
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous flow extraction of lithium from brine using silica-coated LMO beads
Jialun
Su†
a,
Qiyue
Fan†
a,
Xiangting
Hu
a,
Yue
Sun
a,
Jie
Lin
a,
Jiayue
Xu
a,
Bingcai
Pan
 bc and
Zhenda
Lu
*ac
bc and
Zhenda
Lu
*ac
aCollege of Engineering and Applied Sciences, State Key Laboratory of Analytical Chemistry for Life Science, and Jiangsu Key Laboratory of Artificial Functional Materials, Nanjing University, Nanjing 210093, China. E-mail: luzhenda@nju.edu.cn
bState Key Laboratory of Pollution Control and Resource Reuse, School of the Environment, Nanjing University, Nanjing 210023, China
cResearch Center for Environmental Nanotechnology (ReCENT), Nanjing University, Nanjing 210023, China
First published on 27th February 2025
Abstract
The increasing demand for lithium-ion batteries in the electric vehicle market has intensified the demand for efficient lithium extraction from salt lake brine. This study presents a novel approach using silica-coated lithium manganese oxide (LMO) adsorbents embedded in millimeter-sized sodium alginate (SA) beads (LMO@SiO2/SA beads). By replacing expensive and environmentally detrimental tetraethyl orthosilicate (TEOS) with low-cost, eco-friendly sodium metasilicate (Na2SiO3), we have developed a more sustainable and cost-effective lithium extraction method. Continuous flow adsorption–desorption experiments demonstrated the excellent performance of the LMO@SiO2/SA beads, maintaining a lithium adsorption capacity of 6.22 mg g−1 and a consistent manganese dissolution ratio of 1.26% per cycle after 50 cycles. These results highlight the potential of this approach for large-scale lithium extraction from salt lakes, providing a sustainable and economical option to support the growing electric vehicle industry.
Introduction
The global increase in electric vehicles has intensified the demand for lithium, a critical component in lithium-ion batteries.1–7 While traditional ore extraction methods face limitations in meeting this growing demand, salt lakes offer a more abundant and environmentally friendly source of lithium.8–15 However, extracting lithium from salt lake brine presents challenges due to the low concentration and high magnesium-to-lithium ratio.16–19Various methods have been proposed for lithium extraction, including lime soda evaporation,20 electrochemical recovery,21–27 and selective adsorption.28–36 Among these methods, selective adsorption offers several advantages, such as simplicity, low cost, and fast kinetics. While previous studies have explored lithium manganese oxide (LMO) adsorbents, their susceptibility to acid dissolution during desorption limits their practical applications.
To address this issue, we introduce a novel approach using silica-coated LMO adsorbents embedded in millimeter sized sodium alginate beads (LMO@SiO2/SA). In this method, sodium metasilicate (Na2SiO3) was applied as a more sustainable and cost-effective silicon source compared to traditional TEOS coating methods. Additionally, sodium alginate is employed to make millimeter-sized composite beads, enabling continuous flow extraction, a crucial aspect for industrial applications. Our results demonstrate excellent lithium adsorption performance, with an adsorption capacity of 6.22 mg g−1 and a minimal manganese dissolution ratio of 1.26% per cycle after 50 cycles. These findings highlight the potential of our approach to meet the growing demand for lithium while addressing the challenges associated with traditional extraction methods.
Materials and methods
Experimental materials
Lithium manganese oxide (LMO, Taiyuan Lizhiyuan Technology Co., Ltd), deionized water (DIW), hydrochloric acid (HCl, Sinopharm Chemical Reagent Co., Ltd), ethanol (Sinopharm Chemical Reagent Co., Ltd), polyacrylic acid (PAA, Macklin Biochemical Co., Ltd), sodium metasilicate nonahydrate (Na2SiO3·9H2O, AR, Sinopharm Chemical Reagent Co., Ltd), DT-135 defoamer (Xushi Chemical Technology Co., Ltd), sodium alginate ((C6H7O6Na)n, Aladdin Biochemical Technology Co., Ltd), anhydrous calcium chloride (CaCl2, Macklin Biochemical Co., Ltd), magnesium chloride hexahydrate (MgCl2·6H2O, AR, Aladdin Biochemical Technology Co., Ltd), lithium chloride (LiCl, Aladdin Biochemical Technology Co., Ltd), potassium chloride (KCl, 99.5%, Macklin Biochemical Co., Ltd), and analytical grade sodium chloride (AR, Sinopharm Chemical Reagent Co., Ltd) were used.Synthesis of LMO@SiO2/SA beads
Synthesis of LMO@SiO2
12 g of pretreated LMO was added into 200 mL of deionized water and the mixture was stirred for 1 hour and ultrasonically treated for 15 minutes to ensure good dispersion of lithium manganese oxide (LMO). 1.2 g of polyacrylic acid (PAA) was added into above mixture solution and continued stirring for an additional 8 hours. Subsequently, 30 g of nonahydrate sodium silicate were added into a 150 mL glass beaker, which was then set in a water bath and heated for 30 minutes until the sodium silicate was completely dissolved. This solution was poured into the pre-treated LMO solution and stirred for another 24 hours. After this, the mixture was transferred to a vacuum oven and dried at 240 °C to remove excess water. Eight drops of an antifoaming agent was added and then the dry mixture was transferred to a crucible and calcined at 500 °C for 4 hours in a muffle furnace. The crucible was removed, and the solidified mass was ground using a mortar to produce a fine LMO@SiO2 powder.Synthesis of HMO@SiO2
0.1 g of LMO@SiO2 was weighed and added into a 250 mL beaker. The mixture was washed with 200 mL of hydrochloric acid at a pH of 1 for one hour. After acid washing, the mixture was centrifuged and oven dried, and the sample HMO@SiO2 powder was obtained.Synthesis of LMO@SiO2/SA beads
40 g of LMO@SiO2 was weighed and added into a 1 L beaker. 700 mL of deionized water was added and stirred for 30 minutes followed by ultrasonication for 10 minutes. Then, 20 g of sodium alginate (SA) was added into the beaker and stirring was continued. The resulting LMO/SA solution was stored in a refrigerator to prevent aggregation of large molecular chains. The LMO/SA solution was loaded into a 50 mL syringe, the infusion pump was set at a rate of 1.5 mL min−1 with a total volume of 50 mL per injection, and a mixed solution of calcium chloride and PAA was used as a coagulation bath for granulation.Continuous flow adsorption–desorption cycle experiments
Preparation of simulated brine and extraction solution
To prepare models or simulated salt lake water, 57.75 g of anhydrous calcium chloride, 352.5 g of hexahydrate magnesium chloride, 4.585 g of lithium chloride, 52.2 g of potassium chloride, and 69.57 g of sodium chloride were added into a 2-liter bottle. The bottled was filled to the mark with deionized water to prepare a solution with a lithium chloride content of 1000 ppm. 444.44 mL of this solution was taken and transferred to a 5-liter plastic container and then 4 L of deionized water and 3 drops of 12 M hydrochloric acid were added to adjust the pH to 6.8, resulting in simulated salt lake water with a lithium chloride content of 100 ppm.For the acidic lithium extraction solution, 4 L of deionized water was taken in a 5-liter plastic cylinder and 50 mL of 12 M hydrochloric acid was added to achieve a solution with a pH of 1 for acid extraction.
Continuous flow extraction cycle
The extraction system of the continuous flow operates in two main stages:(i) Acid-washing stage: after filling the column with LMO@SiO2/SA beads, 800 mL of pH = 1 HCl solution was used for lithium desorption, and the material was transformed into HMO@SiO2/SA beads. The concentrations of Li+ and Mn2+ were then measured using ICP-OES to calculate the desorption ratio of Li+ and the leakage ratio of Mn2+. The formulas are as follows:16

where Ce1 and Ce2 are the concentrations of desorbed Li+ and Mn2+, respectively, V is the volume of the acidic lithium-extracting solution, and m is the mass of the adsorbent.

(ii) Adsorption stage: 800 mL of simulated salt lake water was pumped into the column containing HMO@SiO2/SA beads for the adsorption experiment, after which the material was converted to LMO@SiO2/SA beads. The concentration of Li+ was determined using ICP-OES to calculate the Li+ adsorption capacity using the formula:

Characterization techniques
The surface morphology of samples was revealed by SEM (SEM, Ultra 55, Zeiss, Germany). TEM characterization was used to demonstrate the crystal structure, and the element analysis of nanoparticles was obtained using an FEI Tecnai F20 microscope equipped with an energy-dispersive spectroscopy detector operating at 200 kV (TEM, Tecnai G2 F20 X-TWIN, FEI, USA). The concentration of the Li+ and Mn2+ in the solution was determined using an inductively coupled plasma optical-emission spectrometer (ICP-OES, iCAP 7400, Thermo Scientific, USA). Nitrogen adsorption measurements at 77 K were performed to study the porosity and specific surface area of the material (Brunauer–Emmett–Teller, Micromeritics TriStar 3000, USA). XRD (X-ray diffraction) patterns were used to analyze the phase change and crystalline structures of samples using an X-ray diffractometer (Ultima III, Rigaku, Japan).Results and discussion
Fig. 1a schematically illustrates the preparation process of LMO@SiO2. As shown in Fig. 1b, X-ray diffraction (XRD) analysis was conducted on both the ball-milled LMO powder and the calcined LMO@SiO2 powder. The diffraction peaks of the LMO@SiO2 powder at 18.76°, 36.34°, and 44.18° closely align with those of the ball-milled LMO powder, corresponding to the spinel (111), (311), and (400) planes, respectively.39–41 These planes are formed by lithium in the tetrahedral sites and manganese in the octahedral centers.42,43 The XRD results confirm that our LMO powder is composed of spinel-structured LiMn2O4, a key characteristic of lithium ion sieves. This structure enables the lithium ion sieve (LIS) to maintain vacancies left after lithium extraction in acidic solutions, thereby selectively adsorbing lithium ions.44 | ||
| Fig. 1 (a) Schematic illustration of the preparation process of LMO@SiO2. (b) XRD patterns of LMO before and after coating (LMO/LMO@SiO2). (c)–(f) SEM images of LMO at various sizes before and after coating (LMO/LMO@SiO2). (g) TEM images of LMO@SiO2. (h)–(i) The EDS characterization of Mn and Si elements in the TEM image (g). (j)–(k) In situ TEM line scan of Mn and Si signals in LMO@SiO2, and the corresponding element distribution along the red line from left to right. | ||
As depicted in Fig. 1c–f, we examined uncoated LMO and calcined LMO@SiO2 powders using scanning electron microscopy (SEM). The SEM images reveal that the surface of uncoated LMO is relatively smooth. After coating and calcination with sodium silicate, a uniform silicon dioxide (SiO2) layer is observed to encapsulate the LMO surface at both the 5 μm and 500 nm scales, indicating effective encapsulation by sodium silicate calcination.
To further analyze the SiO2 layer formed after sodium silicate calcination, transmission electron microscopy (TEM) was employed to study the encapsulation of LMO@SiO2, as shown in Fig. 1g–j. Fig. 1g demonstrates a well-defined silicon dioxide coating with distinct contrasts. In situ energy-dispersive X-ray spectroscopy (EDS) analysis in Fig. 1h–i clearly reveals that the Si signal map overlaps with the Mn signal map, indicating a uniform element distribution. Additionally, line-scan TEM analysis performed across the dashed line in Fig. 1j, and as illustrated in Fig. 1k, further confirms that the silicon dioxide layer primarily encases the LMO after sodium silicate calcination. This encapsulation is expected to significantly reduce manganese dissolution in acidic solutions.
Severe manganese dissolution occurs during lithium elution in acidic solutions, leading to the destruction of the lithium ion sieve framework and poor adsorption–desorption cycle performance.31,45 This hinders the industrial application of lithium ion sieves. Fig. 2a illustrates the manganese dissolution process. During acid washing, the delithiation of LMO is accompanied by the disproportionation reaction of Mn3+, where Mn3+ transforms into Mn2+ and Mn4+ in acidic solutions. Mn2+ tends to dissolve in the solution, disrupting the LMO spinel structure, as shown in the following reaction:46,47
| 4Li[Mn3+Mn4+]O4 + 8H+ → 3[Mn24+]O4 + 4Li+ + 2Mn2+ + 4H2O |
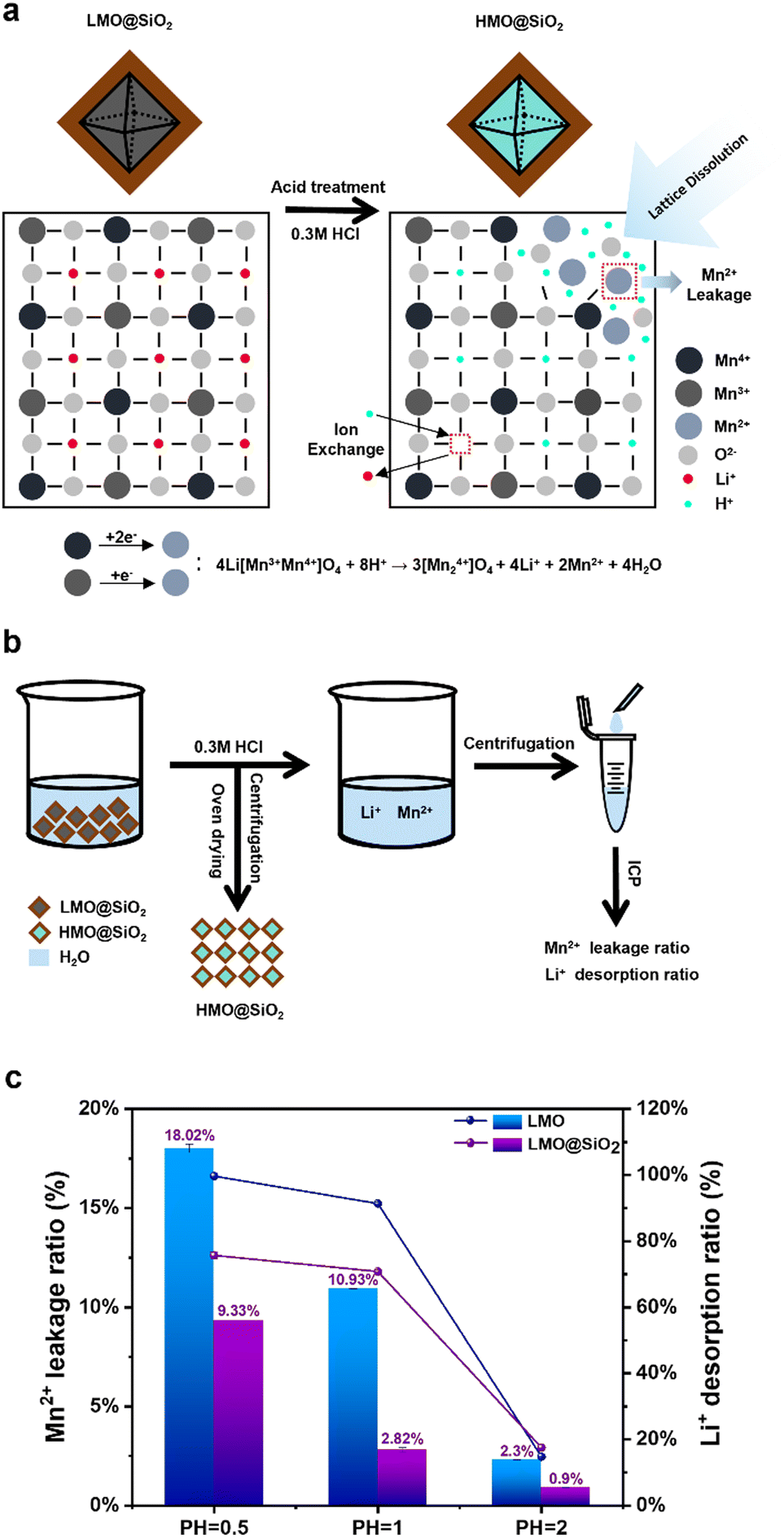 | ||
| Fig. 2 (a) Schematic representation of the mechanism and dissolution process from LMO@SiO2 to HMO@SiO2 acid treatment. (b) Schematic diagram of Li+ and Mn2+ adsorption–desorption processes and ICP characterization of treated solutions. (c) Li+ desorption and Mn2+ leakage ratio of LMO and LMO@SiO2 at different pH values. | ||
In this study, we coated LMO with sodium silicate and obtained coated LMO@SiO2 after calcination. This significantly reduces the ratio of manganese dissolution in acidic solutions and greatly enhances the cycle life due to the excellent protective properties of the SiO2 coating layer. Compared with the typical silicon coating source TEOS, the affordability and availability of sodium silicate enable great potential for industrial applications. Fig. 2b describes the adsorption–desorption process of the LMO@SiO2 adsorbent. The LMO@SiO2 powder transforms into HMO@SiO2 (HMO, the Li+ sites in LMO were occupied by H+) in the acidic solution. After centrifugation and drying, the HMO@SiO2 powder is collected and used in the subsequent lithium adsorption process. The acidic solution contains certain amounts of Mn2+ and Li+. Further centrifugation, titration, and ICP-OES analysis allow us to calculate the dissolution ratio of Mn2+ and the desorption ratio of Li+.
Control experiments were conducted with uncoated LMO and coated LMO@SiO2 powders in acidic solutions of different pH values. As shown in Fig. 2c, under various acidic conditions, the coated LMO@SiO2 material exhibits a lower manganese dissolution ratio and a higher lithium desorption ratio. In the acid washing process to recover lithium, the SiO2 coating reduced the manganese ion dissolution ratio from 10.93% to 2.82% at pH = 1, a decrease of approximately 74%. However, the SiO2 coating slightly impacted the lithium ion desorption ratio. At pH = 0.5 and pH = 1, the uncoated lithium ion desorption ratios were 99.67% and 91.35%, respectively, while after coating, these ratios dropped to 75.70% and 70.82%. At pH = 2, both the manganese ion dissolution and lithium ion desorption ratios for coated and uncoated LMO materials were below 3% and 20%, respectively, indicating that lithium ions could not be effectively extracted from the LMO material at this pH level. These results suggest that the SiO2 coating is an effective way to improve the selectivity of lithium ion recovery from spent lithium-ion batteries. In acid washing environments with pH = 0.5 and pH = 1, the manganese ion dissolution ratio significantly decreased after the SiO2 layer coating, with the most notable reduction observed at pH = 1, where the ratio dropped from 10.93% to 2.82%, a decrease of approximately 74%. However, the SiO2 coating slightly impacted the lithium ion desorption ratio. At pH = 0.5 and pH = 1, the uncoated lithium ion desorption ratios were 99.67% and 91.35%, respectively, while after coating, these ratios dropped to 75.70% and 70.82%. At pH = 2, although the manganese ion dissolution ratio for coated and uncoated LMO materials were below 3%, both the lithium ion desorption ratios were below 20%, indicating that lithium ions could not be effectively extracted from the LMO material at this pH level. Therefore, at low pH values (0.5 and 1), the SiO2 layer effectively prevents the leakage of manganese ions from the LMO spinel structure, thereby maintaining the integrity of the LMO structure and enhancing its adsorption–desorption cycle performance. Although the lithium desorption ratio is lower than that of the uncoated material due to the silica layer also affecting the free movement of Li+, this is considered acceptable. Specifically, while the silica coating moderately decreased lithium desorption efficiency (by ∼20% at pH 0.5 and 1), it significantly enhanced adsorbent stability, reducing the manganese dissolution ratio by over 70%.
In industrial applications, the operation of powdered lithium extraction materials can be challenging due to significant losses during adsorption and desorption. These materials often wash out with the flow of brine, leading to issues such as system blockages, poor fluidity, and low material recovery rates.17 To address these problems, it is important to process the materials into granules. This paper utilizes environmentally friendly and renewable sodium alginate to granulate LMO@SiO2 using a sol–gel templating method, as illustrated in Fig. 3a. Sodium alginate rapidly forms a gel under mild conditions. When divalent cations like Ca2+ are present, an ion exchange reaction occurs between the Na+ on the G units and the divalent cations, leading to the accumulation of G units and the formation of a cross-linked network structure, resulting in a hydrogel. However, the mechanical strength of sodium alginate is relatively poor, so we enhanced the mechanical strength of the microspheres by crosslinking polyacrylic acid (PAA) and sodium alginate (SA) with Ca2+. The addition of PAA facilitated crosslinking with both SA and Ca2+, introducing additional hydrogen bonds and ionic interactions between PAA–PAA, PAA–Ca2+, and PAA–SA, thereby forming a double-cross-linked network composed of PAA, SA, and Ca2+. Additionally, during the preparation of the cyclic adsorption simulated brine solution, we maintained the pH around 6.8. Under these neutral conditions, sodium alginate was less prone to dissolution.48–50 Due to the affordability and rapid sphere formation of sodium alginate, granulation can be extended to a large scale.
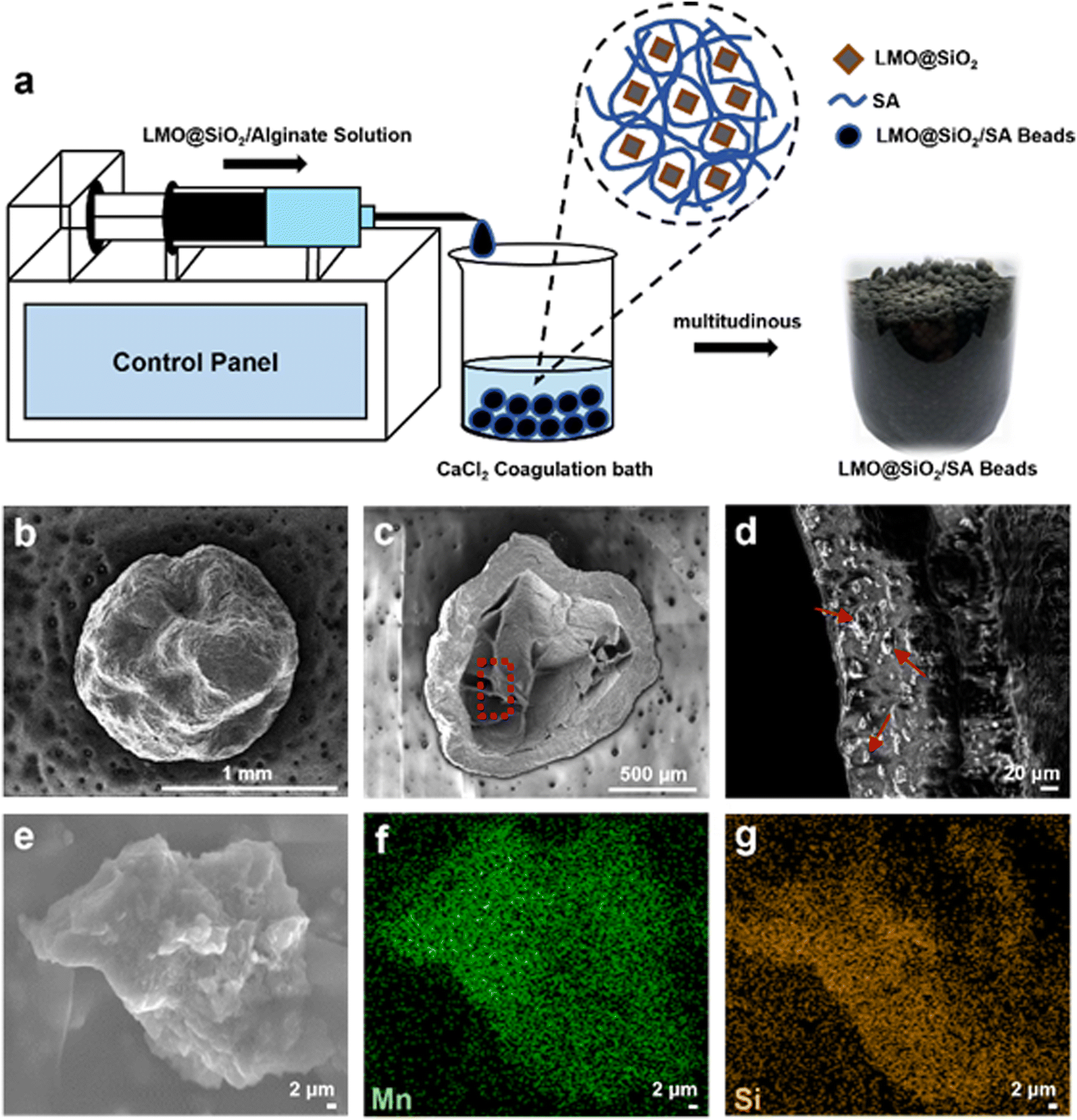 | ||
| Fig. 3 (a) Schematic of granulation of LMO@SiO2 and actual images of LMO@SiO2/SA beads. (b)–(d) SEM characterization of whole, cross-sectional, and internal venation of LMO@SiO2/SA beads. (e)–(g) High magnified SEM image and the corresponding EDS characterization of Mn and O elements of the embedded LMO@SiO2 powder. | ||
We characterized the morphology of the LMO@SiO2/SA beads using SEM, as shown in Fig. 3b–d. Before freeze-drying, the material is spherical, as depicted in the right image of Fig. 3a, with a smooth surface. After freeze-drying, the overall appearance remains, but the surface becomes relatively rough. The cross-section of the material, shown in Fig. 3c, reveals a loose interior with a dense shell, which further protects the material from dissolution. Fig. 3d shows an enlarged view from the red circle in Fig. 3c, where the distribution of the LMO@SiO2 powder (indicated by red arrows) is clearly visible. High-magnified SEM and corresponding EDS analysis on this powder are shown in Fig. 3e–g. The figures clearly display the presence of Mn and Si elements, indicating that LMO@SiO2/SA beads contain structures of SiO2, LMO, and the sodium alginate molecular framework, with uniform distribution throughout the composite beads.
Surface area analyses of sodium alginate millimeter beads of LMO@SiO2/SA and HMO@SiO2/SA were conducted using a BET analyzer, as shown in Fig. 4. Before acid washing, the LMO beads appeared black, with a surface area measured by BET of 54.58 m2 g−1 and a pore volume of 9.25 cm3 g−1, indicating that it functions as a macroporous adsorbent material. The acid wash resulted in the partial dissolution of Mn3+/Mn4+ to Mn, changing the bead's color from black LMO to brownish-red HMO, as shown in the inset of Fig. 4. BET measurements showed a slight decrease in the surface area and pore volume of the HMO beads to 46.56 m2 g−1 and 8.87 cm3 g−1, respectively, confirming that the overall structure remained intact after acid washing.51
 | ||
| Fig. 4 BET analysis of the adsorption pore volume for LMO@SiO2/SA beads and HMO@SiO2/SA beads. Insets are the corresponding single beads before (LMO) and after (HMO) acid treatment. | ||
To evaluate the industrial potential of HMO@SiO2/SA beads for lithium extraction, we established a continuous flow extraction system as depicted in Fig. 5a, with the actual setup shown in Fig. 5b. We simulated a brine solution resembling that of Da Qaidam Salt Lake in Qinghai Province for cyclic testing (Fig. 5c), the specific ion concentrations are given in Table 1. Due to the high concentration of Mg2+ in the simulated brine, which could interfere with subsequent ICP analyses, we diluted the solution ten-fold for the adsorption experiments.
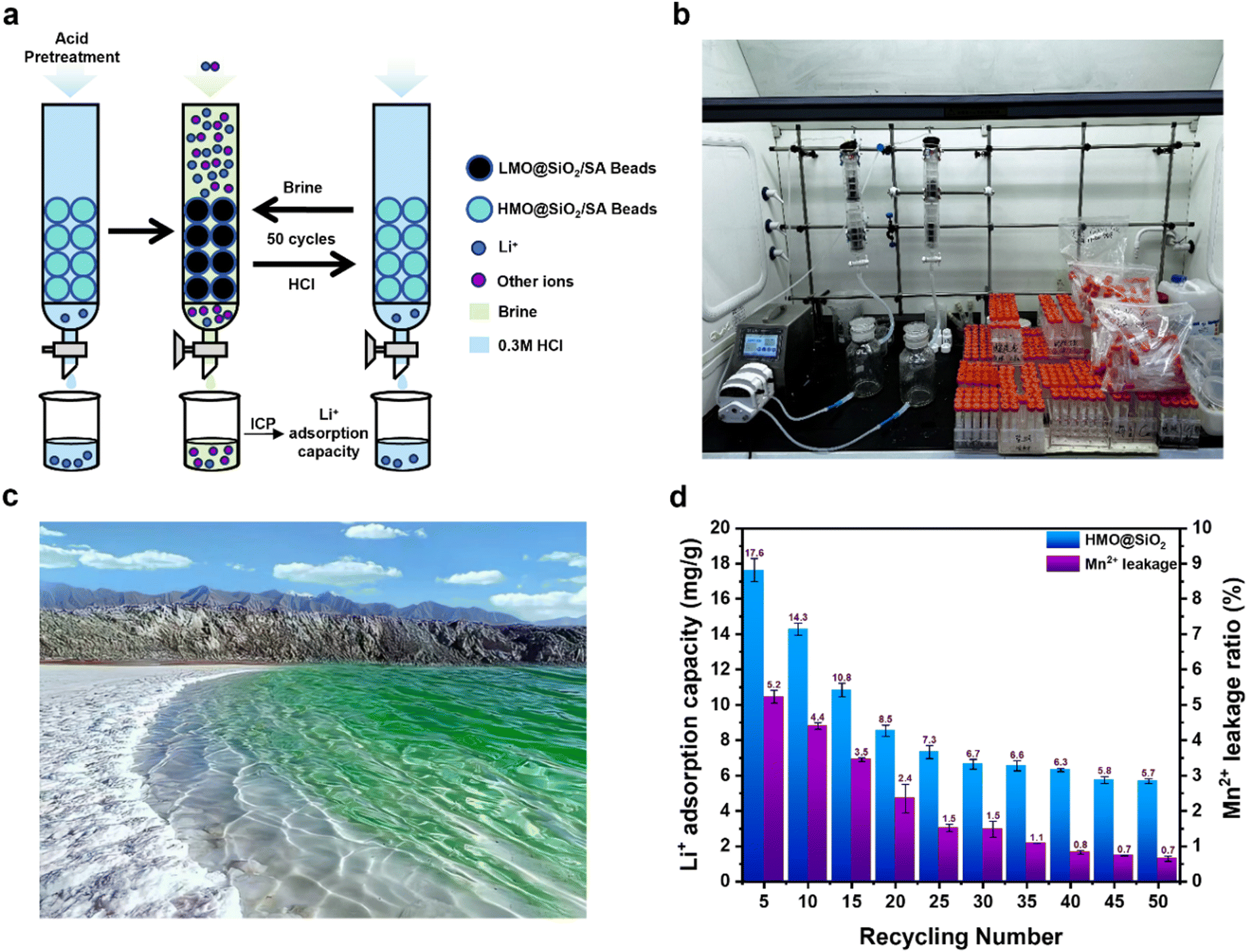 | ||
| Fig. 5 (a) and (b) Schematic and actual images of the continuous flow adsorption–desorption cycle system. (c) Photograph of Da Qaidam Salt Lake in Qinghai Province. (d) ICP analysis of the Li+ adsorption capacity and Mn2+ dissolution ratio over 50 cycles. | ||
| Composition | Li | Na | K | Ca | Mg |
|---|---|---|---|---|---|
| Concentration (mg L−1) | 330.6 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 882.5 882.5 |
11737.5 |
9613.3 | 18825 |
The adsorption and desorption results of Li+ from the simulated brine are presented in Fig. 5d. In the initial five cycles, HMO@SiO2/SA beads exhibited an impressive adsorption capacity of 17.6 mg g−1 for Li+, with a Mn2+ dissolution ratio of 5.2%. However, over the course of 1–25 cycles, the adsorption capacity and the Mn2+ dissolution ratio gradually decreased to 7.30 mg g−1 and 2.40%, respectively. This decline is likely attributable to the structural damage of the spinel caused by repeated acid washes of the internal HMO. From cycles 30 to 50, the adsorption capacity for Li+ stabilized at approximately 6.22 mg g−1, with an average Mn2+ loss ratio of 1.26%. These findings demonstrate that HMO@SiO2/SA beads can effectively extract Li+ from simulated salt lake brine, exhibiting robust stability and lithium extraction capabilities over 50 adsorption–desorption cycles.
And the stability trend observed after 30 cycles in Fig. 5d may be attributed to a combination of surface passivation, manganese dissolution equilibrium, and the formation of stable Li+ transport channels. During the initial cycling stage, the material undergoes a gradual surface passivation process. A small amount of surface and near-surface manganese ions may dissolve until reaching a dynamic equilibrium. Concurrently, the SiO2 coating layer likely forms a more compact and stable surface structure in the acidic environment, providing continuous protection against further manganese dissolution and structural degradation. Moreover, the repeated insertion and extraction of lithium ions may create stable transport pathways within the material, which reach a relatively fixed state after approximately 30 cycles. This optimization of the internal structure facilitates efficient and stable lithium-ion transport in subsequent cycles. The synergistic effect of these processes results in the observed stability trend post 30 cycles.
Conclusions
Our research represents a significant advancement in the field of direct lithium extraction from brine, addressing the growing demand for lithium in electric vehicle batteries. By utilizing sodium silicate as the silicon source, we successfully synthesized LMO@SiO2 composites, yielding a powdered lithium-ion sieve with enhanced structural stability. Notably, in a highly acidic environment (pH = 0.5), the manganese dissolution ratio for LMO@SiO2 was significantly reduced to 9.33%, compared to 18.02% for uncoated LMO, showing the protective effectiveness of the SiO2 coating. To simulate real-world industrial applications of this lithium-ion adsorbent in direct lithium extraction from brine, we encapsulated LMO@SiO2 in sodium alginate beads (LMO@SiO2/SA beads). The results demonstrate that, in a multi-ion environment with a high Mg/Li ratio, these beads initially exhibited a high adsorption capacity of 17.6 mg g−1, which stabilized at 6.22 mg g−1 after 50 cycles, while maintaining a low average manganese dissolution ratio of 1.26% per cycle. These findings highlight the potential of LMO@SiO2/SA beads as a promising material for efficient and sustainable lithium extraction from brine, contributing to the development of advanced energy storage solutions.Author contributions
Jialun Su: investigation, data curation, formal analysis, software, validation, visualization, and writing – original manuscript draft; Qiyue Fan: investigation, data curation, formal analysis, software, validation, visualization, and writing – original manuscript draft; Xiangting Hu: formal analysis, methodology, and software; Yue Sun: formal analysis, methodology, and software; Jie Lin: methodology and software; Jiayue Xu: methodology and software; Bingcai Pan: validation; Zhenda Lu: funding acquisition, conceptualization, methodology, project administration, supervision, and writing – review and editing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Technologies R&D Program of China (2022YFA1205602), the grant from the National Natural Science Foundation of China (22075128), the grant from the State Key Laboratory of Analytical Chemistry for Life Science (5431ZZXM2409), and the National Science Fund for Distinguished Young Scholars (21925602). We also acknowledge the assistance from the Technical Center of Nano Fabrication and Characterization, Nanjing University, for the TEM characterization.Notes and references
- H. Li, J. Pan, Y. Ping, J. Su, M. Fang, T. Chen, B. Pan and Z. Lu, Nano Lett., 2023, 23, 10458–10465 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- S. E. Kesler, P. W. Gruber, P. A. Medina, G. A. Keoleian, M. P. Everson and T. J. Wallington, Ore Geol. Rev., 2012, 48, 55–69 CrossRef.
- A. Alessia, B. Alessandro, V.-G. Maria, V.-A. Carlos and B. Francesca, J. Cleaner Prod., 2021, 300, 126954 CrossRef CAS.
- J.-M. Tarascon and M. Armand, Nature, 2010, 414, 359–367 CrossRef PubMed.
- L. V. Garcia, Y.-C. Ho, M. M. Myo Thant, D. S. Han and J. W. Lim, Processes, 2023, 11, 418–441 CrossRef CAS.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 RSC.
- M. Zhang, H. Tian, H. Wei, P. Tian, C. Yu and Z. Zhao, Rare Met. Cem. Carbides, 2018, 46, 11–19 CAS.
- M. L. Vera, W. R. Torres, C. I. Galli, A. Chagnes and V. Flexer, Nat. Rev. Earth Environ., 2023, 4, 149–165 CrossRef CAS.
- Y. Zeng, W. Li, Z. Wan, S. Qin, Q. Huang, W. Cai, Q. Wang, M. Yao and Y. Zhang, Adv. Funct. Mater., 2024, 2400416 CrossRef CAS.
- J. Li, S. Zhu and Z. Li, Sci. Sin.: Chim., 2017, 47, 1273–1283 Search PubMed.
- R. Xie, Z. Zhao, X. Tong, X. Xie, Q. Song and P. Fan, Particuology, 2024, 87, 46–53 CrossRef CAS.
- T. Kundu, S. S. Rath, S. K. Das, P. K. Parhi and S. I. Angadi, Powder Technol., 2023, 415, 118142 CrossRef CAS.
- M. Anik Hasan, R. Hossain and V. Sahajwalla, Process Saf. Environ. Prot., 2023, 178, 976–994 CrossRef CAS.
- Y. Hu, H. Su, Z. Zhu, T. Qi and Q. Pang, Environ. Chem. Lett., 2024, 22, 105–120 CrossRef CAS.
- M. Zhao, C. Zhao, Y. Zhang, J. Luan, G. Pan, C. Han, H. Peng, H. Yu, G. Zhao, G. Tang, Y. Li, X. Zhang and Y. Liu, J. Environ. Chem. Eng., 2023, 11, 110859 CrossRef CAS.
- H. Wang, J. Cui, M. Li, Y. Guo, T. Deng and X. Yu, Chem. Eng. J., 2020, 389, 124410 CrossRef CAS.
- J. Yang, Y. Wang, M. Zhang, P. Wang, X. He, H. Zhou and P. He, Angew. Chem., Int. Ed., 2024, e202411957 CAS.
- R. Zhu, Environ. Chem. Lett., 2023, 21, 1611–1626 CrossRef CAS.
- A. H. Hamzaoui, A. Mnif, H. Hammi and R. Rokbani, Desalination, 2003, 158, 221–224 CrossRef CAS.
- M. Pasta, A. Battistel and F. La Mantia, Energy Environ. Sci., 2012, 5, 9487–9491 RSC.
- C. P. Lawagon, G. M. Nisola, R. A. I. Cuevas, H. Kim, S.-P. Lee and W.-J. Chung, Chem. Eng. J., 2018, 348, 1000–1011 CrossRef CAS.
- J. M. Tarascon and D. Guyomard, J. Electrochem. Soc., 1991, 138, 2864–2868 CrossRef CAS.
- S. Kim, J. Lee, J. S. Kang, K. Jo, S. Kim, Y.-E. Sung and J. Yoon, Chemosphere, 2015, 125, 50–56 CrossRef CAS PubMed.
- J. Lee, S.-H. Yu, C. Kim, Y.-E. Sung and J. Yoon, Phys. Chem. Chem. Phys., 2013, 15, 7690–7695 RSC.
- J. Wang and G. M. Koenig, Chem. – Eur. J., 2024, 30, 2776–2798 Search PubMed.
- A. Battistel, M. S. Palagonia, D. Brogioli, F. La Mantia and R. Trócoli, Adv. Mater., 2020, 32, 1905440–1905462 CrossRef CAS.
- Y. Chen, F. Zhang, Y. Wang, C. Yang, J. Yang and J. Li, Microporous Mesoporous Mater., 2018, 258, 170–177 CrossRef CAS.
- L. Zhang, D. Zhou, G. He, Q. Yao, F. Wang and J. Zhou, Mater. Lett., 2015, 145, 351–354 CrossRef CAS.
- R. Chitrakar, Y. Makita, K. Ooi and A. Sonoda, Dalton Trans., 2014, 43, 8933–8939 RSC.
- R. Chitrakar, H. Kanoh, Y. Miyai and K. Ooi, Ind. Eng. Chem. Res., 2001, 40, 2054–2058 CrossRef CAS.
- K. Ooi, Y. Miyai and S. Katoh, Sep. Sci. Technol., 1986, 21, 755–766 CrossRef CAS.
- X. Luo, K. Zhang, J. Luo, S. Luo and J. Crittenden, Environ. Sci. Technol., 2016, 50, 13002–13012 CrossRef CAS PubMed.
- S. Zhang, X. Wei, X. Cao, M. Peng, M. Wang, L. Jiang and J. Jin, Nat. Commun., 2024, 15, 238–246 CrossRef CAS PubMed.
- R. Trócoli, C. Erinmwingbovo and F. La Mantia, Chem. Electro. Chem., 2017, 4, 143–149 Search PubMed.
- M. J. Park, G. M. Nisola, A. B. Beltran, R. E. C. Torrejos, J. G. Seo, S.-P. Lee, H. Kim and W.-J. Chung, Chem. Eng. J., 2014, 254, 73–81 CrossRef CAS.
- H. Li, T. Chen, J. Jiang, G. Gao, C. Wang and Z. Lu, Chem. Eng. J., 2023, 451, 138662 CrossRef CAS.
- H. Li, T. Chen, W. Wang, G. Gao and Z. Lu, Chem. Nano. Mater., 2022, 8, 324–330 Search PubMed.
- B. Ammundsen, D. J. Jones, J. Rozière, H. Berg, R. Tellgren and J. O. Thomas, Chem. Mater., 1998, 10, 1680–1687 CrossRef CAS.
- A. Gao, Z. Sun, S. Li, X. Hou, H. Li, Q. Wu and X. Xi, Dalton Trans., 2018, 47, 3864–3871 RSC.
- J. Xiao, X. Nie, S. Sun, X. Song, P. Li and J. Yu, Adv. Powder Technol., 2015, 26, 589–594 CrossRef CAS.
- Z. Xie, H. Eikhuemelo, J. Zhao, C. Cain, W. Xu and Y. Wang, J. Electrochem. Soc., 2015, 162, 1523–1529 CrossRef.
- W. Xiao, C. Xin, S. Li, J. Jie, Y. Gu, J. Zheng and F. Pan, J. Mater. Chem. A, 2018, 6, 9893–9898 RSC.
- H.-J. Hong, T. Ryu, I.-S. Park, M. Kim, J. Shin, B.-G. Kim and K.-S. Chung, Chem. Eng. J., 2018, 337, 455–461 CrossRef CAS.
- Z.-Y. Ji, M.-Y. Zhao, J.-S. Yuan, J. Wang, J.-Q. Zhou, H.-B. Yin and B.-Y. Sun, Solvent Extr. Ion Exch., 2016, 34, 549–557 CrossRef CAS.
- J. C. Hunter, J. Solid State Chem., 1981, 39, 142–147 CrossRef CAS.
- B. Ammundsen, D. J. Jones, J. Roziere and G. R. Burns, Chem. Mater., 1995, 7, 2151–2160 CrossRef CAS.
- R. Abka-khajouei, L. Tounsi, N. Shahabi, A. K. Patel, S. Abdelkafi and P. Michaud, Mar. Drugs, 2022, 20, 364–381 CrossRef CAS PubMed.
- S. Kumar and D. Sahoo, Algal Res., 2017, 23, 233–239 CrossRef.
- S. Jothisaraswathi, B. Babu and R. Rengasamy, J. Appl. Phycol., 2006, 18, 161–166 CrossRef CAS.
- I. P. S. Fernando, W. Lee, E. J. Han and G. Ahn, Chem. Eng. J., 2020, 391, 123823 CrossRef CAS.
Footnote |
| † J. Su and Q. Fan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |