
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene inks for printing based on thermoresponsive ABC triblock terpolymer gels†
Xu
Liu‡
,
Bailin
Feng‡
,
Stefano
Tagliaferri
 ,
Anna P.
Constantinou
,
Alexandra E.
Porter
,
Cecilia
Mattevi
and
Theoni K.
Georgiou
*
,
Anna P.
Constantinou
,
Alexandra E.
Porter
,
Cecilia
Mattevi
and
Theoni K.
Georgiou
*
Department of Materials, Imperial College London, Royal School of Mines, Exhibition Road, SW7 2AZ, London, UK. E-mail: t.georgiou@imperial.ac.uk
First published on 2nd June 2025
Abstract
Additive manufacturing has revolutionized the fabrication of complex 3D materials. Hydrogels are commonly used as “inks” in 3D printing and offer easy mixing and processing of many materials. Here, the synthesis and characterization of a new library of thermoresponsive ABC triblock copolymers based on oligo(ethylene glycol) methyl ether methacrylate (OEGMA, Molar Mass, MM = 300 g mol−1, A block), 2-phenylethyl methacrylate (PhEMA, B block) and di(ethylene glycol) methyl ether methacrylate (DEGMA, C block) is reported. Polymers of different comonomer compositions were fabricated and investigated in terms of their aqueous solution properties and their ability to form thermogels. The most promising polymer was then used to fabricate a graphene-containing ink, and graphene constructs were successfully printed and characterized in terms of the electrical conductivity properties.
Introduction
Additive manufacturing techniques, also called 3-Dimensional (3D) printing techniques have revolutionized fabricating methods over the last decades.1–3 In 3D printing, the designed structures/objects are manufactured layer-by-layer with computer-controlled translation stages following the data of the computer models. The digital assembly allows rapidly turning computer-aided designs into complex 3D prototypes without wasting excess materials, that is an advantage compared to conventional methods that require dies, molds, or lithographic masks.1–3 Consequently, due to this and the ability to rapidly fabricate products on demand, 3D printing technology has contributed to both industrial and academic research production. 3D printing has been used to fabricate constructs for energy, robotics, aerospace, and healthcare,1–11 while at the same time, it has strengthened the influence of polymers in our society.Many different polymeric materials have been used for 3D printing including thermoplastics, thermosets, elastomers, functional polymers, polymer blends, composites and gels.1–4,7–19 Gels offer the advantage that they can be easily mixed with other ingredients, thus producing more intricate inks, and assisting the printing of conventionally non-printable materials. It is a prerequisite that all “inks” must exhibit non-Newtonian shear-thinning behavior that is a critical property to be considered in 3D extrusion printing.
Many different types of gels have been used in 3D printing but one of the most common components is Poloxamers, which are commercially available and have excellent rheological properties. Poloxamers are nonionic ABA triblock copolymers based on poly(ethylene glycol) (PEG, A block) and poly(propylene glycol) (PPG, B block) and have many trade names including Pluronic, Kolliphor and Synperonic. For example, Pluronic® F127 has been used to print graphene and copper inks to fabricate electrodes for electrochemical energy storage.20 Pluronic® F127 was incorporated in the inks to provide stabilization of the copper and graphene by preventing agglomeration. These inks were characterized by high storage modulus (in the range of 100 kPa), and stable structures were successfully printed.20
Pluronic® F127 solution was also used as a support gel for the printing of an end functionalized dimethacrylate Pluronic® F127, which was crosslinked while printing.21 Graphene oxide and multiwalled carbon nanotubes were incorporated into the ink and were successfully printed.
In some studies, Pluronic® F127 was used as the sacrificial material, i.e., the component which is discarded after the final product is fabricated.22,23 For example a Pluronic® F127 gel was printed in pillar shape and the printed structure was immersed in agarose solution, which was gelled by heating to 35–45 °C, at which both samples were in the gel phase.22 Removal of the Pluronic® F127 construct was achieved by cooling the structure to 4 °C (liquid phase), thus a vascular structure was revealed.22 Similarly, in another study, Pluronic® F127 was used to create channels to mimic vascular structures.23 In this study, Pluronic® F127 gel was successfully printed in 30 layers. The same gel was printed on top of an alginate/gelatin block, and the resulting structure was covered by another alginate/gelatin block. After crosslinking the alginate component, the construct was then cooled down to 4 °C, so Pluronic® F127 gel was liquified and removed from the resulting vascular structure.23
In addition to mixing additives with the pre-synthesized copolymers, copolymers can also be synthesized directly in the presence of additives.24 For example, poly(glycerol monomethacrylate-block-hydroxypropyl methacrylate) (PGMA–PHPMA) graphene oxide gels have been fabricated using two distinct approaches. The first method included the incorporation of graphene oxide into pre-synthesized PGMA–PHPMA copolymers. In contrast, the second method involved the in situ synthesis of HPMA through reversible addition-fragmentation chain transfer (RAFT) polymerization, where PGMA functions as the macromolecular chain transfer agent, with graphene oxide being present throughout the process. Both resulting structures demonstrated good mechanical strength properties and were successfully printed.24
In-house synthesized block copolymers were also used for 3D printing. For example, an ABA triblock copolymer based on isopropyl glycidyl ether (A block) and PEG (B block) was successfully printed in 8 layers.25 A solution of an AB diblock copolymer based on 2-methyl-2-oxazoline and 2-n-propyl-2-oxazine was mixed with fibroblast cells, and the mixture was successfully printed at room temperature.26
Graphene, a single-layer atomic material,27 exhibits extraordinary mechanical strength28 and outstanding conductivity.29 These properties make it highly popular in conductive applications. However, its inherently non-polar nature results in poor dispersibility in aqueous solutions, which makes it difficult to be printed by 3D printer. Consequently, graphene is often functionalized or chemically modified prior to use,30 which in turn affects the conductivity of the final product. Therefore, we developed a series of polymers that contain an aromatic ring, which interact with graphene to function as inks for printing. Therefor here, to the best of our knowledge, we report the first study where ABC triblock copolymer solutions are used as inks for printing. The ABC architecture was chosen, as our group has previously proven than ABC triblock copolymers with the B block as the hydrophobic one show promise as thermoresponsive gels when compared to other terpolymer achitectures.31–37 The AB, BC architecture were chosen for comparison with the ABC architecture. In this study, a new library of thermoresponsive ABC triblock copolymers and two diblock copolymers (AB, CB) based on oligo(ethylene glycol) methyl ether methacrylate (OEGMA, Molar Mass, MM = 300 g mol−1, A block), 2-phenylethyl methacrylate (PhEMA, B block) and di(ethylene glycol) methyl ether methacrylate (DEGMA, C block) were synthesized via group transfer polymerization (GTP). The PhEMA monomer was chosen because it contains benzyl rings and it was hypothesized that it would assist with the stabilization of the graphene-based inks via π–π interactions, while DEGMA and OEGMA were chosen for their relatively low (∼30 °C) and high (∼75 °C) cloud point temperatures (Tcps), respectively,38 which were used to tailor the thermogelling abilities of the polymer. Polymers of varying compositions were fabricated because it is well established that the composition has a key role in the thermogelling properties,19,31–37 and their aqueous solution and thermoresponsive properties were studied. Furthermore, the most promising polymer was chosen to print a graphene containing ink.
Experimental
Materials
Di(ethylene glycol) methyl ether methacrylate (DEGMA, 95%), oligo(ethylene glycol) methyl ether methacrylate (OEGMA, MM = 300 g mol−1, 95%), 2-phenylethyl methacrylate (PhEMA, 99%), 1-methoxy-1-(trimethylsiloxy)-2-methylpropene (MTS, 95%), tetrahydrofuran (THF, ≥99.9%), 2,2-diphenyl-1-picrylhydrazyl (DPPH), calcium hydrate (CaH2, ≥90%), deuterated chloroform (CDCl3, 99.8%), acetone, n-hexane (>95%), tetrahydrofuran (THF, inhibitor-free, high-performance liquid chromatography (HPLC) grade, ≥99.9%), basic aluminum oxide and of poly(methyl methacrylate) (PMMA) standard samples (MM = 2000, 4000, 8000, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 50000, 100000 g mol−1) for the calibration of size exclusion chromatography (SEC) were bought from Sigma-Aldrich. The precursors of the catalyst, tetrabutylammonium bibenzoate (TBABB), were purchased from Acros Organics, U.K.: benzoic acid and tetrabutylammonium hydroxide (40% in water). Graphene (graphene nanoplatelets, 5 μm particle size) was purchased from Sigma Aldrich. The chemical structures of monomers used in this study are shown in Fig. 1.
000, 50000, 100000 g mol−1) for the calibration of size exclusion chromatography (SEC) were bought from Sigma-Aldrich. The precursors of the catalyst, tetrabutylammonium bibenzoate (TBABB), were purchased from Acros Organics, U.K.: benzoic acid and tetrabutylammonium hydroxide (40% in water). Graphene (graphene nanoplatelets, 5 μm particle size) was purchased from Sigma Aldrich. The chemical structures of monomers used in this study are shown in Fig. 1.
 | ||
| Fig. 1 Chemical structures of the monomers OEGMA, PhEMA and DEGMA. | ||
Monomers purification
The low MM monomers such as DEGMA and PhEMA were purified in a similar manner. Firstly, the monomer was based twice through basic alumina to remove any acidic impurities and collected in a round-bottom flask. Then, the free-radical inhibitor DPPH was added into the flask to prevent any unwanted polymerization. Consequently, the drying agent, CaH2, was added to the monomer and it was stirred for at least 4 hours under argon atmosphere. The monomers were then kept in the fridge and were distilled prior use. On the other hand, the OEGMA monomer, a high MM monomer, was firstly mixed with anhydrous THF (50 vol%/50 vol%) and then based through the basic alumina columns twice. No DPPH was added and only CaH2 to dry the monomer solution. This was left to stir for at least 4 hours under argon. The OEGMA monomer was not distilled and was directly syringed into the polymerization flask using a 0.45 μm PTFE filter to remove the solids, similar to the procedure established by Vamvakaki et al.39 MTS, the GTP initiator, was distilled directly under vacuum before polymerization. The catalyst TBABB was synthesized from benzoic acid and tetrabutylammonium hydroxide according to Dicker et al.40 All glassware were dried in the oven at 140 °C overnight prior use.ABC triblock terpolymer synthesis
The amphiphilic ABC triblock terpolymers were synthesized via sequential GTP. A detailed example of a polymer synthesis follows. 53 mL of the polymerization solvent (THF) and 0.5 mL of the initiator (MTS, 0.43 g, 2.5 mmol) were syringed in a 250 mL round-bottom flask that was under argon and contained ∼10 mg of the TBBA catalyst. Then the monomers were added sequentially as shown in Fig. 2. First 15.6 mL of OEGMA solution (7.06 g, 0.02 mol) were syringed and an exotherm was observed. After ∼15 minutes, and when the temperature depleted 1 mL of the solution was withdrawn for Size Exclusion Chromatography (SEC) and Proton Nuclear Magnetic Resonance Spectroscopy (1H-NMR) analyses. Then, 5.2 mL of the second monomer, PhEMA (5.05 g, 0.03 mol) were injected into the polymerization flask. Again, after the temperature came back to room temperature a sample was taken for SEC and NMR. Finally, the last monomer was added. Specifically, 7.9 mL of DEGMA (8.07 g, 0.04 mol) were added and after 15 minutes a sample was again taken for SEC and NMR analyses. The reaction was terminated with 1 mL of methanol. All polymers were synthesized using a similar methodology and what was varied were the monomer ratios. The polymers were precipitated in cool n-hexane and were left to dry in a vacuum oven at room temperature for at least a week before use.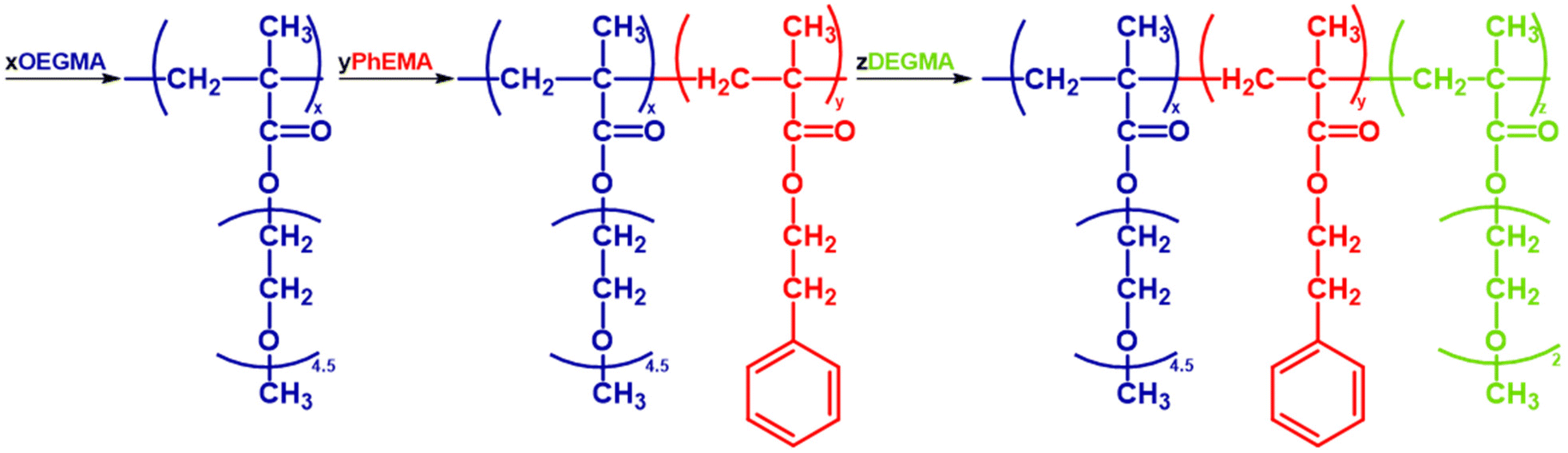 | ||
| Fig. 2 The sequential GTP synthesis of an OEGMA-b-PhEMA-b-DEGMA triblock copolymer. TBABB, THF and MTS represent the polymerization catalyst, solvent, and initiator respectively. | ||
Characterization in organic solvents
For the sample preparation ∼0.2 mL of the polymer solution (directly sampled from the polymerization flask) was added in a glass 2 mL vial. Then around 0.9 mL of solvent was added, and the sample was filters using a 0.45 μm PTFE filter before being analyzed.
Characterization in aqueous solutions
The dhs, as determined by DLS, are compared to the theoretical (calculated) diameters, which assume the formation of classical core–shell spherical micelles. The equation used for all ABC triblock terpolymers is the following: dh (nm) = (DPPhEMA + 2 × DPOEGMA or DPDEGMA) × 0.254, depending on whether DPOEGMA > DPDEGMA or DPOEGMA < DPDEGMA, respectively, where DP stands for degree of polymerization.36,41 For diblock copolymers, the equation is: dh (nm) = (DPhydrophobic block + 2 × DPhydrophilic block) × 0.254 (hydrophobic is always PhEMA while the hydrophilic block is either OEGMA or DEGMA).42 The DPs used in the calculations are experimentally determined, as resulted from 1H NMR spectroscopy, and the Mn values, as resulted from SEC analysis.
Visual tests of graphene/polymer mixtures in water were also performed. The graphene was kept constant at 10 w/w% while the polymer, P10: OEGMA10-b-PhEMA17-b-DEGMA11, concentration varied from 1 to 20 w/w%.
Printing polymer/graphene aqueous solutions
The graphene inks (P10 10 w/w% + 10 w/w% graphene) were sonicated for 5 minutes and loaded into 3 mL polypropylene syringes with blunt nozzles (400 μm inner diameter) for Direct Ink Writing. The Direct Ink Writing of the inks was performed using a customized 3D printer (3D Inks, LLC), consisting of a Cartesian coordinate robot with a nominal precision of 1 μm. The syringes were mounted onto the three-axis stage of the 3D printer and connected to a displacement-controlled plunger, which provided a feed rate of 6 mm s−1. RoboCAD 5 was used to design the CAD model and convert it into G-code. P10: OEGMA10-b-PhEMA17-b-DEGMA11/graphene was printed onto glass substrates heated to 40 °C using a hotplate.Conductivity measurements
The electrical conductivity of the printed structure was tested utilizing a four-electrode measurement. Conductive silver paint (RSPRO conductive paint) was used to create four equally spaced contacts, each spaced 1.2 cm apart, on a printed line. A Gamry interface 1000 galvanostat was used to obtain the I–V curve of the printed structure through a 4-probe measurement. The electrical conductance G was determined by fitting the obtained I–V curve with a linear fitting. Subsequently, the electrical conductivity σ was calculated from the conductance G, the equation of which is:where l is the distance between the contacts and A is the cross-sectional area of the printed line.

Results and discussion
Polymer synthesis
All polymers were fabricated via GTP, a living polymerization method, that specializes on methacrylate polymers.44–46 Two diblock copolymers (AB, BC) and a series of ABC triblock copolymers based on OEGMA, PhEMA and DEGMA of varying composition were fabricated. PhEMA was chosen to be the B block of the ABC triblock copolymers, because our group has established that this architecture has better sol–gel thermogelling properties in triblock copolymers based on different monomers.31,33–36,47Molar mass and composition
The target MM of all copolymers was 8000 g mol−1, based on previous studies indicating that copolymers with MMs in the range of 7000 to 10000 g mol−1 exhibit optimal thermoresponsive behavior, including a clear sol–gel transition and good mechanical properties.41,48 In this study, four groups with four different compositions of PhEMA were investigated: 25 w/w% (P1–P3), 30 w/w% (P4–P6), 35 w/w% (P7–P9), and 40 w/w% (P10–P14). The ratio of the OEGMA and DEGMA units was varied to tailor the thermoresponsive property of triblock polymers OEGMA-b-PhEMA-b-DEGMA in each group. The structures of triblock polymers and diblock polymers are schematically illustrated in Fig. 3. OEGMA, PhEMA and DEGMA units are represented in blue, red, and green, respectively.
 | ||
| Fig. 3 Schematic illustration of the synthesized OEGMA-b-PhEMA-b-DEGMA copolymers. OEGMA, PhEMA and DEGMA units are represented in blue, red, and green, respectively. | ||
The MM characteristics and dispersity (Đ) of diblock and triblock polymers and their corresponding precursors were determined by SEC. The SEC traces of the precursors of P1 and the final synthesized polymer, OEGMA8-b-PhEMA11-b-DEGMA20, are shown in Fig. 4. The precursors, OEGMA8 and OEGMA8-b-PhEMA11 are denoted with blue and red curves respectively, while the ABC triblock copolymer OEGMA8-b-PhEMA11-b-DEGMA20 is in green. It can be seen from the SEC traces that the MM increased at each polymerization step and that no other peaks or shoulders are presented confirming a successful sequential GTP. This was similar for all synthesized copolymers (see Fig. S1 in ESI†).
 | ||
| Fig. 4 The SEC traces of P1: OEGMA8-b-PhEMA11-b-DEGMA20 and its precursors OEGMA8 and OEGMA8-b-PhEMA11 are shown in blue, red and green, respectively. | ||
The theoretical polymer structure, theoretical MM, experimental determined number average MM (Mn), dispersity (Đ), theoretical and experimental composition of all synthesized polymers are listed in Table 1. To maintain clarity in the main text, detailed data for the polymer precursors are provided separately in Table S1 in the ESI.† The Mns and Đs were obtained from SEC measurement, while the experimental compositions were determined by NMR. The data confirms the successful synthesis of all polymers, each exhibiting a narrow MM distribution (MMD) with Đ values ranging from 1.10 to 1.20.
| No. | Theoretical polymer structurea | Target MM (g mol−1) |
M
n
(g mol−1) |
Đ | w/w% OEG-b-PhE-b-DEG | |
|---|---|---|---|---|---|---|
| Theoretical | 1H NMR | |||||
| a OEG, PhE and DEG are further abbreviations of oligo(ethylene glycol) methyl ether methacrylate, 2-phenylethyl methacrylate and di(ethylene glycol) methyl ether methacrylate, respectively. b Number-average molar mass (Mn) and dispersity (Đ) were determined by SEC. The SEC was calibrated by using poly(methyl methacrylate) (PMMA) standard samples. | ||||||
| P1 | OEG8-b-PhE11-b-DEG20 | 8200 | 9700 | 1.20 | 30-25-45 | 31-24-45 |
| P2 | OEG10-b-PhE11-b-DEG17 | 8200 | 9870 | 1.17 | 35-25-40 | 38-25-37 |
| P3 | OEG11-b-PhE11-b-DEG15 | 8200 | 10100 |
1.15 | 40-25-35 | 43-24-33 |
| P4 | OEG12-b-PhE13-b-DEG11 | 8200 | 9550 | 1.12 | 45-30-25 | 47-29-24 |
| P5 | OEG14-b-PhE13-b-DEG9 | 8200 | 9550 | 1.13 | 50-30-20 | 52-30-18 |
| P6 | OEG15-b-PhE13-b-DEG7 | 8200 | 9810 | 1.11 | 55-30-15 | 59-28-13 |
| P7 | OEG11-b-PhE15-b-DEG11 | 8200 | 10500 |
1.14 | 40-35-25 | 43-33-24 |
| P8 | OEG12-b-PhE15-b-DEG9 | 8200 | 11300 |
1.13 | 45-35-20 | 47-33-20 |
| P9 | OEG14-b-PhE15-b-DEG7 | 8200 | 8490 | 1.13 | 50-35-15 | 51-34-15 |
| P10 | OEG10-b-PhE17-b-DEG11 | 8200 | 8200 | 1.16 | 35-40-25 | 36-39-25 |
| P11 | OEG11-b-PhE17-b-DEG9 | 8200 | 8600 | 1.15 | 40-40-20 | 42-39-19 |
| P12 | OEG12-b-PhE17-b-DEG7 | 8200 | 9000 | 1.14 | 45-40-15 | 47-38-15 |
| P13 | OEG16-b-PhE17 | 8200 | 8300 | 1.17 | 60-40-0 | 61-39-0 |
| P14 | DEG26-b-PhE17 | 8200 | 10400 |
1.17 | 0-40-60 | 0-40-60 |
As shown in Table 1, the Đ value of each polymer ranges from 1.10 to 1.20, which is satisfactory low for polymers obtained via GTP when a OEG(PEG)-based methacrylate macromonomer was used in the polymerisation.31,33–36,39,47–50 This is because the OEGMA monomer is a macromonomer and has its own MMD and also it cannot be purified as well as the other monomers, resulting to broader MMD.
When comparing the theoretical MM with the experimental number average MM, Mn values in Table 1, it can be concluded that some deactivation was presented for all polymerizations, thus resulting to higher MM than theoretically predicted. This was expected and observed before in other GTP studies when using the OEGMA monomer.31,33–36,39,49,50 Specifically, the Mns for all polymers ranged from 8 to 11 kg mol−1.
The experimental compositions determined by 1H NMR, along with theoretical compositions, are also provided in Table 1 for the final polymers and the same information for both the precursors to the polymers and the copolymers is provide in Table S1.†Fig. 5 shows the 1H NMR spectra of P1: OEGMA8-b-PhEMA11-b-DEGMA20 and its linear precursors. It can be observed there are many overlapping peaks between OEGMA and DEGMA because of similar chemical structures, while the PhEMA has one distinct peak at 2.8–3.0 ppm in the NMR spectrum. Therefore, the ratio of OEGMA/PhEMA/DEGMA could not be directly calculated via the NMR spectrum of triblock polymer Fig. 5(c). Therefore, the NMR spectrum of the precursors (diblock polymer) Fig. 5(b) was used to obtain the ratio of OEGMA/PhEMA and the ratio of (OEGMA + DEGMA)/PhEMA was calculated via the NMR spectrum of the triblock polymer Fig. 5(c). Then, the DEGMA/PhEMA was calculated by using the ratio of (OEGMA + DEGMA)/PhEMA minus the ratio of OEGMA/PhEMA. Thus, the ratio of OEGMA/PhEMA/DEGMA was obtained. All the determined ratios are presented in Table 1 and Table S1,† and it can be concluded that the experimental compositions are sufficiently close to the theoretical compositions, thus confirming a successful synthesis.
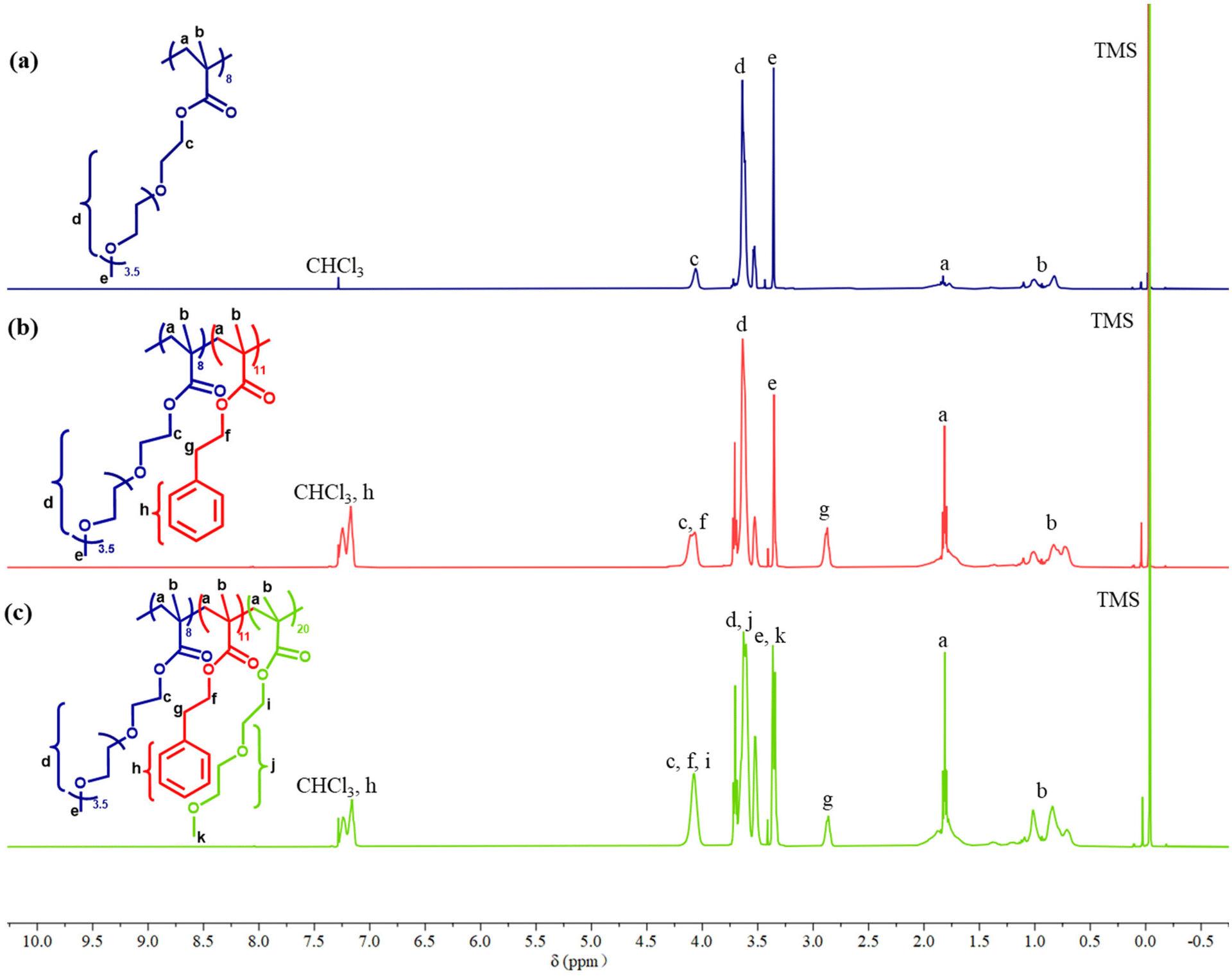 | ||
| Fig. 5 1H NMR spectra of (a) homopolymer OEGMA8, (b) diblock polymer OEGMA8-b-PhEMA11, and (c) ABC triblock terpolymer OEGMA8-b-PhEMA11-b-DEGMA20. The chemical structures of the corresponding polymers are also shown and the different 1H are labelled in the spectra. | ||
Aqueous properties
The aqueous and thermoresponsive properties of the ABC triblock polymers (1 w/w%) in DI water were investigated by DLS and UV-Vis and the results are presented in Table 2. All polymers are soluble in water, except for P1 and P14. This is due to the high hydrophobicity of these two copolymers.| No. | Theoretical polymer structurea | w/w% PhEMA | Hydrodynamic diameter (dh, nm) | Cloud point Temperature (Tcp, °C) | ||
|---|---|---|---|---|---|---|
| Theor.b | Experim.c | PDI | UV-vis (± 1) | |||
| a OEG, PhE and DEG are further abbreviations of oligo(ethylene glycol) methyl ether methacrylate, 2-phenylethyl methacrylate and di(ethylene glycol) methyl ether methacrylate respectively. b The calculation of theoretical diameter is based on the polymer structure of this polymer series and experimental degree of polymerization (DP). For ABC triblock polymers, two equations are applied: dh (nm) = (DPB + 2 × DPA) × 0.254 or dh (nm) = (DPB + 2 × DPC) × 0.254. It depends on if DPA > DPC or DPC > DPA.36,41 For AB diblock polymers, the equation is: dh (nm) = (DPB + 2 × DPA) × 0.254.42 For all polymers in this series, A, B and C represent OEGMA, PhEMA and DEGMA respectively. c The experimental diameters shown are the average values of the values of peaks by intensity in the DLS curves. d P1 (OEG8-b-PhE11-b-DEG20) and P14 (DEG26-b-PhE17) are insoluble at 1 w/w% in DI water. | ||||||
| P1 | OEG8-b-PhE11-b-DEG20 | 25 | — | —d | —d | —d |
| P2 | OEG10-b-PhE11-b-DEG17 | 25 | 11.9 | 13.5 | 0.053 | 48 |
| P3 | OEG11-b-PhE11-b-DEG15 | 25 | 12.7 | 13.5 | 0.040 | 54 |
| P4 | OEG12-b-PhE13-b-DEG11 | 30 | 11.9 | 15.7 | 0.056 | 56 |
| P5 | OEG14-b-PhE13-b-DEG9 | 30 | 13.0 | 18.2 | 0.065 | 55 |
| P6 | OEG15-b-PhE13-b-DEG7 | 30 | 13.7 | 18.2 | 0.123 | 57 |
| P7 | OEG11-b-PhE15-b-DEG11 | 35 | 12.7 | 21.0 | 0.059 | 56 |
| P8 | OEG12-b-PhE15-b-DEG9 | 35 | 13.5 | 24.4 | 0.104 | 59 |
| P9 | OEG14-b-PhE15-b-DEG7 | 35 | 12.4 | 24.4 | 0.112 | 52 |
| P10 | OEG10-b-PhE17-b-DEG11 | 40 | 9.8 | 18.2 | 0.054 | 45 |
| P11 | OEG11-b-PhE17-b-DEG9 | 40 | 10.6 | 18.2 | 0.039 | 52 |
| P12 | OEG12-b-PhE17-b-DEG7 | 40 | 11.7 | 21.0 | 0.037 | 49 |
| P13 | OEG16-b-PhE17 | 40 | 12.9 | 24.4 | 0.077 | 55 |
| P14 | DEG26-b-PhE17d | 40 | — | —d | —d | —d |
Hydrodynamic diameters
The hydrodynamic diameters were measured by DLS. To compare with experimental data, theoretical hydrodynamic diameters were calculated based on the polymer structures and experimentally determined DP, using the equation described in the Characterization in aqueous solution section. These calculations are based on the assumption that the copolymers self-assemble into spherical micelles with a core-shell structure in aqueous solution.A spherical micelle with the core-shell structure of P1: OEGMA8-b-PhEMA11-b-DEGMA20 is schematically illustrated in Fig. 6. In this illustration, the units of OEGMA, PhEMA and DEGMA are represented in blue, red and green spheres respectively. The core of the spherical micelle consists of PhEMA units, and the shell of the spherical micelle is made from OEGMA and DEGMA units. All other copolymers self-assemble in a similar way to form spherical micelles with the core-shell structure as shown in Fig. 6.
 | ||
| Fig. 6 A schematic illustration of a spherical micelle with the core-shell structure of P1 OEGMA8-b-PhEMA11-b-DEGMA20. The units of OEGMA, PhEMA and DEGMA are represented in blue, red, and green spheres respectively. | ||
It can be seen from Fig. 6 that the size of the micelle depends on the length of hydrophilic part and hydrophobic part. The hydrodynamic diameter (dh) of the copolymers synthesized in this paper varied from 13.5–24.4 nm. The experimental diameters are slightly higher than the theoretical diameters in all samples as can be observed in Table 2 (DLS histograms are provided in Fig. S3 in the ESI†). Similar results have been reported in our previous study.30,31,36,39,51 This is because the theoretical calculations do not consider the long OEG side chain of OEGMA300 and assume complete overlap of the hydrophobic part (PhEMA block) of the polymer.44 In addition, the PDI values range from 0.040 to 0.123 in Table 2, thus are satisfactory low and no other peaks were detected by intensity. This suggests that the size of the self-assembled particles in polymer solutions is uniform.
Cloud points temperatures
The Tcps by turbidimetry of 1 w/w% polymer solutions in DI water are presented in Table 2 and the transmittance versus temperature graphs are provided in Fig. S4 in the ESI.† The Tcps range from 45 to 59 °C and different trends are observed as the hydrophilic OEGMA component is increased. Specifically, for the 25 w/w%, 30 w/w% and 40 w/w% of PhEMA, the Tcp decreases as the hydrophilic OEGMA component decreases, as it was observed before.33,35,37,48,52,53 P10 (OEG10-b-PhE17-b-DEG11) exhibits the lowest Tcp as expected, which is 45 °C. This is because P10 is the most hydrophobic polymer among the 13 copolymers studied, with 40 w/w% PhEMA content. However, for the 35 w/w% PhEMA copolymers the opposite trend is observed, i.e., the Tcps decrease with increasing the hydrophilic OEGMA content. Even more surprising, at the intermediate hydrophobic content of 35 w/w% a maximum is observed as the hydrophilic content is increased. These opposite trends could be explained by the fact that all the polymers are in a self-assembled structure and what could have key role in the Tcp is the corona of the micelle that is consisted mostly by the OEGMA content. Thus, it is believed that the size of the corona will have the biggest influence and as this is increased, the Tcp will increase, as the polymer will be able to stabilize themselves in solution better (for higher temperatures). Therefore, P8 (OEG12-b-PhE15-b-DEG9) has the highest Tcp because of the biggest micelle size. In addition, it should also be pointed out that this the theoretical DP and MM differences will also affect the Tcp as it has established that the MM also has a significant effect on the Tcp.37,48,51,54–56Gelation points by visual tests
The gelation points of polymer P1–P13 OEGMAx-b-PhEMAy-b-DEGMAz were investigated at different concentrations (1, 2, 5, 10, 15, 20 w/w%) in DI water by visual tests. The representative optical images illustrating the various states of the polymer solutions are shown in Fig. S5 (ESI†), and the phase diagrams of these polymers are presented in Fig. 7. The composition of OEGMA decreases from left to right and the composition of PhEMA increases from top to bottom. Four different phases can be observed: runny solution shown in the transparent shapes (square: clear solution, triangle: slightly cloudy solution and circle: cloudy solution), viscous solution shown in the red shapes (triangle: transparent viscous solution, circle: cloudy viscous solution), gel shown in the blue shapes (triangle: transparent gel, circle: cloudy gel) and two-phases shown in the green shapes (circle: gel syneresis, square: precipitation). | ||
| Fig. 7 Phase diagrams of polymers P1–P13 OEGMAx-b-PhEMAy-b-DEGMAz with different concentrations (1, 2, 5, 10, 15, 20 w/w%) in DI water. The composition of DEGMA increases from left to right and the composition of PhEMA increases from top to bottom. Four different phases can be observed: runny solution shown in the transparent shapes (square: clear solution, triangle: slightly cloudy solution and circle: cloudy solution), viscous solution shown in the red shapes (triangle: transparent viscous solution, circle: cloudy viscous solution), gel shown in the blue shapes (triangle: transparent gel, circle: cloudy gel) and two-phases shown in the green shapes (circle: gel syneresis, square: precipitation). | ||
From all ABC triblock copolymers only five polymers with the higher PhEMA content (35 w/w% and 40 w/w%) were able to form thermogels (shown in blue triangles and circles). The need to have at least ∼35 w/w% hydrophobic content to form thermogels have been previously observed by our group in OEGMA based ABC triblock copolymers with different hydrophobic block.33,36,47,51 When the PhEMA content is 35 w/w%, P8 and P9 can form thermogels at a critical gelation concentration (CGC) of 20 and 10 w/w% copolymer in solution respectively. The gels are observed at 48 and 49 °C for P8 and P9 at the CGC, respectively. When the PhEMA content is 40 w/w%, all the copolymers can form gels. It is found that as the hydrophilicity of the copolymers increases, the gel region of the copolymers decreases, which means the copolymers formed gels at higher concentration and higher temperature, while P10 has the largest gel area, and P13 has the smallest one. This tendency is due to the inhibited hydrophobic effect. Moreover, it is observed that P10 has two gel regions, which may be due to possibly a change in the self-assembled polymer structure as the temperature increases. As the primary objective of this study was to identify the gelation temperature necessary for establishing optimal conditions for subsequent 3D printing, the underlying mechanisms of the two distinct gel regions were not investigated further. It was also observed, as expected, that when the concentration of the copolymer increases, the gelation temperature decreases, indicating that it can be tenably controlled by the polymer concentration. P10 is the best performing polymer that forms gels even at 31 °C at a concentration of 10 w/w%.
Gelation points by rheology tests
The rheological properties of the samples that visually formed a thermogel were investigated over a temperature range of 20 °C to 60 °C. The results of P9 and P10 are shown in Fig. 8, while the rheological data for the other polymers are provided in Fig. S6 (ESI†). The storage modulus (G′) and loss modulus (G′′) are denoted in blue and red circles, respectively. In rheology the formation of a gel is defined as the point where the storage modulus exceeds the loss modulus, i.e., when G′ > G′′.36,57 For all samples the gelation point agrees with the visual observations. | ||
| Fig. 8 Changes of moduli (storage modulus G′ is denoted by blue circles and the loss modulus G′′ is denoted by red circles) with temperature for the polymer solutions of P9 and P10 that can form the gel at 15 w/w% and 20 w/w% in DI water. | ||
Specifically, for P9 and P10 at concentrations of 15 w/w% and 20 w/w%, there are two intersecting areas for each concentration. For P9, the gelation points at 15 w/w% and 20 w/w% are 46 °C and 45 °C, respectively, and they agree with the visual results within experimental error. The second crossovers of these two concentrations appear at 50 °C and 51 °C as the temperature increases. They also match with results obtained from the visual tests that both precipitations are observed at 53 °C. However, there is an interesting trend for P9 that the storage modulus still exceeds the loss modulus after the second crossover. It does not indicate the gel still exists because both magnitudes of two moduli decrease obviously compared with the maximum of two moduli. The magnitude of the modulus represents the strength and viscosity of the gel, and the phase separation usually happens when the magnitude decreases dramatically. It means there is a two-phase solution after the second crossover of both concentrations that is supported by visual observations. It is worth mentioning that the gel must exhibit sufficient strength to prevent flow upon tube inversion during the visual test. However, rheological test may still classify a very soft material as a gel, provided that the energy stored within the system exceeds the energy dissipated as heat. Thus, it is not surprising that the results of the rheological measurements do not perfectly align with those obtained by visual tests. In contrast, the storage modulus of P10 at the second intersecting area remains the same or a little higher than the storage modulus of the first intersecting area, which indicates that P10 can form gels twice. This phenomenon was also found in the visual test. Moreover, P10 has the maximum storage moduli among the tested copolymers, which means P10 forms the strongest gels and as expected the strength of the gel increased by increasing the polymer concentration.
Application in 3D printing
The most promising polymer, P10 OEGMA10-b-PhEMA17-b-DEGMA11 was chosen to be trialed as an “ink” for 3D printing based on the visual and rheological results. The hypothesis was that the benzyl ring in the PhEMA units will assist in stabilizing graphene. To identify what graphene/polymer ratio could be used to print, the phase diagram of P10 was constructed again in the presence of 10 w/w% graphene and the result is presented in Fig. 9. Representative optical images of the ink in various states as the temperature increases are provided in Fig. S7 (ESI†) | ||
| Fig. 9 A phase diagram of polymers P10 OEGMA10-b-PhEMA17-b-DEGMA11 with different concentrations (1, 2, 5, 10, 15, 20 w/w%) mixed with 10 w/w% of graphene in DI water. | ||
According to the Fig. 9, the gelation points of P10 at 10, 15 and 20 w/w% with 10 w/w% graphene are 30 °C, 28 °C and 27 °C respectively. When compared with the corresponding gelation points of P10 in Fig. 7, the gelation points in the presence of graphene are lower. Thus, it can be concluded that graphene has assisted the formation of a gel at lower temperatures, as was expected since it forms π–π interactions with the polymer. This observation was similar to a study where an oligolysine end functionalized Pluronic® F127 was mixed with graphene oxide (GO) and GO reduced the gelation point of Pluronic® F127 through electrostatic interactions.58
To minimize the copolymer content, a solution mixture containing 10 w/w% of P10 and 10 w/w% of graphene was selected as the ink. Initially, the shear-thinning behavior of the ink was examined at 40 °C, which is shown in Fig. 10. It can be seen from Fig. 10 that when the shear rate increases, the viscosity of the ink decreases significantly, indicating the expected shear-thinning property of the ink. This property enhances the extrudability of the ink through nozzles during the printing process, facilitating the fabrication of the desired printed structures.6
 | ||
| Fig. 10 Viscosity-shear rate curve for the ink (10 w/w% P10 and 10 w/w% graphene in DI water) at 40 °C. | ||
In addition, the rheological property of the ink has also been investigated by the rheology test. The result is shown in Fig. 11. From Fig. 11, it can be concluded that the ink formed gels at 30 °C and precipitated at 41 °C, which matches with the observations from the visual test.
 | ||
| Fig. 11 Changes of moduli (storage modulus G′ is denoted by blue circles and the loss modulus G′′ is denoted by red circles) with temperature for the ink (10 w/w% P10 and 10 w/w% graphene in DI water). | ||
Both the visual test observations and the rheological data of the ink support the conclusion that the interaction between hydrophobic graphene particles and the amphiphilic block copolymer significantly influences the self-assembly behavior and gelation properties of the system. The triblock terpolymer OEGMA10-b-PhEMA17-b-DEGMA11 self-assembles in aqueous solution due to its amphiphilic architecture: at lower temperatures, the hydrophilic OEGMA and DEGMA blocks form the corona, while the hydrophobic PhEMA block aggregates to form the micellar core. As temperature increases, the thermoresponsive DEGMA block becomes increasingly hydrophobic,59 contributing to micelle core formation and triggering gelation. Upon the addition of graphene, the polymer solution exhibits earlier gelation, a broader gelation temperature window, and an increased G′, indicating enhanced mechanical strength.
This enhancement is attributed to specific π–π stacking interactions between the benzene rings of the PhEMA block and the π-conjugated graphene surface. These non-covalent interactions drive graphene to preferentially localize in the hydrophobic micelle core during self-assembly. Rather than disrupting micelle formation, the graphene particles integrate into the micellar structures, reinforcing the micellar network and enhancing intermicellar connectivity. In this system, graphene acts as a physical crosslinking or reinforcing agent within the gel. This phenomenon is consistent with previous reports showing that graphene or graphene oxide, when incorporated into amphiphilic polymer systems, can improve viscoelastic properties and gel stability via physical entanglement and π–π interactions.60–62
Therefore, an aqueous solution of 10 w/w% P10 and 10 w/w% graphene was selected as the ink because it presents the highest graphene/polymer ratio, and it is still able to form a thermogel. The polymer/graphene ink was prepared and processed at ambient temperature, followed by being printed using a 3D printer onto a heated substrate maintained at 40 °C to achieve stable serpentine structures. After printing, the polymer-based structures were air-dried overnight to remove any residual solvent. The dried samples were then annealed in a tubular furnace at 350 °C for 30 minutes to promote the carbonisation of the terpolymer,63,64 ensuring proper stabilisation of the printed structures prior to the SEM and four-point probe conductivity tests.
The successfully printed structure is shown in Fig. 12. Fig. 12 shows that the edges of the printed structure are well defined, which indicates the high potential of this kind of ink. The electrical conductivity of this printed structure was tested via a four-electrode measurement (Fig. 13a), exhibiting an electrical conductivity of ∼2760 S m−1. This conductivity exceeds that of recently reported graphene structures fabricated using Direct Ink Writing (Fig. 13b),65–67 attributed to the utilization of graphene nanoplatelets instead of reduced graphene oxide and the minimal requirement of polymeric binder during the printing procedure. This kind of structure might find application as sensors and energy storage systems owing to the conductive and capacitive nature of graphene platelets.63 This shows that with further studies and optimization this type of ABC triblock copolymer could be used to print graphene containing inks.
 | ||
| Fig. 12 SEM images of the printed structures based on 10 w/w% of P10 OEGMA10-b-PhEMA17-b-DEGMA11 with 10 w/w% of graphene. (a) A two-layer cross-grid pattern, (b) a single-layer serpentine pattern, (c and d) surface morphology of the printed structure. | ||
 | ||
| Fig. 13 (a) I–V curve of the printed structure; (b) electrical conductivity of the printed structure compared with three different DIW inks.65–67 | ||
It is important to note that this study focused on a single graphene addition (10 w/w%), and this study did not systematically investigate the effect of graphene concentration. However, previous studies have observed that polymer-graphene systems can exhibit concentration-sensitive rheological and structural properties, where excessive graphene content may lead to micelle disruption or phase separation.61,68 Future work may focus on optimizing the graphene ratio to further tailor gelation behavior and mechanical properties. Despite this limitation, this study clearly demonstrates that introducing 10 w/w% graphene enhances the thermoresponsive gelation and mechanical performance of the triblock terpolymer system and this type of ABC triblock copolymer could be used to print graphene containing inks.
Conclusions
In this study, a new series of ABC triblock copolymers and two diblock copolymers (AB, CB) were successfully synthesized via GTP based on OEGMA (A), PhEMA (B) and DEGMA (C). The targeted MMs for all polymers were the same but thirteen polymers of varying composition were fabricated and their thermoresponsive and aqueous solution properties were investigated. These properties were strongly affected by the composition of the ABC triblock terpolymers and five out of thirteen polymers were found to form thermogels at elevated temperatures. The most promising thermogel, P10 OEGMA10-b-PhEMA17-b-DEGMA11, was then mixed with (10 w/w%) graphene and the gelation temperature was decreased as graphene assisted with the formation of the physical gel. Finally, a 10 w/w% graphene and 10 w/w% OEGMA10-b-PhEMA17-b-DEGMA11 “ink” was used to successfully print a stable structure, which has an excellent electrical conductivity of ∼2760 S m−1.Author contributions
BF and XL carried most of the polymer synthesis and characterization and wrote the first draft. ST did the printing of graphene inks. APC helped with polymer synthesis, data analysis and the cloud point temperature determination. AEP assisted with the supervision of the project. CM supervised the graphene ink printing. TKG had the idea, supervised, coordinated the project and edited the manuscript.Abbreviations
| CP | Cloud point |
| DEGMA | Di(ethylene glycol) methyl ether methacrylate (DEGMA) |
| DLS | Dynamic light scattering |
| SEC | Gel permeation chromatography |
| GTP | Group transfer polymerization |
| MTS | Methyltriemthylsilyl dimethylketene acetal |
| MM | Molar mass |
| NMR | Nuclear magnetic resonance |
| OEGMA | Oligo(ethylene glycol) methyl ether methacrylate |
| PhEMA | 2-Phenylethyl methacrylate |
| PTFE | Poly(tetrafluoroethylene) |
| TBABB | Tetrabutylammonium bibenzoate |
| THF | Tetrahydrofuran. |
Data availability
The data supporting this article has been included in the ESI of this manuscript.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. P. C. acknowledges the Engineering and Physical Sciences Research Council (EPSRC) for the Doctoral Prize Fellowship (EP/M506345/1). C. M. would like to acknowledge the award of funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (Grant Agreement No. 819069) and the award of a Royal Society University Research Fellowship (UF160539).References
- D. Bhardwaj, R. Singhmar, M. Garg, D. Gupta, A. Dhiman, S. S. Han and G. Agrawal, Designing advanced hydrogel inks with direct ink writing based 3D printability for engineered biostructures, Eur. Polym. J., 2024, 205, 112736, DOI:10.1016/j.eurpolymj.2023.112736.
- Z. Jiang, B. Diggle, M. L. Tan, J. Viktorova, C. W. Bennett and L. A. Connal, Extrusion 3D Printing of Polymeric Materials with Advanced Properties, Adv. Sci., 2020, 7(17), 2001379, DOI:10.1002/advs.202001379.
- T. Huang, W. F. Liu, C. L. Su, Y. Y. Li and J. Y. Sun, Direct ink writing of conductive materials for emerging energy storage systems, Nano Res., 2022, 15(7), 6091–6111, DOI:10.1007/s12274-022-4200-2.
- N. Parvin, V. Kumar, S. W. Joo and T. K. Mandal, Cutting-Edge Hydrogel Technologies in Tissue Engineering and Biosensing: An Updated Review, Materials, 2024, 17(19), 4792, DOI:10.3390/ma17194792.
- S. Tajik, C. N. Garcia, S. Gillooley and L. Tayebi, 3D Printing of Hybrid-Hydrogel Materials for Tissue Engineering: a Critical Review, Regener. Eng. Transl. Med., 2023, 9(1), 29–41, DOI:10.1007/s40883-022-00267-w.
- S. Tagliaferri, A. Panagiotopoulos and C. Mattevi, Direct ink writing of energy materials, Mater. Adv., 2021, 2(2), 540–563, 10.1039/d0ma00753f.
- J. Kapr, L. Petersilie, T. Distler, I. Lauria, F. Bendt, C. M. Sauter, A. R. Boccaccini, C. R. Rose and E. Fritsche, Human Induced Pluripotent Stem Cell-Derived Neural Progenitor Cells Produce Distinct Neural 3D In Vitro Models Depending on Alginate/Gellan Gum/Laminin Hydrogel Blend Properties, Adv. Healthcare Mater., 2021, 10(16), 2100131, DOI:10.1002/adhm.202100131.
- T. Göckler, S. Haase, X. Kempter, R. Pfister, B. R. Maciel, A. Grimm, T. Molitor, N. Willenbacher and U. Schepers, Tuning Superfast Curing Thiol-Norbornene-Functionalized Gelatin Hydrogels for 3D Bioprinting, Adv. Healthcare Mater., 2021, 10(14), 2100206, DOI:10.1002/adhm.202100206.
- T. Distler, C. Polley, F. Shi, D. Schneidereit, M. D. Ashton, O. Friedrich, J. F. Kolb, J. G. Hardy, R. Detsch, H. Seitz and A. R. Boccaccini, Electrically Conductive and 3D-Printable Oxidized Alginate-Gelatin Polypyrrole:PSS Hydrogels for Tissue Engineering, Adv. Healthcare Mater., 2021, 10(9), 2001876, DOI:10.1002/adhm.202001876.
- J. Gao, X. Ding, X. Yu, X. Chen, X. Zhang, S. Cui, J. Shi, J. Chen, L. Yu, S. Chen and J. Ding, Cell-Free Bilayered Porous Scaffolds for Osteochondral Regeneration Fabricated by Continuous 3D-Printing Using Nascent Physical Hydrogel as Ink, Adv. Healthcare Mater., 2021, 10(3), 2001404, DOI:10.1002/adhm.202001404.
- A. Dobos, J. Van Hoorick, W. Steiger, P. Gruber, M. Markovic, O. G. Andriotis, A. Rohatschek, P. Dubruel, P. J. Thurner, S. Van Vlierberghe, S. Baudis and A. Ovsianikov, Thiol–Gelatin–Norbornene Bioink for Laser-Based High-Definition Bioprinting, Adv. Healthcare Mater., 2020, 9(15), 1900752, DOI:10.1002/adhm.201900752.
- K. B. C. Imani, J. Park and J. Yoon, 3D printable and mechanically tunable hydrogels achieved through hydrophobic and ionic interactions, Soft Matter, 2024, 20(29), 5781–5787, 10.1039/d4sm00602j.
- D. Merckle, E. Constant, Z. Cartwright and A. C. Weems, Ring Opening Copolymerization of Four-Dimensional Printed Shape Memory Polyester Photopolymers Using Digital Light Processing, Macromolecules, 2021, 54(6), 2681–2690, DOI:10.1021/acs.macromol.0c02401.
- Q. Liu, T. Jain, C. Peng, F. Peng, A. Narayanan and A. Joy, Introduction of Hydrogen Bonds Improves the Shape Fidelity of Viscoelastic 3D Printed Scaffolds While Maintaining Their Low-Temperature Printability, Macromolecules, 2020, 53(10), 3690–3699, DOI:10.1021/acs.macromol.9b02558.
- J. Zhang, K. Launay, N. S. Hill, D. Zhu, N. Cox, J. Langley, J. Lalevée, M. H. Stenzel, M. L. Coote and P. Xiao, Disubstituted Aminoanthraquinone-Based Photoinitiators for Free Radical Polymerization and Fast 3D Printing under Visible Light, Macromolecules, 2018, 51(24), 10104–10112, DOI:10.1021/acs.macromol.8b02145.
- S. Ban, H. Lee, J. Chen, H. S. Kim, Y. H. Hu, S. J. Cho and W. H. Yeo, Recent advances in implantable sensors and electronics using printable materials for advanced healthcare, Biosens. Bioelectron., 2024, 257, 116302, DOI:10.1016/j.bios.2024.116302.
- A. H. Williams, S. Roh, A. R. Jacob, S. D. Stoyanov, L. Hsiao and O. D. Velev, Printable homocomposite hydrogels with synergistically reinforced molecular-colloidal networks, Nat. Commun., 2021, 12(1), 2834, DOI:10.1038/s41467-021-23098-9.
- X. Shi, J. Zhang, N. Corrigan and C. Boyer, Controlling mechanical properties of 3D printed polymer composites through photoinduced reversible addition–fragmentation chain transfer (RAFT) polymerization, Polym. Chem., 2022, 13(1), 44–57, 10.1039/D1PY01283E.
- M. A. da Silva, P. Haddow, S. B. Kirton, W. J. McAuley, L. Porcar, C. A. Dreiss and M. T. Cook, Thermoresponsive Triblock-Copolymers of Polyethylene Oxide and Polymethacrylates: Linking Chemistry, Nanoscale Morphology, and Rheological Properties, Adv. Funct. Mater., 2022, 32(9), 2109010, DOI:10.1002/adfm.202109010.
- V. G. Rocha, E. García-Tuñón, C. Botas, F. Markoulidis, E. Feilden, E. D'Elia, N. Ni, M. Shaffer and E. Saiz, Multimaterial 3D Printing of Graphene-Based Electrodes for Electrochemical Energy Storage Using Thermoresponsive Inks, ACS Appl. Mater. Interfaces, 2017, 9(42), 37136–37145, DOI:10.1021/acsami.7b10285.
- A. Basu, A. Saha, C. Goodman, R. T. Shafranek and A. Nelson, Catalytically Initiated Gel-in-Gel Printing of Composite Hydrogels, ACS Appl. Mater. Interfaces, 2017, 9(46), 40898–40904, DOI:10.1021/acsami.7b14177.
- M. Müller, J. Becher, M. Schnabelrauch and M. Zenobi-Wong, Printing Thermoresponsive Reverse Molds for the Creation of Patterned Two-component Hydrogels for 3D Cell Culture, J. Visualized Exp., 2013, 77, 50632, DOI:10.3791/50632.
- R. E. B. Fitzsimmons, M. S. Aquilino, J. Quigley, O. Chebotarev, F. Tarlan and C. A. Simmons, Generating vascular channels within hydrogel constructs using an economical open-source 3D bioprinter and thermoreversible gels, Bioprinting, 2018, 9, 7–18, DOI:10.1016/j.bprint.2018.02.001.
- Q. Yue, Z. D. Luo, X. Y. Li and L. A. Fielding, 3D printable, thermo-responsive, self-healing, graphene oxide containing self-assembled hydrogels formed from block copolymer wormlike micelles, Soft Matter, 2023, 19(34), 6513–6524, 10.1039/d3sm00737e.
- M. Zhang, A. Vora, W. Han, R. J. Wojtecki, H. Maune, A. B. A. Le, L. E. Thompson, G. M. McClelland, F. Ribet and A. C. Engler, et al., Dual-Responsive Hydrogels for Direct-Write 3D Printing, Macromolecules, 2015, 48(18), 6482–6488, DOI:10.1021/acs.macromol.5b01550.
- T. Lorson, S. Jaksch, M. M. Lübtow, T. Jüngst, J. Groll, T. Lühmann and R. Luxenhofer, A Thermogelling Supramolecular Hydrogel with Sponge-Like Morphology as a Cytocompatible Bioink, Biomacromolecules, 2017, 18(7), 2161–2171, DOI:10.1021/acs.biomac.7b00481.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Two-dimensional gas of massless Dirac fermions in graphene, Nature, 2005, 438(7065), 197–200, DOI:10.1038/nature04233.
- N. Savage, MATERIALS SCIENCE Super carbon, Nature, 2012, 483(7389), S30–S30, DOI:10.1038/483S30a.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Ultrahigh electron mobility in suspended graphene, Solid State Commun., 2008, 146(9–10), 351–355, DOI:10.1016/j.ssc.2008.02.024.
- G. J. Huang, Z. G. Chen, M. D. Li, B. Yang, M. L. Xin, S. P. Li and Z. J. Yin, Surface Functional Modification of Graphene and Graphene Oxide, Acta Chim. Sin., 2016, 74(10), 789–799, DOI:10.6023/a16070360.
- A. P. Constantinou, H. Zhao, C. M. McGilvery, A. E. Porter and T. K. Georgiou, A comprehensive systematic study on thermoresponsive gels: Beyond the common architectures of linear terpolymers, Polymers, 2017, 9(1), 31, DOI:10.3390/polym9010031.
- A. P. Constantinou, L. Z. Wang, S. B. Wang and T. K. Georgiou, Thermoresponsive block copolymers of increasing architecture complexity: a review on structure-property relationships, Polym. Chem., 2023, 14(3), 223–247, 10.1039/d2py01097f.
- A. P. Constantinou and T. K. Georgiou, Thermoresponsive gels based on ABC triblock copolymers: Effect of the length of the PEG side group, Polym. Chem., 2016, 7(11), 2045–2056, 10.1039/c5py02072g.
- A. P. Constantinou, N. F. Sam-Soon, D. R. Carroll and T. K. Georgiou, Thermoresponsive Tetrablock Terpolymers: Effect of Architecture and Composition on Gelling Behavior, Macromolecules, 2018, 51(18), 7019–7031, DOI:10.1021/acs.macromol.8b01251.
- A. P. Constantinou, T. Lan, D. R. Carroll and T. K. Georgiou, Tricomponent thermoresponsive polymers based on an amine-containing monomer with tuneable hydrophobicity: Effect of composition, Eur. Polym. J., 2020, 130, 109655, DOI:10.1016/j.eurpolymj.2020.109655.
- A. P. Constantinou, B. Zhan and T. K. Georgiou, Tuning the Gelation of Thermoresponsive Gels Based on Triblock Terpolymers, Macromolecules, 2021, 54(4), 1943–1960, DOI:10.1021/acs.macromol.0c02533.
- A. P. Constantinou, G. Patias, B. Somuncuoğlu, T. Brock, D. W. Lester, D. M. Haddleton and T. K. Georgiou, Homo- and co-polymerisation of di(propylene glycol) methyl ether methacrylate – a new monomer, Polym. Chem., 2021, 12(24), 3522–3532, 10.1039/D1PY00444A.
- M. Shibata, T. Terashima and T. Koga, Thermoresponsive Gelation of Amphiphilic Random Copolymer Micelles in Water, Macromolecules, 2021, 54(11), 5241–5248, DOI:10.1021/acs.macromol.1c00406.
- M. Vamvakaki, N. C. Billingham and S. P. Armes, Synthesis of water-soluble statistical copolymers and terpolymers containing pendent oligo(ethylene glycol derivatives), Polymer, 1999, 40(18), 5161–5171, DOI:10.1016/S0032-3861(98)00718-6.
- I. B. Dicker, G. M. Cohen, W. B. Farnham, W. R. Hertler, E. D. Laganis and D. Y. Sogah, Oxyanions Catalyze Group-Transfer Polymerization To Give Living Polymers, Macromolecules, 1990, 23(18), 4034–4041, DOI:10.1021/ma00220a002.
- A. P. Constantinou, H. Zhao, C. M. McGilvery, A. E. Porter and T. K. Georgiou, A Comprehensive systematic study on thermoresponsive gels: Beyond the common architectures of linear terpolymers, Polymers, 2017, 9(1), 31 CrossRef PubMed.
- L. Wang, A. P. Constantinou, Y. Li and T. K. Georgiou, A library of thermoresponsive diblock and statistical copolymers: Unravelling the effect of molar mass, Eur. Polym. J., 2024, 207, 112810 CrossRef CAS.
- S. Wang and T. K. Georgiou, Temperature-and Time-dependent Evolution of Hydrogel Network Formed by Thermoresponsive BCACB Pentablock Terpolymers: Effect of Composition, Polymer, 2025, 128426 CrossRef.
- M. Rikkou-Kalourkoti, O. W. Webster and C. S. Patrickios, Group Transfer Polymerization, in Encyclopedia of Polymer, Science and Technology, 2002.
- O. W. Webster, W. R. Hertler, D. Y. Sogah, W. B. Farnham and T. V. RajanBabu, Group-Transfer Polymerization., 1. A New Concept for Addition Polymerization with Organosilicon Initiators, J. Am. Chem. Soc., 1983, 105(17), 5706–5708, DOI:10.1021/ja00355a039.
- O. W. Webster, Group transfer polymerization: Mechanism and comparison with other methods for controlled polymerization of acrylic monomers, Adv. Polym. Sci., 2004, 167, 1–34 CAS.
- M. A. Ward and T. K. Georgiou, Multicompartment thermoresponsive gels: Does the length of the hydrophobic side group matter?, Polym. Chem., 2013, 4(6), 1893–1902, 10.1039/c2py21032k.
- M. A. Ward and T. K. Georgiou, Thermoresponsive triblock copolymers based on methacrylate monomers: effect of molecular weight and composition, Soft Matter, 2012, 8(9), 2737–2745, 10.1039/C2SM06743A.
- S. N. Georgiades, M. Vamvakaki and C. S. Patrickios, Synthesis and Characterization of Double-Hydrophilic Model Networks Based on Cross-linked Star Polymers of Poly(ethylene glycol), Methacrylate and Methacrylic Acid, Macromolecules, 2002, 35(13), 4903–4911, DOI:10.1021/ma011822z.
- M. S. Kyriacou, S. C. Hadjiyannakou, M. Vamvakaki and C. S. Patrickios, Synthesis, Characterization, and Evaluation as Emulsifiers of Amphiphilic−Ionizable Aromatic Methacrylate ABC Triblock Terpolymers, Macromolecules, 2004, 37(19), 7181–7187, DOI:10.1021/ma049325g.
- Q. Li, A. P. Constantinou and T. K. Georgiou, A library of thermoresponsive PEG-based methacrylate homopolymers: How do the molar mass and number of ethylene glycol groups affect the cloud point?, J. Polym. Sci., 2021, 59(3), 230–239, DOI:10.1002/pol.20200720.
- C. Diehl and H. Schlaad, Thermo-Responsive Polyoxazolines with Widely Tuneable LCST, Macromol. Biosci., 2009, 9(2), 157–161, DOI:10.1002/mabi.200800213.
- J. A. Jones, N. Novo, K. Flagler, C. D. Pagnucco, S. Carew, C. Cheong, X. Z. Kong, N. A. D. Burke and H. D. H. Stöver, Thermoresponsive copolymers of methacrylic acid and poly(ethylene glycol) methyl ether methacrylate, J. Polym. Sci., Part A: Polym. Chem., 2005, 43(23), 6095–6104, DOI:10.1002/pola.21099.
- N. S. Ieong, M. Hasan, D. J. Phillips, Y. Saaka, R. K. O'Reilly and M. I. Gibson, Polymers with molecular weight dependent LCSTs are essential for cooperative behaviour, Polym. Chem., 2012, 3(3), 794–799, 10.1039/C2PY00604A.
- R. Hoogenboom, H. M. L. Thijs, M. J. H. C. Jochems, B. M. van Lankvelt, M. W. M. Fijten and U. S. Schubert, Tuning the LCST of poly(2-oxazoline)s by varying composition and molecular weight: alternatives to poly(N-isopropylacrylamide)?, Chem. Commun., 2008, 5758–5760, 10.1039/B813140F.
- V. Bütün, S. P. Armes and N. C. Billingham, Synthesis and aqueous solution properties of near-monodisperse tertiary amine methacrylate homopolymers and diblock copolymers, Polymer, 2001, 42(14), 5993–6008, DOI:10.1016/S0032-3861(01)00066-0.
- G.-E. Yu, H. Li, J. P. A. Fairclough, A. J. Ryan, N. McKeown, Z. Ali-Adib, C. Price and C. Booth, A Study of Lyotropic Mesophases of Concentrated Solutions of a Triblock Copolymer of Ethylene Oxide and 1,2-Butylene Oxide, E16B10E16, Using Rheometry, Polarized Light Microscopy, and Small-Angle X-ray Scattering, Langmuir, 1998, 14(20), 5782–5789, DOI:10.1021/la9709453.
- P. H. Li, X. Y. Dai, Y. L. Sui, R. Li and C. L. Zhang, Thermally induced and physically cross-linked hydrogel doped with graphene oxide for controlled release, Soft Matter, 2021, 17(13), 3664–3671, 10.1039/d1sm00151e.
- T. A. Shmool, A. P. Constantinou, A. Jirkas, C. Zhao, T. K. Georgiou and J. P. Hallett, Next generation strategy for tuning the thermoresponsive properties of micellar and hydrogel drug delivery vehicles using ionic liquids, Polym. Chem., 2022, 13(16), 2340–2350 RSC.
- Q. Yue, Z. Luo, X. Li and L. A. Fielding, 3D printable, thermo-responsive, self-healing, graphene oxide containing self-assembled hydrogels formed from block copolymer wormlike micelles, Soft Matter, 2023, 19(34), 6513–6524 RSC.
- Q. Yue, S.-P. Wen and L. A. Fielding, Preparation and characterisation of graphene oxide containing block copolymer worm gels, Soft Matter, 2022, 18(12), 2422–2433 RSC.
- L. Shen, J. E. Low, S. Y. Wong, X. Li, W. C. Tjiu, Y. Liu and C. B. He, Multi-walled carbon nanotube/polyimide composite film fabricated through electrophoretic deposition, Polymer, 2010, 51(10), 2155–2160 Search PubMed.
- S. Tagliaferri, G. Nagaraju, A. Panagiotopoulos, M. Och, G. Cheng, F. Iacoviello and C. Mattevi, Aqueous inks of pristine graphene for 3D printed microsupercapacitors with high capacitance, ACS Nano, 2021, 15(9), 15342–15353 Search PubMed.
- H. Ding, S. Barg and B. Derby, Direct 3D printing of graphene using capillary suspensions, Nanoscale, 2020, 12(21), 11440–11447 Search PubMed.
- Y. Q. Jiang, Z. Xu, T. Q. Huang, Y. J. Liu, F. Guo, J. B. Xi, W. W. Gao and C. Gao, Direct 3D Printing of Ultralight Graphene Oxide Aerogel Microlattices, Adv. Funct. Mater., 2018, 28(16), 1707024, DOI:10.1002/adfm.201707024.
- W. B. Li, Y. H. Li, M. Su, B. X. An, J. Liu, D. Su, L. H. Li, F. Y. Li and Y. L. Song, Printing assembly and structural regulation of graphene towards three-dimensional flexible micro-supercapacitors, J. Mater. Chem. A, 2017, 5(31), 16281–16288, 10.1039/c7ta02041d.
- X. W. Tang, H. Zhou, Z. C. Cai, D. D. Cheng, P. S. He, P. W. Xie, D. Zhang and T. X. Fan, Generalized 3D Printing of Graphene-Based Mixed-Dimensional Hybrid Aerogels, ACS Nano, 2018, 12(4), 3502–3511, DOI:10.1021/acsnano.8b00304.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, The chemistry of graphene oxide, Chem. Soc. Rev., 2010, 39(1), 228–240 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lp00071h |
| ‡ These authors contributed to the work equally and should be regarded as co-first authors. |
| This journal is © The Royal Society of Chemistry 2025 |