charges remain unchanged at ca. 0.948e, which is also evidence of the formation of the ferryl group. It is useful to note that the energy of these structures only differs by 1.44 eV per Fe atom, an energy value that is compatible with visible light. The computed energy difference suggests that once the ferryl groups are formed, they can be stabilized in the material, and thus they can be available as alternative active sites for photocatalysis. The calculated band gap of the ferryl group bearing Fe-ZIF-7-III is 2.116 eV, indicating that the material retains the ability to absorb light in the visible region (below 586 nm). The degradation experiments shown above indicated that light activation is a key point in the catalytic process. We acknowledge that the elucidation of the mechanism is a difficult task, as it would be needed to consider excited state reactions. Thus, the mechanism of the photocatalytic reactions involved in the degradation of the caffeine molecule in the presence of the ferryl ion anchored in ZIF-7-III is beyond this study and will be treated in a separate study.
 | ||
| Fig. 8 Fe-ZIF-7-III in the presence of H2O2 and H2O molecules (left) and the ferryl group and two water molecules (right). Relevant distances between Fe and O atoms and also O atoms connected in H-bonds are shown. | ||
To check the reusability of the catalyst, we used it in three consecutive cycles of photocatalytic degradation of caffeine. After 90 min of irradiation, the initial conditions were reset by adding the amount of caffeine and H2O2 consumed. To examine better the changes among cycles, caffeine degradation was fitted to a first-order kinetics and pseudo-rate constants (k) were calculated in each case (Fig. 9). For the first cycle, a k value of 0.0073 min−1 was obtained; there was a decrease in the second cycle to 0.047 min−1 and then remained constant in the third cycle (0.049 min−1). Despite a slight initial loss of efficiency, the material appeared to maintain its activity as a photocatalyst, an important result in terms of its re-use.
 | ||
| Fig. 9 Pseudo-first order kinetic adjustment of the 3 cycles carried out under simulated solar light under the following conditions: pH 6.4, 4 mg L−1 caffeine, 200 mg L−1 Fe-ZIF-7-III and 20 mg L−1 hydrogen peroxide. | ||
Conclusions
A biomimetic approach has allowed the stabilization of hypervalent ferryl ions in a MOF as the first result of successful photocatalytic water treatment by this route. For this purpose, pure phase Fe-ZIF-7-III (UPO-4) was synthesized using a novel mild route. The zeolitic material retains the 2D structure after the incorporation of Fe, as shown by X-ray diffraction, with only a slight decrease in the crystallinity of the layers due to the incorporation of Fe. At the atomic level, it is caused by slight local distortions around the Fe atoms, as suggested by DFT calculations. Although the amount of iron incorporated was less than one ppm unit, the electronic study showed that the photocatalytic performance that can be expected from the material would be adequate to extend the light absorption to the visible region and efficiently use solar energy.The activity of the synthesized material as a photocatalyst was demonstrated through the successful degradation in water of the model contaminant caffeine using simulated sunlight irradiation. The presence of a specific ferryl ion scavenger clearly affected the behaviour of the process indicating a mechanism without the major intervention of hydroxyl radicals, supporting the hypothesis of the presence of ferryl ions. To support this point, DFT calculations were performed. Innovations include the successful stabilization of hypervalent iron within the MOF, marking a novel approach in the design of photocatalysts for water treatments as there is no previous work in this area using a hypervalent metal MOF. Therefore, Fe-ZIF-7-III (UPO-4) represents a significant advancement by effectively leveraging solar energy for environmental remediation, while maintaining catalytic activity across multiple cycles. Future work should focus on the effect of photo-Fenton parameters on degradation kinetics, optimizing iron incorporation, broadening the scope of target pollutants, and elucidating the mechanisms behind photocatalytic activity, ultimately contributing to sustainable water treatment solutions. Further work is underway to design and test other hypervalent metals and MOFs for water decontamination and disinfection.
Author contributions
Lucas Santos-Juanes: conceptualization, investigation, methodology, validation, visualization, and writing – original draft; Noelia Rodriguez-Sanchez: investigation, validation, writing – original draft, and writing – review & editing; Salvador R. G. Balestra: investigation, validation, and writing – review & editing; Nuria O. Núñez: investigation, validation, and writing – review & editing; Antonio Arques: funding acquisition and writing – review & editing; A. Rabdel Ruiz-Salvador: conceptualization, funding acquisition, investigation, methodology, project administration, supervision, visualization, writing – original draft, and writing – review & editing; and Menta Ballesteros: conceptualization, funding acquisition, methodology, project administration, supervision, writing – original draft, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Spanish Ministerio de Ciencia e Innovación (PID2021-126400OB-C31, AquaEnAgri Project) and the European Commission (HORIZON.1.2 – Marie Skłodowska-Curie Actions VALZEO Project 101086354). Salvador R. G. Balestra also thanks Consejería de Universidades, Investigación e Innovación, Junta de Andalucía (grant number: POSTDOC_21_00069). We are grateful to C3UPO for providing high performance computing facilities.References
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O’Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2018, 4, eaat9180 CrossRef CAS PubMed.
- S. Mukherjee and N. R. Champness, Nat. Rev. Chem., 2024, 8, 6–7 CrossRef PubMed.
- B. Mohan, Virender, R. Kadiyan, S. Kumar, V. Gupta, B. Parshad, A. A. Solovev, A. J. L. Pombeiro, K. Kumar and P. K. Sharma, Microporous Mesoporous Mater., 2024, 366, 112932 CrossRef CAS.
- D. E. Jaramillo, A. Jaffe, B. E. R. Snyder, A. Smith, E. Taw, R. C. Rohde, M. N. Dods, W. DeSnoo, K. R. Meihaus, T. D. Harris, J. B. Neaton and J. R. Long, Chem. Sci., 2022, 13, 10216–10237 RSC.
- M.-Y. Gao, S.-Q. Wang, A. A. Bezrukov, S. Darwish, B.-Q. Song, C. Deng, C. R. M. O. Matos, L. Liu, B. Tang, S. Dai, S. Yang and M. J. Zaworotko, Chem. Mater., 2023, 35, 10001–10008 CrossRef CAS PubMed.
- W. Li, Z. Zhu, Q. Chen, J. Li and M. Tu, Cell Rep. Phys. Sci., 2023, 4(12), 101679 CrossRef CAS.
- Q. Wan, M. Wakizaka and M. Yamashita, Inorg. Chem. Front., 2023, 10, 5212–5224 RSC.
- L. Jiao, Y. Wang, H.-L. Jiang and Q. Xu, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- D. Y. Osadchii, A. I. Olivos-Suarez, Á. Szécsényi, G. Li, M. A. Nasalevich, I. A. Dugulan, P. S. Crespo, E. J. M. Hensen, S. L. Veber, M. V. Fedin, G. Sankar, E. A. Pidko and J. Gascon, ACS Catal., 2018, 8, 5542–5548 CrossRef CAS.
- D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planas, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocellà, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590–595 CrossRef CAS PubMed.
- P. Melix and R. Q. Snurr, J. Phys. Chem. C, 2023, 127, 15227–15238 CrossRef CAS.
- F. Saiz and L. Bernasconi, Catal. Sci. Technol., 2023, 13, 5689–5701 RSC.
- Á. Szécsényi, G. Li, J. Gascon and E. A. Pidko, Chem. Sci., 2018, 9, 6765–6773 RSC.
- P. Verma, K. D. Vogiatzis, N. Planas, J. Borycz, D. J. Xiao, J. R. Long, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2015, 137, 5770–5781 CrossRef CAS PubMed.
- K. Hou, J. Börgel, H. Z. H. Jiang, D. J. SantaLucia, H. Kwon, H. Zhuang, K. Chakarawet, R. C. Rohde, J. W. Taylor, C. Dun, M. V. Paley, A. B. Turkiewicz, J. G. Park, H. Mao, Z. Zhu, E. E. Alp, J. Zhao, M. Y. Hu, B. Lavina, S. Peredkov, X. Lv, J. Oktawiec, K. R. Meihaus, D. A. Pantazis, M. Vandone, V. Colombo, E. Bill, J. J. Urban, R. D. Britt, F. Grandjean, G. J. Long, S. DeBeer, F. Neese, J. A. Reimer and J. R. Long, Science, 2023, 382, 547–553 CrossRef CAS PubMed.
- G. Deng, Z. Wang, J. Ma, J. Jiang, D. He, X. Li, A. Szczuka and Z. Zhang, Environ. Sci. Technol., 2023, 57, 18586–18596 CrossRef CAS PubMed.
- L. Li, Z. Yin, M. Cheng, L. Qin, S. Liu, H. Yi, M. Zhang, Y. Fu, X. Yang, X. Zhou, G. Zeng and C. Lai, Chem. Eng. J., 2023, 454, 140126 CrossRef CAS.
- S. Malato, M. I. Maldonado, P. Fernández-Ibáñez, I. Oller, I. Polo and R. Sánchez-Moreno, Mater. Sci. Semicond. Process., 2016, 42, 15–23 CrossRef CAS.
- X. Lei, Y. Lei, X. Zhang and X. Yang, J. Hazard. Mater., 2021, 408, 124435 CrossRef CAS PubMed.
- V. I. Parvulescu, F. Epron, H. Garcia and P. Granger, Chem. Rev., 2022, 122, 2981–3121 CrossRef CAS PubMed.
- V. K. Sharma, X. Ma and R. Zboril, Chem. Soc. Rev., 2023, 52, 7673–7686 RSC.
- W. Yang, S. Guo and J. Chen, Cailiao Daobao, 2020, 34, 11022–11028 Search PubMed.
- A. Kirchon, P. Zhang, J. Li, E. A. Joseph, W. Chen and H.-C. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 9292–9299 CrossRef CAS PubMed.
- Y. Mou, X. Yuan, H. Chen, Y. Yang, H. Dai, J. Bai, J. Chen, J. W. Chew, H. Wang and Y. Wu, J. Mater. Chem. A, 2023, 11, 22631–22655 RSC.
- S. Rojas and P. Horcajada, Chem. Rev., 2020, 120, 8378–8415 CrossRef CAS PubMed.
- M. Jafarzadeh, ACS Appl. Mater. Interfaces, 2022, 14, 24993–25024 CrossRef CAS PubMed.
- Y. Chen, C. J. Miller and T. D. Waite, Environ. Sci. Technol., 2022, 56, 1278–1288 CrossRef CAS PubMed.
- M. A. Ehudin, L. B. Gee, S. Sabuncu, A. Braun, P. Moënne-Loccoz, B. Hedman, K. O. Hodgson, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2019, 141, 5942–5960 CrossRef CAS PubMed.
- A. Ghosh, E. Tangen, H. Ryeng and P. R. Taylor, Eur. J. Inorg. Chem., 2004, 4555–4560 CrossRef CAS.
- D. W. Lewis, A. R. Ruiz-Salvador, A. Gómez, L. M. Rodriguez-Albelo, F.-X. Coudert, B. Slater, A. K. Cheetham and C. Mellot-Draznieks, CrystEngComm, 2009, 11, 2272 RSC.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- S. Zhang, J. Wang, Y. Zhang, J. Ma, L. Huang, S. Yu, L. Chen, G. Song, M. Qiu and X. Wang, Environ. Pollut., 2021, 291, 118076 CrossRef CAS PubMed.
- K.-Y. A. Lin and H.-A. Chang, J. Taiwan Inst. Chem. Eng., 2015, 53, 40–45 CrossRef CAS.
- W. Ren, J. Gao, C. Lei, Y. Xie, Y. Cai, Q. Ni and J. Yao, Chem. Eng. J., 2018, 349, 766–774 CrossRef CAS.
- B. Hashemzadeh, H. Alamgholiloo, N. N. Pesyan, E. Asgari, A. Sheikhmohammadi, J. Yeganeh and H. Hashemzadeh, Chemosphere, 2021, 281, 130970 CrossRef CAS PubMed.
- F. M. Amombo Noa, M. Abrahamsson, E. Ahlberg, O. Cheung, C. R. Göb, C. J. McKenzie and L. Öhrström, Chem, 2021, 7, 2491–2512 CAS.
- P. Zhao, G. I. Lampronti, G. O. Lloyd, M. T. Wharmby, S. Facq, A. K. Cheetham and S. A. T. Redfern, Chem. Mater., 2014, 26, 1767–1769 CrossRef CAS PubMed.
- G. J. Leonel, C. B. Lennox, J. M. Marrett, T. Friščić and A. Navrotsky, Chem. Mater., 2023, 35, 7189–7195 CrossRef CAS PubMed.
- B. Reif, C. Paula, F. Fabisch, M. Hartmann, M. Kaspereit and W. Schwieger, Microporous Mesoporous Mater., 2019, 275, 102–110 CrossRef CAS.
- R. Grau-Crespo, A. Aziz, A. W. Collins, R. Crespo-Otero, N. C. Hernández, L. M. Rodriguez-Albelo, A. R. Ruiz-Salvador, S. Calero and S. Hamad, Angew. Chem., Int. Ed., 2016, 55, 16012–16016 CrossRef CAS PubMed.
- S. García-Ballesteros, J. Grimalt, S. Berto, M. Minella, E. Laurenti, R. Vicente, M. F. López-Pérez, A. M. Amat, A. B. Prevot and A. Arques, ACS Omega, 2018, 3, 13073–13080 CrossRef PubMed.
- I. Vallés, I. Sciscenko, M. Mora, P. Micó, A. M. Amat, L. Santos-Juanes, J. Moreno-Andrés and A. Arques, Appl. Catal., B, 2023, 331, 122708 CrossRef.
- M. He, J. Yao, Q. Liu, Z. Zhong and H. Wang, Dalton Trans., 2013, 42, 16608 RSC.
- T. D. Kühne, M. Iannuzzi, M. D. Ben, V. V. Rybkin, P. Seewald, F. Stein, T. Laino, R. Z. Khaliullin, O. Schütt, F. Schiffmann, D. Golze, J. Wilhelm, S. Chulkov, M. H. Bani-Hashemian, V. Weber, U. Borštnik, M. Taillefumier, A. S. Jakobovits, A. Lazzaro, H. Pabst, T. Müller, R. Schade, M. Guidon, S. Andermatt, N. Holmberg, G. K. Schenter, A. Hehn, A. Bussy, F. Belleflamme, G. Tabacchi, A. Glöβ, M. Lass, I. Bethune, C. J. Mundy, C. Plessl, M. Watkins, J. VandeVondele, M. Krack and J. Hutter, J. Chem. Phys., 2020, 152, 194103 CrossRef PubMed.
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, J. Phys. Chem. Lett., 2020, 11, 8208–8215 CrossRef CAS PubMed.
- R. Sabatini, T. Gorni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 041108 CrossRef.
- J. Ning, M. Kothakonda, J. W. Furness, A. D. Kaplan, S. Ehlert, J. G. Brandenburg, J. P. Perdew and J. Sun, Phys. Rev. B, 2022, 106, 075422 CrossRef CAS.
- P. Zhao, G. I. Lampronti, G. O. Lloyd, M. T. Wharmby, S. Facq, A. K. Cheetham and S. A. T. Redfern, Chem. Mater., 2014, 26, 1767–1769 CrossRef CAS PubMed.
- R. G. Parr and W. Yang, J. Am. Chem. Soc., 1984, 106, 4049–4050 CrossRef CAS.
- C. Morell, A. Grand and A. Toro-Labbé, J. Phys. Chem. A, 2005, 109, 205–212 CrossRef CAS PubMed.
- C. Morell, A. Grand and A. Toro-Labbé, Chem. Phys. Lett., 2006, 425, 342–346 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- S. Ling and B. Slater, J. Phys. Chem. C, 2015, 119, 16667–16677 CrossRef CAS.
- K. L. Svane, J. K. Bristow, J. D. Gale and A. Walsh, J. Mater. Chem. A, 2018, 6, 8507–8513 RSC.
- C. Campañá, B. Mussard and T. K. Woo, J. Chem. Theory Comput., 2009, 5, 2866–2878 CrossRef PubMed.
- S. Hamad, S. R. G. Balestra, R. Bueno-Perez, S. Calero and A. R. Ruiz-Salvador, J. Solid State Chem., 2015, 223, 144–151 CrossRef CAS.
- S. Park, K. Y. Cho and H.-K. Jeong, J. Mater. Chem. A, 2020, 8, 11210–11217 RSC.
- R. Tatewaki, T. Yamaki, M. Yoshimune, H. Negishi, T. Imura, H. Sakai and N. Hara, Colloids Surf., A, 2020, 603, 125157 CrossRef CAS.
- L. Xiang, D. Liu, H. Jin, L.-W. Xu, C. Wang, S. Xu, Y. Pan and Y. Li, Mater. Horiz., 2020, 7, 223–228 RSC.
- N. Buğday, S. Altın and S. Yaşar, Int. J. Energy Res., 2022, 46, 795–809 CrossRef.
- F. Zhang, Y.-H. Li, J.-Y. Li, Z.-R. Tang and Y.-J. Xu, Environ. Pollut., 2019, 253, 365–376 CrossRef CAS PubMed.
- W. Wang, G. Huang, J. C. Yu and P. K. Wong, J. Environ. Sci., 2015, 34, 232–247 CrossRef CAS PubMed.
- X. Wang, X. Zhang, Y. Zhang, Y. Wang, S.-P. Sun, W. D. Wu and Z. Wu, J. Mater. Chem. A, 2020, 8, 15513–15546 RSC.
- X. Luo, J. Liang, W. Han, J. Wang and Z. Zhou, J. Mol. Struct., 2023, 1291, 135910 CrossRef CAS.
- W. Wei, Y. Dai, K. Lai, M. Guo and B. Huang, Chem. Phys. Lett., 2011, 510, 104–108 CrossRef CAS.
- S. Mao, C. Liu, Y. Wu, M. Xia and F. Wang, Chemosphere, 2022, 291, 133039 CrossRef CAS PubMed.
- E. Dhivya, S. Saravanan and N. Aman, Russ. J. Inorg. Chem., 2022, 67, S141–S149 CrossRef.
- L. C. Chuang, C. H. Luo, S. W. Huang, Y. C. Wu and Y. C. Huang, Adv. Mater. Res., 2011, 214, 97–102 CAS.
- B. Czech and M. Hojamberdiev, J. Photochem. Photobiol. Chem., 2016, 324, 70–80 CrossRef CAS.
- R. Bedre Jagannatha, S. Rani Ramu, M. Padaki and R. G. Balakrishna, Nanochemistry Res., 2017, 2, 86–95 Search PubMed.
- D. Palma, A. Bianco Prevot, M. Brigante, D. Fabbri, G. Magnacca, C. Richard, G. Mailhot and R. Nisticò, Materials, 2018, 11, 1084 CrossRef PubMed.
- T. A. Fernandes, S. G. Mendo, L. P. Ferreira, N. R. Neng, M. C. Oliveira, A. Gil, M. D. Carvalho, O. C. Monteiro, J. M. F. Nogueira and M. J. Calhorda, Environ. Sci. Pollut. Res., 2021, 28, 17228–17243 CrossRef CAS PubMed.
- L. Santos-Juanes, A. A. Amat and A. Arques, Curr. Org. Chem., 2017, 21, 1074–1083 CrossRef CAS.
- I. Gladich, S. Chen, H. Yang, A. Boucly, B. Winter, J. A. Van Bokhoven, M. Ammann and L. Artiglia, J. Phys. Chem. Lett., 2022, 13, 2994–3001 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01217h |
| This journal is © The Royal Society of Chemistry 2025 |