
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
YAP/TAZ cytoskeletal remodelling is driven by mechanotactic and electrotactic cues†
Bernadette
Basilico‡
a,
Maddalena
Grieco‡
 b,
Stefania
D’Amone
c,
Ilaria Elena
Palamà
c,
Clotilde
Lauro
a,
Pamela
Mozetic
c,
Alberto
Rainer
d,
Simone
de Panfilis
e,
Valeria
de Turris
e,
Giuseppe
Gigli
cf and
Barbara
Cortese
*b
b,
Stefania
D’Amone
c,
Ilaria Elena
Palamà
c,
Clotilde
Lauro
a,
Pamela
Mozetic
c,
Alberto
Rainer
d,
Simone
de Panfilis
e,
Valeria
de Turris
e,
Giuseppe
Gigli
cf and
Barbara
Cortese
*b
aDepartment of Physiology and Pharmacology, Sapienza University, 00185 Rome, Italy
bNational Research Council – Institute of Nanotechnology (CNR Nanotec), c/o Department of Physics “E. Fermi”, University Sapienza, Pz.le Aldo Moro 5, 00185, Rome, Italy. E-mail: barbara.cortese@nanotec.cnr.it
cNational Research Council – Institute of Nanotechnology (CNR Nanotec), c/o Ecotekne, University of Salento, Via Monteroni, 73100, Lecce, Italy
dDepartment of Engineering, University Campus Bio-Medico of Rome, 00128, Rome, Italy
eIstituto Italiano di Tecnologia (IIT), 00185, Rome, Italy
fDepartment of Medicina Sperimentale, University of Salento, Campus Ecotekne, Via Monteroni, 73100 Lecce, Italy
First published on 26th November 2024
Abstract
Cells respond dynamically to multiple cues in complex microenvironments, which influence their behaviour, function, and molecular pathways. Despite recent advances, understanding cell interactions in such environments remains challenging. While biophysical cues are recognized for interacting with mechano-transduction proteins like YAP/TAZ, their role under glioblastoma electrotaxis is unclear. Our study investigates the functional role of mechano-transduction proteins under a physiological electric field (EF) with different rigidities. EF exposure highlights rigidity-dependent responses involving focal adhesion, cytoskeletal remodelling and YAP/TAZ coactivators relocation, showing to induce a shuttling in a rigidity-dependent manner. Further inhibition of PI3K/Akt and pharmacologically disrupting YAP/TAZ-TEAD interaction was shown to induce marked cytoskeletal remodelling under EFs. Our work characterises the therapeutic opportunities and limitations of EFs and uncovers the intricate interplay of physical cues and molecular signalling pathways in glioblastoma, offering potential insights for the development of therapeutic interventions in the future.
Introduction
Cells respond to various stimuli from the extracellular microenvironment, ranging from physical cues and constraints in forces and mechanics to chemical and electrical cues.1–3 Yes-associated protein (YAP) and its transcriptional coactivator with PDZ-binding motif (TAZ, also called WWTR1), are well-renowned transcriptional coactivator effectors that connect cellular mechanics and signalling cascades underlying gene expression, cell proliferation, and differentiation.4 YAP localisation is crucial for its function and becomes transcriptionally active when it is nuclear.5,6 From a canonical biochemical point of view, YAP is regulated by the Hippo signalling pathway.7 Core components of the Hippo pathway, such as MST1/2 and LATS1/2, promote YAP phosphorylation at serine 127 (Ser127), leading to its cytoplasmic sequestration and inhibition of coactivation with TAZ. Moreover, AKT-mediated phosphorylation of YAP at Ser127 facilitates its binding to 14-3-3, thereby retaining it in the cytoplasm. YAP/TAZ activity is also regulated by various upstream factors, including changes in the cytoskeletal tension, and cell shape.8,9 From a mechanical perspective, in a neoplastic setting, alterations in the microenvironment can induce aberrant mechanical signals that impact YAP/TAZ activity, primarily mediated by the tension and organisation of the F-actin cytoskeleton through Rho family small GTPases.6,10 Actin polymerisation induced by F-actin nucleator overexpression correlates with YAP/TAZ activation.11 Myosin contractility and actin-severing and -capping proteins respectively enhance and reduce YAP nuclear localisation,6,12 while force-induced unfolding of talin promotes YAP nuclear translocation.13,14 Despite these findings, the specific contributions of these molecular elements and the precise mechanisms by which mechanical signals regulate YAP is far from understood.Aberrant YAP localisation has been linked to glioblastoma (GBM) tumorigenesis.4,15 GBMs prominent infiltrative behaviour has prompted focus of research into arresting or slowing its motility, to facilitate more successful localised treatment. GBM migration is a tightly regulated process involving dynamic cytoskeletal rearrangements mainly engaging focal adhesion kinases (FAK) and integrins16 while recruiting arrays of intracellular proteins, (i.e., PXN, paxillin and VCL, vinculin).17 In addition, the GBMs invasive behaviour is closely linked to its ability to perceive and respond to various cues within the extracellular microenvironment.18,19 Integral to the GBM microenvironment is the interaction with the synaptic and electrical activity of neural circuits which have been shown to generate small physiological electric fields (EF). EFs in the range of 0.1 V cm−1 are predicted to arise in the developing amphibian brain regions,20 while EFs up to 2 V cm−1 have been measured in the injured cornea.21 EFs also have been shown to occur physiologically due to ionic and voltage gradients, which are established through the spatial distribution of ion channels, pumps, and leaks.22 EF Studies reported on GBM have primarily focused on the mechanistic aspects of electrotaxis, with relatively few investigations into its broader implications. The influence of the EF (from 1–5 V m−1) on the migration direction, migration rate, and orientation showed to be closely related to EF strength, species, cell types, or microenvironments.3,22 For example, neural lineage and GBM cells prefer to migrate from anode to cathode,23,24 whereas hiPSCs25 and chicken Schwann cells report an anodic response to EFs.26 However, research on the electrotaxis of GBM remains limited and the underlying mechanisms driving this EF response were not explored further. The occurrence that substrate or plating conditions can significantly alter the electrotactic response underscores the need for more comprehensive studies.
Herein, we investigated the application of low field-strength direct current, a regime in which electrotaxis occurs, to evaluate GBM cells’ response within substrates of different stiffnesses showing that the electrotactic directional response of cells can be regulated in a rigidity-dependent manner. Exanimation of various aspects of cell behaviour, such as viability, morphology, adhesion, motility, and mitochondrial dynamics related to the mechanosensitive proteins, identified the contribution of the EF in regulating YAP/TAZ localisation and activation by the modulation of actin stress fibre assembly. Exploration of genes linked to the cytoskeleton, extracellular matrix, and focal adhesions (FAs) reveals that the EF plays a pivotal role in hindering TEAD expression, overriding the mechanotactic effects. The results of this research will lead to an increased understanding of how electric fields interact with mechanical signalling, offering potential insights for the development of GBM therapeutic interventions in the future.
Results
Electric and mechanotactic cues regulate cytoskeletal and FA remodelling
To examine the overriding effects of the mechanotactic and electric cues, we cultured GBM cell lines (U251-MG and GL15) onto custom galvanotaxis chips with different substrate stiffness, using Petri dishes (referred herein to as stiff, with a Young's Modulus of 2.28–3.28 GPa19,27) and PDMS with a crosslinking ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of 5–10 kPa,18,19,28 obtained as previously described1,18,19 (referred to as soft). First, we investigated whether the variation of rigidity and/or exposure to an EF of 200 mV mm−1 affected either cell morphology which was quantified at the beginning and at the end of each experiment (i.e., after 3 h of EF exposure) to verify the influence of EF on each cell. A substantial change in the whole cell morphology in response to a change of substrate rigidity was observed (in Fig. S1A and B, ESI†). The area of both cell lines was smaller on soft substrates compared to stiff. However, after EF stimulation, both cell lines cultivated on soft substrates reported an increased whole cell area (Fig. S1A and B, ESI†). Moreover, we observed that exposure to an EF of the cells on soft substrates led to an increased circularity (Fig. S1C and D, ESI†). Of note, the change of morphology and shape of cells cultivated on stiff substrates was cell line dependent. Viability, assessed through a live/dead assay (Fig. S1E and F, ESI†) showed no changes regardless of the rigidities of the substrates and either with or without exposure to an EF.
:1 of 5–10 kPa,18,19,28 obtained as previously described1,18,19 (referred to as soft). First, we investigated whether the variation of rigidity and/or exposure to an EF of 200 mV mm−1 affected either cell morphology which was quantified at the beginning and at the end of each experiment (i.e., after 3 h of EF exposure) to verify the influence of EF on each cell. A substantial change in the whole cell morphology in response to a change of substrate rigidity was observed (in Fig. S1A and B, ESI†). The area of both cell lines was smaller on soft substrates compared to stiff. However, after EF stimulation, both cell lines cultivated on soft substrates reported an increased whole cell area (Fig. S1A and B, ESI†). Moreover, we observed that exposure to an EF of the cells on soft substrates led to an increased circularity (Fig. S1C and D, ESI†). Of note, the change of morphology and shape of cells cultivated on stiff substrates was cell line dependent. Viability, assessed through a live/dead assay (Fig. S1E and F, ESI†) showed no changes regardless of the rigidities of the substrates and either with or without exposure to an EF.
As FAs play crucial roles in mechano-transduction and regulate processes such as spreading, proliferation, differentiation, and motility and considering the association of electrically induced morphological changes with actin cytoskeleton reorganisation,29 it was questioned whether EF would affect FAs formation using PXN-GFP expressed cells. Confocal imaging (Fig. 1(A)) showed that the number of FAs and adhesion area for U251-MG noticeably increased on soft substrates when exposed to an EF (Fig. 1(B) and (C)). On the contrary, GL15 cells exposed to an EF showed an increase in the number of FAs as well as an increased adhesion area, on substrates of higher rigidity (Fig. S2A–C, ESI†).
 | ||
| Fig. 1 Mechanotactic and electric cues compete in regulating morphology and FA formation. (A) Representative confocal analysis of U251 cells expressing PXN-GFP (green) plated onto substrates of increasing stiffness with and without the effect of a 3 h EF treatment. Scale bar 20 μm. (B) and (C) Bar graphs showing quantification of the number of FAs per cell (FA number) and (F) total FA area. (Control: n = 12 for stiff, n = 11 for soft substrates, EF: n = 10 for stiff, n = 10 for soft substrates for 3 different technical replicates). (D)–(F) PXN, FAK, and ITGAV expression in U251-MG cells were validated by qPCR. Values are expressed as means ± S.E.M., for at least 3 different technical replicates. Significance was determined using two-way ANOVA followed by Tukey's multiple comparisons test with *p < 0.05, **p < 0.01, ***p < 0.005. | ||
Quantitative PCR (RT-qPCR) analysis of the FA genes in U251-MG cells revealed a rigidity dependence of PXN and FAK which were downregulated on soft substrates upon EF exposure (Fig. 1(G) and (H)). As different integrins are involved in adhesion and migration we further investigated expression levels of ITGAV on all substrates, observing a further decrease in the expression levels regardless of stiffness, when applying an EF, (Fig. 1(I)). This is consistent with FAK downregulation, as FAK activation typically occurs upon integrin binding to the extracellular matrix, initiating downstream signal transduction. Also, the downregulation of VCL upon EF exposure only on soft substrates was noted (Fig. S3A, ESI†). However, GL15 cells didn’t report significant changes of the FA genes levels (Fig. S3B, ESI†).
Overall, our data suggested that EF inhibited new FAK gene expression, yielding a change of cytoskeletal tension through regulation of FA formation and integrin signalling in a cell-dependent manner.
Electric fields regulate cell motility in response to substrate stiffness
As the decreased gene expression suggested a restrained turn-over in actin filament rearrangements, intrinsic in cell migration, to further study the influence of the microenvironment GBM cells electrotaxis, we tracked and analysed the motility of cells in response to the different electro/mechano-stimuli. Exposure to an EF led to a decrease of cell motility, regardless of substrate stiffness (Fig. 2(A) and Fig. S4A, ESI†) for both cell lines. Moreover, consistent with previous reports,30 both cells changed directionality in accordance to substrate stiffness, showing a cathodic response on stiff substrates, whereas an anodic directional response was observed on soft substrates (Fig. 2(B) and Fig. S4B, ESI†). | ||
| Fig. 2 The EF regulates cell migration overriding mechanotactic cues. (A) The EF influences cell motility leading to a reduced motility in U251-MG cells on all substrates. (B) Individual U251-MG cell's FMI distribution under EF exposure shows rigidity dependency under EF exposure with cells being cathodic on stiff substrates and anodic on soft, (untreated: n = 79 for stiff, n = 83 for soft substrates; EF: n = 114 for stiff, n = 105 for soft substrates). (C)–(E) Normalised motility values of U251-MG-PI3K-inhibited cells to untreated cells on their respective substrates, showing path length (C) and motility (D), with (+) and without (−) an EF, and their FMI distribution under an EF (E) showing that PI3K-inhibited cells reverted their directionality on stiff substrates from cathodic to anodic (PI3K treated cells without EF: n = 97 for stiff, n = 97 for soft substrates; PI3K treated cells with EF: n = 114 for stiff, n = 74 for soft substrates). (F)–(H) Gene expression levels of PXN (F), FAK (G) and ITGAV (H) were determined using qPCR. Data are from at least three independent experiments, two-way ANOVA followed by Tukey's multiple comparisons test, ***p < 0.001, and unpaired two-tailed Mann–Whitney U test was run between each pair of groups, *p < 0.05. Values are expressed as means ± S.E.M. | ||
As the phosphoinositide-3-kinase (PI3K)–Akt pathway has been shown to act as key player in the regulation of GBM motility31 and electrotactic responses,32–34 we used the inhibitor (wortmannin) to investigate the interdependency of intracellular signalling mechanisms on the mechano/electrotaxis of the microenvironment. When we examined cell movement of PI3K-inhibited U251-MG cells without EF exposure, we observed a reduced path length (Fig. 2(C)) and cell migration (Fig. 2(D)) by approximately 50% on stiff and about 20% on soft substrates compared to untreated cells (100%). Upon EF exposure, a further reduction in motility was observed only on soft substrates, with no significant difference on stiff. Conversely, GL15 cells solely treated with the PI3K inhibitor and in the absence of an EF, showed a more pronounced decrease in path length and motility on soft substrates compared to stiff. In contrast, exposure to an EF further decreased motility on stiff substrates (Fig. S4C and D, ESI†). These data indicate that the effects of wortmannin on cell motility differ depending on cell type as well as the substrate rigidity. Surprisingly, both cell lines PI3K-inhibited reverted their migration direction from cathode to anode on stiff substrates, indicating that PI3K regulates the direction of migration under EF (Fig. 2(E) and Fig. S4E, ESI†).
As the PI3K–Akt pathway contributes to FAs and stress fibre formation mediated by actomyosin contractility,35 we checked mRNA expression levels of the genes involved in the regulation of movement. Exposure of both PI3K-inhibited cell lines to an EF, conveyed a tendency of the PXN expression to be downregulated, whereas FAK was upregulated (Fig. 2(F)–(G) and Fig. S4F–G, ESI†). However, inhibition of PI3K highlighted a cell type-dependent effect on ITGAV gene expression. Specifically, ITGAV showed a tendency to be upregulated in U251-MG cells (Fig. 2(H)) and downregulated in GL15 cells (Fig. S4H, ESI†), regardless of substrate rigidity, suggesting that EF exposure might trigger activation and recruitment of cell-type specific alternative pathways in modulating migration.
These results suggested that the EF modulates the expression of cell adhesion proteins by activating the PI3K signalling pathway necessary for triggering a cell-dependent transcriptional feedback mechanism which modulates cytoskeletal tension and FA dynamics to enable anodic migration.
YAP localisation and phosphorylation are regulated by exposure to an EF
As increasing evidence has identified the transcriptional coactivators, YAP and TAZ, as cytoskeletal tension-activated regulators of gene expression,6 we next determined whether the EF could influence YAP/TAZ localisation in U251-MG and GL15 cells. Without EF exposure, cell lines displayed canonical YAP localisation, showing to be nuclear on stiff substrates and cytoplasmatic on soft.36 Interestingly, exposure to an EF triggered YAP shuttling showing opposite effects with a higher YAP distribution throughout the cytoplasm within cells on stiff substrates, whereas it appeared predominantly localised to the nuclei on soft (Fig. 3(A), (B) and Fig. S5A, B, ESI†). These results reveal that the EF acts on the contractile stress of the cell on both soft and stiff substrates, with YAP relocalisation. As YAP localisation has been shown to be regulated by nuclear compression,37 we then questioned if the electro/mechano-stimuli impacted cell nuclei. We, thus, quantified the nuclear volume using confocal Z-stacks for cells on the different substrates and w/wo EF exposure. We observed an increase of the nuclear volume for both cells on stiff substrates (Fig. 3(C) and Fig. S5C, S6, S7, ESI†), whereas EF led to an inverse relationship on soft substrates, showing an increased nuclear volume for U251-MG and a decreased volume for GL15. | ||
| Fig. 3 YAP nuclear shuttling is determined by EF and substrate rigidity. (A) Representative confocal images of U251-MG cells cultured on stiff and soft substrates depicting YAP localisation with/without exposure to an EF of 2 V cm−1. Cells were stained with anti-YAP (green). Scale bar 25 μm. (B) Quantification of the YAP nuclear/cytoplasmic ratio in response to substrate rigidity and exposure to an EF. (Untreated: n = 40 for stiff, n = 41 for soft substrates, EF: n = 38 for stiff, n = 28 for soft substrates, from at least 3 technical replicates). (C) Quantification of nuclear volume of U251-MG cells on substrates of different stiffness and w/wo EF (untreated: n = 63 for stiff, n = 64 for soft substrates, EF: n = 64 for stiff, n = 66 for soft substrates, from at least 3 technical replicates). (D) Representative western blot membranes of YAP, TAZ, p-YAPSer127 and GAPDH protein expression of U251-MG cells with (+) and without (−) an EF and with (+) and without (−) treatment with the PI3K inhibitor (Wortm = wortmannin), cultured on substrates of different rigidity. (E)–(H) Bar graphs showing the protein quantification of p-YAPSer127 (E) and YAP (F) of protein expression in U251-MG cells untreated cells cultured on different substrates and w/wo EF and p-YAPSer127 (G) and YAP (H) of protein expression in U251-MG wortmannin-treated cells. Data are normalised to GAPDH expression from at least 3 technical replicates, p-values calculated with the unpaired two-tailed Mann–Whitney U test, *p < 0.05; **p < 0.01. (I) RT-qPCR quantification of the canonical YAP target gene CTGF in U251-MG cells with and without PI3K-inhibition cultured on substrates of different rigidity and w/wo exposure to an EF of 2 V cm−1. Gene expression levels were normalised to GAPDH in all RT-qPCR experiments from at least 3 technical replicates. Three-way ANOVA followed by Tukey's post hoc test for multiple comparisons. Data are expressed as means± S.E.M. | ||
Since YAP localisation is also determined by its phosphorylation state, we investigated whether YAP phosphorylation varied after EF exposure. Western blots showed that U251-MG cells exposed to an EF reported enhanced p-YAPSer127 expression on all substrates, especially on stiff (Fig. 3(D)–(F)). In line with this observation, we noted that YAP expression was significantly reduced upon EF exposure on stiff (Fig. 3(F)). Similarly, GL15 cells upon EF exposure showed YAP reduction on stiff substrates (Fig. S5E and F, ESI†).
Our data suggested that the EF specifically affects the activity and distribution of YAP/TAZ with changes of the nuclear volume, in glioma cell lines in vitro in a cell type-dependent manner leading to changes in YAP mechanotransduction.
YAP/TAZ activity modulation by the EF is independent of the PI3K pathway
Crosstalk between the Hippo and PI3K-TOR pathways has identified YAP activation/dephosphorylation as a downstream target of PI3K.38 Thus, we examined whether the observed changes in YAP expression levels induced by an EF are PI3K-dependent. Exposure to an EF was shown to increase p-YAPSer127 expression within PI3K-inhibited cells (Fig. 3(D), (G) and Fig. S5D, E, ESI†), especially on soft substrates and a tendency to decrease YAP/TAZ protein expression (Fig. 3(D), (H) and Fig. S5D, F, ESI†).To investigate if the PI3K pathway modulated a feedback loop via transcriptional targets of YAP, we checked CTGF mRNA expression compared with control treatment, as determined by RT-PCR. Without inhibition, the effect of the EF was a significant downregulation of the CTGF levels in U251-MG cells. However, under PI3K inhibition we observed a tendency to increase mRNA levels of CTGF regardless of the substrate rigidity and the presence of EF of both inhibited cell lines (Fig. 3(I) and Fig. S5G, ESI†). Taken together, these results suggest that the YAP/TEAD transcriptional activity is activated through pathways dependent from the PI3K, independent from EF, reinforcing the hypothesis that exposure to an EF increases strain in the cell cytoskeletal structure.
YAP inhibition reduces GBM motility and is dependent on substrate rigidity
Since the nuclear YAP/TAZ binds to the TEAD family of transcription factors to activate the expression of pro-proliferative and survival-enhancing gene programs, we assessed the effect of EF exposure on the YAP/TAZ-TEAD transcriptional activation. We pharmacologically inhibited the YAP/TAZ-TEAD axis by treating cells with SuperTdu (ST) and Verteporfin (VP), and analysed cell responses with and without EF application. ST is a polypeptide which mimics the function of a previously identified YAP antagonist (VGLL4) to disrupt the YAP-TEAD interaction. VP, instead, is a porphyrinic photosensitizer which binds to the conserved TEAD interaction domain in YAP, disrupting YAP-TEAD binding, and inducing YAP/TAZ protein degradation without light activation.39 Thus, exposure of VP-treated cells to light was minimized and the motility of cells was analysed only under ST treatment to explore its potential role in cell invasiveness (Fig. 4(A)–(C)). Without EF exposure, we observed reduced migration trajectories in both ST-inhibited cell lines, more specifically only on stiff substrates, with decreased path length (Fig. 4(A)) and speed (Fig. 4(B)) of U251-MG cells (∼60%) cultured on stiff, and no effect on soft substrates. Conversely, EF exposure showed no further influence on the invasiveness parameters on stiff for U251MG (Fig. 4(A) and (B)), while it decreased them (∼80%) on soft. Moreover, directional movement of U251-MG cells under an EF, remained cathodic on stiff, and anodic on soft (Fig. 4(C)). GL15 behaved similarly (Fig. S8A and B, ESI†) but upon EF, GL15 acquired an anodic behaviour regardless of rigidity (Fig. S8C, ESI†). This suggested that the effect of ST was rigidity- and cell-dependent. | ||
| Fig. 4 YAP–TEAD interaction is required for GBM motility. (A)–(C) Motility parameters of U251-MG cells treated with ST. Data are normalised to their respective untreated cells, seeded on substrates of different rigidity and subjected to an EF of 2 V cm−1. Bar graphs showing the quantification of (A) path length, (B) instantaneous speed and (C) directionality of migration. Inhibited cells without EF: n = 75 for stiff, n = 67 for soft substrates; EF: n = 58 for stiff, n = 75 for soft substrates; from at least 3 different technical replicates, two-way ANOVA followed by Tukey's multiple comparisons test, *p < 0.05, **p < 0.01, ***p < 0.001, and unpaired two-tailed Mann–Whitney U test, **p < 0.01. (D) Representative western blot membranes of the indicated proteins after inhibition of U251-MG cells with ST and VP, and w/wo an EF. (E)–(G) Normalised quantification of protein levels, specifically p-YAPSer127 (E), YAP (F) and TAZ (G) compared to untreated cells on their respective substrates. Data are normalised to GAPDH expression from at least 3 technical replicates, two-way ANOVA followed by Tukey's post hoc test for multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001. Data are expressed as means ± S.E.M. (H) Heatmap visualisation of RT-qPCR analysis of mRNA levels of CYR61, CTGF, PXN, FAK, VCL and ITGAV expression in TEAD-inhibited U251-MG cells with (+) and without (−) EF. Relative quantification of each gene expression level was normalised according to the GAPDH gene expression. Columns represent treatments with (+) and without (−) EF and, of untreated (−), ST-inhibited (+) and VP-inhibited (+) U251-MG cells. Green represents downregulation, and red represents upregulation. Colour intensity represents the mRNA expression values. Rows, mRNA of the genes. | ||
Next, we examined the protein expression profiles of YAP and TAZ following the disruption of YAP-TEAD interaction. Western blot analysis of U251-MG inhibited cells showed that the effect of an EF was to increase the expression of p-YAPSer127 of inhibited cells (Fig. 4(D), (E) and Fig. S8D, E, ESI†). YAP and TAZ levels were decreased considerably under VP treatment with respect to ST (Fig. 4(D), (F), (G) and Fig. S8D, F, ESI†) especially on soft substrates.
mRNA levels of ST-treated U251-MG cells (Fig. 4(H)), without EF exposure, showed a reduction in the target genes CYR61 and CTGF genes on soft substrates. Exposure to an EF of ST-treated cells appeared to not affect inhibited U251-MG cells on stiff, but slightly affected those on soft, with a tendency to increase CTGF, FAK and ITGAV expression. VP treatment decreased CTGF, VCL and ITGAV gene levels without EF exposure regardless of the rigidity of the substrate (Fig. S8H, ESI†). Exposure to an EF, did not affect VP treated cells significantly. No significant variations were observed in GL15-inhibited cells (Fig. S8H, ESI†). These data indicate that the EF was not able to rescue YAP/TAZ transcriptional activity.
Electrotaxis overrides mechanotactic effects on cell mitochondria
Accumulating evidence suggests a potential correlation between YAP and mitochondrial structure, reporting an increased expression of YAP1 associated with decreased fragmentation of mitochondria40 and an increase in mitochondrial fusion.41 To investigate whether the presence of an EF could alter mitochondrial organisation and functions, we examined the spatial arrangement of the mitochondrial structure, quantifying individual and network size, fragment length and mitochondrial footprint of mitochcondria as previously described1,42 (Fig. 5(A)–(F)) Our analyses revealed EF exposure induced a more fragmented mitochondrial network of U251-MG cells (Fig. 5(B) and (C)) on softer substrates, and a structural change characterized by a decreased length of branching network and individual fragments (Fig. 5(D) and (E)) regardless the rigidity. In contrast, GL15 cells exhibited a longer length of branching network and fragments under the influence of an EF, suggesting a more fused and branched mitochondrial structure (Fig. S9A–E, ESI†). Further analysis of the overall mitochondrial organisation/structure highlighted stiffness-dependent changes in the mitochondrial footprint when cells were subjected to the EF. Specifically, on stiff substrates, EF reduced the size of the mitochondrial footprint, whereas it increased on soft for both cell lines (Fig. 5(F) and Fig. S9F, ESI†). | ||
| Fig. 5 Mitochondria are affected by mechanotactic and electrotactic cues. (A) Representative mitochondrial skeletonised structures, obtained from U251-MG cells cultured on substrates of different rigidity w/wo 3 h of EF (2 V cm−1). Scale bar 25 μm. (B)–(F) Bar graphs showing the quantification of (B) number of individuals, (C) number of networks, (D) mean summed branch lengths, (E) mean network branches and (F) mitochondrial footprint (control: n = 26 for stiff, n = 29 for soft substrates, EF: n = 10 for stiff, n = 29 for soft substrates for at least 10 different confocal acquisitions). (G) Analysis of mitochondrial membrane potential using JC-1 staining. Cells were exposed to EF on substrates of different rigidity and fluorescence transition from red to green was evaluated (control: n = 114 for stiff, n = 131 for soft substrates, EF: n = 133 for stiff, n = 176 for soft substrates). Data are from at least 3 independent experiments, two-way ANOVA followed by Tukey's multiple comparisons test, *p < 0.05, **p < 0.01, ***p < 0.001. Values are expressed as means ± S.E.M. | ||
Considering the close connection between the morphology of mitochondrial networks and the mitochondrial membrane potential (MMP), we analysed the effect of the EF on the MMP of cells cultured on substrates with different rigidities. We observed that on stiff, the EF induced the depolarisation of the mitochondrial membrane in U251-MG cells, whereas on soft substrates, the MMP increased upon EF application (Fig. 5(G)). In contrast, GL15 cells showed the opposite outcome, with the EF triggering mitochondrial depolarisation on soft substrates and an increased MMP on stiff (Fig. S9G, ESI†).
Overall, these results suggest that EF plays a regulatory role in both YAP and mitochondrial dysfunction, influencing changes in mitochondrial membrane potential and fragmentation. Importantly, these effects are also cell-type dependent.
Discussion
Different studies have examined how the EF application can affect a variety of cellular processes, including migration, proliferation, and directionality.3,43 It's worth noting that the differing migration directions—cathodic, for example for keratinocytes, and anodic, for example for fibroblasts— are well-documented behaviours and several hypotheses have been proposed to explain the directional choice of these cells, including calcium signalling pathways, PI3K, transforming growth factor-β3 (TGF-β3), Golgi polarization, and integrin expression.44–46 However, a clear consensus on the underlying mechanisms remains elusive. Although a few findings have provided insights into the molecular mechanism responsible for the modulation of galvanotaxis in the presence of different migration cues, related to FAs,47 matrix stiffness48 and the surface charge of the substrate,49 we show for the first time the interrelation between YAP/TAZ along with their correlation with direct EFs (Fig. 6). | ||
| Fig. 6 Model depicting the effect of mechanotactic and electrotactic cues on YAP/TAZ function. Without exposure to EFs, the canonical YAP/TAZ pathway shows that YAP/TAZ are activated by enhanced substrate stiffness and translocate to the nucleus. Upon exposure to EFs, the Hippo signalling pathway is activated, TEAD transcription is inhibited and a nuclear/cytoplasm shuttling of YAP/TAZ is observed with promotion of dephosphorylation phosphorylation at Ser127. Grey mechanistic elements, dotted lines and circles, serve to suggest possible connections and represent mechanisms which may interfere in the scenario of YAP/TAZ regulation. | ||
Moreover, prior studies are mainly based on electrotactic chambers on Petri or glass substrates. Here we show a significant difference between stiff (Petri) and soft substrates, highlighting the importance of the cell–substrate interface, rather than considering substrate compliance alone.
In general cells on soft substrates have YAP mostly localised in the cytoplasm, whereas cells on stiff substrates mediate YAP nuclear translocation, as we observed. Yet, exposure to an EF, caused an increase of dephosphorylation of the YAP protein, regardless of the stiffness, with the disruption of the YAP/TEAD interaction and the transcriptional dysregulation. While we observed that exposure to EFs was shown to reduce the cell area on stiff substrates and to increase it on soft substrates, it appeared, also, to increase the circularity of cells, with an elevation of FAs under EF, especially on soft substrates (Fig. 1). This phenomenon is likely associated with the maturation of newly formed focal contacts anchored to the substrate. These mature focal contacts impede cell retraction and facilitate the decoupling of YAP activation from cell spreading. We also detected that exposure to an EF caused YAP to translocate from the cytoplasm to the nucleus on soft substrates, whereas, on stiffer substrates, we noted a marked cytoplasmic redistribution of nuclear YAP (Fig. 2). Our results align with previous findings investigating YAP localisation in relation to nuclear compression induced by cell strain energy or extracellular forces. Nuclear flattening caused by external mechanical forces augmented increased nuclear entry of YAP.13,37 Moreover, the external stimuli were shown to induce a matrix remodelling involving dephosphorylation of the YAP/TAZ and shuttling from the cytoplasm to the nucleus.6 Phosphorylated YAP located in the cytoplasm was shown to interact with the cytoskeleton, counteracting its entry into the nucleus and gradually getting degraded in the cytoplasm.50 We hypothesise that the change in protein location we observed under EF was attributed to the remodelling of the cytoskeleton, suggesting that this translocation was mediated by forces transmitted to the nucleus.
Furthermore, YAP/TAZ has been shown to transcriptionally regulate the architecture of the actin cytoskeleton, facilitating persistent cell motility. Previous studies showed that knockdown of YAP decreases proliferation and migration in human GBM cell lines in vitro.51 This effect may be linked to YAP's regulatory role in actin cytoskeletal dynamics, achieved through its impact on the transcription of the Rho GTPase-activating protein (Rho-GAP).52 This regulation is characterized by an increase in stress fibres and FA maturation.53 Our results corroborate these findings, as we observed that the migration behaviour of GBM cells upon EF exposure was decreased regardless of the stiffness, with a decreased YAP/TAZ protein expression.
Low-frequency EFs were shown to activate YAP and associated pathways, potentially by changing the cell membrane environment or cytoskeletal organization.54–56 For example, Jia and co-workers investigated YAP/TAZ expression of macrophages using alternating current (AC) EFs.54 They observed a higher YAP/TAZ expression under the effect of the EF, suggesting that AC-EFs might promote M2 polarisation in macrophages by activating YAP/TAZ-mediated signalling pathways. Abasi and his group reported an increased YAP nuclear localisation in HUVEC cells due to the application of pulsed electric fields, which mimic mechanical forces like shear stress.55 This nuclear partitioning indicated YAP activation, which influenced cellular responses like proliferation and tight junction formation and was similar to the behaviour observed under mechanical stresses, such as those involving substrate stiffness or fluid shear. Colciago56 reported the use of electromagnetic fields showing a YAP relocation from nuclear to the cytoplasmatic and its downregulation, consistent with our findings. Since EFs are also linked to cytoskeletal reorganization and the regulation of actin dynamics, they create conditions that reinforce YAP's active, growth-promoting role in the nucleus. Thus, EFs can mimic mechanical stresses like shear stress or substrate stiffness, activating YAP by altering cellular localization. Key components in the formation of FAs and protruding lamellipodia, typically involved in the adhesion/migration process, are FAK and PXN, while VCL is a cytoskeletal scaffold protein which plays an essential role in the regulation of FAs assembly and disassembly57 and ITGAV is necessary for the maturation of focal adhesions.14 We observed a reduced expression of target transcription genes such as ITGAV, PXN and FAK expression, upon exposure to an EF. Downregulation of FAK and PXN, which are typically implicated in FA formation, suggests that EF regulates FA formation through an alternative pathway that bridges integrin clustering. Thus, the EF may aid in decreasing subsequent substrate stiffness potentially through the regulation of FAs and CTGF reduction. Besides YAP, TAZ has also been identified as a significant oncogenic factor. Our data show that exposure to EFs could block YAP and TAZ-mediated transcription activation with downregulation of YAP-dependent gene expression levels, such as CTGF and Cyr61. TEAD inhibition markedly suppresses cell migration, as we observed under EF exposure. Follow-up studies would provide information as to whether YAP/TAZ inactivation induces expression of a subset of genes which can lead to AKT activation and cell survival or may be due to non-specific effects.
The PI3 kinase (PI3K)–Akt axis has been implicated with the invasion ability of GBM cells, and it is known to be tightly associated with the electrotactic response and directionality.32–34 Studies in literature have demonstrated that the PI3K–Akt signalling pathway is a downstream effector of adhesive interactions critical for galvanotaxis,34 reporting an induced asymmetric distribution of PIP3 and cytoskeletal proteins. From our results, it appears that in response to an EF, YAP and TAZ act to dissipate cytoskeletal tension through a cell-intrinsic feedback mechanism, which is also dependent on PI3K activation. Under exposure to an EF, the directionality of movement was contingent on the stiffness of the substrate, with a cathodic or anodic preference respectively on stiff and soft substrates.48 Suppression of the PI3K activity led to an increased cellular anodic response under EF, accompanied by reduced cell motility, especially on soft substrates, consistent with previous studies.34 Our findings further suggested the PI3K/Akt pathway mediated the EF directionality, and cytoskeleton rearrangement as we observed a tendency of PI3K-inhibited cells to increase the expression of target genes, as well as integrin and focal adhesion-related genes (i.e., FAK, ITGAV) upon EF exposure.58
Reorganisation of the cytoskeletal network may regulate mitochondrial structures. Different studies have reported a link between the Hippo pathway and mitochondrial function.41 Overexpression of YAP has been reported to promote the expression of genes that regulate mitochondrial fusion, leading to fused mitochondria. Our study identifies YAP's involvement in regulating mitochondrial structure remodelling under exposure to an EF. This implies that mitochondrial changes are likely linked to the localisation of YAP in GBM cells and are cell- and context-specific.
The YAP–TEAD complex controls the transcription of various oncogenes to promote tumour cell proliferation, transformation, and invasion. We observed that both inhibitors ST and VP disrupted the YAP–TEAD interaction and were not rescued under EF. Specifically, we found that selective inhibition of YAP/TAZ-TEAD interaction with VP increased cytoskeletal tension, suggesting either additional transcriptional targets or potentially complementary roles for YAP and TAZ. While further research is necessary to dissect potentially distinct co-effector interactions or transcriptional targets of YAP and TAZ, these data contribute to the emerging evidence of crosstalk between transcriptional activity and cytoskeletal dynamics.
Together, our novel findings clarify a link between oncogenic YAP and EFs, assigning an important role to EF in regulating focal adhesion dynamics, and collectively show that EFs regulate the nuclear translocation of YAP/TAZ, thus dictating their roles as transcriptional co-activators and highlighting their potential application in electroceutical treatments for regenerative medicine. We believe that understanding the mechanisms of this transduction and the biochemical pathways involved will open up innovative approaches for treatment and improve our knowledge of how external factors interact with cancer biology.
Future work will adopt a more comprehensive approach, utilising 3D substrates and sequencing methods to globally assess gene expression under the influence of electrical stimulation.
In summary, our study seeks to bridge the gap between external physical cues and critical cancer signalling pathways, offering insights that could lead to innovative treatment strategies and a deeper understanding of how environmental factors impact glioblastoma.
Materials and methods
Cell cultures
Human glioma cell lines U251-MG and GL15 were grown in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco), 100 unit per ml penicillin G sodium and 100 mg ml−1 streptomycin sulfate at 37 °C in humidified air with 5% CO2.Electrotaxis and electrotactic chambers
Electrotaxis experiments were carried out using a modified customized device, devised with standard microfabrication techniques. Standard 35mm culture dishes (Falcon brand, Corning Life Sciences) were used as the basis for the substrates. In brief Sylgard 184 elastomer (PDMS, Dow Corning), was used in a mixing ratio PDMS:heptane (50:1):1 to create soft substrates. Once mixed, the PDMS was poured into 35 mm diameter Petri dishes to create ∼2 mm thick films and cured at 65 °C overnight (12–24 hours) as previously described.1,18 Subsequently, a micro-channel of 170 μm high, 4 mm wide and 15 mm long in PDMS (10:1) was bonded via oxygen plasma treatment to Petri dishes and soft PDMS. Once assembled, all substrates were washed with 70% ethanol and DI water and sterilised with UV before seeding. GBM cells were seeded in the central microchannel for 24hours before use. In each experiment, direct current was applied through (2%) agar-salt bridges connecting with silver/silver chloride electrodes held in place by stainless steel alligator clips into the two beakers containing Steinberg's solution (consisting 60 mM NaCl, 0.7mM KCl and 0.3mM Ca(NO3)2, 0.8mM MgSO4 and 1.4mM Tris base, pH 7.4). The agar bridges were connected to a pooled medium on either side of the channel. Cells were exposed to 2 V/cm direct current EF for 3 hours.
Cell viability assay
Cell viability was measured using a commercially available live–dead double staining kit (04511-1KT-F, Merck, St. Louis, MO, USA). Briefly, the cells with or without EF treatment were exposed to a 0.25μM staining solution of calcein and propidium iodide (PI). Following the viability staining protocol, the constructs were imaged by an Olympus iX73 microscope equipped with an X-Light V3 spinning disc head (Crest Optics), an LDI laser illuminator (89 North), a Prime BSI sCMOS camera (Photometrics). Images were acquired as a single stack with a 10x/NA 0.25 objective (Olympus). The captured images were subsequently analysed using a built-in script in ImageJ, which uses particle segmentation to pick out the live and dead cells, respectively.
Morphological quantification
Whole cell area was obtained drawing a ROI around each cell before and after EF exposure, to measure the effect of an EF on the cells. To measure nuclear volume Z stacks of the single GBM cells seeded on substrates of different stiffness and w/wo exposure to an EF, nuclei were visualised by DAPI (4′,6′-diamidino-phenylindole) staining, applied at 1 μg ml−1 directly before the microscopic measurement using Ibidi μ Dish (35 mm). For each condition, z-stacks were acquired with a z-step size of 0.1 μm using a ZEISS LSM980 confocal microscope with PLN APO 40×/1.3 OIL DICIII immersion objective lens and a pinhole of 1 AU. The 3D visualisation and quantification were performed with Fiji software as briefly described. Nuclear volume of the segmented cells were calculated using the 3D Object Counter plugin (ImageJ)59 and quantified using 3D Image Suite.60 Results are based upon the 3D Region of Interest (ROI) Manager from 3D Image Suite.Cell tracking
Cell migration was observed mounting the dishes onto an Olympus IX73 inverted microscope, equipped with a QImaging OptiMOS sCMOS camera (Crisel, QImaging, Surrey, BC, Canada) with MetaVue program (Molecular Devices, Sunnyvale, CA, USA) and a stage-mounted incubator with CO2 and temperature control (H201; Okolab, Pozzuoli, Italy) using 10× (Plan N, NA = 0.25, Ph1) or 20× (LUCPlan FLN, NA = 0.45, Ph2) magnification. Cell migration was analysed to determine directedness (cosθ) path length and track speed by using ImageJ Software (NIH, Bethesda, MA, USA) with MTrackJ. Briefly, individual cell tracks from time-lapse microscopy were recorded over 3 hours and set to a common origin for spatial comparison. Results from a minimum of three separate experiments with individual tracked cells were pooled for data analysis. The trajectories and parameters such as track lengths, displacement (Euclidean distance at each time point), cell instantaneous speed, directionality (track length [the last position minus the initial position] divided by the total displacement of the cell), were plotted using Prism Software. The forward migration index was calculated by dividing the displacement along the DCEF vector by the total (x,y)-displacement between the initial and final positions of the cells yields the directedness. Anodal migration was considered to have a negative directedness value, whereas cathodal directedness was positive.
GBM cell infection with Paxillin-GFP
U251-MG and GL15 cells were seeded at a density of 10000 cm−2 in complete medium to reach 40% confluence at the time of infection. The medium was aspirated and changed with DMEM containing viral particles at MOI of 20. Cells were infected with GFP-Paxillin lentiviral particles (pLV[Exp]-Neo-EF1A > hPXN3xGGGGS:EGFP, VectorBuilder, Chicago, IL, USA) by using Polybrene (5 μg ml−1, Santa Cruz Biotechnology). The day after infection, cells were washed several times with PBS and complete medium was added. After selection by neomycin resistance for 1 week, cells were expanded and used for further experiments. FAs were calculated as previously described.61 In brief, images acquired at the same magnification and resolution were analysed with ImageJ where first a subtracted background was applied with a sliding paraboloid and rolling ball radius of 25 pixels. Subsequent enhancement of images, using also CLAHE plug-in, images were binarized using automatic THRESHOLD command and particles analysed (size = 0.30–15; circularity = 0.00–0.99).
Chemical inhibitors
Wortmannin (Merck, St. Louis, MO, USA), chemical grade verteporfin (VP; Merck, St. Louis, MO, USA) and Supertdu (ST; Merck, St. Louis, MO, USA) were used in cell culture. Stock solutions of these compounds (10 mM) were prepared in dimethylsulphoxide (DMSO) and stored at −20 °C. Cells treated with VP were processed in the dark to limit related effects due to exposure to light (no lights in the tissue culture hood, no microscopy-light exposure of treated cells, and culture plates and cell pellets were covered and protected from direct light exposure). To assess the effects of kinase inhibitors on the response of cells to electric fields and avoid that high dose or prolonged PI3K inhibition could lead to cell cycle arrest and/or apoptosis, cultures were treated with wortmannin reagent immediately before EF exposure whereas ST and VP were treated for 24 hours.Measurement of mitochondrial mass and membrane potential
Mitochondrial mass localisation was obtained by loading cells with MitoTracker Green FM MitoTracker Green FM (M7510 Thermofisher, 200 nM), for 30 min. After treatment, cells were fixed and imaged under a laser scanning confocal microscope (Nikon A1R+). To quantitatively assess cellular mitochondrial networks, a semi-automated ImageJ plug-in Mitochondrial Network Analysis (MiNA) toolset (available at https://github.com/stuartlab) was used. In brief, images were processed to 8-bit images on grayscale and subsequently by applying unsharp mask, enhance local contrast CLAHE, and median filtering and skeletonised to identify and count/measure mitochondrial morphology, such as mean length of branches, mean network size (the mean number of branches per network), and mitochondrial footprints (mitochondrial coverage area). Subsequent analysis through the MiNA macro plugin generated mitochondria footprint, and branch length parameters.Mitochondrial membrane potential (Δψm) was evaluated with the potentiometric dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolo-carbocyanine iodide; Carlo Erba) with and without EF. Cells were seeded on the different substrates and allowed to attach for 24 h. After 3 h of EF exposure, the media was then replaced with fresh media containing JC-1 (5 μg ml−1) and cells were incubated for an additional 15 min at 37 °C in the dark. Following loading, cells were washed and imaged immediately in PBS. Fluorescent images of JC-1 staining were acquired on the Olympus iX73 microscope equipped with an X-Light V3 spinning disc head (Crest Optics), an LDI laser illuminator (89 North), a Prime BSI sCMOS camera (Photometrics) and a MetaMorph Software (Molecular Devices). Images were acquired with a 20×/NA 0,45 objective (Olympus). The fluorescence was measured from 5 random fields with ImageJ Software (NIH, Bethesda, MD, USA). A region of interest (ROI) around each cell along with background readings, was drawn and the area, mean fluorescence and integrated density were measured. Thus, the CTFC was calculated as integrated density − (area of selected cell × mean fluorescence of background readings). The fraction of the red, attributed to a potential-dependent aggregation, and the green, reflecting the monomeric form of JC-1, fluorescence was calculated.
Immunofluorescence and quantification of YAP/TAZ
Immunofluorescence staining was performed by fixing cells w/wo EF exposure in 4% paraformaldehyde in PBS for 15min at RT and permeabilised with 0.1% Triton X-100 for 5min. Cells were then labelled with primary antibodies and with DYE-Light-conjugated secondary antibodies against goat IgG at a dilution of 1:500 for 1 h at 37 °C. Nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (Merck, St. Louis, MO, USA), 1 mg ml−1 in PBS 1× for 5 min. Subsequently, samples were embedded in ProLong Gold antifade reagent (Thermo Fisher Scientific) and visualised on either an Olympus confocal microscopy system equipped with a 20× (UPlan FLN, NA 0.50), 40× (UPlanFLN, NA 1.30, oil) and 60× (UPlanSApo, NA 1.35, oil) with a resolution of 1024 × 1024 pixels or a Nikon confocal microscopy system equipped with 20× (UPlanFLN, NA 1.30, oil) and 60× (UPlanSApo, NA 1.35, oil) lenses.
Total YAP signal intensity was then determined, and YAP nucleus/cytoplasm ratio was calculated using the following eqn (1):
 | (1) |
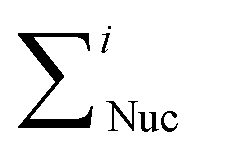 and
and  represent the sum of the intensity values for the pixels in the nuclear and cytoplasmic region respectively, and ANuc and ACyto are the area of the corresponding nuclear and cytoplasmic regions.
represent the sum of the intensity values for the pixels in the nuclear and cytoplasmic region respectively, and ANuc and ACyto are the area of the corresponding nuclear and cytoplasmic regions.
Western blot
Cells treated were lysed in 100 μl/106 cells of RIPA buffer (Thermo Scientific) with 1 mM Na3VO4, 1 mM PMSF, 1% NP-40 and protease inhibitors added. Lysed samples were incubated on ice for 30 min and centrifuged at 12000 rpm for 15 min at 4 °C. Supernatants were collected and the protein amount was quantified using a DC Protein Assay (BioRad, California USA). In total, 20 μg of proteins for each sample, added with Laemmli buffer (BioRad, California USA) supplemented with a reducing agent such as 2-mercaptoethanol, were boiled for 5 min at 90 °C and loaded onto a 10% (w/v) polyacrylamide SDS gels. Proteins were transferred onto nitrocellulose membranes using a Mini Trans-Blot Electrophoretic Transfer Cell (BioRad, California USA). Membranes were blocked with 5% Bovine serum albumin (BSA) (w/v) (Merck, St. Louis, MO, USA) in tris-buffered saline (TBS)-Tween (0.1% (v/v)) for 1 h, and then incubated at 4 °C overnight with the rabbit polyclonal IgG anti Phospho-YAP (Ser127) (1:1000 Cell Signaling) and the rabbit polyclonal IgG anti-YAP/TAZ (1:1000 Cell Signaling). After the washing steps with TBS-Tween (0.1% (v/v)), membranes were incubated with Goat Anti-Rabbit IgG (H + L)-HRP Conjugate (1:5000) (BioRad, California USA) for 1 hr at RT. The protein band intensity was identified with Clarity Western ECL Substrate (BioRad, California USA) using ChemiDoc (MolecularImager® ChemiDoc™mod. MP System—BioRad Laboratories) and densitometric analyses were performed with ImageLab Software (Biorad) and normalised to the corresponding GAPDH (Millipore Burlington, Massachusetts, Stati Uniti) controls.
RT-qPCR
Specific primer sequences belong to the company (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Chromosomal location of the primers, amplicon lengths and design details are shown in Table 1. Gene expression was quantified by quantitative polymerase chain reaction (qPCR) on a CFX96 Connect RT-PCR instrument (BioRad, California USA) in at least three technical replicates per gene. Briefly, total RNA was extracted from cells lysed in Trizol reagent (Invitrogen). The quality and yield of RNAs were verified using NANODROP One (Thermo Fisher Scientific). For RT-qPCR, reverse transcription reaction was performed in a thermocycler using iScript™ cDNA Synthesis Kit (BioRad, California USA) under the following conditions: priming, 25 °C, 5 min, reverse transcription, 46 °C, 20 min, RT inactivation, 95 °C, 1 min. The PCR protocol consisted of 40 cycles at 95 °C, 5′′ and 60 °C, 30′′. For quantification analysis, the comparative Threshold Cycle (Ct) method was used. The Ct values from each gene were normalised to the Ct value of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) used as a housekeeping gene, in the same cDNA samples and was calculated by the equation 2−ΔCT.| Gene | Chromosome location | Product size (bp) | Detected coding transcript(s) | Primer design |
|---|---|---|---|---|
| bp: base pair; CYR61: cysteine-rich, angiogenic inducer, 61; CTGF: connective tissue growth factor; PXN: paxillin; FAK: PTK2 protein tyrosine kinase 2; VCL: vinculin; ITGAV: integrin, alpha V (vitronectin receptor, alpha polypeptide, antigen CD51). | ||||
| CYR61 | 1:86047790-86047902 | 83 | ENST00000451137, ENST00000536321, ENST00000360431 | Exonic |
| CTGF | 6:132269858-132269975 | 88 | ENST00000367976 | Exonic |
| PXN | 12:120650302-120651731 | 139 | ENST00000458477, ENST00000228307, ENST00000424649, ENST00000536957, | Intron-spanning |
| ENST00000331257, ENST00000267257, ENST00000541856, ENST00000397506 | ||||
| FAK | 8:141727697-141727807 | 81 | ENST00000522684, ENST00000519654, ENST00000519465, ENST00000517887, | Exonic |
| ENST00000521059, ENST00000523539, ENST00000340930, ENST00000519419, | ||||
| ENST00000430260, ENST00000521986, ENST00000522424, ENST00000521562, | ||||
| ENST00000523388, ENST00000395214, ENST00000535192, ENST00000395218, | ||||
| ENST00000354438, ENST00000395221, ENST00000538769 | ||||
| VCL | 10:75863626-75864902 | 125 | ENST00000372755, ENST00000436396, ENST00000211998, ENST00000537043, | Intron-spanning |
| ENST00000415462 | ||||
| ITGAV | 2:187503136-187506162 | 130 | ENST00000261023, ENST00000374907, ENST00000433736, ENST00000544640 | Intron-spanning |
Statistical analysis
All experiments were conducted with a minimum of 3 independent experiments. ANOVA followed by Tukey-HSD post hoc tests were performed on all data sets. Error is reported in bar graphs as the standard error of the mean unless otherwise noted. Significance was indicated by *, **, or *** corresponding to P < 0.05, 0.01, or 0.001.Author contributions
All authors contributed to the study. In particular: Bernadette Basilico: data curation, formal analysis, investigation, methodology, writing – original draft, writing – review & editing. Maddalena Grieco: data curation, formal analysis, investigation, methodology, writing – original draft, writing – review & editing. Stefania D’Amone: investigation, methodology. Ilaria Elena Palamà: writing – review & editing. Clotilde Lauro: investigation, methodology. Pamela Mozetic: investigation, methodology. Alberto Rainer: investigation. Simone de Panfilis: investigation. Valeria de Turris: investigation. Giuseppe Gigli: supervision. Barbara Cortese: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, writing – original draft, writing – review & editing. All authors read and approved the final manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no potential conflicts of interest.Acknowledgements
The research leading to these results has received funding from AIRC under IG 2021 – ID. 26328 project – P.I. Cortese Barbara and AIRC under MFAG 2015 – ID. 16803 project – “P.I. Cortese Barbara”. The authors are also grateful to the “Tecnopolo per la medicina di precisione” (TecnoMed Puglia) – Regione Puglia: DGR n.2117 del 21/11/2018, CUP: B84I18000540002 and “Tecnopolo di Nanotecnologia e Fotonica per la medicina di precisione” (TECNOMED) – FISR/MIUR-CNR: delibera CIPE n.3449 del 7-08-2017, CUP: B83B17000010001. We also thank Irene Iacuitto, Giovanna Loffredo and Manuela Marchetti for practical administrative support.References
- B. Basilico, I. E. Palamà, S. D’Amone, C. Lauro, M. Rosito, M. Grieco, P. Ratano, F. Cordella, C. Sanchini, S. Di Angelantonio, D. Ragozzino, M. Cascione, G. Gigli and B. Cortese, Front. Oncol., 2022, 12, 983507 CrossRef CAS.
- S. Garofalo, A. Porzia, F. Mainiero, S. Di Angelantonio, B. Cortese, B. Basilico, F. Pagani, G. Cignitti, G. Chece, R. Maggio, M. E. Tremblay, J. Savage, K. Bisht, V. Esposito, G. Bernardini, T. Seyfried, J. Mieczkowski, K. Stepniak, B. Kaminska, A. Santoni and C. Limatola, eLife, 2017, 6, e33415 CrossRef.
- B. Cortese, I. E. Palamà, S. D’Amone and G. Gigli, Integr. Biol., 2014, 6, 817–830 Search PubMed.
- T. Panciera, L. Azzolin, M. Cordenonsi and S. Piccolo, Nat. Rev. Mol. Cell Biol., 2017, 18, 758–770 Search PubMed.
- S. Piccolo, M. Cordenonsi and S. Dupont, Clin. Cancer Res., 2013, 19, 4925–4930 CrossRef CAS PubMed.
- S. Dupont, L. Morsut, M. Aragona, E. Enzo, S. Giulitti, M. Cordenonsi, F. Zanconato, J. Le Digabel, M. Forcato, S. Bicciato, N. Elvassore and S. Piccolo, Nature, 2011, 474, 179–183 CrossRef CAS.
- Z. Meng, T. Moroishi and K. L. Guan, Genes Dev., 2016, 30, 1–17 CrossRef CAS.
- A. Das, R. S. Fischer, D. Pan and C. M. Waterman, J. Biol. Chem., 2016, 291, 6096–6110 CrossRef CAS.
- M. Fischer, P. Rikeit, P. Knaus and C. Coirault, Front. Physiol., 2016, 7, 41 Search PubMed.
- B. Zhao, L. Li, L. Wang, C. Y. Wang, J. Yu and K. L. Guan, Genes Dev., 2012, 26, 54–68 CrossRef CAS PubMed.
- L. Sansores-Garcia, W. Bossuyt, K. Wada, S. Yonemura, C. Tao, H. Sasaki and G. Halder, EMBO J., 2011, 30, 2325–2335 CrossRef CAS PubMed.
- M. Aragona, T. Panciera, A. Manfrin, S. Giulitti, F. Michielin, N. Elvassore, S. Dupont and S. Piccolo, Cell, 2013, 154, 1047–1059 Search PubMed.
- A. Elosegui-Artola, R. Oria, Y. Chen, A. Kosmalska, C. Pérez-González, N. Castro, C. Zhu, X. Trepat and P. Roca-Cusachs, Nat. Cell Biol., 2016, 18, 540–548 CrossRef CAS PubMed.
- E. M. Morandi, R. Verstappen, M. E. Zwierzina, S. Geley, G. Pierer and C. Ploner, Sci. Rep., 2016, 6, 28889 CrossRef CAS PubMed.
- M. Castellan, A. Guarnieri, A. Fujimura, F. Zanconato, G. Battilana, T. Panciera, H. L. Sladitschek, P. Contessotto, A. Citron, A. Grilli, O. Romano, S. Bicciato, M. Fassan, E. Porcù, A. Rosato, M. Cordenonsi and S. Piccolo, Nat. Cancer, 2021, 2, 174–188 CrossRef CAS.
- K. Jiang, G. Yao, L. Hu, Y. Yan, J. Liu, J. Shi, Y. Chang, Y. Zhang, D. Liang, D. Shen, G. Zhang, S. Meng and H. Piao, Cell Death Dis., 2020, 11, 230 CrossRef CAS.
- I. Thievessen, N. Fakhri, J. Steinwachs, V. Kraus, R. S. McIsaac, L. Gao, B. C. Chen, M. A. Baird, M. W. Davidson, E. Betzig, R. Oldenbourg, C. M. Waterman and B. Fabry, FASEB J., 2015, 29, 4555–4567 CrossRef CAS PubMed.
- I. E. Palamà, S. D’Amone, P. Ratano, A. Donatelli, A. Liscio, G. Antonacci, M. Testini, S. Di Angelantonio, D. Ragozzino and B. Cortese, Cancers, 2019, 11(5), 643 CrossRef.
- B. Cortese, G. Gigli and M. Riehle, Adv. Funct. Mater., 2009, 19, 2961–2968 CrossRef CAS.
- C. D. McCaig, A. M. Rajnicek, B. Song and M. Zhao, Physiol. Rev., 2005, 85, 943–978 CrossRef PubMed.
- R. Nuccitelli, Curr. Top. Dev. Biol., 2003, 58, 1–26 CrossRef.
- C. D. McCaig, B. Song and A. M. Rajnicek, J. Cell Sci., 2009, 122, 4267–4276 CrossRef CAS PubMed.
- F. Li, T. Chen, S. Hu, J. Lin, R. Hu and H. Feng, PLoS One, 2013, 8, e61195 CrossRef CAS.
- K. I. Nakajima, K. Zhu, Y. H. Sun, B. Hegyi, Q. Zeng, C. J. Murphy, J. V. Small, Y. Chen-Izu, Y. Izumiya, J. M. Penninger and M. Zhao, Nat. Commun., 2015, 6, 8532 CrossRef CAS PubMed.
- J. Park, H. Yang, D. Woo, S. Jeon, H. Do, S. Huh, N. Kim, J. Kim and K. Park, Biomaterials, 2012, 33, 7300–7308 CrossRef CAS.
- M. J. McKasson, L. Huang and K. R. Robinson, Exp. Neurol., 2008, 211, 585–587 CrossRef.
- S. Acevedo-Acevedo and W. C. Crone, J. Negat. Results Biomed., 2015, 14, 22 CrossRef PubMed.
- A. M. Smith, D. G. Inocencio, B. M. Pardi, A. Gopinath and R. C. Andresen Eguiluz, ACS Appl. Polym. Mater., 2024, 6, 2405–2416 CrossRef CAS PubMed.
- I. Titushkin and M. Cho, Biophys. J., 2009, 96, 717–728 CrossRef CAS PubMed.
- Y. J. Huang, G. Hoffmann, B. Wheeler, P. Schiapparelli, A. Quinones-Hinojosa and P. Searson, Sci. Rep., 2016, 6, 21583 CrossRef CAS.
- J. Langhans, L. Schneele, N. Trenkler, H. von Bandemer, L. Nonnenmacher, G. Karpel-Massler, M. D. Siegelin, S. Zhou, M. E. Halatsch, K. M. Debatin and M. A. Westhoff, Oncogenesis, 2017, 6, 398 CrossRef.
- M. Zhao, B. Song, J. Pu, T. Wada, B. Reid, G. Tai, F. Wang, A. Guo, P. Walczysko, Y. Gu, T. Sasaki, A. Suzuki, J. V. Forrester, H. R. Bourne, P. N. Devreotes, C. D. McCaig and J. M. Penninger, Nature, 2006, 442, 457–460 CrossRef CAS PubMed.
- A. Guo, B. Song, B. Reid, Y. Gu, J. V. Forrester, C. A. Jahoda and M. Zhao, J. Invest. Dermatol., 2010, 130, 2320–2327 CrossRef CAS.
- L. Yao, Y. Li, J. Knapp and P. Smith, J. Cell. Physiol., 2015, 230, 1515–1524 CrossRef CAS.
- Z. Sun, S. S. Guo and R. Fässler, J. Cell Biol., 2016, 215, 445–456 CrossRef CAS.
- R. Yagi, L. F. Chen, K. Shigesada, Y. Murakami and Y. Ito, EMBO J., 1999, 18, 2551–2562 CrossRef CAS PubMed.
- N. Koushki, A. Ghagre, L. K. Srivastava, C. Molter and A. J. Ehrlicher, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2301285120 CrossRef CAS.
- Z. Lin, P. Zhou, A. von Gise, F. Gu, Q. Ma, J. Chen, H. Guo, P. R. van Gorp, D. Z. Wang and W. T. Pu, Circ. Res., 2015, 116, 35–45 CrossRef CAS PubMed.
- F. Gibault, F. Bailly, M. Corvaisier, M. Coevoet, G. Huet, P. Melnyk and P. Cotelle, ChemMedChem, 2017, 12, 954–961 CrossRef CAS PubMed.
- S. Patrick, P. Gowda, K. Lathoria, V. Suri and E. Sen, J. Cell Sci., 2021, 134(22), jcs259188 CrossRef CAS PubMed.
- R. Nagaraj, S. Gururaja-Rao, K. T. Jones, M. Slattery, N. Negre, D. Braas, H. Christofk, K. P. White, R. Mann and U. Banerjee, Genes Dev., 2012, 26, 2027–2037 CrossRef CAS.
- A. J. Valente, L. A. Maddalena, E. L. Robb, F. Moradi and J. A. Stuart, Acta Histochem., 2017, 119, 315–326 CrossRef CAS PubMed.
- C. Chen, X. Bai, Y. Ding and I. S. Lee, Biomater. Res., 2019, 23, 25 CrossRef PubMed.
- J. Pu and M. Zhao, J. Cell Sci., 2005, 118, 1117–1128 Search PubMed.
- H. Clancy, M. Pruski, B. Lang, J. Ching and C. D. McCaig, Exp. Cell Res., 2021, 406, 112736 CrossRef CAS.
- M. Yamashita, Commun. Biol., 2023, 6, 680 CrossRef CAS.
- K. Katoh, Life, 2022, 12(4), 531 CrossRef CAS PubMed.
- U. Ahmed, S. N. Iwasa, L. Poloni, J. E. Ahlfors, C. Yip, M. R. Popovic and C. M. Morshead, Bioelectricity, 2020, 2, 229–237 CrossRef PubMed.
- E. I. Finkelstein, P. H. Chao, C. T. Hung and J. C. Bulinski, Cell Motil. Cytoskeleton, 2007, 64, 833–846 Search PubMed.
- S. Huang, X. Wang, X. Wu, J. Yu, J. Li, X. Huang, C. Zhu and H. Ge, Am. J. Physiol.: Cell Physiol., 2018, 315, C474–C484 CrossRef CAS PubMed.
- B. J. Thompson, BioEssays, 2020, 42, e1900162 Search PubMed.
- Y. Qiao, J. Chen, Y. B. Lim, M. L. Finch-Edmondson, V. P. Seshachalam, L. Qin, T. Jiang, B. C. Low, H. Singh, C. T. Lim and M. Sudol, Cell Rep., 2017, 19, 1495–1502 Search PubMed.
- D. E. Mason, J. M. Collins, J. H. Dawahare, T. D. Nguyen, Y. Lin, S. L. Voytik-Harbin, P. Zorlutuna, M. C. Yoder and J. D. Boerckel, J. Cell Biol., 2019, 218, 1369–1389 CrossRef CAS PubMed.
- Y. Jia, J. Xu, Q. Shi, L. Zheng, M. Liu, M. Wang, P. Li and Y. Fan, Med. Nov. Technol. Devices, 2022, 16, 100142 CrossRef.
- S. Abasi, A. Jain, J. P. Cooke and A. Guiseppi-Elie, Front. Mol. Biosci., 2023, 10, 1161191 CrossRef CAS PubMed.
- A. Colciago, S. Melfi, G. Giannotti, V. Bonalume, M. Ballabio, L. Caffino, F. Fumagalli and V. Magnaghi, Cell Death Discovery, 2015, 1, 15021 Search PubMed.
- S. V. Plotnikov, A. M. Pasapera, B. Sabass and C. M. Waterman, Cell, 2012, 151, 1513–1527 CrossRef CAS.
- N. Borreguero-Muñoz, G. C. Fletcher, M. Aguilar-Aragon, A. Elbediwy, Z. I. Vincent-Mistiaen and B. J. Thompson, PLoS Biol., 2019, 17, e3000509 CrossRef PubMed.
- S. Bolte and F. P. Cordelières, J. Microsc., 2006, 224, 213–232 CrossRef CAS PubMed.
- J. Ollion, J. Cochennec, F. Loll, C. Escudé and T. Boudier, Bioinformatics, 2013, 29, 1840–1841 CrossRef CAS PubMed.
- G. Nardone, J. Oliver-De La Cruz, J. Vrbsky, C. Martini, J. Pribyl, P. Skládal, M. Pešl, G. Caluori, S. Pagliari, F. Martino, Z. Maceckova, M. Hajduch, A. Sanz-Garcia, N. M. Pugno, G. B. Stokin and G. Forte, Nat. Commun., 2017, 8, 15321 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00891j |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |