
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInnovative dissolving microneedles for enhanced delivery of alpha arbutin and ascorbic acid: a novel LC–MS quantification approach
Ola
Tarawneh†
 *a,
Sara
Almasri†
a,
Ala A.
Alhusban
a,
Mohammad
Hailat
a,
Lama
Hamadneh
b,
Juhaina M.
Abu Ershaid
c,
Zeyad
Hailat
d and
Yahia F.
Makableh
e
*a,
Sara
Almasri†
a,
Ala A.
Alhusban
a,
Mohammad
Hailat
a,
Lama
Hamadneh
b,
Juhaina M.
Abu Ershaid
c,
Zeyad
Hailat
d and
Yahia F.
Makableh
e
aFaculty of Pharmacy, Al-Zaytoonah University of Jordan, Amman, Jordan. E-mail: ola.tarawneh@zuj.edu.jo
bFaculty of Medicine, Al-Balqa Applied University, As-Salt, Jordan
cDepartment of Applied Pharmaceutical Sciences and Clinical Pharmacy, Faculty of Pharmacy, Isra University, Jordan
dDepartment of Information Systems, Yarmouk University, Irbid 21163, Jordan
eInstitute of Nanotechnology, Jordan University of Science and Technology, Irbid, Jordan
First published on 16th December 2024
Abstract
This study presents a novel dissolving microneedles (MN) formulation designed to deliver alpha arbutin (AA) and ascorbic acid for hyperpigmentation treatment. Utilizing hydroxypropyl methylcellulose (HPMC) and polyvinylpyrrolidone (PVP-K90), we successfully prepared and characterized MN arrays with high drug loading efficiencies of 89.01% for AA and 94.09% for ascorbic acid. A pioneering liquid chromatography–tandem mass spectrometry (LC–MS) technique was developed for simultaneous drug quantification, achieving precision and accuracy as per FDA guidelines. Our MN demonstrated excellent mechanical properties, with effective skin penetration and dissolution within 5 minutes and permeation rates of 4.28% h−1 for AA and 3.61% h−1 for ascorbic acid. This innovative approach offers a promising platform for the transdermal delivery of active compounds, highlighting significant advancements in drug delivery technology.
Introduction
Hyperpigmentation is a common issue that may cause social embarrassment and can be treated by laser, physical peeling, or using medications, for example, alpha arbutin (AA) and ascorbic acid.1 In recent years, there has been a rise in innovative cosmetic products for skin whitening. These products utilize active ingredients designed to improve their performance. The stratum corneum (SC), which acts as a natural protective barrier, limits the passive penetration of active ingredients, limiting the performance of conventional methods.2Microneedles (MN) arrays are promising drug delivery systems that require careful formulation to deliver the drug through the skin successfully. MNs are organized in an array pattern on a base patch and consist of MN tips with height ranging from 25 to 1000 μm.3 Upon administration, MNs create reversible micrometer-sized channels within the skin layers that enable the penetration of drugs into the skin, possessing sufficient length to effectively pierce the stratum corneum (SC) while not extending deep enough to access the nerve terminals underneath it.3–5 Thereby, MNs enhance therapeutic efficacy through rapid onset of action, facilitate self-administration, improve bioavailability, and significantly enhance patient compliance, all while causing minimal pain to the patient.6 MNs are categorized into five distinct types based on their design and functional mechanisms: solid, hollow, coated, hydrogel-forming, and dissolving polymeric MNs.7 The fabrication of dissolving polymeric MNs involves using a centrifuge to concentrate drugs in the microtips of MNs. However, this method increases the manufacturing complexity due to the increased viscosity of the polymer blend.8 Various approaches have been used to improve the desirable characteristics of MNs, such as increasing polymer concentration during production.9 Different hydrophilic, biocompatible, and biodegradable polymers were used for MN fabrication that successfully encapsulate the drugs inside the matrix, for example, hydroxypropyl methylcellulose (HPMC) and polyvinylpyrrolidone (PVP-K90).1 Developing an appropriate transdermal drug delivery system with exceptional permeability properties and facilitating efficient drug interaction with the skin has been achieved using the MNs.10
The selection of polymers in the fabrication of MNs is an area of special challenge where one should meet the art requirement in fabricating a robust 3-dimensional structure, and the designated structure is demanded to deliver the drug by penetrating the skin without triggering an enormous level of pain. These unique characteristics may require using a single polymer or more to adjust the designated properties.11,12
PVP is a synthetic water-soluble polymer available in several grades and is characterized by various molecular weights and viscosities. PVP has excellent mechanical strength and biocompatibility, making it widely used in the fabrication of MNs.13 The hydroxypropyl methylcellulose (HPMC) synthetic polymer is derived from cellulose, a swelling, gelling, and thickening agent; the polymer exhibits solubility in water, biocompatibility, biodegradability, non-toxicity, and rheological characteristics.14
He et al. fabricated dissolving MNs loaded with propranolol hydrochloride for the treatment of infantile hemangioma, employing two water-soluble polymers, hyaluronic acid (HA) and PVP-K90 using a micro molding technique and two casting methods involving centrifuging at 4000 rpm.15
Wang and colleagues aimed to formulate dissolving MNs by combining AA and ascorbic acid and investigated their combined effect on dark brown guinea pigs, specifically assessing their impact on tyrosinase activity, melanin production, and mechanical properties of MNs where hyaluronic acid was used as a matrix.16,17
Aung et al. aimed to improve skin lightening by fabricating AA MNs. The first MN formulation was created by incorporating the polymer Gantrez™ S-97, and another MN formulation was designed by using the combination polymers HPMC and 40% w/w PVP K-90 (HPMC/PVP) by centrifugation at 4500 rpm.18
Sawutdeechaikul et al. fabricated dissolvable MNs for treating post-acne hyperpigmentation pimples loaded with ascorbic acid and glutathione. Using the micro-molding process, the needle base material was created, which consisted of a combination of hyaluronic acid, polyvinyl alcohol (PVA), and sucrose in a weight ratio of 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.19
:1:1.19
Xing et al. attempted to fabricate dissolving MNs loaded with azelaic acid and co-drug the alkaloid matrine for anti-acne treatment utilizing the polymers sodium carboxymethyl cellulose (CMC), PVP, and trehalose.20
Yang et al. attempted to overcome the limitation associated with lidocaine cream by creating dissolving MNs using the HA polymer to create a solution; the mixture droplets were then centrifuged for 10 s at 5000 rpm.21
The novelty of this research revolves around the ability to produce MNs comprising AA and ascorbic acid without centrifugation. The MN molds were washed before casting with water. Water increases the hydrophilicity of the silicone mold that is anticipated to receive the hydrophilic polymers. The modified tested method was deemed successful upon evaluation of the MNs by Drelich et al.22 The wet molds would have a strong affinity for the polymer mixture. In addition, the structure of ascorbic acid, which incorporates multiple proton donor and acceptor oxygen groups, would enhance diverse intramolecular interactions during conformational changes.23 This facilitates the manufacture of MNs and eliminates the need for centrifugation in the fabrication process.
Additionally, a vital property of MNs is the ability to penetrate the stratum corneum layer, where, in real life, it is anticipated that the patient would press the MN patches on the skin to obtain the mechanical force required to cause skin micro perforation. Nevertheless, the lack of appropriate MN guidelines would make the pressing time undetermined. Hence, an appropriate experiment addresses the time required for pressing. Herein, we propose an experiment where three layers of parafilm M® (PF) are exposed to MNs and subjected to pressing force for different two-time intervals: 30 seconds and 1 minute to investigate the proper advice given to the patient upon dispensing MNs as qualitative finding determine the optimal contact time required to achieve effective skin penetration and drug delivery. Quantifying combined medicines in a pharmaceutical formulation requires an accurate procedure to guarantee no interference of the medications and lead to false readings. Different techniques can quantify combined medications, where the technique selection depends mainly on the accuracy, precision, reproducibility, and rate of obtaining accurate results. The LC–MS method provides facile, accurate separation of AA and ascorbic acid. It could be employed in potential in vivo experiments where the possible interference from the biological matrix would raise a concern if the non-discriminative technique were used, as shown elsewhere.24
Materials and instrumentation
Materials
Alpha arbutin, molecular weight (MW) = 272.25 Da, was purchased from TCI (Tokyo, Japan). L-Ascorbic acid (MW = 176.1 Da) and methylene blue (MW = 319.85 Da) were purchased from Sigma-Aldrich® (Dorset, UK). Hydroxypropyl methylcellulose (HPMC) was purchased from Acros Organics (New Jersey, USA), with an average molecular weight = 1261.45 Da. Polyvinylpyrrolidone K90 (PVP-K90) (MW = 360000 Da) was obtained from TCI (Tokyo, Japan). Poly(vinyl alcohol) with a grade 40–88 was procured from Sigma Aldrich. Phosphate buffered saline (PBS) pH 7.4 tablets were purchased from Sigma Aldrich Co., USA. LC–MS grade water, methanol, ethanol, and chloroform were obtained from J.T. Baker®, UK. Ammonium formate was purchased from Honeywell Burdick and Jackson, USA. The internal standard vitamin B6 was obtained from Sigma-Aldrich®, UK.
Instrumentation
The adjustable LED digital microscope MX200-B with magnification up to 1000× was obtained from T TAKMLY (US) for imaging of the dissolvable MNs. Elmasonic S40(H) from Elma in Germany was utilized for sonication. Analytical balances Explorer® was obtained from OHAUS corporation in the United States. The laboratory Centrifuge Z326K from Hermle in Germany centrifugated the MNs. The TA-XTplus Texture Analyzer from Stable Micro Systems in the United Kingdom was used to evaluate MNs' mechanical properties. The estimation of pH was determined by the digital pH meter Jenway 3510, given by Antylia in the USA. Stirring Hot Plate from Antylia's SHP-200-S Stuart was used for heating and mixing. The Autovortex Blender SA1 from Antylia was purchased in the United States. Genlab™ Classic Oven obtained from Gallenkamp & Co. Ltd in England. For analysis, a UV-1800 spectrometer from Shimadzu in Japan was utilized. A Shimadzu LCMS-8030 Triple Quad MS system was obtained from Shimadzu, Japan. The vertical Franz diffusion cell system with 8 mL receptor volume and orifice diameter of 11.28 mm and with a volatile donor chamber that has a 7/25 Teflon stopper, flat ground (ground O-ring) joint, and a stainless-steel clamp and 1000 μL pipette tip assemblies were obtained from PermeGear, Inc, USA. The polydimethylsiloxane (PDMS) microneedles mold MPatchTM composed of 225 (15 × 15) pyramidal-shaped arrays were acquired from Micropoint Technologies Pte Ltd, Singapore. The Fourier transform infrared (FTIR) spectrometer was obtained from PerkinElmer, USA. A nylon syringe filter of pore size one μm had a diameter of 25 mm.Methods
Fabrication of the microneedles
To prepare the HPMC solution, 4 g of HPMC was added to 50 mL water and dissolved using a magnetic stirrer until a clear mixture was obtained; then 10 g of PVP-K90 was added to 50 mL water and stirred until a clear mixture was obtained. Then, an equal volume of the two polymers was prepared (5 mL from each and appropriately mixed until homogeneous). The PDMS MNs (15 × 15) pyramidal-shaped arrays were washed with water for 1 hour, and combination of drug AA: ascorbic acid was loaded in polymer solution in ratios of 2.5%:2.5%:95% w/w were used where 50 mg of AA and 50 mg of ascorbic acid were added to 1900 mg of a readily prepared mixture of HPMC (8% w/v) and PVP-K90 (20% w/v). The mixture was sonicated (Elmasonic S40 (H), Germany) for 10 min. The trapped air bubbles were removed by centrifugation at 1500 rpm for 10 min, and 50 μg was cast carefully in the MN mold. After weighing the designated volume, the procedure was repeated with every 50 μL (equivalent to 48.3 mg). After one hour, another volume of 100 μL was added, and the MNs were left to air-dry for 48 h.
Morphology
A digital microscope (MX200-B, T TAKMLY), was exploited to examine the tips and their height, and they were subsequently analyzed using ImageJ® software (US National Institutes of Health, Bethesda, Maryland, USA) for a precise measurement of the MNs after they were dried entirely and to ascertain that the tips were not altered during displacement from the molds.Determining the pH of MNs
The pH was measured by dissolving the blank MNs, AA-loaded MNs, ascorbic acid-loaded MNs, and the designated AA and ascorbic acid-loaded MNs in 100 mL of deionized water, were then sonicated for 30 min. The pH was recorded by placing the electrode of the digital pH meter 3510 (Jenway, Cole-Parmer Ltd, Antylia, USA) on the surface (n = 3) and left for 30 min until equilibrium and the pH reading was recorded.25Fourier transform infrared (FTIR) analysis
Fourier transform infrared (FTIR) spectroscopy (PerkinElmer, Waltham, MA, USA) was utilized for analyzing the functional groups of the investigated polymers, drugs, and the MN formulation to detect any potential chemical interaction, including pure AA powder, ascorbic acid, PVP-K90, HPMC. FTIR was also conducted on the physical mixture of the components fabricating MNs and the drug-loaded MNs. Samples were prepared using cryogenic grinding and liquid nitrogen to attain powder. The FTIR spectra of the samples were obtained throughout the wavenumber range of 400 to 4000 cm−1.26Mechanical properties of MNs (compression test)
Using the TA-XTplus Texture Analyzer (Stable Micro Systems, UK) in compression mode, the mechanical strength of the MNs was evaluated (the ability of the MNs to withstand compression). The MNs were attached to the movable cylinder probe (length 5 cm, cross-sectional area 1.5 cm2) with an adhesive double-face tap. The probe was assigned to move vertically downward at a test speed of 0.50 mm s−1, while the pre-and post-test rates were one mm s−1 against a flat aluminum block as described27,28 with alterations. A force of 32 N per patch (the trigger force was 0.1422 N per needle) was investigated. Moreover, 32 N was reported as a person's mean force when applying an MN.29 The tips and morphology of MNs were assessed using a digital microscope before and after the compression test and using the software ImageJ® (US National Institutes of Health, Bethesda, Maryland, USA). The percentage of the change in height of a set of needles was calculated (n = 3) using the following equation:30| Height reduction percentage = (original height − new height)/original height × 100% | (1) |
Insertion properties into Parafilm M®
Parafilm M® (PF, Bemis Company Inc., Soignies, Belgium) was validated formally in previous work by ref. 31 and 32 as a model skin simulant for MN penetration studies. The tips of the MNs were applied against a five-sheet PF piercing through the layers using thumb pressure for two time intervals: 30 seconds and 1 minute. Before and after insertion, MN tips were visualized by the digital microscope for morphological changes, then each sheet of PF was delicately removed, and the number of punctures was counted relative to the thickness of the PF layers.33 The thickness of each PF sheet was stated to be 130 μm.Skin penetration assessment into total thickness rat skin
An excised full-thickness rat skin (IRB: 01/08/2022–2023) was employed to estimate the insertion depth of the MNs into the skin. The skin was cut with surgical blades, and the hair was shaved with an electric razor and a hair removal cream, then washed with PBS (pH 7.4) for 30 min. The excised skin was then encased in aluminum foil and stored at −20 °C until needed.33Methylene blue (MB) gel was fabricated by mixing two drops of methylene blue dye solution with 1 mL of the successfully formulated MN blank formulation using vortex (SA2 Vortex Mixer, Stuart Scientific, UK) and sonicating for 10 min until a homogenous gel was formed, then cast slowly in the molds. The molds were centrifuged twice at 1500 rpm, 25 °C for 10 min. A total volume of 250 μL was cast, and the molds were left to dry for 24 h in an ambient environment. The dried MB-MNs were then pressed manually into the edge of the skin for 30 s and fixated with forceps before employing the digital microscope to obtain the image and running the software ImageJ to measure the depth of the MNs arrays into the skin.28 Moreover, to evaluate the ability of the formulated MNs to create micro-holes on rat skin, the successful MNs arrays loaded with AA and ascorbic acid were administered into the skin using thumb pressure for 30 seconds. Then, the MN array was detached, and the skin was stained with two drops of MB dye for 10 min. The leftover MB were gently cleaned using a tissue soaked in PBS. Afterward, the blue spots on the skin were observed using a digital microscope.34 The thickness of the rat skin was estimated to be approximately 1.31 mm.
Dissolution evaluation of drug-loaded MNs
To determine the dissolution time of the MNs, which is the time required for the patch to dissolve upon contact with an aqueous medium, the pre-stored excised skin was defrosted and left to melt in a beaker, then washed with PBS (pH 7.4) for 15 min and placed on tissue paper to dry; the MN patch was then manually inserted into the skin with a firm thumb pressure.26 The MNs were carefully detached at predetermined time intervals (0, 0.5, 1, 2, 4, and 5 min) and analyzed at each time interval using a digital microscope to visualize the MN arrays at those intervals.Liquid chromatography–tandem mass spectrometry (LC–MS): Method development for the separation between AA and ascorbic acid
Preparation of the mobile phases. The concentration of 20 mM ammonium formate for mobile phases was obtained by dissolving 1.276 g of ammonium formate in 1 L of water and methanol. The solution was vortexed and sonicated for 5 min. A programmatic approach was used to vary solvent compositions with a 0.40 mL min−1 flow rate, as shown in Table 1.
| Time (min) | Flow (mL min−1) | % A | % B |
|---|---|---|---|
| 0–2 | 0.40 | 95 | 5 |
| 2–2.5 | 0.40 | 25 | 75 |
| 2.5–4 | 0.40 | 5 | 95 |
| 4–6 | 0.40 | 95 | 5 |
Preparation of the stock solution. Stock solution (1 mg mL−1) of AA and ascorbic acid was prepared individually by weighing 100 mg of each drug and placing it in 100 mL of LC–MS water grade inside a volumetric flask. Then, it was subjected to vortexing until it completely dissolved. Serial dilutions of the stock solution took place to give a concentration for AA 0.003, 0.01, 0.05, 0.08, 0.12, and 0.2 mg mL−1 and for ascorbic acid 0.003, 0.009, 0.05, 0.09, 0.14, and 0.2 mg mL−1.
Sample pretreatment preparation. To extract analytes from the drug release, skin deposition, and content uniformity samples for LC–MS analysis, a volume of 250 μL from the sample was added to a volume of 250 μL ethanol and 1500 μL chloroform, then vortexed for 30 s until a well-mixed solution was achieved. The sample was subjected to a two-step extraction process by centrifuging at 4000 rpm for 5 min; the supernatant was collected, and an additional 1500 μL of chloroform was added to the extract before subjecting it to another round of centrifugation at 4000 rpm for 5 min. The resultant supernatant was subjected to evaporation under a stream of nitrogen flow. The residue acquired through this process was mixed with a volume of 1500 μL of methanol and 10 μL of the IS before vortexing for 30 s. The sample was collected through a 1 μm nylon syringe filter, after which it was transferred into the 2 mL vials. Finally, a 5 μL aliquot from the resulting combination was injected into the LC system as described in ref. 35 with some changes.
Limit of detection and limit of quantification. The limit of detection (LOD) and limit of quantification (LOQ) were established as the minimum concentrations that produced signal-to-noise ratios of ≥3 and ≥10, respectively, while implementing the analytical procedure.36 The LOD and LOQ were obtained experimentally.
Accuracy and precision and recovery. Accuracy and precision are crucial in analytical techniques, determining the degree of agreement among measurements.36 Accuracy is determined by analyzing three quality control samples at different levels, with acceptance criteria for each level being ±15% of nominal concentrations. Precision is quantified by calculating the coefficient of variation (CV), with a value of lower or equal to 15% being considered acceptable. To evaluate the efficiency of the extraction technique, the extraction recovery (Rec%) was assessed for AA and ascorbic acid by comparing peak areas of the QC samples before adding to a blank and after spiking at the same concentrations.
The following equation was used for calculating the accuracy values:
| Accuracy = Conccalc/Conctheo × 100 | (2) |
The following equation was used for calculating the precision values: SD is the standard deviation, and mean is the average of the three separate measurements.35,36
| % CV = SD/mean × 100 | (3) |
In vitro permeation studies and kinetics of drug permeation. The study investigated the diffusion of MNs using vertical Franz cells and the gel formulation loaded with the drugs AA and ascorbic acid. An entirely excised rat skin was used, which was cleaned and defrosted. The excised skin was precisely cut to fit the Franz cell donor chamber (11.28 mm, 8 mL receptor volume, surface area of 1 cm2), and MN arrays were applied to it using thumb pressure for 30 s. The receiver compartment was filled with 8 mL of PBS (pH 7.4), and the skin with the pressed MNs was attached to the top. The same MN formulation was placed in its gel form for in vitro permeation study as a control against the dried MN, where 144.9 mg of the gel has been added containing AA:ascorbic acid loaded in polymer solution in ratios of (2.5%:
2.5%:95% w/w).37 The donor compartment was positioned on the Franz cell and securely fastened. The Franz cell was stirred continuously at 600 rpm and maintained at 37 °C. A volume of 3 mL was withdrawn from the receptor chamber at 0.5, 1, 2, 3, 4, 6, and 24 h to assess the amount of AA and ascorbic acid released. An equivalent volume was immediately put back into the receptor compartment to maintain the sink condition.38 AA and ascorbic acid were quantified using the validated LC–MS method. The flux (Jss, μg cm−2 h−1) was estimated from the slope of the linear segment of the plot, showing the cumulative amount of AA or ascorbic acid permeated per unit area (Q/A) vs. the time plot A. Kinetic models are mathematical representations that describe the behavior and interactions of components or systems in motion.34,39| Zero order: Qt = Q0 + K0t | (4) |
| First order: lnQt = lnQ0 + K1 | (5) |
 | (6) |
| Korsmeyer–Peppas: Qt = Ktn | (7) |
| Hixson–Crowell: Q01/3 − Qt1/3 = Kst | (8) |
In vitro determination of drug accumulation in rat skin studies. This investigation was carried out after an in vitro permeation study to determine the accumulation of AA and ascorbic acid in rat skin. After 24 h, the excised rat skin with the attached MN was collected from the Franz cell. To extract the drugs from the skin, the sample was added to 20 mL of PBS (pH 7.4), vortexed for 1 min and sonicated for one h, and then filtered and collected for further analysis by using LC–MS as described with modification.40
Drug loading determination and loading capacity. To quantify the AA and ascorbic acid drug content in the formulated MNs, the MNs were weighed, placed inside a beaker filled with 25 mL of PBS (pH 7.4), vortexed for 30 s, and sonicated for one h for complete dissolution and a clear solution was obtained.41 A volume of 3 mL of the solution was subjected to filtration and collected for further analysis using LC–MS; this procedure was repeated five times (n = 5). The calculation of the loading efficiency and loading capacity percentage was performed using the following equations:42
| % Loading efficiency = amount of drug (mg)/initial amount of drug (mg) × 100 | (9) |
| % Loading capacity = amount of drug (mg)/amount of polymer (mg) × 100 | (10) |
Stability study of the MNs. To assess the stability of the drug content AA and ascorbic acid in the MNs, 10 MNs were stored inside a beaker separately, sealed with PF to minimize moisture and air exposure. The beaker was also wrapped with aluminum foil to protect it from light, reducing the photodegradation. The samples were placed inside a controlled environment at 2–8 °C and were stored for 30 days. After 30 days, the stability testing was conducted by dissolving 5 MN loaded with AA and ascorbic acid in 20 mL PBS (pH 7.4), and further analysis of the drug content took place using LC–MS. The mechanical strength of the MNs was also assessed. TA-texture analyzer was used in compression mode, where 5 MNs were subjected to a force of 32 N, and the height of the MN tips was measured before and after the compression test to assess the deformation and determine the structural integrity of the MNs.
Statistical methods. The study used GraphPad Prism software and Microsoft Excel for statistical analysis, employing t-tests, ANOVAs, and Tukey–Kramer tests to identify significant differences between groups. Data were presented as mean ± standard deviation (p < 0.05).
Results
Evaluation of drug-loaded MNs
The successful formulation containing AA and ascorbic acid showed robust and complete formation of the needle tips, as illustrated in Fig. 1. A digital microscope powered by the ImageJ software was employed to measure the dimensions and height of the MN tips. Micrographs in Fig. 1 showed excellent formation of the MN tips; the average height of the MNs was claimed to be 518.8 ± 10.2 μm. The weight of the dried MNs was measured (n = 5), and it was found that the average weight was 37.08 ± 1.49 mg.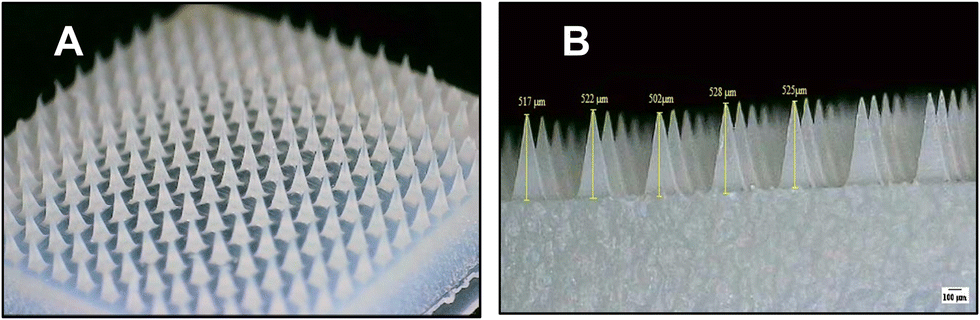 | ||
| Fig. 1 (A) Digital microscope image of the fully formed MN tips and (B) digital microscope images of the prepared MNs, the height was measured using ImageJ where the height of the microtips was 518.8 ± 10.2 μm (US National Institutes of Health, Bethesda, Maryland, USA). | ||
pH measurements
The pH of the blank polymers containing HPMC (8% w/v) and PVP-K90 (20% w/v), without including ascorbic acid and AA, was 8.53 ± 0.31. While the pH of the polymers with the addition of AA was 6.21 ± 0.19, the inclusion of ascorbic acid in the polymers showed a pH of 3.69 ± 0.34. However, the pH of the designated MNs, which includes the combination of AA and ascorbic acid to the polymers, was 4.78 ± 0.57. Fig. 2 summarizes the pH value of the blank polymer alone, with the addition of AA, ascorbic acid, and both drugs.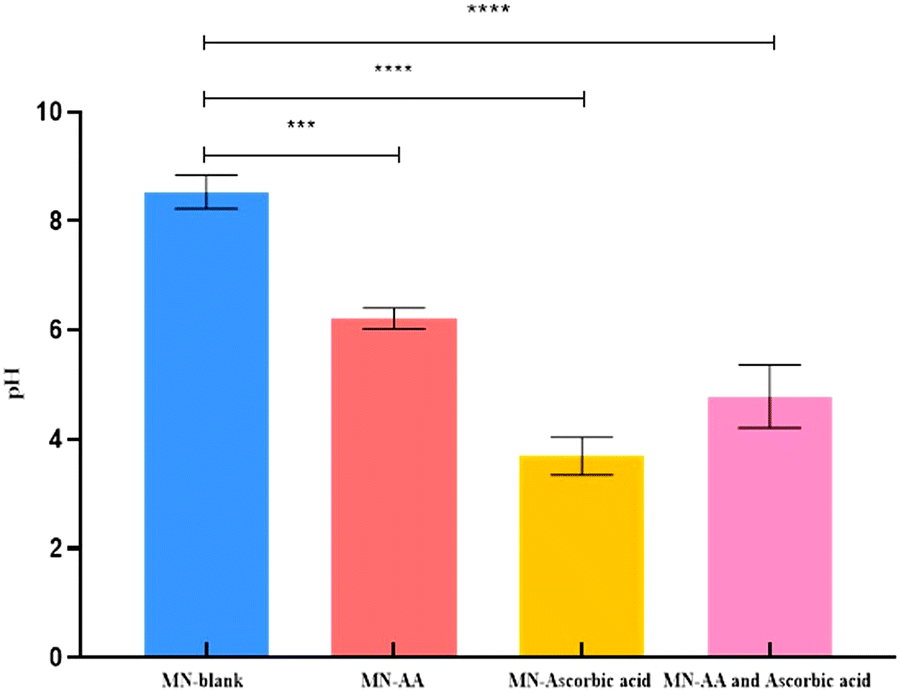 | ||
| Fig. 2 pH value of MNs, which are composed of HPMC 8% (w/v) and PVP-K90 20% (w/v) blank, MN-loaded with AA, MN-loaded with ascorbic acid, and MN-loaded with AA and ascorbic acid. Data presented as mean ± SD (n = 3). ****p-value < 0.0001 significant differences between MN-blank and MN-ascorbic acid, MN-blank and MN-AA and ascorbic acid. ***p-value < 0.001 significant difference between MN-blank and MN-AA. | ||
FTIR spectroscopy
The study examined the infrared transmission spectra of AA, ascorbic acid, HPMC, and PVP in powder and freeze-dried MN-loaded formulations to characterize intermolecular interactions.
Fig. 3A represents the FTIR spectra of pure AA showed distinct absorption peaks at 3406 cm−1 and 3218 cm−1, corresponding to the hydroxyl group. The aromatic nucleus C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations were detected at 1607, 1513, 1455, and 1401 cm−1. The wavenumber 2972 to 2898 cm−1 corresponded to the CH functional group's aliphatic stretching vibrations. The presence of ether bonds in the AA was detected by bands from 1227 to 1033 cm−1. The absorption peak at 1033 cm−1 indicated a C–OH group stretching mode.
C stretching vibrations were detected at 1607, 1513, 1455, and 1401 cm−1. The wavenumber 2972 to 2898 cm−1 corresponded to the CH functional group's aliphatic stretching vibrations. The presence of ether bonds in the AA was detected by bands from 1227 to 1033 cm−1. The absorption peak at 1033 cm−1 indicated a C–OH group stretching mode.
 | ||
| Fig. 3 FTIR spectra of the materials used in the MN formulation: (A) AA spectrum, (B) ascorbic acid, (C) HPMC, (D) PVP-K90, (E) MN loaded AA and ascorbic acid. | ||
The FTIR spectrum of ascorbic acid in Fig. 3B shows four peaks, including four hydroxyl groups, at 3524, 3407, 3312, and 3212 cm−1. CH band stretching is detected at 2988 and 2902 cm−1, while the lactone CO bond vibrational stretching at 1752 cm−1, CC bond vibrational stretching at 1658 cm−1, and COC stretching at 1111 cm−1 aligns with previous studies.
The FTIR analysis of HPMC revealed a characteristic peak at 3445 cm−1, indicating O–H stretching, as shown in Fig. 3C. A distinct peak at 2982 cm−1 corresponds to the vibration of the C–H group. A band at 1377 cm−1 is due to the bending vibration of the hydroxyl group (OH). The broad spectrum at 1056 cm−1 corresponds to vibrations from C–O stretches. These measured peaks were comparable to those of previous research.
The FTIR analysis of PVP-K90 reveals hydrophilic properties, with a sharp band at 3676 cm−1 corresponding to O–H bond stretching vibration, as shown in Fig. 3D. Other peaks include 1659 cm−1 for CO stretching, 1409 cm−1 for C–H bending of CH2, and 1251 cm−1 for C–N stretching. Significant peaks at 2988 cm−1 were observed for C–H bond stretching, consistent with previous research.43–45
Fig. 3E shows the IR spectrum of the formulated MNs, showing a shift in the CO of PVP and the vibrational stretching of ascorbic acid to an intense peak at 1650 cm−1. AA's aromatic nucleus CC stretching was shifted to 1495, 1440, 1425, and 1422 cm−1. Combination bands associated with the C–O stretching bond in HPMC were detected at 1057 cm−1. Broadband around 3394 cm−1 indicated hydrogen bonding between the polymers PVP and HPMC and the AA and ascorbic acid drugs. The presence of absorption bands suggests successful material integration.
Mechanical properties of MNs
 | ||
| Fig. 4 Compression effect: presentation of images photographed by digital microscope before and after compression test at 32 N. (A) blank MN before compression, (B) blank MN after compression, (C) MN-loaded AA and ascorbic acid before compression, and (D) MN-loaded AA and ascorbic acid after compression. | ||
 | ||
| Fig. 5 Insertion depth on PF of the two-time intervals (A) 30 s-time intervals with the corresponding number of layers, (B) 1 min and the associated number of layers. | ||
 | ||
| Fig. 6 (A) Image of the MB-MN arrays punctured into full-thickness rat skin, (B) image of the rat skin punctured by MN-loaded AA and ascorbic acid before immersing with the MB dye, (C) image of the rat skin punctured after immersing with two drops of MB dye. | ||
 | ||
| Fig. 7 Dissolution profile of the MN tips: 0, 0.5, 1, 2, 4 and 5 min. | ||
LC–MS
 | ||
| Fig. 8 LC–MS chromatogram of AA and ascorbic acid loaded MNs with IS (vitamin B6) at optimized chromatographic conditions. Column: Atlantis Premier BEH C18 AX Column (2.1 mm × 100 mm, particle size 1.7 μm); mobile phase (A) 20 mM ammonium formate in water, mobile phase (B) 20 mM ammonium formate in methanol with gradient elution. The flow rate is 0.40 mL min−1. | ||
| Parameters | AA | Ascorbic acid | ||||
|---|---|---|---|---|---|---|
| L (QC) 0.004 mg mL−1 | M (QC) 0.08 mg mL−1 | H (QC) 0.15 mg mL−1 | L (QC) 0.004 mg mL−1 | M (QC) 0.08 mg mL−1 | H (QC) 0.15 mg mL−1 | |
| Mean | 0.0040 | 0.0799 | 0.1499 | 0.0040 | 0.0802 | 0.150 |
| SD | 1.52 × 10−6 | 4.42 × 10−7 | 4.62 × 10−7 | 9.59 × 10−7 | 9.86 × 10−7 | 1.07 × 10−6 |
| Accuracy | 100.01 ± 0.0375 | 100 ± 0.0006 | 100 ± 0.0003 | 99.77 ± 0.0240 | 100.28 ± 0.0012 | 100 ± 0.0007 |
| CV | 0.0375 | 0.0006 | 0.0003 | 2.40 × 10−2 | 1.23 × 10−3 | 7.14 × 10−4 |
 | ||
| Fig. 9 The permeation profile of (A) AA and (B) ascorbic acid from the MNs and gel formulation means ± SD (n = 5). | ||
 | ||
| Fig. 10 Mechanical compression stability testing on MNs loaded AA and ascorbic acid after 30 days: presentation of images photographed by digital microscope before and after compression test at 32 N. (A) Before the compression, (B) after the compression. | ||
Discussion
The employment of MNs in drug delivery attracts attention from healthcare providers, especially pharmacists, and the scientific community in multidisciplinary research. The proof of concept of the efficacy of that delivery system will be based on, firstly, evidence of the formation of the microtips, secondly, examination of the successful compatibility of the formulation, and finally, characterization of the unique properties of the formed microtips and their efficacy.In this regard, the formulated MNs were composed of AA, ascorbic acid, PVP-K90, and HPMC and were successful with fully formulated microtips (Fig. 1). The designated MNs were based on trial and error in selecting proper ratios to avoid using a centrifuge and on ascorbic acid's ability to form the needed hydrogen bonding and strong MNs. FTIR studies revealed hydrogen bonding between the components of MNs, indicating the interaction between the OH functional groups. The FTIR spectrum of the formulated MNs closely matches the combined spectra of individual components, confirming the strength and compatibility of the MNs. The proton acceptor PVP and proton donor HPMC withstand forces due to hydrogen bonding formation, indicating an interaction between the materials.49 The polymer's ability to create hydrogen bonding interactions with drugs promotes molecular stability, potentially reducing recrystallization tendency.50 The functional group interaction indicates that the microtips in the MNs are strong and capable of resisting applied force, resulting in a compression mode, reflecting the rigidity and toughness of the MNs.
To prove the strength of MNs, a skin insertion experiment was conducted to examine rat skin. Employing PVP-K90 (MW of 360 kDa) exhibits appropriate compression strength, consistent with previous research.51 Adding the HPMC polymer enhances the strength of the MN formulation due to its rheology attributes, as previously reported.52 The combination of PVP-K90 and HPMC enhances the ability of microtips to resist the applied compression force. In addition, the investigated drugs to the polymeric matrix of MNs did not affect their mechanical properties (Fig. 4). The geometric features of the MNs, such as the rectangular pyramid shape, play a significant role in dictating their mechanical qualities. A study by Li et al. found that the rectangular pyramid shape provided greater mechanical strength than the cone cylinder design.53
The skin is the targeted organ to deliver drugs and has to be penetrated as the principal concept of MNs as transdermal drug delivery. The proper simulated skin model is essential for developing insertion tests. Artificial membranes like PF can assess the depth of MNs.5,54 Five PF sheets were used with two-time intervals (30 s and 1 min), puncturing approximately 260 μm and 390 μm, respectively.55 Longer application times resulted in increased puncture depth, as shown in Fig. 5. This helps in understanding the challenges and properties of drug penetration in the skin.
Furthermore, it highlights the need to establish dispensing guidelines for patient education by different healthcare providers: pharmacists, physicians, and nurses. Cleaning the area where the MN patch is applied and pressing the time upon application is necessary. Based on our findings, we recommend 1 min based on our polymer type and our model. However, we highlight the need to establish proper experiments based on different combinations of ingredients and to consider the limited strength of specific populations, such as geriatric and rheumatoid patients whose trigger force is estimated to be lower than the average human force, estimated at 32 N.56
The medical application of MNs requires their ability to be inserted into the skin without deformation. Understanding the mechanical properties and composition of the skin is crucial for improving MN insertion. The effectiveness of MNs in penetrating the skin depends on factors such as the viscoelastic nature of the skin, the physicochemical characteristics of the drugs, and the dimension of the MNs.57,58 Full-thickness rat skin was used for this study. The maximum length of MNs punctured into rat skin was comparable to the 30 s PF two-layer insertion test, estimated at 260 μm. The needles penetrate the skin, reaching the basal layer of the epidermis, which is full of melanocytes.5 This allows the drugs to target the desired areas for skin depigmentation directly.59 MNs are inserted into the dermis, where they are assumed to be in direct contact with the interstitial fluid and initiate a binding mechanism with water molecules; this results in the hydration of hydrophilic terminal groups on the polymers, causing swelling and exposing the hydrophobic backbone; this interaction with water is through van der Waals forces, resulting in more swelling.60,61 The chosen polymers influence the dissolving rates of MNs. PVP-K90 and HPMC, due to their hydrophilicity, took 5 min to completely dissolve MN tips on full-thickness rat skin, a fast process as shown in Fig. 7.
The chromatographic technique was developed to achieve satisfactory compound separation and desirable peak shape while minimizing the fronting and tailing of peaks, which can disrupt the accurate and precise quantification of analytes.62 The reverse-phase C18 column was chosen for its compatibility and versatility for various analytes. The ionization mode for LC–MS detection was ESI, which is suitable for investigating polar and moderately polar substances due to its soft ionization nature.63 The IS minimizes the impact of interfering matrix elements, such as other substances that are not the analyte of interest, decreases sample processing errors, and limits detection variability. Vitamin B6 was selected based on the structure of compounds AA and ascorbic acid. Linearity is crucial in determining the linear range of the technique.6,64
The correlation coefficients R2 for both drugs were over 0.99, which is acceptable since R2 should be above 0.95.65,66 To reduce matrix effects, specialized sample pretreatment is necessary. The liquid–liquid extraction (LLE) method, which involves separating components from a mixture using two immiscible liquids, was used for sample pretreatment with chloroform and methanol as organic solvents.67 The method was accurate and precise for AA and ascorbic acid, with extraction recovery estimated at 88% and 92%, respectively. A study by Wang et al. employed an HPLC-UV method for separating nine skin whitening agents in one injection, using a reversed-phase C18 column and a mobile phase consisting of water and methanol, both containing 0.1% acetic acid.68
The effectiveness of MNs in penetrating the skin depends on several factors, including the dense nature of the skin, which makes passive diffusion difficult unless a large amount of drug is supplied. The physicochemical characteristics of AA and ascorbic acid also affect drug departure and penetration. The type and dimension of MNs also affect drug penetration effectiveness. The continuous dissolution of polymers should ease drug transfer. The MN insertion into the skin can cause defects in skin integrity, leading to increased permeation due to hydration, swelling, and fluidization.69 The full-thickness rat skin is often used as the primary model for in vitro percutaneous permeation studies due to its accessibility, cost-effectiveness, and similar structural features to human skin.70
The study evaluated the release of AA and ascorbic acid from a designated MN formulation using Franz cells, representing human biological systemic circulation. The release was determined over 24 h, with the receptor chamber maintained at a sink condition to maintain drug concentration within 10–30% of the maximum solubility.
The initial burst release was attributed to the rapid dissolution of the drug from the dissolving polymers PVP-K90 and HPMC. This phenomenon is observed in most drug delivery systems and is influenced by a variety of factors,71 including the high solubility, and the physicochemical properties of AA and ascorbic acid also contribute to the drug's release from MNs and the initial burst, the partition coefficient where the skin's lipophilic membrane SC enhances drug permeation through the skin, with increased lipophilicity potentially enhancing drug permeation, the polymer and drug interaction as observed from the FTIR analysis revealing a broad hydrogen-bonding band around 3394 cm−1, which is indicative of interactions between the hydroxyl groups of the polymers PVP and HPMC, and the functional groups of the AA and ascorbic acid. Furthermore, the polymer:solvent ratio and the drying and storage stages contribute to burst release through drug migration within the polymer matrix. These properties influence the driving forces behind drug release, impacting the rate and the extent of the initial burst AA permeated more than ascorbic acid, with a higher flux through the skin membranes. Franz cell studies often consider that drug diffusion occurs at a steady state under “the ideal sink” conditions.72 The permeation profile is shown in Fig. 9.
Aung et al. compared AA permeation in MNs loaded with PVP-K90 and HPMC, with AA permeating 1.260 mg within 12 h. The current study estimated AA delivery of 3.21 mg within six h, with a flux value of 120.48 μg cm−2 h−1. The increased viscosity of the MNs in the Aung et al.34 investigation could explain the entrapped AA, indicating a lower AA content. The enhanced delivery of AA and ascorbic acid through the skin led to the low accumulation of AA and ascorbic acid in the skin. The hydrophilic characteristics of PVP-K90 and the moisture retention properties of HPMC resulted in the observed behavior. The results highlight the importance of using MN arrays for effective transdermal delivery of AA and ascorbic acid to ensure more effective penetration into the skin. The pH of the formulated MNs, which incorporate AA and ascorbic acid, aligns with the skin's natural pH range, ensuring their suitability for skin preparations. Wang et al. conducted a study on AA and ascorbic acid, focusing on their synergistic effects in pigmented brown guinea pigs while utilizing HPLC-UV to assess drug loading. Our study extends this work by incorporating thorough ex vivo permeation studies to investigate the behavior of the drugs during skin penetration. We employed the developed method of LC-MS, which allowed for a detailed assessment of the cumulative drug permeation over 24 hours.
Although the study successfully described the successful preparation and characterization of dissolving MN arrays, it had some limitations. First, polymer selection challenges: selecting appropriate polymers is critical and demanding. The polymers must be strong enough to form a three-dimensional structure that can penetrate the skin without causing severe pain. Second, ex vivo permeation studies demonstrated rapid and effective transport of AA and ascorbic acid, but these findings may only partially apply to in vivo conditions. Biological variability in live skin may have an impact on MN performance.
Conclusions
Herein, the development of commonly used whitening agents, AA, and ascorbic acid, was carried out in MN arrays, where this study successfully produced a formulation at a safe pH level for the skin. The drug-loaded AA and ascorbic acid MNs demonstrated sufficient mechanical properties at the force of 32 N, exhibiting an 8.481 ± 1.14% reduction of the tips, and could penetrate the third layer of the skin within one minute. The FTIR results provided strong evidence of the success of the formulation where broadband is observed around 3394 cm−1, which signifies the presence of hydrogen bonding between the polymers PVP and HPMC, and the drugs AA and ascorbic acid. The assessment of MB-MNs loading was conducted through visual inspection to confirm that the MN met the design specifications required for effective skin penetration. A complete dissolution of the MNs on the rat skin was documented at 5 min, indicating rapid dissolving of the MN arrays. The LC–MS method used for assessment was precise and accurate, and the evaluation was precise, offering a more reliable approach than traditional methods. Ex vivo permeation studies confirmed the fast and effective transport of AA and ascorbic acid within 24 h, where the cumulative amount was estimated to be 3.28 ± 0.13 mg for AA and 2.95 ± 0.11 mg for ascorbic acid. Overall, the study confirmed the successful development of a properly designed MN formulation for skin whitening.Data availability
This article includes all of the data used in its preparation. However, the authors make all of the data available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research grants for this study were awarded by the Scientific Research and Innovation Support Fund, Ministry of Higher Education, Jordan, MPH/1/17/2018 and Al-Zaytoonah University of Jordan (2023–2022/17/49).References
- Y. C. Boo, Antioxidants, 2021, 10, 1129 CrossRef CAS PubMed.
- W. Y. Jeong, M. Kwon, H. E. Choi and K. S. Kim, Biomater. Res., 2021, 25(1), 24 CrossRef PubMed.
- W. Zhang, W. Zhang, C. Li, J. Zhang, L. Qin and Y. Lai, Int. J. Mol. Sci., 2022, 23(5), 2401 CrossRef CAS PubMed.
- T. Liu, G. Jiang, G. Song, J. Zhu and Y. Yang, Biomed. Microdevices, 2020, 22, 1–11 CrossRef PubMed.
- J. C. Joyce, H. E. Sella, H. Jost, M. J. Mistilis, E. S. Esser, P. Pradhan, R. Toy, M. L. Collins, P. A. Rota, K. Roy, I. Skountzou, R. W. Compans, M. S. Oberste, W. C. Weldon, J. J. Norman and M. R. Prausnitz, J. Controlled Release, 2019, 304, 135 CrossRef CAS PubMed.
- Salwa, N. T. Chevala, S. R. Jitta, S. M. Marques, V. M. Vaz and L. Kumar, J. Drug Delivery Sci. Technol., 2021, 65, 102711 CrossRef CAS.
- K. Ahmed Saeed AL-Japairai, S. Mahmood, S. Hamed Almurisi, J. Reddy Venugopal, A. Rebhi Hilles, M. Azmana and S. Raman, Int. J. Pharm., 2020, 587, 119673 CrossRef CAS PubMed.
- K. Ahmed Saeed AL-Japairai, S. Mahmood, S. Hamed Almurisi, J. Reddy Venugopal, A. Rebhi Hilles, M. Azmana and S. Raman, Int. J. Pharm., 2020, 587, 119673 CrossRef CAS PubMed.
- A. Tucak, M. Sirbubalo, L. Hindija, O. Rahić, J. Hadžiabdić, K. Muhamedagić, A. Čekić and E. Vranić, Micromachines, 2020, 11(11), 961 CrossRef PubMed.
- X. Huang, Q. Chang, J. H. Gao and F. Lu, Tissue Eng., Part B, 2023, 29, 190–202 CrossRef CAS PubMed.
- D. Kulkarni, D. Gadade, N. Chapaitkar, S. Shelke, S. Pekamwar, R. Aher, A. Ahire, M. Avhale, R. Badgule, R. Bansode and B. Bobade, Sci. Pharm., 2023, 91, 27 CrossRef CAS.
- O. Tarawneh, A. M. Hammad, H. Abu Mahfouz, L. Hamadneh, R. Hamed, I. Hamadneh, A. Rasheed Al-Assi, H. A. Mahfouz, L. Hamadneh, R. Hamed, I. Hamadneh and A. R. Al-Assi, Cellul. Chem. Technol., 2023, 57, 117–124 CrossRef CAS.
- P. Franco and I. De Marco, Polymers, 2020, 12, 1114 CrossRef CAS PubMed.
- L. L. Tundisi, G. B. Mostaço, P. C. Carricondo and D. F. S. Petri, Eur. J. Pharm. Sci., 2021, 159, 105736 CrossRef CAS PubMed.
- J. He, Z. Zhang, X. Zheng, L. Li, J. Qi, W. Wu and Y. Lu, Pharmaceutics, 2021, 13, 579 CrossRef CAS PubMed.
- Y. S. Wang, W. H. Yang, W. Gao, L. Zhang, F. Wei, H. Liu, S. Y. Wang, Y. Y. Li, W. M. Zhao, T. Ma and Q. Wang, Eur. J. Pharm. Sci., 2021, 160, 105749 CrossRef CAS PubMed.
- Y. S. Wang, W. H. Yang, W. Gao, L. Zhang, F. Wei, H. Liu, S. Y. Wang, Y. Y. Li, W. M. Zhao, T. Ma and Q. Wang, Eur. J. Pharm. Sci., 2021, 160, 105749 CrossRef CAS PubMed.
- N. N. Aung, T. Ngawhirunpat, T. Rojanarata, P. Patrojanasophon, P. Opanasopit and B. Pamornpathomkul, AAPS PharmSciTech, 2020, 21, 1–13 CrossRef PubMed.
- P. Sawutdeechaikul, S. Kanokrungsee, T. Sahaspot, K. Thadvibun, W. Banlunara, B. Limcharoen, T. Sansureerungsikul, T. Rutwaree, M. Oungeun and S. Wanichwecharungruang, Sci. Rep., 2021, 11(1), 24114 CrossRef CAS PubMed.
- M. Xing, S. Zhang, G. Yang, Z. Zhou and Y. Gao, Eur. J. Pharm. Sci., 2021, 165, 105935 CrossRef CAS PubMed.
- H. Yang, G. Kang, M. Jang, D. J. Um, J. Shin, H. Kim, J. Hong, H. Jung, H. Ahn, S. Gong, C. Lee, U. W. Jung and H. Jung, Pharmaceutics, 2020, 12, 1067 CrossRef CAS PubMed.
- J. Drelich, E. Chibowski, D. D. Meng and K. Terpilowski, Soft Matter, 2011, 7, 9804–9828 RSC.
- N. Kanlayakan, K. Kerdpol, C. Prommin, R. Salaeh, W. Chansen, C. Sattayanon and N. Kungwan, New J. Chem., 2017, 41, 8761–8771 RSC.
- J. J. Pitt, Clin. Biochem. Rev., 2009, 30, 19–34 Search PubMed.
- S. Abdella, F. Afinjuomo, Y. Song, R. Upton and S. Garg, Pharmaceutics, 2022, 14(3) DOI:10.3390/PHARMACEUTICS14030542.
- K. Peng, L. K. Vora, I. A. Tekko, A. D. Permana, J. Domínguez-Robles, D. Ramadon, P. Chambers, H. O. McCarthy, E. Larrañeta and R. F. Donnelly, J. Controlled Release, 2021, 339, 361–380 CrossRef CAS PubMed.
- S. Rojekar, L. K. Vora, I. A. Tekko, F. Volpe-Zanutto, H. O. McCarthy, P. R. Vavia and R. F. Ryan, Eur. J. Pharm. Biopharm., 2021, 165, 41–51 CrossRef CAS PubMed.
- I. A. Tekko, G. Chen, J. Domínguez-Robles, R. R. S. Thakur, I. M. N. Hamdan, L. Vora, E. Larrañeta, J. C. McElnay, H. O. McCarthy, M. Rooney and R. F. Donnelly, Int. J. Pharm., 2020, 586, 119580 CrossRef CAS PubMed.
- E. Sjöholm and N. Sandler, Int. J. Pharm., 2019, 564, 117–123 CrossRef PubMed.
- E. Larrañeta, R. E. M. Lutton, A. D. Woolfson and R. F. Donnelly, Mater. Sci. Eng., R, 2016, 104, 1–32 CrossRef.
- E. Larrañeta, J. Moore, E. M. Vicente-Pérez, P. González-Vázquez, R. Lutton, A. D. Woolfson and R. F. Donnelly, Int. J. Pharm., 2014, 472, 65–73 CrossRef PubMed.
- L. K. Vora, R. F. Donnelly, E. Larrañeta, P. González-Vázquez, R. R. S. Thakur and P. R. Vavia, J. Controlled Release, 2017, 265, 93–101 CrossRef CAS PubMed.
- U. Nagra, K. Barkat, M. U. Ashraf and M. Shabbir, Dose-Response, 2022, 20, 1–21 CrossRef PubMed.
- N. N. Aung, T. Ngawhirunpat, T. Rojanarata, P. Patrojanasophon, P. Opanasopit and B. Pamornpathomkul, AAPS PharmSciTech, 2020, 21, 1–13 CrossRef PubMed.
- D. Krnáč, K. Reiffová and B. Rolinski, J. Chromatogr. B:Anal. Technol. Biomed. Life Sci., 2019, 1128, 121772 CrossRef PubMed.
- M. Hailat, I. Al-Ani, Z. Zakareia, R. Al-Shdefat, O. Al-Meanazel, M. K. Anwer, M. Hamad, W. Abu Rayyan, R. Awad and W. Abu Dayyih, Separations, 2022, 9, 303 CrossRef CAS.
- I. A. Tekko, G. Chen, J. Domínguez-Robles, R. R. S. Thakur, I. M. N. Hamdan, L. Vora, E. Larrañeta, J. C. McElnay, H. O. McCarthy, M. Rooney and R. F. Donnelly, Int. J. Pharm., 2020, 586, 119580 CrossRef CAS PubMed.
- J. Chen, X. Liu, S. Liu, Z. He, S. Yu, Z. Ruan and N. Jin, Drug Dev. Ind. Pharm., 2021, 47, 1578–1586 CrossRef CAS PubMed.
- A. Zaid Alkilani, B. Musleh, R. Hamed, L. Swellmeen and H. A. Basheer, J. Funct. Biomater., 2023, 14, 57 CrossRef CAS PubMed.
- Q. K. Anjani, A. H. Bin Sabri, E. Utomo, J. Domínguez-Robles and R. F. Donnelly, Mol. Pharmaceutics, 2022, 19, 1191–1208 CrossRef CAS PubMed.
- C. Soujanya, B. Lakshmi Satya, M. Lokesh Reddy, K. Manogna, P. Ravi Prakash and A. Ramesh, Int. J. Pharmacol. Pharm. Sci., 2014, 6, 282–286 CAS.
- K. M. El-Say, Drug Des., Dev. Ther., 2016, 10, 825–839 CrossRef CAS PubMed.
- K. Suknuntha, D. S. Jones and V. Tantishaiyakul, ScienceAsia, 2012, 38, 188–195 CAS.
- S. Biswal, J. Sahoo and P. N. Murthy, AAPS PharmSciTech, 2009, 10, 329 CrossRef CAS PubMed.
- S. Bhatnagar, N. G. Bankar, M. V. Kulkarni and V. V. K. Venuganti, Int. J. Pharm., 2019, 556, 263–275 CrossRef CAS PubMed.
- U. Chiriac, H. Rau, O. R. Frey, A. C. Röhr, S. Klein, A. L. Meyer and B. Morath, Antibiotics, 2022, 11(5), 541 CrossRef CAS PubMed.
- Z. Alkather, M. Hailat, R. Al-Shdefat and W. Abu Dayyih, Curr. Pharm. Anal., 2020, 17, 1263–1271 CrossRef.
- A. Guiraldelli, Ich, 2018, 1 Search PubMed.
- J. S. LaFountaine, L. K. Prasad, C. Brough, D. A. Miller, J. W. McGinity and R. O. Williams, AAPS PharmSciTech, 2015, 17, 120 CrossRef PubMed.
- J. Brouwers, M. E. Brewster and P. Augustijns, J. Pharm. Sci., 2009, 98, 2549–2572 CrossRef CAS PubMed.
- I. C. Lee, Y. C. Wu, S. W. Tsai, C. H. Chen and M. H. Wu, RSC Adv., 2017, 7, 5067–5075 RSC.
- H. Kathuria, D. Lim, J. Cai, B. G. Chung and L. Kang, ACS Biomater. Sci. Eng., 2020, 6, 5061–5068 CrossRef CAS PubMed.
- Y. Li, X. Hu, Z. Dong, Y. Chen, W. Zhao, Y. Wang, L. Zhang, M. Chen, C. Wu and Q. Wang, Eur. J. Pharm. Sci., 2020, 151, 105361 CrossRef CAS PubMed.
- M. I. Nasiri, L. K. Vora, J. A. Ershaid, K. Peng, I. A. Tekko and R. F. Donnelly, Drug Delivery Transl. Res., 2022, 12, 881–896 CrossRef CAS PubMed.
- E. Larrañeta, J. Moore, E. M. Vicente-Pérez, P. González-Vázquez, R. Lutton, A. D. Woolfson and R. F. Donnelly, Int. J. Pharm., 2014, 472, 65–73 CrossRef PubMed.
- E. Sjöholm and N. Sandler, Int. J. Pharm., 2019, 564, 117–123 CrossRef PubMed.
- A. Prabhu, J. Jose, L. Kumar, S. Salwa, M. Vijay Kumar and S. M. Nabavi, AAPS PharmSciTech, 2022, 23, 1–12 CrossRef PubMed.
- S. J. Kirkpatrick, R. K. Wang, D. D. Duncan, M. Kulesz-Martin and K. Lee, Opt. Express, 2006, 14, 9770 CrossRef PubMed.
- M. Cario, C. Pain, P. Kaulanjan-Checkmodine, D. Masia, G. Delia, V. Casoli, P. Costet, J. F. Goussot, V. Guyonnet-Duperat, A. Bibeyran, K. Ezzedine, C. Reymermier, V. Andre-Frei and A. Taieb, Pigm. Cell Melanoma Res., 2020, 33, 435–445 CrossRef CAS PubMed.
- Q. K. Anjani, A. H. Bin Sabri, E. Utomo, J. Domínguez-Robles and R. F. Donnelly, Mol. Pharmaceutics, 2022, 19, 1191–1208 CrossRef CAS PubMed.
- G. Yao, G. Quan, S. Lin, T. Peng, Q. Wang, H. Ran, H. Chen, Q. Zhang, L. Wang, X. Pan and C. Wu, Int. J. Pharm., 2017, 534, 378–386 CrossRef CAS PubMed.
- F. Eugelio, S. Palmieri, F. Fanti, L. Messuri, A. Pepe, D. Compagnone and M. Sergi, Molecules, 2023, 28, 1531 CrossRef CAS PubMed.
- F. Farouk, D. Wahba, S. Mogawer, S. Elkholy, A. Elmeligui, R. Abdelghani and S. Ibahim, Eur. J. Drug Metab. Pharmacokinet., 2020, 45, 89–99 CrossRef CAS PubMed.
- S. Imre, A. Tero-Vescan, M. T. Dogaru, L. Kelemen, D. L. Muntean, A. Curticǎpean, N. Szegedi and C. E. Vari, J. Chromatogr. Sci., 2019, 57, 243–248 CAS.
- K. T. K. Reddy, K. Gandla, P. V. Babu, M. V. K. Chakravarthy, P. Chandrasekhar and R. Sagapola, J. Pharm. Negat. Results, 2022, 13, 16–27 CAS.
- U. Chiriac, H. Rau, O. R. Frey, A. C. Röhr, S. Klein, A. L. Meyer and B. Morath, Antibiotics, 2022, 11(5), 541 CrossRef CAS PubMed.
- O. López-fernández, R. Domínguez, M. Pateiro, P. E. S. Munekata, G. Rocchetti and J. M. Lorenzo, Antioxidants, 2020, 9(6), 479 CrossRef PubMed.
- Y. H. Wang, C. Avonto, B. Avula, M. Wang, D. Rua and I. A. Khan, J. AOAC Int., 2015, 98, 5–12 CrossRef CAS PubMed.
- R. Neupane, S. H. S. Boddu, J. Renukuntla, R. J. Babu and A. K. Tiwari, Pharmaceutics, 2022, 12(2), 152 CrossRef PubMed.
- E. Abd, S. A. Yousef, M. N. Pastore, K. Telaprolu, Y. H. Mohammed, S. Namjoshi, J. E. Grice and M. S. Roberts, Clin. Pharmacol., 2016, 8, 163 CAS.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS PubMed.
- S. F. Ng, J. J. Rouse, F. D. Sanderson, V. Meidan and G. M. Eccleston, AAPS PharmSciTech, 2010, 11, 1432–1441 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |