
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning solvent strength can fractionate PVC into ultra-low molecular weight material with low dispersity†
Ali
Al Alshaikh
 a,
Jaewoo
Choi
a,
Feranmi V.
Olowookere
a,
Caira
McClairen
b,
Owen G.
Lubic
a,
Pravin S.
Shinde
a,
C. Heath
Turner
a and
Jason E.
Bara
*a
a,
Jaewoo
Choi
a,
Feranmi V.
Olowookere
a,
Caira
McClairen
b,
Owen G.
Lubic
a,
Pravin S.
Shinde
a,
C. Heath
Turner
a and
Jason E.
Bara
*a
aDepartment of Chemical & Biological Engineering, The University of Alabama, Tuscaloosa, AL 35487-0203, USA. E-mail: jbara@eng.ua.edu
bSchool of Chemical and Biomolecular Engineering, Georgia Institute of, Technology, Atlanta, GA 30332, USA
First published on 31st January 2025
Abstract
The drive towards a circular economy in plastic materials has become a worldwide goal. It is apparent that conventional recycling alone falls well short of achieving circularity in plastic materials due to the complex formulations of commercial products. Poly(vinyl chloride) (PVC) is a post-consumer plastic that is especially challenging to recycle mechanically. However, compared to other commodity plastics, PVC is potentially well-suited for chemical recycling, especially via dissolution processes that selectively remove additives. Solvent-based recycling of PVC would circumvent thermomechanical processes that cause degradation of the polymer backbone. Yet, solvent-based recycling has its own set of considerations. Recycling a “Katamari” of mixed products of unknown origins (and potentially widely varying molecular weight distributions) might yield a purified PVC product that is of low value and/or without obvious utility. Thus, solvent fractionation of the feed into two or more products of relatively narrow molecular weight distributions may be required instead of bulk dissolution of the entire mass of polymer. In this work, we demonstrate solvent-based fractionation of PVC as both single-step and sequential processes. Two solvent systems were considered: acetone–methanol and tetrahydrofuran–methanol. The content of methanol in the solvent systems was varied to adjust the overall “strength” of the solvent system, thus controlling the molecular weight of the recovered soluble and insoluble fractions of PVC. Sequential fractionation proved capable of producing PVC fractions with dispersities (Đ) as low as 1.14. Further, sequential fractionation of commercial PVC, containing additives, was highly promising for removing additives from the bulk (76.9%) of recovered PVC.
1. Introduction
The undeniable impacts of plastic pollution on the planet has charged world governments with renewed enthusiasm in dealing with end-of-life plastics. The United Nations Environment Programme1 and many governments around the world2–4 have already planned or taken measures toward managing plastic waste. Industry is playing its role towards a solution as well.5–9 Academic interest in end-of-life plastics has also grown exponentially. Research into the “circular economy” of plastics can be generalized by the development of new recyclable polymers10,11 or products, development of sustainable (biobased) monomer production methods,12–15 and the development of both open- and closed-loop recycling/upcycling methods.16,17True closed-loop recycling is best embodied in the circularity of post-industrial and post-consumer plastic regrind for materials of known origin/composition. Solvent-based recovery is a straightforward closed-loop recycling process that could be applied to plastics where the identity of the plastic is known but the overall composition (plastic + additives) may be complex or unknown. In simple and general terms, solvent-based recovery is the use of solvents for (selective) recovery of plastics from waste.18 This is done using a “strong solvent” for the dissolution of plastic from other materials which may be present (e.g., pigments, fillers). The insoluble materials are removed by filtration and clean plastic(s) is recovered via precipitation into an “anti-solvent”. The same concept can be applied to leaching just the soluble additives (e.g. plasticizers, dyes) from waste plastics, effectively “washing” the plastic.19 Although not a new concept,20 interest in solvent-based recovery has recently grown. This resurgence brought modern technologies21,22 and techniques23 to the field, with an emphasis on the use of existing24,25 or new26,27 “green” solvents in these processes.
Compared to mechanical recycling, solvent recovery not only promises a cleaner and more consistent28 product, but also more options for controlling the form or morphology of the product obtained. Powder and pellets can be obtained using precipitation,29 films can be obtained from blow spinning30 or solvent casting.31,32 However, solvent-based recovery has its own set of considerations that need to be addressed in order to be most effectively employed. Most methods of solvent-based recovery focus on the recovery of the entire mass of plastic (Table S1†) with removal of additives. However, unless “take-back” arrangements are made between producer and end-user(s) for specific products, simple solvent recovery methods are not likely to yield desirable results for mixed products of the same plastic. This is because dissolving two or more products made from the same plastic but with different molecular weight distributions will usually yield a material with a higher dispersity (Đ)33 (also known as polydispersity index, PDI), which may render the recovered “pure” material much less useful. It is best to think of this method as complementary rather than competitive. Ideally, certain plastics would be sorted into their own “best” way of recovery (e.g., chemolysis of PET34 or polyolefins,35 and so on). To save on solvent use, solvent-based recovery can work as a complementary method to mechanical recycling,36 where pure products are desired, or with tough-to-recycle wastes. While solvent-based separation of mixed plastics seems ideal, it can also be coupled with the advances in the centralized37 and automated38 physical sorting of plastics, which can facilitate single plastic recycling/upcycling.
Amongst the plastics that would benefit from isolation from mixed plastics is polyvinyl chloride (PVC). PVC is “anti-synergistic” (i.e., problematic) in mixed plastic recycling,39,40 may contain currently prohibited substances,41 and generally not collected by municipal recycling programs. Special treatment is generally needed42 to prevent complications that may arise from the presence of PVC in mixed waste that is thermomechanically recycled.43,44 Due to these complications and given the common presence of additives in PVC,45 solvent-based recovery is particularly well-suited to PVC and has already been explored for large-scale operation. The VinyLoop process46 was introduced by Solvay in 2003. Despite its closure in 2018 (due to EU phthalate regulation47), VinyLoop showed the potential of recycling by dissolution, which lead to its recent redevelopment under INEOS Inovyn.48 Other new processes from CreaSolv®49 and Polyloop®50 indicate further current interest in solvent-based recycling of PVC. Recent work by Wagner51 demonstrated successful removal of up to 99.4% of diethylhexyl phthalate (DEHP) that was originally present in PVC flooring.
Although rarely mentioned in the academic literature, a major challenge for solvent-based recovery of PVC is not presence of residual additive(s) (which in can be remedied),19 but rather that commercial PVC products will come from various grades (K-values) of PVC resins that are of widely different molecular weights.52 Thus, PVC recovered from recycling of mixed PVC products (using solvents, or mechanically) would likely have a much larger Đ than virgin resins (where typically Đ ∼2), and could make the recovered “pure” PVC less valuable and useful. Hence, a method for a fractional recovery of PVC into two or more molecular weight distributions each with Đ ≤ 2 becomes a very interesting option.
Fractionation is commonly used for lignins,53 however it is less commonly applied to plastics. The still developing, yet mature, field of lignin fractionation gave rise to many fractionation methods, like fractional precipitation, pH dependent precipitation, membrane fractionation, and most commonly solvent extraction (single and sequential).53 Much like lignin products,54 blends of multiple PVC K-values would exhibit a broad molecular weight distribution curve. In works by Pepperl,33,55 it was shown that virgin PVC contains non-negligible amounts of chains with molecular weights as low as 3 kDa (for K-50) and up to 200 kDa or more (for K-60 and K-70) (Fig. 1). Based on observations in our recent work on PVC dehyrochlorination,56 we became interested in how we could selectively obtain low molecular weight chains via dissolution in relatively weak solvents, while also isolating the large molecular weight chains with strong solvents.
 | ||
| Fig. 1 Selected molecular weight distributions for virgin PVC (K-50, 60, and 70) as measured by Pepperl. Adapted with permission from Pepperl.33 Copyright (2000), with permission from John Wiley and Sons. | ||
Herein, we demonstrate that solvent extraction of PVC can be used as a means of obtaining products with targeted molecular weight distributions and can yield fractions with very narrow Đ values (as low as ∼1.1). Solvent extraction was performed in both single-step and multi-step (i.e., sequential) process using a weak solvent (acetone (Ace)), a weak solvent + nonsolvent blend (Ace and methanol (MeOH)), and a strong solvent + nonsolvent blend (tetrahydrofuran (THF) and MeOH). Single-step solvent blend extraction methods show that virgin PVC resin (K-50) can be fractionated to obtain molecular weight ranges that correspond to solvent “strength”. Sequential solvent extractions shows that PVC of low Đ can be fractionated from the bulk polymer, which is conceptually illustrated in Fig. 2. Further, we illustrate the viability of this method in fractionating and cleaning for commercial PVC samples (plasticized PVC and PVC pipe), which shows its potential as a recycling route for PVC. These results suggest that solvent fractionation of PVC can offer unique opportunities as the solubility of the low molecular weight fractions produced using this method can provide well-defined feedstocks for chemical modification and/or depolymerization of PVC56 or back into PVC blends as melt rheology modifiers,33 while the remaining (i.e., insoluble) recovered clean PVC can be readily routed back as feedstock to existing applications.
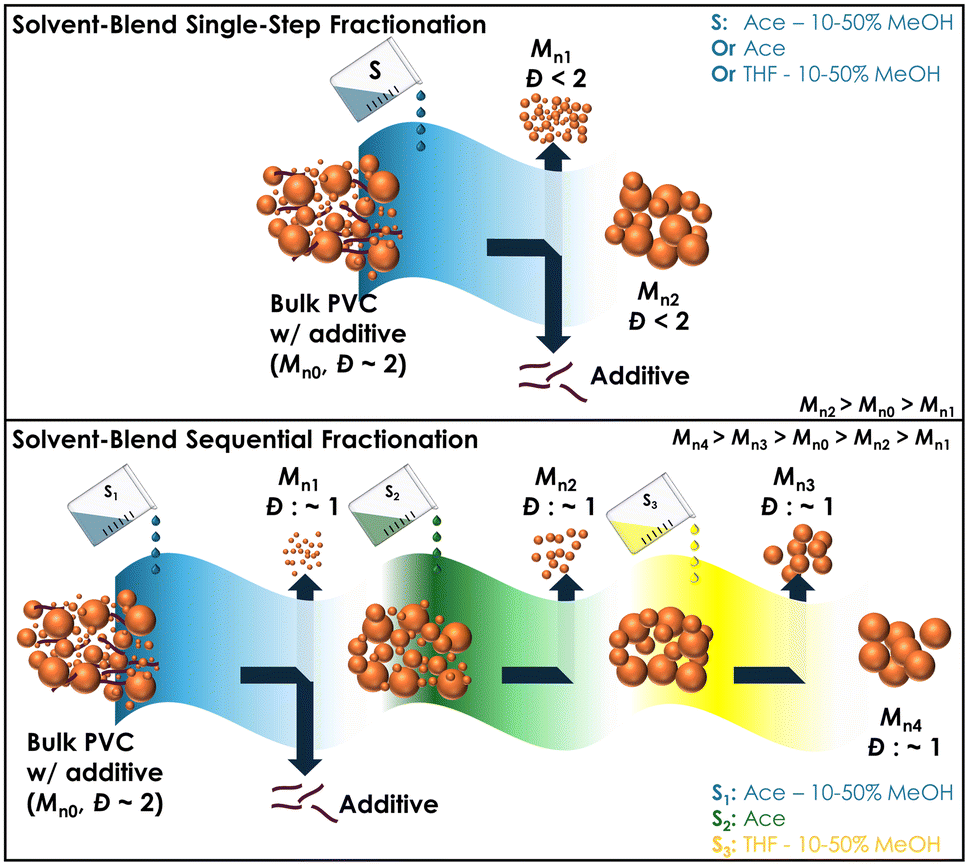 | ||
| Fig. 2 Illustration of the single-step and sequential fractionation of PVC in this work. | ||
2. Experimental
2.1. Materials
Virgin PVC (K-50 and K-65 grades) was generously provided by a major US producer. Plasticized PVC pellets (Sylvin 9684-95) used in this work were purchased in 10 lb batches from eBay seller “The Freight Adoption Agency”, and were received as transparent flexible pellets with a pale purple hue. Schedule 40 PVC pipe was recovered from leftover construction materials.Ethanol (EtOH, ≥99.5%), MeOH (≥99.8%), hexane (mixture of isomers, ≥98.5%), and Ace (≥99.5%) were purchased from VWR through the Chemistry Stockroom at The University of Alabama. THF (≥99.0%) and high-performance liquid chromatography (HPLC) grade THF (Macron, ChromAR®, ≥99.8%) were purchased from VWR.
2.2. Single-step PVC K-50 fractionation
PVC K-50 (1 g) was added to a polypropylene centrifuge tube (15 mL), followed by a solution of Ace–MeOH or THF–MeOH (10 g) prepared according to the pre-determined concentrations (Table 1). Solvent blends are referred to as wt%THF/Ace![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :wt%MeOH going forward (e.g. 50 wt% Ace and 50 wt% MeOH blend is denoted as 50Ace:50MeOH). The contents of the tube were mixed using a vortex mixer (VWR) for 30 s. The tube was then moved to an ultrasonic bath (VWR) and sonicated for 1.5 h, mixed using a vortex mixer, then sonicated again for 1.5 h. Once the sonication period was completed, the tubes were placed in a centrifuge (VWR) and centrifuged for 30 min at 6500 rpm. The liquid phase in the tube was then decanted into a pre-weighed beaker inside a hood and the solvent was allowed to evaporate (∼24–48 h). Once the solvent was evaporated, the beaker was weighed again to determine the yield of the soluble portion of PVC obtained from the fractionation. Each fractionation was concurrently performed in triplicate, and the solutions were combined prior to evaporation.
:wt%MeOH going forward (e.g. 50 wt% Ace and 50 wt% MeOH blend is denoted as 50Ace:50MeOH). The contents of the tube were mixed using a vortex mixer (VWR) for 30 s. The tube was then moved to an ultrasonic bath (VWR) and sonicated for 1.5 h, mixed using a vortex mixer, then sonicated again for 1.5 h. Once the sonication period was completed, the tubes were placed in a centrifuge (VWR) and centrifuged for 30 min at 6500 rpm. The liquid phase in the tube was then decanted into a pre-weighed beaker inside a hood and the solvent was allowed to evaporate (∼24–48 h). Once the solvent was evaporated, the beaker was weighed again to determine the yield of the soluble portion of PVC obtained from the fractionation. Each fractionation was concurrently performed in triplicate, and the solutions were combined prior to evaporation.
| Entry | PVC source | Solvent system | M w (kDa) | M n (kDa) | Đ | T g (°C) | Yielda (%) |
R
ab (MPa1/2) |
|---|---|---|---|---|---|---|---|---|
| a Yield = mass of recovered fraction/mass of starting material. b R a: Hansen solubility parameters (HSPs) distance. c Virgin PVC resins as received from supplier. | ||||||||
| PVC K-65c |
107 | 58.6 | 1.82 | 83.9 | — | |||
| F65 | K-65 | 100% Ace | 35.7 | 23.4 | 1.53 | 76.4 | 17.2 | 2.69 |
| PVC K-50c |
42.8 | 23.3 | 1.83 | 79.1 | — | |||
| F1 | K-50 | 50Ace:50MeOH |
4.10 | 3.38 | 1.21 | 58.0 | 2.90 | 16.2 |
| F2 | K-50 | 60Ace:40MeOH |
6.76 | 5.17 | 1.31 | 69.8 | 6.90 | 14.8 |
| F3 | K-50 | 70Ace:30MeOH |
10.1 | 7.57 | 1.34 | 72.8 | 11.8 | 13.5 |
| F4 | K-50 | 80Ace:20MeOH |
14.7 | 10.1 | 1.46 | 74.8 | 18.9 | 12.1 |
| F5 | K-50 | 90Ace:10MeOH |
20.0 | 13.2 | 1.51 | 76.4 | 29.3 | 9.06 |
| F6 | K-50 | 100% Ace | 26.3 | 17.2 | 1.53 | 74.5 | 44.2 | 2.69 |
| F7 | K-50 | 50THF:50MeOH |
19.6 | 12.5 | 1.57 | 67.8 | 20.0 | 12.7 |
| F8 | K-50 | 60THF:40MeOH |
33.9 | 21.9 | 1.55 | 77.0 | 69.9 | 10.6 |
| — | K-50 | 70THF:30MeOH |
Full dissolution with 70% THF or more | 8.67 | ||||
2.3. Sequential PVC K-50 fractionation
Sequential fractionations were done using similar methods to single-step fractionation. However, once the solution was decanted, another 10 g solution of higher Ace content was added and the extraction process repeated similarly. Ace content was increased by increments of +10 wt%, until an extraction of 100% Ace was finished. The extractions were then repeated using THF–MeOH solutions starting with 50THF:50MeOH, until all the PVC was dissolved using a solution of 70THF:30MeOH.
2.4. Sequential commercial PVC mixture fractionation
PVC pipe was first cut into small trimmings (Fig. S3†). PVC pipe trimmings and pellets were separately cleaned with water and soap, followed by washing with EtOH, and finally soaked in hexanes overnight to remove any freely soluble additives and surface coatings. PVC was then removed from hexane and allowed to dry for 2 h. Washed PVC pipe trimmings (15 g) and PVC pellets (15 g) were then moved into a glass bottle (250 mL) equipped with a stir bar. A 50Ace:50MeOH solution (200 g) was then added to the bottle and stirred for 4 h. The solids were then allowed to settle, and the liquid phase was then carefully decanted into a funnel plugged by cotton to filter any particulates. The solution was then evaporated on a pre-weighed Pyrex dish inside a hood (∼24–48 h). The extraction was then repeated similarly using solutions of increasing Ace content by increments of +10 wt%, until an extraction with 100% Ace was done. The extractions were then repeated using THF–MeOH solutions starting with 50THF:50MeOH, until all the PVC was dissolved using a solution of 90THF:10MeOH.
2.5. Characterization
Gel permeation chromatography multi-angle laser light scattering with simultaneous refractive index (GPC-MALS-RI) was used to determine the absolute molecular weights of the PVC sources as well as the fractionation products. The measurements were performed on DAWN MALS (D8, Waters-Wyatt Technology Corporation, Goleta, CA USA) detector in HPLC grade THF at a flow rate of 1.0 mL min−1 with a polarized single frequency light of 662.9 nm in conjunction with the RI detector (Model 2414, Waters-Wyatt Technology, Goleta, CA USA) using Styragel HR 5E mixed bed column (300 mm × 7.8 mm × 5 μm) for separation. The weight-average molecular weight (Mw), the number-average molecular weight (Mn), the dispersity (Đ = Mw/Mn) and hydrodynamic radius (Rh) of each sample was determined by the Zimm plot using the RI increment (dn/dc) value of 0.1010 mL g−1 (a standard value for PVC in THF). Astra and Empower software programs were used to run and analyze the GPC traces for MALS and RI detectors, respectively. THF was served as a mobile phase as well as solvent for dissolving PVC samples with optimized concertation of 8.0 mg mL−1 and the injection volume of 80 μL. The MALS detector was operated at room temperature while the RI detector, pump oven and column oven were maintained at 40 °C. The GPC system was calibrated by using narrow distribution polystyrene standards.Attenuated total reflection Fourier transform infrared (ATR-FTIR) was used to study the molecular structure of produced fractions, PVC sources, and additive; FTIR spectra were obtained using the PerkinElmer Spectrum 2 instrument. Scans were acquired between 4000–400 cm−1 using a resolution of 4 cm−1, and 64 accumulations.
Differential scanning calorimetry (DSC) measurements were carried out on PVC sources and fractionation products using Discovery DSC250 instrument (TA Instruments, New Castle, DE, USA) in an ultrapure (USP) N2 atmosphere (20 mL min−1) at a ramp rate of 10 °C min−1 with at least three heating/cooling cycles in the temperature range from −40 °C to 150 °C. Trios software (Waters Technology) was used to analyze the DSC traces to determine the thermal properties of samples such as the glass transition temperature (Tg).
The viscosity measurements were obtained using the ViscoQC 300L PTD 100 cone-plate measurement system with a CP40 bob. Viscosity samples were prepared on a 10:90 polymer sample to solvent (THF) weight ratio. The instrument was calibrated with a certified reference standard (Cannon instrument) before use. The measurements were collected using a 0.5 mL polymer solution at a temperature of 20 ± 1 °C and shear rates of 10 or 100 s−1 for 5 min.
2.6. Computational methods
To provide molecular-level insights into the fractionation of PVC in Ace–MeOH and THF–MeOH solvent blends, molecular dynamics (MD) simulations were performed at varying MeOH % and number of PVC chains (nPVC). Table S2† outlines the system compositions used in this study. The PVC120 model was employed, as it has been validated in previous studies57–59 as a reliable chain length (120 repeat units) for atomistic solvated polymer simulations. While the model does not capture the full molecular weight distribution observed experimentally, it has been shown to sufficiently represent the fundamental physics of experimental systems.The MD simulations were conducted using the Gromacs 2021.1 package.60 The OPLS-AA force field61 was applied to represent bonded and nonbonded interactions for PVC and the solvents. Atomic charges for PVC were derived using ab initio Hartree–Fock/STO-3G62 in PolyParGen,63 while solvent charges were obtained using B3LYP/6-31++(d,p).64 van der Waals interactions were captured using the Lennard-Jones potential with a 1.0 nm cutoff, and long-range electrostatics were calculated using the particle mesh Ewald (PME) method65 beyond the 1.0 nm cutoff. Geometric combination rules were applied for cross interactions.
The system configurations were built using the PACKMOL package,66 followed by energy minimization via the steepest descent method. We then equilibrated the systems in the isothermal-isobaric (NPT) ensemble at 1 bar and 300 K for 10 ns with a time step of 1 fs, using the Parrinello–Rahman barostat67 and velocity-rescaling thermostat68 to maintain pressure and temperature respectively. To ensure sufficient exploration of the phase space, an annealing protocol was applied, heating the system to 600 K and cooling to 300 K over 4 cycles of 40 ns. Afterward, the systems were further equilibrated for 20 ns. Production runs were performed for 20 ns, during which configurations were extracted every 30 ps for further analyses. Hydrogen bond lengths were constrained with the LINCS algorithm,69 and periodic boundary conditions were applied in all dimensions. Detailed computational methods and calculations can be found in the ESI, Section S2.1.†
3. Results
3.1. PVC single-step fractionation by solvent-blend extraction
Unlike most solvent-based plastic recovery, fractionation offers the opportunity for selective recovery of certain “grades” of a specific plastic, based on the solvent or solvent-blend used (Fig. 3). However, among commodity plastics which bear resin identification codes (RIC) 1–6 which comprise 1 – PET (a polyester) and vinyl polymers (2 – HDPE, 3 – PVC, 4 – LDPE, 5 – PP, and 6 – PS), only PVC and PS have any appreciable solubility in common organic solvents at or near ambient temperature.18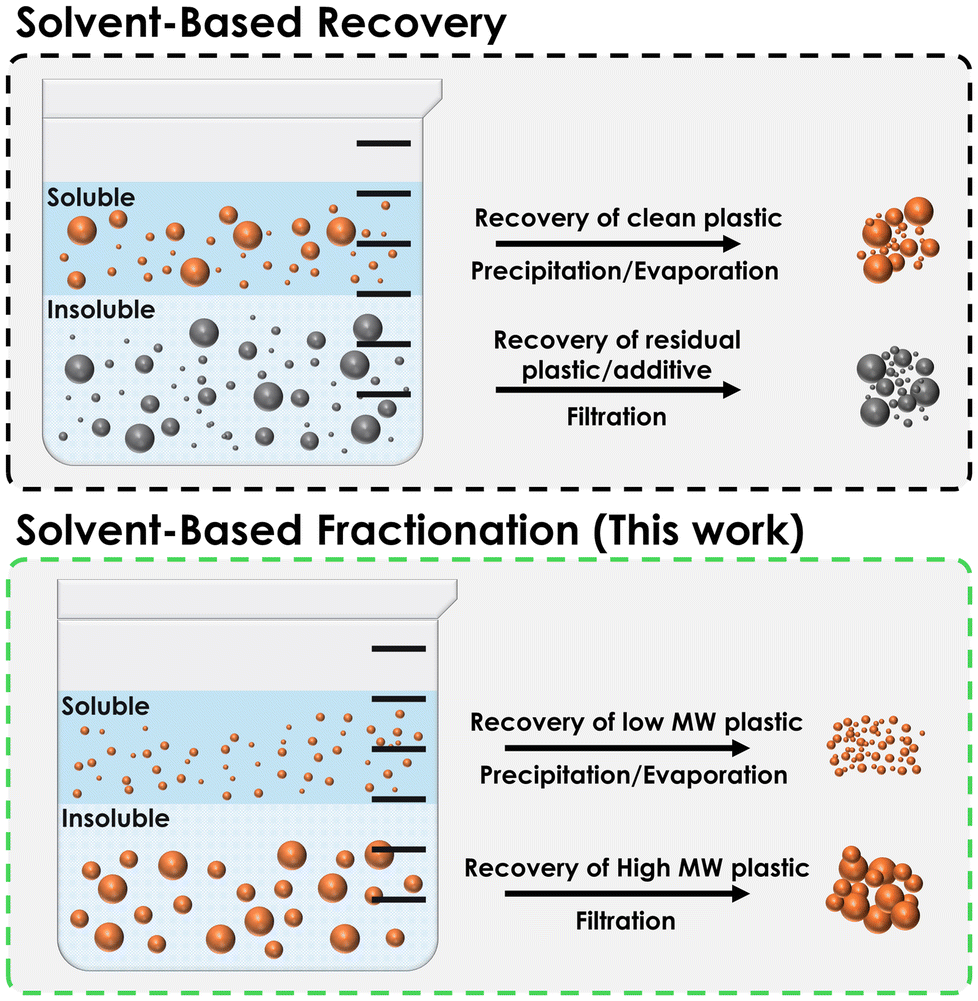 | ||
| Fig. 3 Comparison between solvent-based recovery of “whole” plastic and solvent-based fractionation of specific plastic “grades”. Note that the recovery method can be reversed, with the recovered material being the insoluble (in fractionation terminology, recovery of the soluble = solvent extraction, recovery of the insoluble = fractional precipitation). | ||
Recently, we showed that K-50 and K-65 PVC were readily fractionated using either pure ethyl acetate (EtOAc) or Ace,56 which we consider to be relatively weak solvents for PVC due to their inability to dissolve the entire bulk PVC (in any K-value). This led us to consider how solvent blends could be used to more finely fractionate PVC. To fractionate the lower molecular weight portions that are known to be present in K-50 and K-65 PVC,33,55 extractions were done using mixtures of a weak solvent (Ace) and a non-solvent (MeOH). For higher molecular weights (short of bulk), mixtures of a strong solvent (THF) and a non-solvent (MeOH) were examined. Choice of solvents was motivated by their low boiling point for ease of recovery, and/or their recommendation for green chemistry.70 While methyl ethyl ketone (MEK or 2-butanone) is likely a better choice in the long-term as the strong solvent when considering price and green chemistry recommendations,70 THF was chosen as the strong solvent for this study due to its lower boiling point and is very well-matched to PVC per Hansen solubility parameters (HSPs).71
Solvent extraction can be done as a single-step or sequential process (Fig. 2). Single-step solvent extraction is one of the most common fractionation methods owing to its simplicity. Experiments were conducted with PVC using different solvent systems (Table 1). Fractions showed a gradual increase in Mw, Mn, and Đ with increase of weak solvent or strong solvent (Fig. 4 and Table 1). In all cases PVC K-50 was used, although only a 100% Ace fractionation was performed for PVC K-65.
 | ||
Fig. 4 Weight average molecular weight (Mw, log scale) of ( ) PVC K-50 single-step, ( ) PVC K-50 single-step, ( ) PVC K-50 sequential and (■) commercial PVC sequential fractionations in different solvent systems. Bottom X: content of solvent systems. Top X: Hansen solubility parameters (HSPs) distance Ra (MPa1/2) of corresponding solvent system (Bottom X). ) PVC K-50 sequential and (■) commercial PVC sequential fractionations in different solvent systems. Bottom X: content of solvent systems. Top X: Hansen solubility parameters (HSPs) distance Ra (MPa1/2) of corresponding solvent system (Bottom X). | ||
Perhaps as expected, the weight-average molecular weight of the extracts of K-50 (Mw = 26.3 kDa) and K-65 (Mw = 35.7 kDa) for the 100% Ace were different, corresponding to the molecular weight distributions within the individual source material (Table 1). This seems reasonable, as source PVC with higher molecular weight would contain fewer low molecular weight chains soluble in Ace, thus shifting the average molecular weight of the associated Ace extracts. The yields of the Ace extracts reflect this as well (K-50 > K-65). Using Ace as the only solvent, 44.2% by mass was extracted from K-50 with Đ = 1.53, while only 17.2% by mass was extracted from K-65 with similar value of Đ = 1.53. Experiments on Ace extractions of K-50 and K-65 blends followed similar trends (Table S4†). The Ace extracts of the 80% K-50:20% K-65 blend (Mw = 29.9 kDa, Đ = 1.67) yielded 29.9% extracts; the 50% K-50:50% K-65 blend (Mw = 31.9 kDa, Đ = 1.52) yielded 27.2%; the 20% K-50:80% K-65 (Mw = 38.0 kDa, Đ = 1.85) yielded 15.4%. In contrast to single grade extractions, the extracts of 80% K-50 and 80% K-65 blends exhibited a relatively higher Đ due to the wider and asymmetrical range in molecular weight of the source material (Table S4†). Regardless, a Đ value <2 for the blend extracts show the promise of this method for processing of mixed PVC products, regardless of source molecular weight.
3.2. PVC sequential fractionation by solvent-blends extraction
While single-step fractionation with Ace could be efficient for bulk recovery of the lower MW PVC chains present in a given sample, sequential fractionation could offer the ability to obtain low Đ fractions (<1.5), which can be blended into specific grades when needed.33 Previous sequential fractionation methods of PVC have focused on fractional precipitation.72 Sequential solvent extraction fractionation was chosen here for rather logistical reasoning. In fractional precipitation, extra care needs to be placed while processing the solvent, to avoid any solvent loss, which could cause an inconsistency in further solvent blend ratios. Further, recovery of pure solvents may be complicated during fractional precipitation, especially for Ace–MeOH blends due to the presence of an azeotrope.73 Hence the choice of sequential solvent extraction, as those issues are non-existent, making it a more convenient choice for processing large volumes of PVC.In the methods presented here, PVC is contacted with the weakest solvent mixture first (here, 50Ace:50MeOH). The mixture is then separated into its liquid and solid components. The residual undissolved PVC is then contacted by an incrementally stronger solvent mixture in increments of –10% MeOH until it is 100% Ace. The residual PVC is then extracted in THF–MeOH blends similarly.
Similar to single-step solvent extraction, fractions see a gradual rise in the average molecular weight with increase of weak/strong solvent content (Table 2 and Fig. 4). However, the Đ values of the fractions were remarkably lower, and consistently <1.4, except S10 which had a Đ of 1.73, but contained the remaining PVC as the last fraction.
| Entry | Solvent system | M w (kDa) | M n (kDa) | Đ | T g (°C) | Yield (%) | R a (MPa1/2) |
|---|---|---|---|---|---|---|---|
|
a Virgin PVC or commercial products as received.
b Full dissolution.
c 50:50 mixture by weight of: PVC pellet: Mw = 105 kDa, Mn = 57.1 kDa, Đ = 1.84, Tg = 81.9 °C. PVC pipe: Mw = 115 kDa, Mn = 64.8 kDa, Đ = 1.78, Tg = 84.7 °C.
d Only additives extracted using less Ace content.
|
|||||||
| PVC K-50a |
42.8 | 23.3 | 1.83 | 79.1 | — | ||
| S1 | 50Ace:50MeOH |
4.22 | 3.36 | 1.26 | 49.7 | 1.20 | 16.2 |
| S2 | 60Ace:40MeOH |
9.84 | 8.51 | 1.16 | 67.4 | 5.47 | 14.8 |
| S3 | 70Ace:30MeOH |
14.5 | 12.7 | 1.14 | 75.0 | 3.98 | 13.5 |
| S4 | 80Ace:20MeOH |
23.8 | 19.4 | 1.22 | 78.1 | 6.32 | 12.1 |
| S5 | 90Ace:10MeOH |
29.6 | 23.7 | 1.25 | 78.6 | 9.63 | 9.06 |
| S7 | 100% Ace | 33.7 | 26.0 | 1.29 | 76.6 | 14.1 | 2.69 |
| S8 | 50THF:50MeOH |
34.3 | 24.4 | 1.40 | 77.3 | 3.24 | 12.7 |
| S9 | 60THF:40MeOH |
44.4 | 31.5 | 1.41 | 76.9 | 23.1 | 10.6 |
| S10b |
70THF:30MeOH |
67.4 | 39.1 | 1.73 | 76.5 | 32.4 | 8.67 |
| Commercial PVC Mixa,c |
109 | 60.5 | 1.80 | N/A | — | ||
| C1d |
80Ace:20MeOH |
12.1 | 8.28 | 1.46 | 49.6 | 1.0 | 12.1 |
| C2 | 90Ace:10MeOH |
16.7 | 10.6 | 1.57 | 63.2 | 1.4 | 9.06 |
| C3 | 100% Ace | 21.6 | 15.4 | 1.40 | 74.0 | 2.1 | 2.69 |
| C4 | 50THF:50MeOH |
26.8 | 21.3 | 1.26 | 70.4 | 0.6 | 12.7 |
| C5 | 60THF:40MeOH |
31.7 | 23.3 | 1.36 | 73.3 | 1.9 | 10.6 |
| C6 | 70THF:30MeOH |
96.8 | 62.0 | 1.56 | 80.7 | 46 | 8.67 |
| C7 | 80THF:20MeOH |
123 | 76.4 | 1.61 | 85.0 | 18 | 6.69 |
| C8b |
90THF:10MeOH |
170 | 98.0 | 1.73 | 83.0 | 11 | 2.57 |
Commercial samples of PVC (pipe and plasticized pellets) were also sequentially fractionated to simulate these methods on end-of-life plastics. Both products had relatively high, but slightly different molecular weights, with PVC pipe and plasticized pellets having an Mw = 115 and 105 kDa, with an Mn = 64.8 and 57.1 kDa respectively, measured after removing additives (Table 2). Since the starting materials had relatively high molecular weights, no fractions were expected to be recovered for the Ace blends with 50%–30% MeOH. Nonetheless, these fractionations were attempted, and acted as selective additive extraction steps, which prevented significant contamination in subsequent fractions. The fractions produced here showed a similar trend to K-50 experiments, however, they did not have similar molecular weights. This can be attributed to the difference in molecular weight of the source materials, but also the product form. PVC pipe and pellets were extracted either as-is or after cutting to small cubes (Fig. S3†), which would interfere with the extraction process. Further, the fused/gelled behavior of commercial extruded PVC74 leads to even less surface area for dissolution. PVC K-50 was received as a fine powder, which increased the surface area during the extraction. During the Ace phase of extractions, commercial PVC only gradually swelled, and only broke into powders, likely also breaking fused particles, during the THF phase of extractions. This aligns well the relation between swelling/surface area and dissolution as explained by the Flory–Huggins theory (Section 3.6, Fig. 7).
To show the utility of the sequential fractionation process, fractions were used to formulate PVC K-50 and K-65 products by blending them with the virgin resins. For example, C3 was blended with PVC K-65 to produce a K-50 like product (Table S5†). The blend, 70% C3 and 30% K-65, had an Mw of 45.3 kDa which is 5.5% off from the K-50 resin used in this work, and 4.2% from the calculated average. In another experiment, C7 and C8 were separately blended with K-50 to produce a K-65 like product. In a 1:1 ration of fraction to resin, the C7 blend had an Mw of 94.5 kDa and the C8 blend had an Mw of 99.4 kDa, which were 13% and 7.6% off from K-65 and 12% and 7.0% from the calculated average respectively.
3.3. Structural analysis
From spectroscopic analyses, all fractions produced during this work were identical to virgin PVC resin. For solvent-based recovery methods, the thermal or chemical degradation of the products can be a concern. Since all extractions were run under ambient temperatures and pressures (energy inputs from sonification are likely negligible), it is highly unlikely that any thermal degradation would occur, as the degradation temperature of PVC is >250 °C75 Thermal degradation of PVC typically occurs through dehydrochlorination, which releases HCl and forms conjugated sequences of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds ("polyenes") which can undergo further reactions, including crosslinking76 In all products made in this work, no sign of CC bond formation was observed. In FTIR, CC peaks are observed between 1600–1700 cm−1 (νCC), which are not present in any products (Fig. 5).
C bonds ("polyenes") which can undergo further reactions, including crosslinking76 In all products made in this work, no sign of CC bond formation was observed. In FTIR, CC peaks are observed between 1600–1700 cm−1 (νCC), which are not present in any products (Fig. 5).
 | ||
| Fig. 5 FTIR spectra for (a) PVC K-50 single-step fractionation, (b) PVC K-50 sequential fractionation and (c) sequential commercial fractionation. | ||
Chemical degradation during solvent-based recovery usually occurs as solvolysis or due to presence of additives and contaminants, introducing new structural defects. In this case, the most likely defect would be the formation of double bonds through elimination, or the formation of alcohols or ketones through oxidation. Fortunately, neither degradation routes were observed. Oxidation of PVC would cause –OH or CO peaks to appear in FTIR spectra around 3500 cm−1 (νO–H) and 1700–1780 cm−1 (νCO) respectively. While no –OH peaks were observed in the products, a small broad CO peak (sometimes as two neighboring peaks) can be seen in all products and virgin resin. This CO peak seems to be native to the PVC resin used, and is common in most PVC resins, and can be attributed to many causes as the presence of structural defects, additive, processing agents, or perhaps signals of initiator fragments at the end(s) of PVC chains.
The products made from commercial samples were analyzed for the presence of residual additives. Prior to any extractions, the source commercial PVC was soaked briefly in hexanes and MeOH to extract freely soluble plasticizers, additives, or dyes. This step removes a significant portion of the additives present in the plasticized PVC specifically (30–50%), however, that does not guarantee complete or efficient removal of all additives. So, fractionation started at a 50Ace:50MeOH solvent system, despite it not extracting any PVC, with the goal of removing as much additive as possible prior to recovering PVC. The sequential fractionation methods done with commercial samples allows for most additives to be extracted during the first fractions, with progressively less additive present in subsequent fractions. This was clear by the presence of residual additive in C1 to C4, albeit progressively less each step. This can be seen by the FTIR CO peak around 1740 cm−1 (νCO), indicating the presence of a terephthalate ester (described as “non-ortho phthalate” by the vendor), and another additive peak around 1670 cm−1 (likely νConjugated CO). The intensity and shape of the CH2 peaks around 2780–3000 cm−1(νC–H), for all fractions are PVC-like (unlike PVC pipe or PVC pellet, Fig. S8 and S9†). Which alludes to the existence of only trace amounts of additive in the contaminated fractions. Nonetheless, further purification via dissolution/precipitation may be required for complete removal of additives. Subsequent fractions, accounting for 76.9 wt% of the total weight of the source material, show much cleaner FTIR spectra, which were essentially identical to virgin PVC (Fig. 5c).
3.4. Properties
The glass transition temperatures (Tg) of the fractionation products were measured using DSC. The dependence of Tg on the molecular weight of (linear) polymers is well understood and can be described empirically by the Flory–Fox equation.77 DSC scans of PVC fractions (Section S6†) show that PVC follows this relationship as well, as can be seen in Fig. 6. The results agree well with calculated values obtained using estimates of the Flory–Fox equation parameters of PVC (Tg∞ = 351 K and K = 8 × 104, 16.5 × 104).78 Of note is the agreement of lower molecular weight fractions of commercial PVC, C2 to C4, which were shown to contain amounts of plasticizers. The exception was C1, which had a lower Tg than predicted, or observed in S1, which had a lower Mn (8.28 kDa vs. 3.36 kDa) but similar Tg, this is likely due to presence of additive in C1. This general agreement shows that the leftover plasticizer had little to no effect on the Tg of C2 to C4, thus confirming their presence in trace amounts only.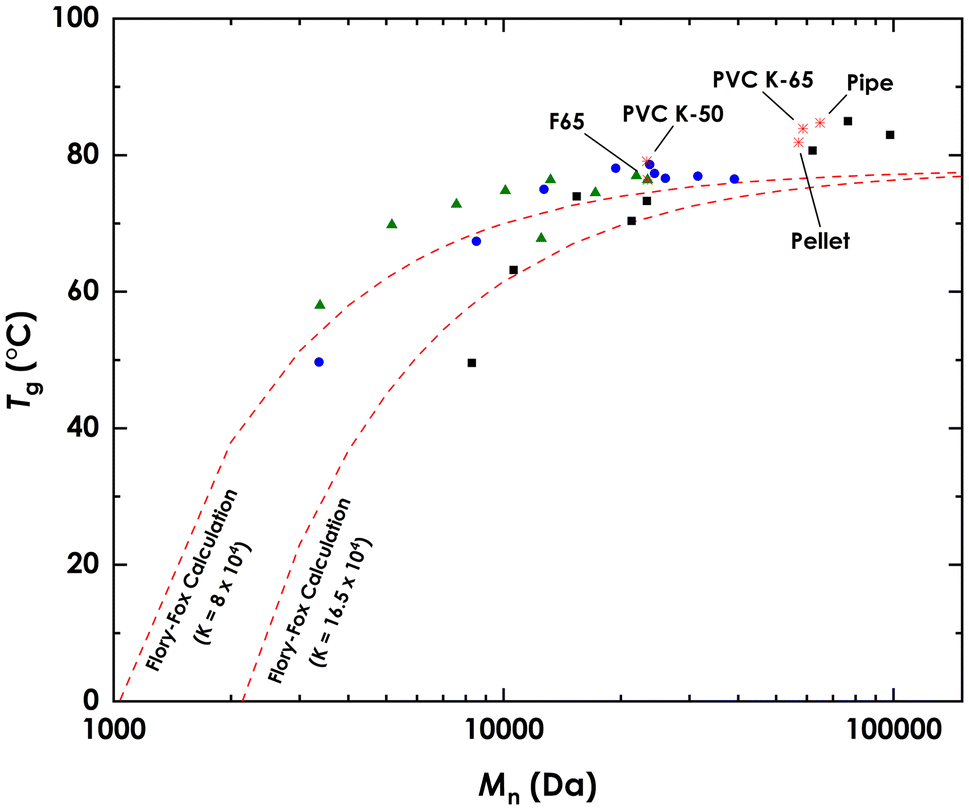 | ||
Fig. 6
T
g values measured using DSC for  single-step PVC K-50 fractions, single-step PVC K-50 fractions,  sequential PVC K-50 fractions, ■ is sequential commercial PVC fractions, sequential PVC K-50 fractions, ■ is sequential commercial PVC fractions,  other samples (individually labeled). other samples (individually labeled).  Predicted Tg values using the Flory–Fox equation. Predicted Tg values using the Flory–Fox equation. | ||
The relationship between polymer molecular weight and polymer solution viscosity is well known.79 This relationship obeys the power law, and is often described by the Mark–Houwink equation.80 Solution viscosity measurements, 100 mg sample per 1 mL THF, of the acetone fractions follow this expected trend (Fig. S15†). F65 and its source PVC K-65, had a solution dynamic viscosity (μ) of 25.33 cP and 134.7 cP respectively; F6 and its source PVC K-50 had μ values of 13.9 cP and 32.27 cP, respectively.
3.5. Computational results
To explore the structural changes of PVC with increasing MeOH present in the solvent blends, the radius of gyration (Rg) of PVC in Ace–MeOH and THF–MeOH was computed (Table S3†). Generally, Rg decreases with higher MeOH concentrations and with increasing nPVC. Additionally, the ratio of surface area (SA) of PVC in solvent mixtures to the SA of bulk PVC was calculated (Fig. 7). A consistent decrease with increasing MeOH was observed. For instance, for nPVC = 5 in Ace–MeOH, the SA ratio drops from 2.45 at 0% MeOH to 1.86 at 80% MeOH. Higher nPVC exhibits lower SA across all MeOH concentrations, indicating that as nPVC increases, PVC chains pack more tightly, reducing solvent accessibility. The addition of MeOH likely leads to polymer chain contraction, further lowering the SA. Of note is the slope change between 20% MeOH and 40% MeOH in the THF/MeOH curves. This slope change corresponds to experimental data (Tables 1 and 2) and the stark change in solubility between 70THF:30MeOH and 60THF:40MeOH in C6 and C5 respectively. Data and discussion about the volume of mixing (VE) and the Flory–Huggins (FH) interaction parameter χ12 are available as in Section S2.2.
 | ||
| Fig. 7 Ratio of surface area for PVC in Ace–MeOH and THF–MeOH blends at different MeOH % and at different nPVC. | ||
The Hansen method (see Section S2.1†) was applied to predict PVC solubility in the mixtures. Molar volumes (Vm) and HSP values are shown in Table 3. The Vm and HSPs of pure Ace, pure THF and bulk PVC are in very good agreement with experimental data at 300 K, validating our simulations. Vm in both mixtures decreases by a factor of ∼2 as MeOH % increases, indicating that THF or Ace blends become more compact with added MeOH. Moreover, the electrostatic term increases while the dispersion term decreases, making both mixtures more polar and less compatible with PVC. Ace–MeOH shows stronger electrostatic interactions and slightly weaker dispersion interactions than THF–MeOH, making it less favorable for dissolving PVC.
| MeOH wt% | V m (Ace–MeOH) |
δ
da (Ace–MeOH) |
δ
eb (Ace–MeOH) |
δ
Hansc (Ace–MeOH) |
V m (THF–MeOH) | δ d (THF–MeOH) | Δ e (THF–MeOH) | δ Hans (THF–MeOH) |
|---|---|---|---|---|---|---|---|---|
| 0 | 72.40 (74.0) | 17.45 (15.5) | 12.91 (12.5) | 21.70 (20.1) | 83.52 (81.7) | 17.73 (16.8) | 8.28 (9.82) | 19.57 (19.4) |
| 10 | 68.932 | 16.65 | 14.49 | 22.07 | 75.52 | 17.4 | 10.48 | 20.31 |
| 20 | 65.12 | 16.46 | 15.94 | 22.91 | 70.16 | 17.01 | 12.36 | 21.03 |
| 30 | 62.45 | 15.94 | 17.15 | 23.41 | 68.36 | 16.12 | 14.28 | 21.54 |
| 40 | 54.92 | 16.14 | 19.06 | 24.97 | 57.91 | 16.44 | 16.95 | 23.61 |
| 50 | 51.73 | 15.81 | 20.31 | 25.74 | 53.73 | 16.07 | 18.75 | 24.69 |
| 60 | 48.95 | 15.47 | 21.48 | 26.47 | 50.36 | 15.65 | 20.35 | 25.67 |
| 80 | 44.05 | 14.78 | 23.69 | 27.92 | 44.59 | 14.86 | 23.2 | 27.55 |
| V m | δ d | δ e | δ Hans | |
|---|---|---|---|---|
|
a δd: energy from dispersion (van der Waals) interactions.
b δe2 = δp2 + δh2. δp: energy from polar (dipole–dipole) interactions, δh: energy from hydrogen bonding interactions.
c δHans: Hansen solubility parameter. δHans2 = δd2 + δp2 + δh2.
|
||||
| Bulk PVC | 5351.23 | 18.04 (18.82) | 7.56 (10.49) | 19.96 (21.54) |
Overall, the total δhans increases with higher % MeOH, indicating reduced compatibility with PVC, as confirmed by heightened Ra values (Fig. S2†). Ra increases with increasing % MeOH, with THF–MeOH showing slightly smaller Ra values than Ace–MeOH.
4. Conclusion
PVC was successfully fractionated using weak solvent/nonsolvent and strong solvent/nonsolvent blends using commodity solvents, two of which (Ace and MeOH) are considered “green”. The fractionations were attempted using both single-step and sequential fractionation methods. Both methods showed a trend in increasing molecular weight of fraction with decrease of nonsolvent, MeOH, in solvent blends. Fractions obtained using sequential fractionation showed remarkable decrease in Đ, with fractions as low as Đ = 1.14. Further, no degradation was observed in any of the extracted samples. Commercial PVC products (e.g., pipe and pellets) were also used to illustrate the capability of this method in recovering clean PVC from formulated materials. Early “light” fractions showed slight contamination, however, the bulk of the recovered polymer, 76.9 wt%, appears identical to the pristine virgin PVC. Tg values for the recovered fractions agreed with theoretical values, constant at high Mn values but reduced greatly at low Mn values. The Tg of the low molecular weight fractions also agreed with theoretical values, proving that the presence of contaminants(plasticizers) was only in trace amounts.The results of PVC fractionation warrant further study. Of interest, is the study and experimentation with green solvents towards fractionation.27 Furthering the inspiration from the lignin fractionation field, sequential fractionation can be attempted using different solvents rather than solvent blends.82 The low Đ of the fractions inspires study of PVC's physical properties, such as determining more accurate Flory–Fox parameters, especially at high molecular weights.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support from the U.S. National Science Foundation (EFMA-2132133). We thank Prof. Steven T. Weinman for the use of his viscometer instrument. A. Alshaikh acknowledges the Saudi Ministry of Education as represented by the Saudi Arabian Cultural Mission (SACM) in the United States for their support. J. E. Bara acknowledges support for C. McClaren from the University of Alabama Graduate School's Strategic Graduate Partnerships Initiative (SGPI). The images within the TOC graphic were created by the authors using AI generative software (Adobe Firefly). The authors’ university has a license for Adobe Firefly and terms of use associated with the software are being followed. All other images/photographs were fully created by the authors or adapted with permission.References
- UNEP , Moving towards the end of plastic pollution, https://www.unep.org/news-and-stories/press-release/moving-towards-end-plastic-pollution, (accessed June, 2024).
- EPA , Circular Economy | US EPA, https://www.epa.gov/circulareconomy, (accessed June, 2024).
- C. Matthews, F. Moran and A. K. Jaiswal, J. Cleaner Prod., 2021, 283, 125263 CrossRef.
- J. Zhu, C. Fan, H. Shi and L. Shi, J. Ind. Ecol., 2019, 23, 110–118 CrossRef.
- PureCycle , PureCycle | The sustainable plastic revolution has arrived, https://www.purecycle.com/, (accessed June, 2024).
- Amcor , Sustainability Packaging Solutions | Amcor, https://www.amcor.com/sustainability, (accessed June, 2024).
- V. Institute , Advancing Circularity in the Vinyl Industry, https://www.vinylinfo.org/circularity/, (accessed June, 2024).
- IBM , Plastic Bank | IBM, https://www.ibm.com/case-studies/plastic-bank-systems-linuxone, (accessed June, 2024).
- Eastman , A Better Circle | Sustainable Company | Innovative Materials | Eastman, https://www.eastman.com/en/sustainability, (accessed June, 2024).
- X. Tang and E. Y. X. Chen, Chem, 2019, 5, 284–312 CAS.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Nature, 2020, 583, 542–547 CrossRef CAS PubMed.
- J.-G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- K. Bhubalan, A. M. Tamothran, S. H. Kee, S. Y. Foong, S. S. Lam, K. Ganeson, S. Vigneswari, A.-A. Amirul and S. Ramakrishna, Environ. Res., 2022, 213, 113631 CrossRef CAS PubMed.
- P. Benyathiar, P. Kumar, G. Carpenter, J. Brace and D. K. Mishra, Polymers, 2022, 14, 2366 CrossRef CAS PubMed.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- S. R. Nicholson, J. E. Rorrer, A. Singh, M. O. Konev, N. A. Rorrer, A. C. Carpenter, A. J. Jacobsen, Y. Román-Leshkov and G. T. Beckham, Annu. Rev. Chem. Biomol. Eng., 2022, 13, 301–324 CrossRef CAS PubMed.
- O. Horodytska, F. J. Valdés and A. Fullana, Waste Manage., 2018, 77, 413–425 CrossRef CAS PubMed.
- Y.-B. Zhao, X.-D. Lv and H.-G. Ni, Chemosphere, 2018, 209, 707–720 CrossRef CAS PubMed.
- S. Ügdüler, K. M. Van Geem, M. Roosen, E. I. P. Delbeke and S. De Meester, Waste Manage., 2020, 104, 148–182 CrossRef PubMed.
- R. J. Sperber and S. L. Rosen, Polym. Eng. Sci., 1976, 16, 246–251 CrossRef CAS.
- L. Wang, Y. Liu, H. Lu and Z. Huang, Chemosphere, 2020, 259, 127402 CrossRef CAS PubMed.
- S. Yousef, M. Tatariants, M. Tichonovas, L. Kliucininkas, S.-I. Lukošiūtė and L. Yan, J. Cleaner Prod., 2020, 254, 120078 CrossRef CAS.
- T. W. Walker, N. Frelka, Z. Shen, A. K. Chew, J. Banick, S. Grey, M. S. Kim, J. A. Dumesic, R. C. Van Lehn and G. W. Huber, Sci. Adv., 2020, 6, eaba7599 CrossRef PubMed.
- C. Samorì, W. Pitacco, M. Vagnoni, E. Catelli, T. Colloricchio, C. Gualandi, L. Mantovani, A. Mezzi, G. Sciutto and P. Galletti, Resour., Conserv. Recycl., 2023, 190, 106832 CrossRef.
- N. D. Gil-Jasso, E. A. Giles-Mazón, G. Soriano-Giles, E. W. Reinheimer, V. Varela-Guerrero and M. F. Ballesteros-Rivas, Fuel, 2022, 307, 121835 CrossRef CAS.
- S. Qian, X. Liu, G. P. Dennis, C. H. Turner and J. E. Bara, Fluid Phase Equilib., 2020, 521, 112718 CrossRef CAS.
- A. Soyemi and T. Szilvási, Ind. Eng. Chem. Res., 2023, 62, 6322–6337 CAS.
- J.-M. Pin, I. Soltani, K. Negrier and P. C. Lee, Polymers, 2023, 15, 4714 CrossRef CAS PubMed.
- J. Cavalcante, R. Hardian and G. Szekely, Sustainable Mater. Technol., 2022, 32, e00448 CrossRef CAS.
- R. Singhal, I. Ishita and P. K. Sow, J. Polym. Environ., 2019, 27, 1240–1251 CrossRef CAS.
- B. A. Pulido, O. S. Habboub, S. L. Aristizabal, G. Szekely and S. P. Nunes, ACS ACS Appl. Polym. Mater., 2019, 1, 2379–2387 CrossRef CAS.
- M. Ramírez-Martínez, S. L. Aristizábal, G. Szekely and S. P. Nunes, Green Chem., 2023, 25, 966–977 RSC.
- G. Pepperl, J. Vinyl Addit. Technol., 2000, 6, 181–186 CrossRef CAS.
- E. Barnard, J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- K. Faust, P. Denifl and M. Hapke, ChemCatChem, 2023, 15, e202300310 CrossRef CAS.
- S. L. Nordahl, N. R. Baral, B. A. Helms and C. D. Scown, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2306902120 CrossRef CAS PubMed.
- C. Cimpan, A. Maul, M. Jansen, T. Pretz and H. Wenzel, J. Environ. Manage., 2015, 156, 181–199 CrossRef CAS PubMed.
- S. P. Gundupalli, S. Hait and A. Thakur, Waste Manage., 2017, 60, 56–74 CrossRef PubMed.
- S. Yan, D. Xia, N.-C. Lai, B. Jiang and X. Liu, J. Hazard. Mater., 2023, 449, 131032 CrossRef CAS PubMed.
- L. Chen, J. B. Moreira, L. C. Meyer and J. Szanyi, Appl. Catal., B, 2023, 335, 122897 CrossRef CAS.
- K. Lewandowski and K. Skórczewska, Polymers, 2022, 14, 3035 CrossRef CAS PubMed.
- P. A. Kots, B. C. Vance, C. M. Quinn, C. Wang and D. G. Vlachos, Nat. Sustain, 2023, 6, 1258–1267 CrossRef.
- M. Sadat-Shojai and G.-R. Bakhshandeh, Polym. Degrad. Stab., 2011, 96, 404–415 CrossRef CAS.
- Z. Ait-Touchente, M. Khellaf, G. Raffin, N. Lebaz and A. Elaissari, Polym. Adv. Technol., 2024, 35, e6228 CrossRef CAS.
- R. Babinsky, Plastics, Additives and Compounding, 2006, vol. 8, pp. 38–40 Search PubMed.
- VinylPlus , PVC recycling technologies, https://www.vinylplus.eu/resources/pvc-recycling-technologies/, (accessed June, 2024).
- VINYLOOP , VINYLOOP: Closure of operation in Italy/Phthalates issue under REACH brings down European PVC recycling project | Plasteurope.com, https://www.plasteurope.com/news/VINYLOOP_t240095/, (accessed June, 2024).
- I. Inovyn , INEOS Inovyn launches next generation recycling pilot plants - to strengthen Europe's PVC recycling, https://www.ineos.com/news/shared-news/ineos-inovyn-launches-next-generation-recycling-pilot-plants–-to-strengthen-europes-pvc-recycling/, (accessed August, 2024).
- CreaSolv , CreaSolv: The safe solution for plastic recycling and a better environment, https://www.creasolv.de/en/, (accessed December, 2024).
- Polyloop , Home - Polyloop, https://polyloop.fr/?lang=en, (accessed December, 2024).
- S. Wagner and M. Schlummer, Resour., Conserv. Recycl., 2024, 211, 107889 CrossRef CAS.
- G. Wypych, in PVC Formulary, ed. G. Wypych, ChemTec Publishing, Toronto Ontario, 3rd edn, 2020, pp. 95–363, DOI:10.1016/B978-1-927885-63-5.50007-0.
- M. Gigli and C. Crestini, Green Chem., 2020, 22, 4722–4746 RSC.
- Q. Ma and X. Zhang, Sci. Rep., 2022, 12, 19136 CrossRef CAS PubMed.
- G. Pepperl, . Vinyl Addit. Technol., 2000, 6, 88–92 CrossRef CAS.
- A. Alshaikh, S. Ezendu, D. Ryoo, P. S. Shinde, J. L. Anderson, T. Szilvási, P. A. Rupar and J. E. Bara, ACS Appl. Polym. Mater., 2024, 6, 9656–9662 CrossRef CAS.
- F. V. Olowookere, A. Al Alshaikh, J. E. Bara and C. H. Turner, Mol. Simul., 2023, 49, 1401–1412 CrossRef CAS.
- F. V. Olowookere and C. H. Turner, Mol. Simul., 2024, 1–9 Search PubMed.
- F. V. Olowookere, G. D. Barbosa and C. H. Turner, Ind. Eng. Chem. Res., 2024, 63, 1109–1121 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1, 19–25 CrossRef.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- J. Valatin, Phys. Rev., 1961, 122, 1012 CrossRef CAS.
- M. Yabe, K. Mori, K. Ueda and M. Takeda, J. Comput. Chem., Jpn., 2019, 5, 2018–0034 Search PubMed.
- J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2008, 4, 297–306 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- S. Nosé and M. Klein, Mol. Phys., 1983, 50, 1055–1076 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- G. Grause, S. Hirahashi, H. Toyoda, T. Kameda and T. Yoshioka, J. Mater. Cycles Waste Manage., 2017, 19, 612–622 CrossRef CAS.
- H. Geerissen, J. Roos and B. A. Wolf, Die Makromol. Chem., 1985, 186, 735–751 CrossRef CAS.
- W. Xu, H. Huo, X. Ma, R. Su, Z. Yuan, X. Liang, H. Wang, T. Wen, Z. Zeng, L. Li and S. Wang, Chem. Eng. J., 2023, 474, 145565 CrossRef CAS.
- J. W. Summers, E. B. Rabinovitch and P. C. Booth, J. Vinyl Technol., 1986, 8, 2–6 CrossRef CAS.
- J. Yu, L. Sun, C. Ma, Y. Qiao and H. Yao, Waste Manage., 2016, 48, 300–314 CrossRef CAS PubMed.
- P. P. R. Cruz, L. C. da Silva, R. A. Fiuza Jr and H. Polli, J. Appl. Polym. Sci., 2021, 138, 50598 CrossRef CAS.
- T. G. Fox Jr and P. J. Flory, J. Appl. Phys., 1950, 21, 581–591 CrossRef.
- X. Lu and B. Jiang, Polymer, 1991, 32, 471–478 CrossRef CAS.
- A. V. Dobrynin, R. Sayko and R. H. Colby, ACS Macro Lett., 2023, 12, 773–779 CrossRef CAS PubMed.
- Compendium of Chemical Terminology, ed. K. Jan and C. Stuart, International Union of Pure and Applied Chemistry (IUPAC), Zürich, Switzerland, 3.0.1 edn, 2019, DOI:10.1351/goldbook.M03706.
- W. Zeng, Y. Du, Y. Xue and H. Frisch, in Physical properties of polymers handbook, ed. J. E. Mark, Springer, New York, NY, 2007, pp. 289–303, DOI:10.1007/978-0-387-69002-5.
- S. Y. Park, J.-Y. Kim, H. J. Youn and J. W. Choi, Int. J. Biol. Macromol., 2018, 106, 793–802 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of recent literature discussing solvent-based recycling of plastics. A discussion about computational methodologies and results. A discussion regarding the additives present in commercial PVC. Experimental spectra for GPC, FTIR, and DSC and viscosity. See DOI: https://doi.org/10.1039/d4lp00313f |
| This journal is © The Royal Society of Chemistry 2025 |