
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monitoring of the dissolution/precipitation behavior of bioglass with simulated body fluid buffered by HEPES
Diana
Horkavcová
 *a,
Eliška
Sedláčková
a,
Petr
Bezdička
b,
Miloslav
Lhotka
c,
Karolína
Pánová
a and
Aleš
Helebrant
a
*a,
Eliška
Sedláčková
a,
Petr
Bezdička
b,
Miloslav
Lhotka
c,
Karolína
Pánová
a and
Aleš
Helebrant
a
aDepartment of Glass and Ceramics, Faculty of Chemical Technology, University of Chemistry and Technology Prague, Technická 5, Prague 166 28, Czech Republic. E-mail: diana.horkavcova@vscht.cz
bInstitute of Inorganic Chemistry of the AV ČR, v.v.i., Husinec-Řež 250 68, Czech Republic
cDepartment of Inorganic Technology, Faculty of Chemical Technology, University of Chemistry and Technology Prague, Technická 5, Prague 16628, Czech Republic
First published on 27th November 2024
Abstract
This research work investigates a 7-day interaction of bioactive glass in the form of grit with simulated body fluid with addition of HEPES buffer (SBF+H). The standard fluid buffered by TRIS (SBF+T) and unbuffered (SBF-Ø) were used for comparison. To understand the process more precisely, the material and the leachates were analyzed at hourly (1H, 2H, 4H) and daily (1D, 2D, 3D, 4D, 7D) intervals. During the static in vitro test the weight and specific surface area of the materials were measured and the surface and volume changes of the material character/composition were monitored by SEM/EDS and XRD. Samples of solution leachates were collected at regular intervals to determine concentrations of calcium, silicon and (PO4)3− and to measure pH. After exposure in SBF+T and SBF+H a new crystalline layer of hydroxyapatite formed on the material surface. The material exposed to SBF+H dissolved less than the one exposed to SBF+T but the hydroxyapatite layer on its surface grew faster. The material exposed only to SBF-Ø without any buffer dissolved much less, while the ions released into the solution very rapidly re-precipitated on the surface. As a result, three amorphous layers containing Si, Ca and P with different thicknesses were gradually formed on the surface. Results of material and solution analyses have clearly shown that both HEPES and TRIS buffers have a significant effect on the formation of hydroxyapatite on the surface of bioactive glass. The behavior of the HEPES buffer with highly bioactive bioglass is very similar to that of the TRIS buffer.
Introduction
New generations of biomaterials focus on improving their interaction with the environment and the ability to respond adaptively to dynamic stimuli. Research on bioactive glasses began in 1969 when Professor Larry Hench introduced a concept of formation of strong bonds between bone and synthetic material caused by reactions on the glass surface. The composition of bioactive glass is very similar to the human body – silicon, sodium, potassium, calcium, etc.1–6 Regeneration and binding to hard and soft tissues depends on various compositions of Na2O, CaO and SiO2. There are several variants of bioactive glasses that differ mainly in their composition and substitutions (silica, phosphorus, boron, magnesium, etc.).7–9 Among the most commonly used is silica glass, known as 45S5 Bioglass (45 wt% SiO2, the letter S represents the crosslinking element and 5 represents the ratio of CaO and P2O5).10–12 When bioglass is implanted close to bone, a critical concentration of Si, Ca, P and Na ions are released on its surface to trigger intra and extracellular reactions that lead to rapid bone formation. A silica-rich gel is then formed on the surface. The gel reacts with ions in body fluids to form hydroxyapatite on the bioglass surface.10,11,13 The ability of a material to bioactively react can be determined based on the time it takes for more than 50% of the surface to be bound to the host tissue.14,15 The so-called bioactivity index IB is used for this purpose.16 Bioactivity can be defined as the action of an active substance on a living organism or tissue. However, a much more precise definition is that a bioactive material is a material designed to have a therapeutic or diagnostic effect by interacting with the living environment.17 The first step in the development and testing of biomaterials is the discovery and characterization of a new material. This is followed by in vitro testing, which consists of exposure of the material to a simulated body environment. If the material is found to be non-toxic then in vivo testing can follow.18 For in vitro testing, a non-cellular simulated body fluid (SBF) test solution has been developed by Kokubo et al. which mimics the inorganic composition of blood plasma. Since 2000, SBF has become the main medium for bioactivity testing with a clearly defined composition according to ISO standard ISO:23317.19,20 One of the biggest challenges of in vitro bioactivity testing of biomaterials (especially glass-ceramics and glass) is maintaining a stable pH of the test medium and the choice of an appropriate buffering agent.21,22 In 1966, 12 buffers were prepared with hydrogen ions in the range of pKa = 6.15–8.35. This range made it possible to use other buffers for testing in biological environments than those that had been used in the past.23–26 Since the TRIS (tris(hydroxymethyl)aminomethane) buffer was demonstrated to significantly affect experimental results of bioactivity tests for glass-ceramic scaffolds in vitro the research focused on finding a suitable alternative by using HEPES (4-(hydroxyethyl)-1-piperazineethanesulfonic acid) buffers, BES (N,N-bis(2-hydroxyethyl)-2-aminoethansulfonic acid), TES (N-[tris(hydroxymethyl)methyl]-2-aminoethansulfonic acid), MOPS (3-morpholinoropanesulfonic acid).27–30The objective of this experimental work was to perform in vitro tests to observe in detail interactions between glass material in the form of grit with modified simulated body fluid buffered by HEPES (SBF+H). In order to have a better understanding of the behavior of this buffer, an in vitro test with a standard solution with buffer TRIS (SBF+T) and a solution without buffer (SBF-Ø) was performed for comparison. Changes on the surface and in the structure of the material were analyzed at hourly (1H, 2H, 4H) and daily (1D, 2D, 3D, 4D, 7D) intervals with optical and electron microscopy, XRD and BET, as well as changes in the weight. Concentrations of Si, Ca and (PO4)3− were measured with AAS, UV-vis and pH values in all types of solutions were measured using a pH meter. This monitoring complements the interesting results of our previous work on HEPES buffer with a highly porous glass-ceramic scaffold.28
Experimental part
Individual raw materials to produce bioglass 48.592 g SiO2 (Sigma-Aldrich), 40.141 g Na2CO3 (Penta s.r.o.), 47.007 g CaCO3 (Lach-Ner s.r.o.) and 14.261 g NaH2PO4·2H2O (Penta s.r.o.) were mixed into a 150 g homogeneous batch. The mixture was melted in an electric laboratory furnace in an oxidizing atmosphere. A part of the batch was placed in a crucible made of PtRh alloy. The crucible was then placed in an oven heated to 1300 °C. Another part of the mixture was added after 15 minutes and then the rest of the batch was added after the same period of melting. The glass was melted in the furnace for 4 hours, during which it was stirred 2 times to homogenize it. After the melting it was poured into a preheated steel mold and placed into a cooling oven heated to 560 °C. After an hour at this temperature the glass was let to cool down in the closed oven until the next day.Preparation of the unbuffered (SBF-Ø) and buffered by TRIS (SBF+T) and HEPES (SBF+H) solutions followed the procedure described in ref. 19, 20, 27, 28 Concentrations of the main ions were the same in all the solutions and they mimicked the inorganic part of blood plasma. The procedures for the preparation of unbuffered and buffered solutions (with the same concentration of buffers) were chosen in order to be comparable with experimental conditions used for testing of another biomaterial, specifically the glass-ceramic scaffold.27,28,30
The tested bioglass (45S5) samples were prepared by crushing pieces of glass. The fraction of 0.3–0.5 mm was separated with a sieve and subsequently cleaned (immersion in ethanol and placed in an ultrasonic bath for 20 min). For each sampling time two parallel samples were prepared, each of them weighing 0.1750 g. The glass grit was immersed into 100 ml of the prepared SBF-Ø, SBF+T and SBF+H solutions. The samples were placed in a thermostat at 36.5 °C. The test lasted 7 days and samples were collected at hourly (1H, 2H, 4H) and daily (1D, 2D, 3D, 4D, 7D) intervals. The measurements at the indicated intervals included pH and analyses of calcium, silicon and (PO4)3−. The sampling process consisted of removal of all SBF from the bottle, the grit was washed repeatedly with ethanol and left to dry in the air.
For SEM/EDS analysis the grit samples were Au–Pd plated for 60 s. A Hitachi S-4700 microscope with a Thomson Scientific Ultradry Silicon Drift Detector energy dispersive analyzer (EDS) at 15 kV was used for the measurements. XRD measurements were performed using a PANanalytical X’Pert PRO diffractometer using a Cu anode at 25 °C. The time of one scanning step was 115.26 s. A change in the specific surface area was determined using the BET method. The specific surface area was determined using an adsorption isotherm on an Micromeritics 3Flex.
The concentration of calcium was determined by atomic absorption spectroscopy using Agilent 280FS OhlAA with flame atomization technique. The atomization was carried out in an acetylene–nitrous oxide flame. The measurements were carried out at the wavelength of λ = 422.7 nm. The data were processed using a calibration curve. The concentration of silicon was determined by AAS using Agilent 280FS with a flame atomization technique, at the wavelength of λ = 256.1 nm in N2O–C2H2 flame. The concentration of phosphate ions was determined spectrophotometrically using a UV-visible Spectrophotometer UV 1601 at the wavelength of λ = 830 nm, according to ČSN EN ISO 6878. Values of pH of the leachates were measured using an inoLab pH meter with a combined glass electrode at 35.5 ± 0.5 °C.
Results
During the 7-day experiment the change in weight was monitored of the original glass sample before the interaction with liquid and the glass sample after the respective exposure time to SBF-Ø, SBF+T and SBF+H solutions (Fig. 1). | ||
| Fig. 1 Weight change (%) of the glass materials during the exposure to SBF-Ø, SBF+T and SBF+H solutions. | ||
The values clearly show that the most significant changes occurred during the first two hours, when there was a significant decrease in the weight. During the interaction, the dissolution of the test material and the precipitation of a new layer on the surface of the material took place simultaneously and therefore the measured weights make it only possible to estimate which of the processes dominates in a given period of time. For the glass grit in SBF+T solution the weight decreased by 5.3% within two hours. This trend suggests a significant dissolution of the material. A dramatic weight loss also occurred from the first to the third day of the experiment. Thus it is possible to conclude that dissolution of the material prevailed. From the third day onwards, the change in the weight was slower, possibly due to precipitation of a new layer on the surface which slows down the dissolution of the material. For the material exposed to the SBF+H solution the weight continually decreased throughout the entire experiment. During the first day the decrease was faster, while from the second day onwards the decrease was less dramatic and finally reached 9.4%. This trend is likely due to the formation of a layer that prevented a higher dissolution rate of the original material. The weight loss of the material exposed to SBF-Ø was also significant (6.3%) during the first two hours. This means that dissolution is the predominant process during that time period. From the first day onwards the weight hardly changes and it is very likely that the material has been coated with a layer that has slowed down the dissolution considerably.
Fig. 2 shows values of the specific surface area before the interaction and after the individual exposure times in the SBF-Ø, SBF+T and SBF+H solutions.
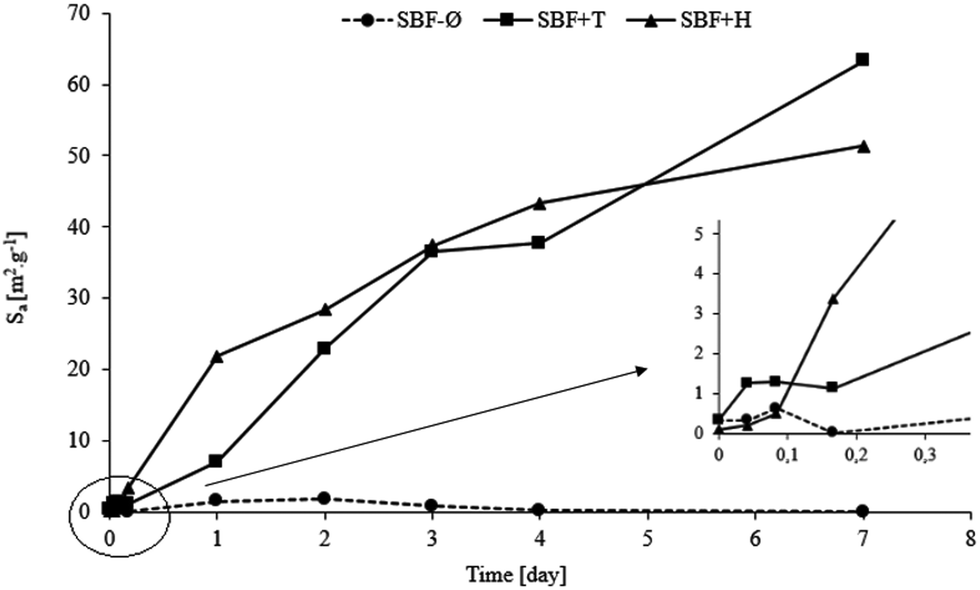 | ||
| Fig. 2 Specific surface area Sa [m2 g−1] of the glass materials during exposure to SBF-Ø, SBF+T and SBF+H solutions (with detail). | ||
The increase of the surface area values of the material in the SBF+T solution was very dramatic. This result confirms that a layer has formed on the original bioglass in interaction with SBF+T which, according to other analyses, is hydroxyapatite (HAp). The cause of the increased surface are is the highly dissected morphology of hydroxyapatite in the form of globules of plate-like crystals, which has been confirmed by scanning electron microscopy (SEM) images. The material also dramatically increased its specific surface area when interacting with the HEPES-buffered solution (SBF+H). Although the increase was less dramatic compared to the material exposed to the TRIS-buffered solution, the trend is very similar. Therefore it is possible to assume that a hydroxyapatite (HAp) phase was formed on the surface of the original bioglass as well. No significant changes of the surface area were observed for the glass exposed to unbuffered SBF-Ø.
The surface of the material before testing is shown in Fig. 3. Surfaces of the grit tested in SBF-Ø, SBF+T and SBF+H solutions are shown in Fig. 4–7.
 | ||
| Fig. 3 Material surface before interaction. | ||
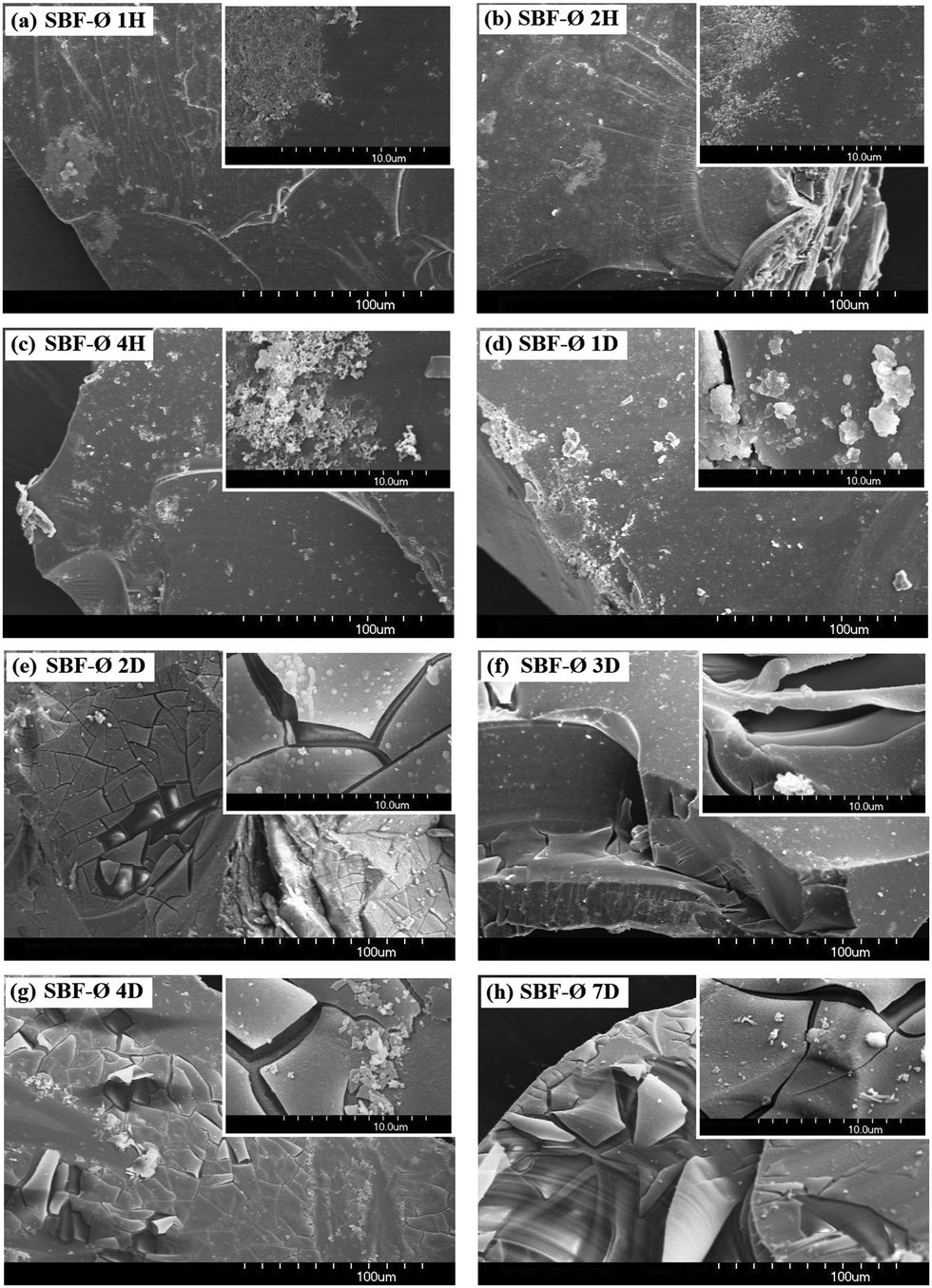 | ||
| Fig. 4 Material surface after the exposure to the SBF-Ø solution: (a) 1H, (b) 2H, (c) 4H, (d) 1D, (e) 2D, (f) 3D, (g) 4D, (h) 7D. | ||
 | ||
| Fig. 5 Material surface after the exposure to the SBF+T solution: (a) 1H, (b) 2H, (c) 4H, (d) 1D, (e) 2D, (f) 3D, (g) 4D, (h) 7D. | ||
 | ||
| Fig. 6 Material surface after the exposure to the SBF+H solution: (a) 1H, (b) 2H, (c) 4H, (d) 1D, (e) 2D, (f) 3D, (g) 4D, (h) 7D. | ||
 | ||
| Fig. 7 EDS (wt%) of material's surface after the exposure to the SBF+T and SBF+H solution: (a) and (b) 4H, (c) and (d) 4D. | ||
The images of the material before the exposure (Fig. 3) show a smooth surface. This is a pure and dense glass material.
After one hour of exposure in non-buffered SBF-Ø solution (Fig. 4a) 100 nm-sized nuclei developed with a tendency to aggregate and form centers on the surface of the material. After two hours (Fig. 4b), more continuous layers are already visible that grow into width and space. The images show both isolated nuclei and their clusters. After the second day (Fig. 4e), the entire surface is already covered with two layers consisting of Si–Ca–P. After the third day (Fig. 4f), another, very thin layer is visible on the surface of the original material, which was falling off during the sample handling. Until the end of the experiment the same trend continued of formation of new layers of different thickness.
Nuclei of a new Si–Ca–P phase, about 100 nm in size, are visible after 1 hour (Fig. 5a) on the material exposed to the SBF+T solution. After two hours (Fig. 5b), a continuous layer is already formed on the surface. The layer starts to fall off after 4 hours (Fig. 5c) but, at the same time, it continuously grows. During the first day (Fig. 5d) new globules are formed on its surface which give rise to a new surface layer. After two days (Fig. 5e) the surface is almost completely covered with globules typical for hydroxyapatite (HAp). As the material was dried for the purposes of the SEM measurements the outer layers are strongly dried and fall off. After seven days (Fig. 5h) the surface of the material is covered with several layers that continue to form globules on the top of each other and tend to grow further.
On the surface of the SBF+H solution material, a relatively thick interlayer (about 2 μm) can be seen after the first hour (Fig. 6a), which cracks when the material is handled in air. Fine globules can be seen on the surface but they do not yet have the structure typical for hydroxyapatite. The elemental composition corresponds to the presence of Si–Ca–P. After two hours (Fig. 6b), a second layer of a similar nature with the thickness of about 4 μm are formed on the first layer. From the fourth day onwards (Fig. 6g), a hydroxyapatite layer consisting of globules is spread over the entire surface. This means that the layers were formed between the second and fourth hour of the experiment. From this moment on, the HAp layer only grows and due to manipulation the material gradually falls off, revealing the underlying layers. Fig. 6f clearly shows the difference between the structure of the upper crystalline layer (thickness around 7–8 μm) and the lower semi-amorphous layer. After 7 days (Fig. 6h) a total of four layers have formed on the material. The last layer grows into the space and binds the granules of the material into aggregates.
The EDS analyses of the particles were performed before and after the test but for capacity reasons only selected results are presented – after 4 hours and 4 days in the buffered solutions. Since the layers were cracking it was possible to perform analyses of both the individual layers and the material tested. Since we are mainly interested in changes in the Si, Ca and P contents these elements are shown preferentially in the figures in wt%. The trend of Si loss (in the material) and Ca and P increase (in the layer) is well visible from the EDS analyses of the material in contact with SBF+T. The Si content in the material decreased from 40 wt% after 4 hours (Fig. 7a) to 29.0 wt% after 4 days (Fig. 7c). The increase of Ca and P contents in the layers precipitated on the surface is also well visible. For element Ca the value increased from 22.4 wt% after 4 hours to 28.8 after 4 days of exposure to SBF+T. For element P the value increased from 12.3 wt% after 4 hours to 14.2 wt% 8 after 4 days of exposure to SBF+T. Between the days 4 and 7 the results of the EDS analyses of the materials exposed to both the buffered solutions remained essentially the same.
The XRD analysis were performed for samples before and after 1, 2, 3, 4 and 7 days of exposure in SBF+T (Fig. 8a), SBF+H (Fig. 8b) and SBF-Ø (Fig. 8c) solutions.
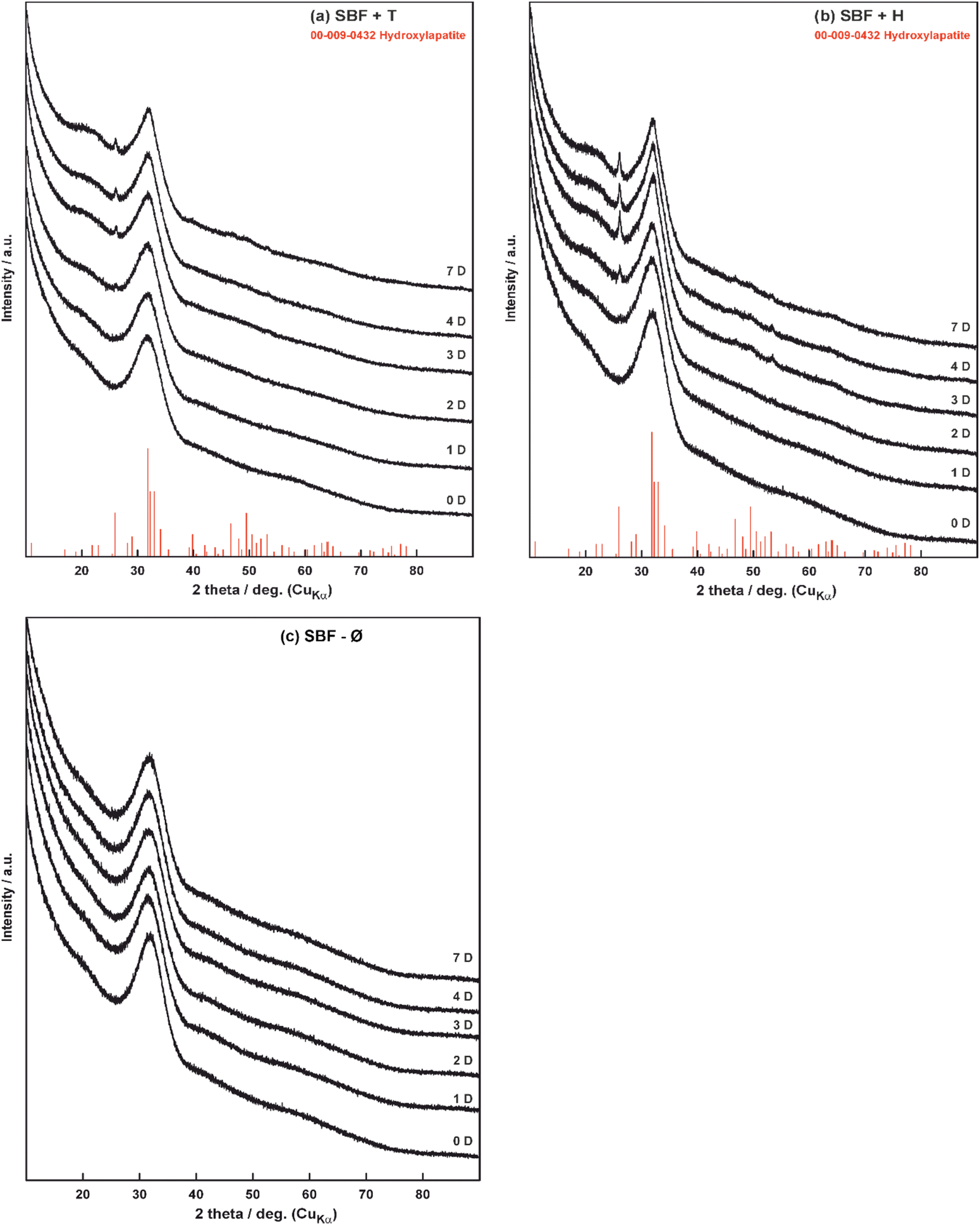 | ||
| Fig. 8 Diffractograms of glass materials before (0D) and during interaction (1–7D) in the solutions: (a) SBF+T, (b) SBF+H, (c) SBF-Ø. | ||
Fig. 8a–c show that the original bioglass is completely amorphous (curve 0D). With the time of interaction of the test material with the TRIS (SBF+T) buffer solution (Fig. 8a) some peaks gradually appear, indicating the increasing presence of the crystalline phase. According to the database, the phase was identified as hydroxyapatite (HAp). Although the other analyses show that HAp starts forming during the first hour, the XRD analysis recorded its presence only from the second day (2D). The XRD analysis was also performed to find crystalline phase on the material that interacted with the HEPES (SBF+H) buffer solution (Fig. 8b). In this case again, a hydroxyapatite crystalline phase was identified from the second day of exposure (2D). As in the previous case, we know that HAp was formed after a few hours but its quantity was very small and it could not be detected by the XRD analysis. The XRD analysis confirmed the results of SEM/EDS observation (Fig. 4) that no crystalline phase was formed on the surface of the samples after exposure in SBF-Ø (Fig. 8c).
The Fig. 9–12 show values of the concentrations of calcium, phosphate and silicon and pH values during the 7 days of interaction of the test material with SBF-Ø, SBF+T and SBF+H solutions. The measured data always correspond to the average of two parallel measurements.
 | ||
| Fig. 9 Values of calcium concentrations in the SBF-Ø, SBF+T and SBF+H solutions (with detail). | ||
 | ||
| Fig. 10 Values of phosphate ion concentration in the SBF-Ø, SBF+T and SBF+H solutions (with detail). | ||
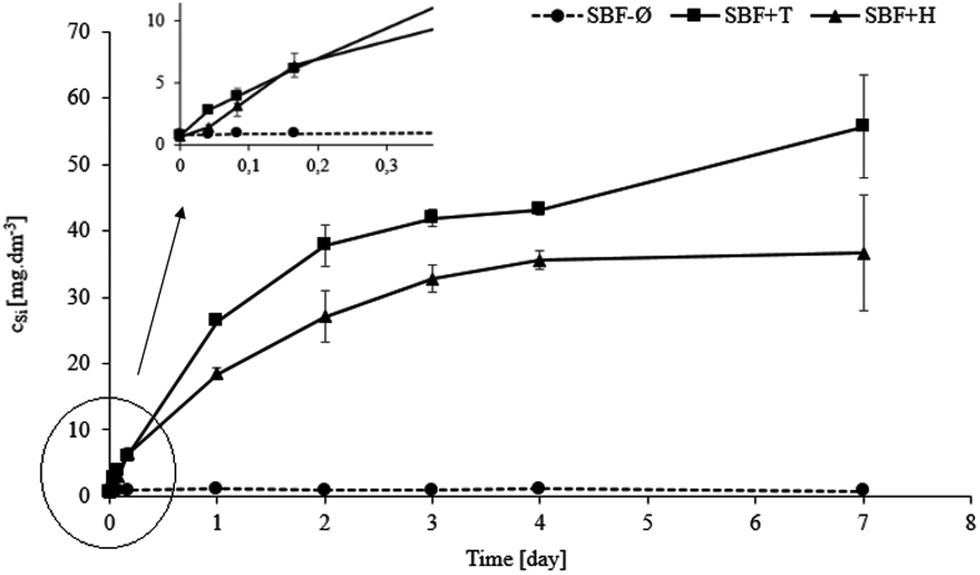 | ||
| Fig. 11 Values of silicon in SBF-Ø, SBF+T and SBF+H solutions (with detail). | ||
 | ||
| Fig. 12 Values of pH values in the SBF-Ø, SBF+T and SBF+H solutions (with detail). | ||
The Fig. 9 shows that there was a dramatic increase in calcium concentration in the SBF+T solution during the first 24 hours. This result again confirms that dissolution of the material occurred within the first few hours. From the first to the third day the increase is only moderate and after the second day the concentration drops sharply. From the third day onwards the concentration becomes stable which is consistent with the conclusion that from about the middle of the experiment the material is covered with a new layer. Throughout the experiment the concentration of calcium is higher than in the original solution. This implies that dissolution of the material is the predominant process but it changes its rate during the test. Even in the case of the SBF+H solution, the concentration of calcium ions is higher than the original value in the course of the whole experiment, indicating a large effect of the dissolution throughout the test. The biggest quantity of calcium are released during the first hours when the strongest dissolution of the material occurs. Between the fourth hour (4H) and the third day (3D) the dissolution still occurs but is already slowed down by the precipitated layer that isolates the material from the solution. From the third day onwards (up to 7D), when the material is already completely covered with the HAp layer, the calcium ion concentration shows little change. In the SBF-Ø solution there is only a slight increase in calcium ions concentration in during the first few hours. These results again correspond to the more significant weight loss at the beginning of the experiment. From the first day onwards, the concentration no longer shows significant changes, which is probably due to the precipitation of a new layer on the surface that makes dissolution of the material more difficult. If we compare the change in concentration in the solution with the original value we can see that the curve is below the original concentration for practically during the entire duration of the experiment. It is possible to assume that the new layer is formed relatively quickly and it gradually grew over the seven days.
In the SBF+T solution there was a dramatic decrease in phosphate ions concentration from the first hour (1H) to the third day (3D) of interaction (Fig. 10). This trend suggests that from the beginning of the experiment (PO4)3− ions were depleted from the solution and they deposited as a new layer on the surface of the material. After the third day (3D) the concentration stabilized, which is consistent with the calcium ion results. The concentration of phosphate ions in SBF+H is also below the original value almost all the time which corresponds with the rapid precipitation of ions from the solution back onto the material surface. During the first four hours the decrease was slow because a certain amount of ions must first be released into the solution before the precipitation can occur. After the fourth hour (4H) the decrease of ions concentration is rapid which is consistent with the previous results of the material analysis. The decrease in the concentration of (PO4)3− ions in unbuffered SBF-Ø is more pronounced from the beginning of the experiment to the first day. After the first day (1D) no more significant change was observed and the ion concentration was relatively stable.
Values of the silicon (Fig. 11) carries important information as it only describes the behavior of the tested material. When the material interacts with SBF+T it shows a dramatic increase of silicon in the solution. During the first three days the measured concentration was almost 42 mg dm−3. From the third day (3D) onwards the increase was slower. This trend is consistent with the previous results showing that the material dissolves significantly at the beginning of the experiment. In the second half of the experiment (after day 3) the dissolution slows down and therefore the increase in the concentration of silicon in the solution is less dramatic. In the SBF+H solution the concentration of silicon during the entire test also increases. This means that the tested material continues to dissolve and it slows down with the time as the precipitated layer grows. Again, it is confirmed here that the dissolution occurs predominantly up to the fourth hour (4H) and then a new layer is formed, blocking the dissolution of the material. In the SBF-Ø solution no significant release of silicon into the solution was observed. The maximum concentration measured was 1.1 mg dm−3. It is possible to conclude that in the SBF-Ø solution the material dissolves only very little and the prevailing process is the layer formation on the surface. This is supported by a very low weight loss of the material. The fact that SBF-Ø solution dissolves the glass significantly less than the buffered ones could be explained in following way, considering the solution chemistry. As reported by,31,32 the organic compounds can retard or accelerate the dissolution of silica. In buffered solutions, the dissolved silicon could be masked in complexes with buffers. Its activity in solution is then low and the driving force of dissolution remains high. On the contrary, in unbuffered environment, the solution layers adjacent to the glass grains are saturated very quickly and the dissolution driving force is negligible.
The Fig. 12 shows that pH values increased in all the solutions, confirming the ion exchange between the test material and the solutions. The sodium and calcium ions contributed the most to the increase in the pH values. Even though TRIS or HEPES buffers were used, the pH could not be kept the same throughout the experiment. As expected, the pH of the SBF-Ø solution is very inconsistent as it does not contain a buffer to keep the pH stable at the same value.
Discussion
The results of the weight change of the glass materials (Fig. 1) show that the greatest dissolution occurred for the grit that interacted with the SBF+T solution and to lesser extent for SBF+H (similar as28). In contrast, the material in the unbuffered SBF-Ø dissolved the least. In this case the TRIS buffer seems to have the most aggressive effect on the material. One of the key reasons for this difference is the stability of the pH in buffered solutions. Buffered solutions, such as TRIS and HEPES, maintain a relatively constant pH throughout the experiment, which controls the rate of glass dissolution by preventing rapid fluctuations in pH levels that would otherwise accelerate ion exchange processes.33 Another important factor is the ion composition in the buffered solutions. Buffers like TRIS are designed not only to stabilize pH but also to prevent interference from unwanted cations, such as Na+ or Ca2+, which could otherwise alter the dissolution kinetics by slowing down ion exchange or causing unwanted precipitation.34 The results of the values in the specific surface area (Fig. 2) indicate that the phases formed on the materials exposed to the buffered solutions (SBF+T, SBF+H) are very similar. The layer with a different morphology was formed on the original bioglass after exposed to unbuffered SBF-Ø but it did not have a significant effect on the surface area of the tested material. The electron microscope images at two different magnifications (Fig. 4–6) suggest that hydroxyapatite formed only on the material after interaction with SBF+T and SBF+H. According to the EDS analysis, an amorphous layer of Si–Ca–P composition formed on the material that was immersed in unbuffered SBF-Ø solution. The results of the EDS analyses (Fig. 7a–d) are only indicative because they are influenced by the complicated shape, orientation and distance of the measured surface of the crushed glass particles. In terms of the dissolution mechanism itself, the interaction between the glass surface and aqueous solutions involves both ion exchange and the formation of surface layers, which are key to controlling corrosion rates in different environments. As highlighted in previous reviews, the kinetics of glass dissolution are controlled by both surface reactions and diffusion processes, particularly in static conditions.35 However, for initial information about changes in the composition of the individual layers, this type of analysis is quite sufficient. It has been demonstrated that the Si content (wt%) in the tested material is decreasing and the contents Ca and P (wt%) in the emerging layers are increasing. It has been shown that the composition of the glass and the medium also plays a crucial role. The presence of calcium in the soaking solution enhances reactivity, leading to faster nucleation and growth of apatite crystals.36These results have been confirmed by results of the XRD analysis (Fig. 8) which confirmed a hydroxyapatite crystalline phase only on the material exposed to the buffered solutions SBF+T and SBF+H from the second day up to seven day of the in vitro test (similar as ref. 28).
The diagram of calcium concentrations in three types of simulated body fluid (Fig. 9) shows that the TRIS-buffered and HEPES-buffered solutions exhibit similar characteristics when interacting with the tested bioglass. The material interacting with SBF+H initially demonstrates a faster dissolution than the one exposed to SBF+T, however, it actually dissolves less. The layer on the original material after the interaction with SBF+H is probably formed faster. There were no significant changes in both buffered solutions after the fourth day (4D) of interaction.
The behavior of the buffered solutions (SBF+T, SBF+H) is very similar (Fig. 10), in both cases there is a relatively rapid release of phosphate ions from the material into the solution and their subsequent precipitation as HAp on the surface of the original material. In contrast, the unbuffered SBF-Ø solution has a relatively high concentration of (PO4)3− ions throughout the experiment. This may indicate that in this case, instead of HAp, an amorphous Si–Ca–P phase is formed that may contain less phosphorus than hydroxyapatite.
When comparing changes in concentration of silicon (Fig. 11) with tests in the SBF+T and SBF+H solutions the values are initially very similar and the divergence occurs around the first day (1D). It can be assumed that the reason for the different dissolution in the buffers may be the different thicknesses or overall character of the hydroxyapatite layers formed on the tested materials. The unbuffered SBF-Ø shows minimal changes.
Conclusions
The subject matter of this work was a 7-day in vitro test of 45S5 bioglass in the form of grit in simulated body fluid buffered with HEPES (SBF+H) and compare with the standard (ISO 23317:2014). The most pronounced decrease in weight was observed for the material exposed to the SBF+T solution. Specific surface area measurements confirmed a dramatic increase in the surface area of the material after interaction with the SBF+T and SBF+H solutions. This clearly indicates that a highly dissected hydroxyapatite formed on the surface of the material in the buffered solutions. The change in the concentration of calcium in the SBF+T and SBF+H solutions indicates that the material was dissolving in them during the entire time of the test. (PO4)3− ions in the buffered solutions were initially rapidly released from the material into the solution and then re-precipitated as HAp once the solution close to the surface was saturated. The silicon provide information on how the material behaves in the solutions. For the buffered solutions the change of pH is slightly smaller. The ISO standard specifies a procedure for static bioactivity testing of compact samples and a test interval of several weeks. It is clear that this standard is not sufficient for detailed monitoring/testing of crushed bioactive glass. The ISO standard does not consider solution analysis which is important for comprehensiveness of the results. Further work should focus on a more detailed understanding of behavior of bioactive glass grit in simulated body fluid at shorter time intervals. Moreover, it might be advisable to adapt the standard to a non-compact highly reactive material, both in the terms of the test method and test duration. Because during the in vitro test the HEPES buffer behaves very similarly to the TRIS buffer in contact with bioglass, last but not least, it would be necessary to test a buffer that would not affect the interaction of the material with the solution and at the same time be able to maintain a physiological pH throughout the experiment.Data availability
https://doi.org/10.5281/zenodo.14100336.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the assistance provided by the Research Infrastructure NanoEnviCz, supported by the Ministry of Education, Youth and Sports of the Czech Republic under Project No. LM2023066.References
- E. T. J. Chong, J. W. Ng and P.-Ch Lee, Classification and Medical Applications of Biomaterials – A Mini Review, BIO Integr., 2023, 4(2), 54–61, DOI:10.15212/bioi-2022-0009.
- A. Hossain, S. Roy and P. S. Guin, The Importance of Advance Biomaterials in Modern Technology. A Review, Asian J. Res. Chem., 2017, 10(4), 441–453, DOI:10.5958/0974-4150.2017.00073.6.
- E. Martin, F. Boschetto and G. Pezzotti, Biomaterials and biocompatibility: An historical overview, J. Biomed. Mater. Res., 2020, 108, 1617–1633, DOI:10.1002/jbm.a.36930.
- X. Liu, P. K. Chu and Ch Ding, Surface modification of titanium, titanium alloys, and related materials for biomedical applications, Mater. Sci. Eng., R, 2004, 47(3), 49–121, DOI:10.1016/j.mser.2004.11.001.
- L. L. Hench and I. Thompson, Twenty-first century challenges for biomaterials, J. R. Soc. Interface, 2010, 7, 379–391, DOI:10.1098/rsif.2010.0151.focus.
- K. H. Karlsson and L. Hupa, Thirty-five years of guided tissue engineering, J. Non-Cryst. Solids, 2008, 354, 717–721, DOI:10.1016/j.jnoncrysol.2007.06.100.
- A. Washio, T. Morotomi, S. Yoshii and C. Kitamura, Bioactive Glass-Based Endodontic Sealer as a Promising Root Canal Filling Material without Semisolid Core Materials, Materials, 2019, 12, 3967, DOI:10.3390/ma12233967.
- S. R. Paital and N. B. Dahotre, Calcium phosphate coatings for bio-implant applications: Materials, performance factors, and methodologies, Mater. Sci. Eng., R, 2009, 66(1), 1–70, DOI:10.1016/j.mser.2009.05.001.
- H. Morikawa, S. Lee, T. Kasuga and D. S. Brauer, Effects of magnesium for calcium substitution in P2O5-CaO-TiO2 glasses, J. Non-Cryst. Solids, 2013, 380, 53–59, DOI:10.1016/j.noncrysol.2013.08.029.
- I. D. Xynos, A. J. Edgar, L. D. K. Buttery, L. L. Hench and J. M. Polak, Gene-expression profiling of human osteoblasts following treatment with the ionic products of Bioglass® 45S5 dissolution, J. Biomed. Mater. Res., 2001, 55, 151–157, DOI:10.1002/1097-4636(200105)55:2%3C151::aid-jbm1001%3E3.0.co;2-d.
- W. Höland, W. Vogel and K. Naumann, Interface reactions between machinable bioactive glass-ceramics and bone, J. Biomed. Mater. Res., 1985, 19, 303–312, DOI:10.1002/jbm.820190311.
- O. Bretcanu, X. Chatzistavrou, K. Paraskevopoulos, R. Conradt, I. Thompson and A. R. Boccaccini, Sintering and crystallisation of 45S5 Bioglass® powder, J. Eur. Ceram. Soc., 2009, 29, 3299–3306, DOI:10.1016/j.jeurceramsoc.2009.06.035.
- A. Washio, T. Morotomi, S. Yoshii and Ch Kitamura, Bioactive glass-based endodontic sealer as a promosing root canal filling material without semisolid core materials, Materials, 2019, 12, 3967, DOI:10.3390/ma12233967.
- S. M. Best, A. E. Porter, E. S. Thian and J. Huang, Bioceramics: Past, present and for the future, J. Eur. Ceram. Soc., 2008, 28(7), 1319–1327, DOI:10.1016/j.jeurceramsoc.2007.12.001.
- R. G. Ribas, V. M. Schatkoski, T. L. D. A. Montanheiro, B. R. C. De Menezes, C. Stegemann, D. M. G. Leite and G. P. Thim, Current advances in bone tissue engineering concerning ceramic and bioglass scaffolds: A review, Ceram. Int., 2019, 45(17, Part A), 21051–21061, DOI:10.1016/j.ceramint.2019.07.096.
- W. Cao and L. L. Hench, Bioactive materials, Ceram. Int., 1996, 22(6), 493–507, DOI:10.1016/0272-8842(95)00126-3.
- D. F. Williams, Biocompatibility pathways and mechanisms for bioactive materials: The bioactivity zone, Bioact. Mater., 2022, 10, 306–322, DOI:10.1016/j.bioactmat.2021.08.014.
- G. Thrivikraman, G. Madras and B. Basu, In vitro/In vivo assessment and mechanisms of toxicity of bioceramic materials and its wear particulates, RSC Adv., 2014, 4(25), 12763–12781, DOI:10.1002/chin.201431279.
- T. Kokubo and H. Takadama, How useful is SBF in predicting in vivo bone bioactivity, Biomaterials, 2006, 27(15), 2907–2915, DOI:10.1016/j.biomaterials.2006.01.017.
- ISO 23317:2014 Implants for surgery – In vitro evaluation for apatite-forming ability of implant materials; 2014, Switzerland.
- L. Varila, S. Fagerlund, T. Lehtonen, J. Tuominen and L. Hupa, Surface reactions of bioactive glasses in buffered solutions, J. Eur. Ceram. Soc., 2012, 32, 2757–2763, DOI:10.1016/j.jeurceramsoc.2012.01.025.
- L. Bingel, D. Groh, N. Karpukhina and D. S. Brauer, Influence of dissolution medium pH on ion release and apatite formation of Bioglass 45S5, Mater. Lett., 2015, 143, 279–282, DOI:10.1016/j.matlet.2014.12.124.
- N. E. Good, G. D. Winget, W. Winter, T. N. Connolly, S. Izawa and R. M. M. Singh, Hydrogen Ion Buffers for Biological Research, Biochemistry, 1966, 5(2), 467–477, DOI:10.1021/bi00866a011.
- I. H. Lelong and G. Rebel, pH drift of “physiological buffers” and culture media used for cell incubation during in vitro studies, J. Pharmacol. Toxicol. Methods, 1988, 39, 203–210, DOI:10.1016/s1056-8719(98)00019-7.
- A. Oyane, H.-M. Kim, T. Furuya, T. Kokubo, T. Miyazaki and T. Nakamura, Preparation and assessment of revised simulated body fluids on bioglass, J. Biomed. Mater. Res., 2003, 65A(1), 188–195, DOI:10.1002/jbm.a.10482.
- H. Takadama, M. Hashimoto, M. Mizuno and T. Kokubo, Round-robin test of SBF for in vitro measurement of apatite forming ability of synthetic materials, Phosphorus Res. Bull., 2004, 17(1), 119–125, DOI:10.3363/prb1992.17.0_119.
- D. Rohanová, A. R. Boccaccini, D. M. Yunos, D. Horkavcová, I. Březovská and A. Helebrant, TRIS buffer in simulated body fluid distorts the assessment of glass-ceramic scaffold bioactivity, Acta Biomater., 2011, 7(6), 2623–2630, DOI:10.1016/j.actbio.2011.02.028.
- D. Rohanová, D. Horkavcová, L. Paidere, A. R. Boccaccini, P. Bozděchová and P. Bezdička, Interaction of HEPES buffer with glass-ceramic scaffold: Can HEPES replace TRIS in SBF?, J. Biomed. Mater. Res., Part B, 2018, 106(1), 143–152, DOI:10.1002/jbm.b.33818.
- D. Horkavcová, D. Rohanová, A. Stříbny, K. Schuhladen, A. R. Boccaccini and P. Bezdička, Interaction of MOPS buffer with glass–ceramic scaffold: Effect of (PO4)3− ions in SBF on kinetics and morphology of formatted hydroxyapatite, J. Biomed. Mater. Res., Part B, 2020, 108(5), 1888–1896, DOI:10.1002/jbm.b.34530.
- D. Horkavcová, A. Stříbny, K. Schuhladen, P. Bezdička, A. R. Boccaccini and D. Rohanová, Effect of buffer in simulated body fluid on morphology and crystallinity of hydroxyapatite precipitated on 45S5 bioactive glass-derived glass-ceramic scaffolds: comparison of Good's buffer systems and TRIS, Mater. Today Chem., 2021, 21, DOI:10.1016/j.mtchem.2021.100527.
- R. K. Iler, in Hydrogen-bonded complexes of silica with organic compounds, ed. Bendz G., Lindqvist I. and Runnström-Reio V., Biochemistry of silicon and related problems, Nobel Foundation Symposia, 1978, vol. 40, Springer, Boston, MA DOI:10.1007/978-1-4613-4018-8_2.
- R. K. Iler, The chemistry of silica: Solubility, polymerization, colloid and surface properties, and biochemistry, A Wiley-Interscience publication, Printed in USA, 1979; ISBN 0-471-02404-X.
- A. Nommeots-Nomm, L. Hupa, D. Rohanová and D. S. Brauer, A review of acellular immersion tests on bioactive glasses-influence of medium on ion release and apatite formation, Int. J. Appl. Glass Sci., 2020, 11, 537–551, DOI:10.1111/ijag.15006.
- N. Stone-Weiss, N. J. Smith, R. E. Youngman, E. M. Pierce and A. Goel, Dissolution kinetics of a sodium borosilicate glass in Tris buffer solution: impact of Tris concentration and acid (HCl/HNO3) identity, Phys. Chem. Chem. Phys., 2021, 23, 16165–16179, 10.1039/d0cp06425d.
- R. Conradt, Chemical durability of oxide glasses in aqueous solution: A review, J. Am. Ceram. Soc., 2008, 91(3), 728–735, DOI:10.1111/j.1551-2916.2007.02101.x.
- V. Cannillo, F. Pierli, I. Ronchetti, C. Siligardi and D. Zaffe, Chemical durability and microstructural analysis of glasses soaked in water and in biological fluids, Ceram. Int., 2009, 35, 2853–2869, DOI:10.1016/j.ceramint.2009.03.029.
| This journal is © The Royal Society of Chemistry 2025 |