
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterface-engineered UiO-66 nanoparticles on porous carbon textiles for reactive protection against toxic 2-chloroethyl ethyl sulfide†
Dimitrios A.
Giannakoudakis
 ab,
Paola S.
Pauletto
a,
Marc
Florent
a and
Teresa J.
Bandosz
*a
ab,
Paola S.
Pauletto
a,
Marc
Florent
a and
Teresa J.
Bandosz
*a
aDepartment of Chemistry and Biochemistry, The City College of New York, 160 Convent Ave, New York, NY 10031, USA. E-mail: tbandosz@ccny.cuny.edu
bInstitute of Chemical Sciences, Faculty of Chemistry, Maria Curie-Sklodowska University, Maria Curie Sklodowska Sq. 3, 20-031 Lublin, Poland
First published on 7th July 2025
Abstract
Porous carbon textile composites with incorporated zirconium-based UiO-66 nanoparticles were synthesized using two approaches: a dip-and-dry post-synthesis deposition method and an in situ synthesis strategy in which UiO-66 nanoparticles were grown directly on the surface of carbon textiles. Pre-oxidation of the carbon textile significantly enhanced UiO-66 deposition and dispersion. The pre-oxidized composite textile synthesized through the in situ approach (CT-O-UiO-i) showed the highest UiO-66 loading of 6.7 wt%, which was more than four times higher than on the oxidized textile modified with pre-synthesized UiO-66 (CT-O-UiO-d). The surface area of CT-O-UiO-d and CT-O-UiO-i was 659 and 430 m2 g−1, respectively. The modified textiles effectively captured the mustard gas simulant, 2-chloroethyl ethyl sulfide (CEES), with weight uptakes reaching up to 396 mg g−1 for CT-O-UiO-d. While the surface area was crucial for physical adsorption, UiO-66 enabled the chemical decomposition of CEES into less toxic compounds such as diethyl disulfide (DEDS) and ethyl vinyl sulfide (EVS). CT-O-UiO-i exhibited the highest reactivity, primarily converting CEES to EVS via dehydrohalogenation. This was attributed to the high dispersion and strong anchoring of UiO-66, increasing the number and accessibility of Lewis acidic sites. Therefore, this study highlights the potential of MOF-modified carbon textiles as functional materials that combine physical and reactive adsorption to ensure effective protection against this chemical warfare agent.
1. Introduction
Bis(2-chloroethyl) sulfide, commonly known as mustard gas, is a potent chemical warfare agent (CWA) that inflicts severe damage to the skin, eyes, and respiratory system, with long-term exposure leading to chronic respiratory issues, tissue necrosis, genetic mutations, and an increased cancer risk.1–3 Its ability to penetrate regular clothing requires a search for personal protective garments that provide effective safety against mustard gas's harmful effects, driving the development of advanced multi-layered fabrics for full-body safeguarding garments and flexible gas mask cartridges.4Activated carbon, a well-established material in filtration technologies, has been widely used in protective garments due to its high surface area and ability to adsorb hazardous organic compounds.4–6 While carbon fabrics are effective in physically adsorbing CWAs, their limited capacity to degrade or neutralize these agents is a major drawback.4,5,7 However, new porous solids, such as metal–organic frameworks (MOFs), have emerged as promising materials for enhancing chemical protection.8–11 Zirconium-based MOFs have gained significant attention due to their easy synthesis, chemical and thermal stability, large surface area, and catalytic activity for degrading CWAs.12–14 It was demonstrated recently that the detoxification of the mustard gas simulant, 2-chloroethyl ethyl sulfide (CEES), on UiO-66 was primarily affected by its Lewis acidity rather than its surface area. The decomposition efficiency ranged from 70% to 84% after 24 hours of exposure, and it increased as the particle size of Zr-based MOF decreased.15 Also, the incorporation of 10 wt% of nano-graphite or oxidized graphitic carbon nitride nanospheres into UiO-66 created new interfaces between the modifiers and MOF units/defects. These interfaces introduced additional active sites, with the graphitic carbon nitride surface groups acting as linkers to the MOF framework, which improved the detoxification performance against CEES.16
Despite their potential, UiO-based MOFs typically exist as powders, which present significant challenges for their direct application onto fabric substrates.8,17,18 Different approaches have been reported to prepare composites that combine the features of powdery MOFs and fabrics substrates. For instance, atomic layer deposition was used to prepare Zr-MOF thin films on polyamide-6 nanofibers for CWAs detoxification; incorporating surfactant assembly agents like β-cyclodextrin and CTAB enhanced both MOF loading and adhesion to the substrate, and the resulting composites such as PA-6@TiO2@UiO-66, UiO-66-NH2, and UiO-67 showed short half-lives and high conversion efficiency of the nerve agent soman.19 A scalable aqueous synthesis of a zirconium-based MOFs coating on polyester textiles resulted in composites with high surface areas and enhanced hydrolysis of nerve agent simulants.20 Zirconium-based MOF-808 was successfully loaded onto a polypropylene nonwoven fabric using a tannic acid and 3-aminopropyltriethoxysilane coating to enhance the MOF attachment. The resulting composite demonstrated rapid catalytic hydrolysis of dimethyl p-nitrophenyl phosphate in a liquid phase, achieving a short half-life of just 1.3 minutes.21
Pre-synthesized UiO-66-NH2 crystals were coated onto nonwoven polypropylene fibrous mats using cetyltrimethylammonium bromide and β-cyclodextrin as surfactant assembly agents at room temperature. The resulting material exhibited the efficient catalytic degradation of dimethyl 4-nitrophenyl phosphate simulant, with a half-life of less than 5 minutes.22 In another study,23 Cu-based MOFs were deposited onto a cotton textile using a simple dip-and-dry process with an ethanolic suspension of the active phase (MOFs). The modified textiles demonstrated enhanced performance for the rapid detection and detoxification of a CWA simulant. An in situ strategy to grow UiO-66-NH2 on electrospun polyacrylonitrile nanofibers was proposed,17 utilizing trifluoroacetic acid to slow down crystallization and generate additional defective sites between the modifiers and MOF units. The combined high porosity and catalytic activity of this material enabled the efficient capture and decomposition of CEES. Zirconium-based MOFs grown on activated carbon textiles via layer-by-layer synthesis showed 40–80% efficiency for detoxification of a nerve agent simulant diisopropyl fluorophosphate reached after 24 hours of exposure. Prior oxidation of the carbon surface increased the amounts of hydroxyl and carboxylate groups, that enhanced metal ion anchoring and promoted MOF growth on the composite.24
Taking advantage of the porous carbon textiles and crystalline UiO-66 framework properties, the objective of this study was to investigate the effects of the conditions of carbon textile composites' synthesis on their surface features and the decomposition efficiency of CEES. The focus was the modification of a porous carbon textile and its oxidized counterpart with MOF particles either by in situ synthesis of an UiO-66 phase or by deposition of pre-synthesized UiO-66 nanoparticles using the dip-and-dry method.23,25 The effect of oxygen functionalities of the carbon surface on the deposition of MOF, contributing to the decomposition of CEES, was also explored. This work aims to advance functional materials capable of providing reliable protection against mustard gas exposure.
2. Experimental
2.1. Materials
Carbon porous textile (Stedcarb, Stedfast Inc.) was supplied by the U.S. Army Natick Soldier Research, Development & Engineering Center and was used after the removal of the polymeric layers, as previously described elsewhere;26 the obtained textile is referred to as CT. This is a particularly robust and flexible CT especially engineered for military purposes. Its resistance upon stretching is demonstrated in Fig. S1.† Zirconium tetrachloride (ZrCl4, ≥99.5%, Alfa Aesar), hydrochloric acid (HCl, 36.5–38%, BDH), N,N-dimethylformamide (DMF, ≥99.7%, Alfa Aesar), terephthalic acid (C8H6O4, ≥99%, Acros Organics), nitric acid (HNO3, 68–70%, VWR), sulfuric acid (H2SO4, 95–98%, VWR), and ethanol (200 Proof, DIY Chemicals) were used as received.2.2. Oxidation of the nanoporous carbon textile
To introduce surface chemistry heterogeneity, CT was oxidized as previously reported.26 Briefly, a CT swatch was immersed in a mixture of concentrated sulfuric acid and nitric acid (75/25 volume ratio) for 5 hours at 10 °C (controlled with an ice bath) with continuous stirring, followed with deionized water washing (until a neutral pH of the filtrate) and finally drying at 150 °C in a vacuum oven overnight. The treatment resulted in the introduction of oxygen- and nitrogen-containing functional groups. The obtained textile is referred to as CT-O.2.3. Modification of porous carbon textiles with UiO-66 nanoparticles
For the deposition of the UiO-66 active phase on the carbon textiles, two protocols were followed. The first one used pre-synthesized UiO-66 particles to decorate the carbon fibers, herein mentioned as a dip-and-dry post-deposition method. In the second protocol, UiO-66 particles were synthesized directly in the presence of the carbon textiles within the synthesis suspension, allowing the in situ growth of the UiO-66 phase on the surface of the carbon textiles.UiO-66 nanoparticles were prepared based on the process previously reported.16,27 Briefly, ZrCl4 (750 mg), DMF (30 mL), and HCl (6 mL) were mixed in a 100 mL reaction vessel and sonicated for 20 minutes. Separately, terephthalic acid (738 mg) was dissolved in DMF (60 mL), sonicated for 5 minutes, and added to the reaction vessel. The mixture was further sonicated for 20 minutes and heated at 80 °C overnight. The resulting powder was filtered, washed with DMF and ethanol, then dried under vacuum at 130 °C for 12 hours (yield 88.3% per mass). Finally, the obtained white UiO-66 powder was activated at 150 °C under high vacuum.
A suspension of UiO-66 particles was prepared after dispersing 25 mg of dried MOF powder in ethanol using ultrasonication (10 min, 40 kHz) for 10 minutes. The dip-and-dry method was employed in the initial step. Approximately 200 mg of the CT swatch (about 5 × 5 cm) was soaked in the suspension for 30 minutes, then sonicated for 5 minutes. Subsequently, the drained textile was placed on a heated plate (∼65 °C) and the remained ethanolic MOF suspension was slowly and homogeneously drop-wise dispersed on the textile's surface in four consecutive steps, allowing for solvent evaporation between each application (at each step, the swatch was turned upside-down). After the entire suspension was used in the modification approach, the swatch was dried in an oven at 100 °C overnight. Following this, the modified textile was subjected to a pressurized airstream for 1 minute with simultaneous mechanical folding/unfolding steps. All textiles retained their structural integrity under these conditions, indicating that the composites are mechanically resistant. Then, the swatch was immersed in ethanol with gentle stirring for 10 minutes to remove loosely attached nanoparticles. Finally, the textile was dried at 110 °C under vacuum overnight. The composite of the initial textile with the deposited UiO-66 particles is designated as CT-UiO-d. The same process was followed for the pre-oxidized carbon textile (CT-O), and the sample obtained is referred to as CT-O-UiO-d.
The in situ approach was based on a scaled-down version of the UiO-66 synthesis protocol. First, 0.1 g of zirconium tetrachloride, 0.8 mL of hydrochloric acid, and 4 mL of DMF were added to a glass reaction vessel (∅ = 3 cm, VWR). The mixture was sonicated for approximately 20 minutes until the chemicals were completely dissolved. In a separate step, 0.1 g of terephthalic acid was dissolved in 8 mL of DMF using sonication and then added to the reaction vessel together with approximately 150 mg of CT (about 3 × 3 cm). The content of the reaction vessel was sonicated for 30 minutes and then heated overnight at 80 °C. The solid phase was recovered by filtration, washed twice with 10 mL of DMF and twice with 10 mL of ethanol. The material was then dried in a vacuum oven (Equatherm) at 130 °C overnight. Following this, the modified textile was subjected to a pressurized airstream for 1 minute with simultaneous mechanical folding/unfolding steps. The resulting sample is referred to as CT-UiO-i. The same procedure was applied to the oxidized carbon textile (CT-O) and the sample is designated as CT-O-UiO-i.
2.4. Characterization techniques
Scanning electron microscopy (SEM) micrographs were acquired using a Quanta 3D FEG microscope (Thermo Fisher Scientific/FEI, USA) with an accelerating voltage ranging from 5 keV to 20 keV.Thermogravimetric analysis was carried out on a thermal analyzer (TA Instruments, SDT Q600). The measurements were recorded over the temperature range of 30 to 1000 °C at a heating rate of 10 °C min−1 under a controlled flow of either air or argon (100 mL min−1). For the latter, the mass spectra of the released gases were simultaneously analyzed using a mass spectrometer (ThermoStar, Pfeiffer Vacuum). The thermal profiles were obtained after prior drying at 150 °C.
X-ray diffraction (XRD) measurements were recorded on a diffractometer (Philips X'Pert X-ray) using CuKα radiation at 40 kV and 40 mA, within the 2θ range from 5° to 70°.
Fourier Transform Infrared (FTIR) spectra were acquired using a Nicolet 380 spectrometer (Thermo) with pellets composed of 10 mg of the composite and 300 mg of KBr. The spectra were recorded in the wavenumber range from 4000 and 400 cm−1 (64 scans and 2 cm−1 resolution).
Nitrogen adsorption and desorption isotherms were obtained at −196 °C using the ASAP 2020 analyzer (Micromeritics). Before analysis, the composites were outgassed overnight at 120 °C under vacuum. The Brunauer–Emmett–Teller (BET) method was used to determine a specific surface area (SBET). A total pore volume (VTot) was obtained from the amount of adsorbed nitrogen at the relative pressure of 0.99. The pore size distribution and micropore volume (Vmic) were estimated using two-dimensional non-local density functional theory (2D-NLDFT).28 The mesopore volume (Vmes) was calculated as the difference between VTot and Vmic.
X-ray photoelectron spectroscopy (XPS) measurements of the interface engineered textiles were conducted using a Multi-Chamber Analytical System (Prevac, Poland) equipped with a monochromatic Al Kα X-ray source (1486.6 eV, 450 W). The analysis chamber was maintained at an ultrahigh vacuum of 8 × 10−9 Pa. The binding energy (BE) scale was calibrated using the C 1s peak at 284.7 eV as a reference.
2.5. CEES decomposition tests
All CEES decomposition experiments were performed in a closed system using a mustard gas surrogate, 2-chloroethyl ethyl sulfide (>97% purity, TCI), referred to as CEES, as the target compound. Prior to the tests, all composite textiles were dried at 150 °C. A vial-in-vial setup was used to estimate the maximum weight uptake of all composites.26 In this setup, a small vial containing 20 mg of composite and another with 40 μL of CEES were enclosed in a 20 mL screw-thread amber vial and securely sealed with a septum cap. The vials were kept in the dark, and the weight uptake (WU) was recorded at different time durations for up to 5 days. Then, desorption experiments were carried out over 2 days by exposing the open vial to ambient conditions inside a fume hood, allowing the release of weakly adsorbed compounds. The exhausted composites were then subjected to thermogravimetric analysis under argon.The interaction between liquid CEES and the composites was evaluated by dropping 3 μL of the toxic compound into the center of a 0.5 inch diameter swatch placed at the bottom of a 20 mL screw-thread amber vial, which was then sealed with a septum cap.26 The vials were kept in the dark for 1 day, and then 40 μL of headspace vapors were sampled through the septum and analyzed by a gas chromatography-mass spectrometer (GCMS). The spent composite was treated with 3 mL of acetonitrile (>99% purity, OmniSolv) to extract remaining chemicals. The extract was filtered through a 0.22 μm nylon membrane, and 2 μL of the filtered solution was automatically injected into the GCMS for analysis. All tests were performed in duplicates, alongside blank tests (without the composite).
GCMS analysis was performed using a GCMS 7820A/5977B system (Agilent Technologies) equipped with an Agilent HP-5MS ultra-inert column (30 m × 0.25 mm × 0.25 μm). The temperature was programmed to increase from 50 °C to 110 °C at 5 °C min−1, then from 110 °C to 270 °C at 40 °C min−1. Helium was the carrier gas at a flow rate of 33.7 mL min−1. Split ratios were set to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 50:1 for vapor and liquid samples, respectively.
:1 and 50:1 for vapor and liquid samples, respectively.
3. Results and discussion
3.1. Characterization of composites
In order to determine the effectiveness of each deposition protocol, the morphological features of all samples were analyzed by a scanning electron microscope (SEM). The initial textile (CT) consists of “helically twisted” cylindrical yarns of fiber bundles (Fig. 1), with the diameter of the fibers ranging from 7 to 11 μm. Oxidation of CT did not markedly alter the textile morphology (CT-O). The only visible effect of oxidation is a slight decrease in the extent of the “helically twisted” assembly, leading to a more parallel arrangement. In addition, the oxidative treatment resulted in the elimination of some impurities of CT.26 | ||
| Fig. 1 SEM micrographs at various magnifications of CT, CT-O, and their composites with UiO-66 nanoparticles. | ||
The deposition of UiO-66 by the dip-and-dry post-synthesis deposition method was successful for both initial (CT-UiO-d) and oxidized (CT-O-UiO-d) samples (Fig. 1). However, the extent of the UiO deposition was significantly higher for the oxidized textile, suggesting the pivotal role of the formed surface functional groups (SFGs) in the oxidation process acting as UiO-attracting sites at the interface.
Based on the amounts of UiO-66 visible on the fibers (Fig. 1), the deposition efficiency from the in situ method is higher compared to that from the dip-and-dry deposition method. This is likely due to surface groups acting as “seeds” for the MOF growth.29 While the sizes of the UiO-66 nanoparticles from the in situ approach are small, reaching 50–110 nm, those obtained in the dip-and-dry process range from 90 to 180 nm, following the sizes of the pre-synthesized UiO-66 nanoparticles (Fig. S2†). The particles form aggregates, which do not surpass 900 nm in size, and are especially visible for CT-UiO-i where the crystals are not homogeneously dispersed. On the contrary, the modified oxidized textile (CT-O-UiO-i) shows the high dispersion of the MOF nanoparticles. These results indicate that the oxidized textiles are good supports for MOF in both dip-and-dry post-synthesis deposition and in situ synthesis methods.
The amount of MOF deposited and the thermal stability of the modified textiles were estimated by thermogravimetric analysis (Fig. S3†). In agreement with the SEM results, the in situ process resulted in more UiO-66 deposited, which further increased on the oxidized sample. The remaining weights at 750 °C for CT-UiO-d, CT-UiO-i, CT-O-UiO-d, and CT-O-UiO-i were 0.6, 2.6, 1.0, and 4.1%, respectively. Based on the thermal decomposition profile of pure UiO-66 that showed 60.9% weight loss at 750 °C, the amount of the UiO-66 loaded per weight was estimated as 1.0, 4.3, 1.6, and 6.7% for CT-UiO-d, CT-UiO-i, CT-O-UiO-d, and CT-O-UiO-i, respectively.
To confirm the UiO-66 nature of the formed crystals, X-ray diffraction was initially performed on the pre-synthesized UiO-66, which was later used in the dip-and-dry protocol, and on the loose UiO-66 particles obtained at the in situ synthesis in the presence of CT and CT-O textiles. The diffractograms (Fig. S4†) show their crystalline structure and structural integrity. The main diffraction peaks at 7.3°, 8.5°, and 25.6° correspond to (111), (002), and (006) reflections, respectively, which are characteristic of the UiO-66 framework.30,31 For the UiO-66 samples synthesized in the presence of the textiles, a slight shift in the two main peaks (2θ < 9°) toward higher angles was observed. This shift could be attributed to minor distortions in the crystal structure, potentially indicating a slight contraction of the lattice parameters.32 Additionally, the UiO-66 sample synthesized in the presence of CT-O exhibited peak broadening at low angles, which might suggest a decrease in the nanoparticle size.33 The sizes of the UiO-66 crystals were calculated from the X-ray diffraction patterns using the Scherrer equation,34 and they were 36, 40, and 15 nm for pure UiO-66, UiO-66 synthesized in presence of CT, and UiO-66 synthesized in presence of CT-O, respectively.
The XRD diffractograms of the composite textiles are shown in Fig. 2a. The broad peak within the 2θ range of 15° to 35° represents amorphous carbon. Sharp peaks in the low 2θ range corresponding to the UiO-66 crystalline phase are visible for all textiles except for CT-O-UiO-d, where the features of the diffraction pattern prevent us from the calculation of the crystallite size. These peaks are more intense in the diffractograms of the textiles prepared via the in situ protocol, compared to those prepared by the post-deposition method. This increase in the peak intensity is primarily attributed to the higher UiO-66 loading in the in situ composites compared to that obtained by the deep-and-dry method, as quantified by the thermogravimetric measurements (Fig. S3†). Interestingly, CT-based composites exhibit the higher peak intensities than the CT-O-based ones, despite having the lower UiO-66 loadings. This is attributed to the larger crystallite size in CT-UiO-i (24 nm) than in CT-O-UiO-i (19 nm), as smaller crystallites produce broader, less intense diffraction peaks.33
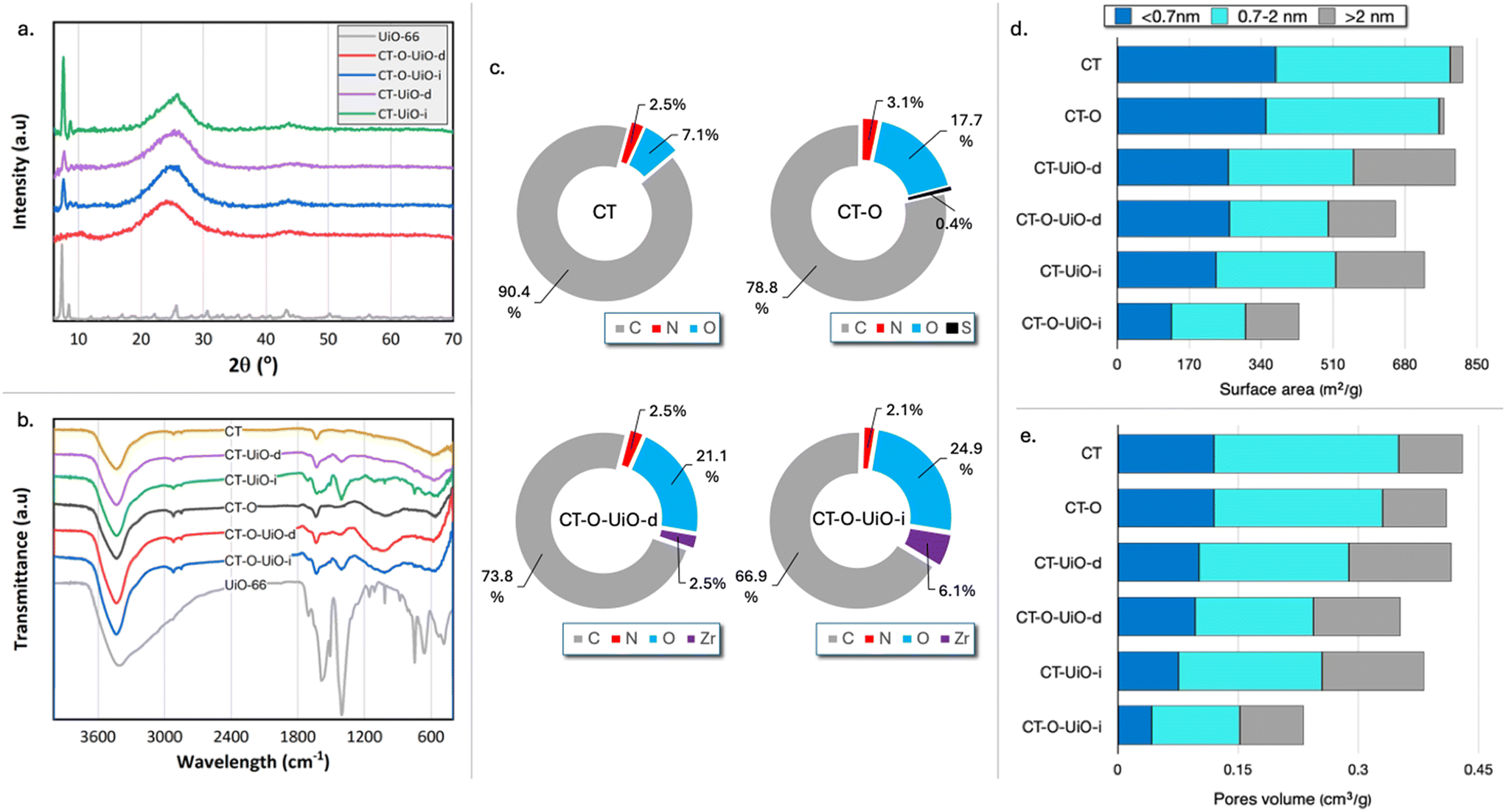 | ||
| Fig. 2 X-Ray diffractogram (a), FTIR spectra (b), XPS results of C, N, O, and Zr atomic (%) concentration26 (c), contributions of surface areas (d), and pore volumes in specific pore sizes (e) derived from N2 adsorption experiments. | ||
The FTIR spectra of the UiO-66-containing samples (Fig. S5†) are consistent with the previously reported spectra of UiO-66, confirming the characteristic absorption bands.31 The bands at 1705 cm−1 and 1656 cm−1 correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the residual DMF solvent. The intense bands at 1586 cm−1 and 1404 cm−1 are attributed to the asymmetric and symmetric stretching modes of the carboxylate (–COO−) groups from the terephthalate linkers. The band at 1018 cm−1 is associated with C–H bending vibrations of the aromatic ring in the linker. At lower frequencies, the bands at 745 cm−1, 660 cm−1, 550 cm−1, and 485 cm−1 correspond to Zr–O stretching vibrations, confirming the presence of the zirconium-based framework.31 The FTIR spectra of the initial and modified textiles are shown in Fig. 2b. CT exhibits characteristic bands associated with carbon-based materials. The broad absorption band around 3450 cm−1 corresponds to the O–H stretching vibrations of hydroxyl groups. Two consecutive small peaks at approximately 2900 cm−1 and 2810 cm−1, observed for all samples, are attributed to asymmetric and symmetric C–H stretching vibrations of a hydrocarbon structure, respectively.35 The band at 1630 cm−1 is associated with either aromatic CC stretching vibrations or conjugated carbonyl and carboxylate groups.35,36 Additionally, a small broad band around 590 cm−1 is linked to the coupled C–H bending vibrations within the carbon structure and adsorbed CO2.37 After oxidation, a new broad band between 1000 and 1300 cm−1 is visible for CT-O, which is attributed to the stretching vibrations of C–O groups, commonly observed in the spectra of oxidized carbons.37 For the modified textiles, the bands about 1580, 1400, 1020, 660, and 550 cm−1 are attributed to the incorporation of UiO-66 into the carbon surface. The intensity of these bands is higher for the textiles prepared via the in situ method compared to those prepared by the post-deposition method, which agrees with the higher UiO-66 loadings in the former sample.
O stretching vibrations of the residual DMF solvent. The intense bands at 1586 cm−1 and 1404 cm−1 are attributed to the asymmetric and symmetric stretching modes of the carboxylate (–COO−) groups from the terephthalate linkers. The band at 1018 cm−1 is associated with C–H bending vibrations of the aromatic ring in the linker. At lower frequencies, the bands at 745 cm−1, 660 cm−1, 550 cm−1, and 485 cm−1 correspond to Zr–O stretching vibrations, confirming the presence of the zirconium-based framework.31 The FTIR spectra of the initial and modified textiles are shown in Fig. 2b. CT exhibits characteristic bands associated with carbon-based materials. The broad absorption band around 3450 cm−1 corresponds to the O–H stretching vibrations of hydroxyl groups. Two consecutive small peaks at approximately 2900 cm−1 and 2810 cm−1, observed for all samples, are attributed to asymmetric and symmetric C–H stretching vibrations of a hydrocarbon structure, respectively.35 The band at 1630 cm−1 is associated with either aromatic CC stretching vibrations or conjugated carbonyl and carboxylate groups.35,36 Additionally, a small broad band around 590 cm−1 is linked to the coupled C–H bending vibrations within the carbon structure and adsorbed CO2.37 After oxidation, a new broad band between 1000 and 1300 cm−1 is visible for CT-O, which is attributed to the stretching vibrations of C–O groups, commonly observed in the spectra of oxidized carbons.37 For the modified textiles, the bands about 1580, 1400, 1020, 660, and 550 cm−1 are attributed to the incorporation of UiO-66 into the carbon surface. The intensity of these bands is higher for the textiles prepared via the in situ method compared to those prepared by the post-deposition method, which agrees with the higher UiO-66 loadings in the former sample.
The XPS analysis, presented in Fig. 2c, revealed the presence of oxygen- and nitrogen-containing surface groups on the initial CT. In particular, the elemental atomic concentration of O and N were 7.1 at% and 2.5 at%, respectively.26 The oxygen presented on carbons was in the form of quinones, carbonyls, and phenols.26 Nitrogen can originate from the synthesis process and/or from the decomposition of residual nylon.38,39 For the oxidized CT-O, the amount of nitrogen was not altered significantly, increasing from 2.5 at% to 3.1 at%, with approximately 0.4 at% of nitrogen in nitrate configurations.26 Even in a very small quantity, the presence of these groups can contribute to defects in the MOF and increase its dispersion. The amount of oxygen-containing group significantly increased to 17.7 at% after oxidation, and all oxygen-containing functional groups significantly increased after oxidation.26 The XPS analysis of CT-O-UiO-d and CT-O-UiO-i showed the Zr atomic concentration of 2.5 at% and 6.1 at%, respectively. For CT-UiO-i and CT-UiO-d textiles, the XPS spectra could not be analyzed due to the differential/intense charging leading to intense shifting and broadening. It is characteristic of heterogeneous samples consisting of separate conducting and insulating phases physically in contact.40–42
Since the intended application of these materials is the protection against CWA compounds, their textural features are of paramount importance. The porosity characteristics obtained from the nitrogen adsorption/desorption isotherms (Fig. S6†) are summarized in Table S1.† The contributions of surface areas and pore volumes in specific pore sizes are presented in Fig. 2e and f, respectively. The initial CT is a microporous material with the surface area (SBET) of 817 m2 g−1 and total pore volume (VTot) of 0.43 cm3 g−1. Oxidation partially blocked some pores, leading to a slight decrease in the textural parameters (SBET: 772 m2 g−1 and VTot: 0.41 cm3 g−1) as shown previously.26 Surface area of CT-UiO-d and CT-UiO-i decreased by 2.1% (800 m2 g−1) and 11.1% (726 m2 g−1), respectively, compared to that of CT. Considering that the UiO-66 particles are highly porous (SBET: 1247 m2 g−1 and VTot: 0.72 cm3 g−1), the decrease in the porosity of the composites is linked to the pore blockage upon the deposition of the UiO-66 particles at the pore entrances of the carbon textiles, as also shown in the SEM images. This blockage limits the access of nitrogen molecules during measurements resulting in a decrease in the surface area of the composite. The decrease in the porosity was even more pronounced for the composites of the oxidized textile. CT-O-UiO-d had a SBET of 659 m2 g−1, which is 14.6% smaller compared to that of CT-O. A decrease in SBET for CT-O-UiO-i was even more noticeable (44.3%; 430 m2 g−1). The initial textiles are predominantly microporous, with the ratio of Vmic/VTot ∼0.8. For all composite textiles, the Vmic/VTot ratio decreased to 0.67 ± 0.02, due to a decrease in the micropore volume, that could be attributed to the blockage of the small pores by UiO-66 deposited inside the larger pores.
Based on the results discussed above, the introduction of SFGs upon the oxidative treatment of CT is important not only for the amount of the MOF phase deposited on the surface of the textiles but also for its homogenous dispersion. Hence, the focus in the evaluation of the composites for CEES detoxification/decomposition is further limited to the analysis of the performance of CT-O-UiO-d and CT-O-UiO-i, where the oxidized textile is used as a support for UiO-66 deposited using the two different approaches.
3.2. Decomposition of CEES vapors
The modified textiles were exposed to vapors of mustard gas surrogate, 2-chloroethyl ethyl sulfide (CEES), using the previously presented vials-in-vial (ViV) approach.26,43 The protection performance was evaluated by recording the weight uptakes (WUs) representing the adsorption capability from the vapor phase. Two different exposure times were arbitrarily chosen: 1 and 5 days, with the results collected in Fig. 3a. The former provided information on the initial adsorption performance, and the maximum weight uptake capacity was estimated after 5 days of exposure, assuming that an equilibrium was reached inside the adsorption systems. | ||
| Fig. 3 The recorded weight uptakes (WUs) after exposure to CEES vapors in the ViV system for 1 and 5 days (max ads.), after 2 days of desorption tests at ambient conditions, and after extraction with acetonitrile (a), desorption profiles expressed as the decrease in the weight uptakes with an increase in the desorption time (b), and the normalized chromatographic peak areas of detected compounds in headspace after exposure to CEES vapors in the ViV system (c). | ||
The results after 1 day of CEES exposure revealed a good performance of CT, reaching the WU of ∼300 mg g−1.26 CT-O showed slightly lower WU (285 mg g−1), which is linked to the decrease in the porosity. The same trend was found for the maximum recorded WUs. On the other hand, the composite textiles, CT-O-UiO-d and CT-O-UiO-i, exhibited contrasting trends in their weight uptakes behavior (Fig. 3a). The weight uptake of CT-O-UiO-d increased, while that of CT-O-UiO-i decreased compared to that of CT-O. After 1 day of exposure, the measured WUs were 293 mg g−1 for CT-O-UiO-d and 265 mg g−1 for CT-O-UiO-i. These values fall between those observed for pure UiO-66 (191 mg g−1) and CT-O (285 mg g−1) and are actually close to the theoretical WUs expected for simple physical mixtures. The latter were estimated to be 284 mg g−1 for CT-O-UiO-d and 279 mg g−1 for CT-O-UiO-i. Similarly, the maximum WU values recorded were 396 mg g−1 for CT-O-UiO-d and 362 mg g−1 for CT-O-UiO-i, which are considered high compared to other results collected under similar conditions.26,43 Nevertheless, they are only slightly above and below the maximum WUs of pure UiO-66 (383 mg g−1) and CT-O (382 mg g−1). While these results do not show much improvement in terms of the weight uptakes of the composites as compared to the starting UiO-66 and CT-O, it has to be noted that the composites have smaller surface area than their pure counterparts, and therefore potentially less physical adsorption sites. Thus, to attain similar weight uptakes with 15% (CT-O-UiO-d) and 44% (CT-O-UiO-i) less surface than CT-O (and much less than UiO-66) implies a positive synergistic effect between the two constituents of the composite materials.
The adsorption strength (stability of weight uptakes) was assessed based on the change in the weight of the spent textile (Fig. 3b) left in open air (under ambient conditions inside the fume hood) to allow the desorption of weakly adsorbed compounds. Compared to CT-O, both composites showed a slower rate of desorption, representing stronger adsorption of species after 2 days of desorption. While CT-O showed a desorption extent (expressed as weight loss) of around 37%, for both CT-O-UiO-d and CT-O-UiO-i, around 30% was measured. The highest retained weight uptake after the desorption tests was for CT-O-UiO-d (∼270 mg g−1). Based on these results, modifying the oxidized textile with UiO-66 positively impacted not only the maximum adsorption capacity but also the strength of adsorption.
Another important marker of the protection efficiency of textiles is their ability to catalytically decompose the toxic CEES molecules into less or non-harmful compounds. To explore this further, the headspaces of the ViV systems were analyzed using GCMS to detect compounds formed as a result of the CEES decomposition. It is important to mention that the as-received CEES sample contains some detectable impurities, specifically diethyl disulfide (DEDS), dichloroethane (2CE), and chloroethane (CE). In the case of CT-O, the amount of these compounds was found to be lower compared to the blank, suggesting that they were also adsorbed on the surface of this textile (Fig. 3c), as shown previously.26,43
Interestingly, the composite textiles exhibit considerable differences in the components of their headspace (Fig. 3c). The remaining CE amount was almost negligible, suggesting its removal by the UiO-66 phase. This compound was detected in the headspace of CT-O.26 In general, the main observed catalytic decomposition pathways for CEES are dehydrohalogenation/elimination leading to ethyl vinyl sulfide (EVS) and hydrolysis/substitution, leading to 2-hydroxyethyl ethyl sulfide (HEES). As presented in Fig. 4, to form both HEES and EVS, a transient cyclic cation needs to be involved.44–47 In addition, HEES can be formed upon the H2O addition to EVS, and vice versa, EVS can be formed from the dehydration of HEES. The detection of diethyl disulfide (DEDS) is indicative of the decomposition of CEES by the cleavage of the S–C bond, and the recombination of specific fragments, a pathway which is different from hydrolysis or dehydrohalogenation.
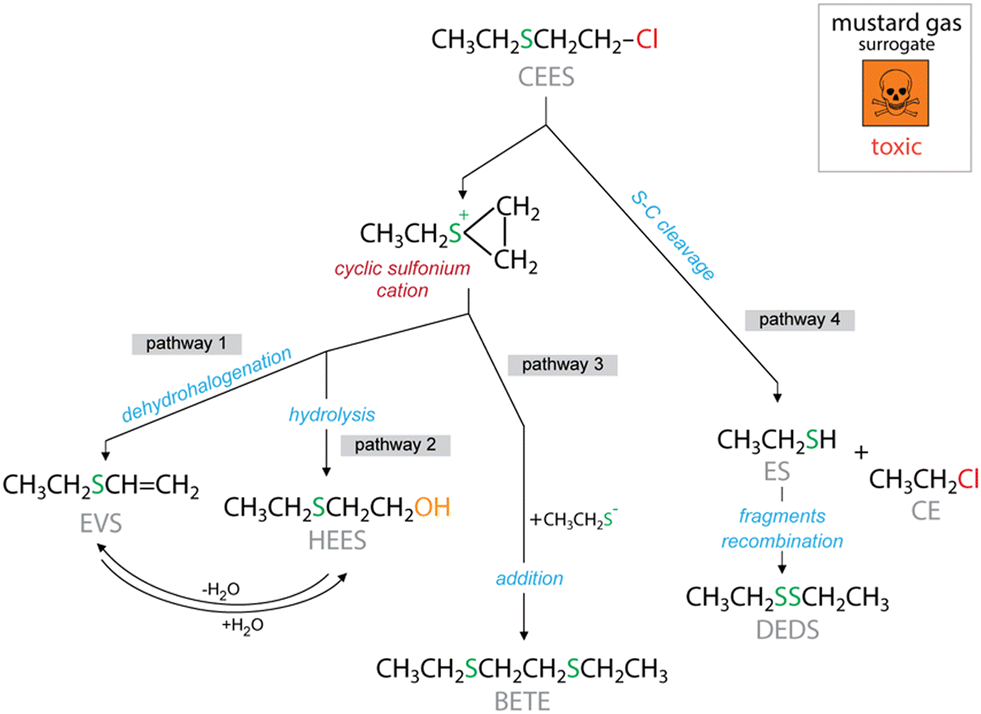 | ||
| Fig. 4 The possible reactive decomposition pathways of 2-chloroethyl ethyl sulfide. | ||
An important finding is that a high amount of EVS was detected only in the case of CT-O-UiO-i (Fig. 3c). The presence of the UiO-66 nanoparticles played a key role in the catalytic decomposition of the CEES molecules into EVS and DEDS. Interestingly, CEES was not hydrolyzed to HEES as reported in various studies of the decomposition of CEES by Zr-MOFs.48–51 It should also be pointed out that no oxidation products were detected in this study. Although oxidation is often reported as a primary detoxification mechanism,52–58 the absence of the oxidation products may be attributed to the simplified experimental conditions. In this study, pure CEES was used in either vapor or liquid form, without presence of solvents, oxidizing agents, heating, or light irradiation, all of which were employed in previous studies that reported CEES oxidation.52–58 A similar absence of oxidation products was also reported when pure CEES vapors were exposed to UiO-66 nanoparticles.16
The thermogravimetric analysis (TGA) under argon was performed on the initial and spent in the ViV experiments CT-O-UiO-d and CT-O-UiO-i samples (Fig. S7†). For the initial samples, the weight loss event below 120 °C represents the release of water and that between 120 °C and 400 °C is linked to both the decomposition of the textile surface oxygen groups and to the onset of the decomposition of terephthalic acid (organic linker of UiO-66).59 The weight loss between 400 °C and 600 °C represents the further decomposition of terephthalic acid.59 For the spent samples, a new weight loss event centered around 150 °C is linked to the decomposition of CEES and its reaction products. Fig. 5 presents the thermal profiles corresponding to selected m/z fragments, including CEES, HEES, and/or BETE (m/z = 47, 61, and 75), DEDS (m/z = 45, 47, 60, and 66), and EVS (m/z = 45, 59, and 73).47,60 The peak maxima at 150 °C are assigned to the release of CEES and the compounds with the same m/z signals (BETE and HEES). In contrast, the DEDS and EVS m/z signals were detected at higher temperatures. It is worth highlighting that m/z 45 may also result from the decomposition of terephthalic acid,59 as evidenced by the thermal profiles of the initial samples shown in Fig. S8.† However, the significantly higher signal intensity observed for the spent samples might indicate the involvement of UiO-66 in the formation of EVS and DEDS.
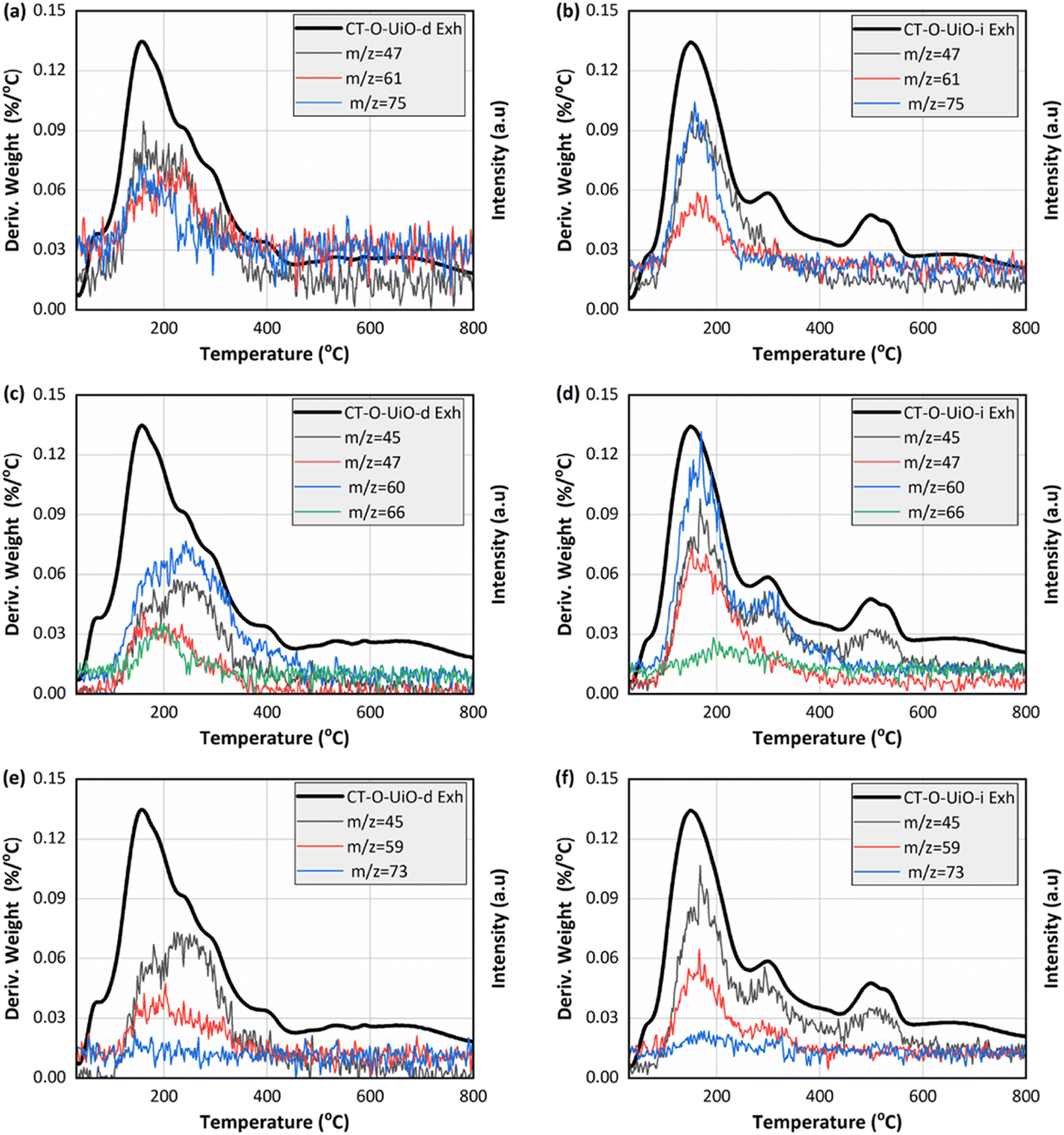 | ||
| Fig. 5 DTG curves in argon (bold lines; left Y axis) and m/z thermal profiles (right Y axis) of spent CT-O-UiO-d and CT-O-UiO-i from the ViV experiments for the fragments related to CEES, HEES, and/or BETE (a and b), DEDS (c and d), and EVS (e and f). | ||
3.3. Decomposition of CEES liquid droplets
The activity of the modified textiles in the decomposition of CEES liquid droplets was assessed after 1 day of exposure in the dark. The amounts of CEES detected in the headspace (Fig. 6a) and extracts (Fig. 6c) were normalized to the total CEES amount measured in the absence of textiles (blank). The content of CEES detected in the headspace of vials was around 4%, 8%, and 17% in the presence of CT-O, CT-O-UiO-d, and CT-O-UiO-i, respectively. These findings demonstrate that the composite textiles effectively adsorbed the liquid CEES and prevented more than 83% of the CEES droplets from vaporizing. The observed trend shows a strong correlation with the surface area of the textiles (Fig. S9†), as evidenced by an R2 value of 0.999, which stresses the importance of physical adsorption of CEES. Interestingly, the CEES content extracted from the textiles was approximately 70%, 69%, and 54% for CT-O, CT-O-UiO-d, and CT-O-UiO-i, respectively. This suggests that the non-extracted portion was either present in the headspace, strongly bonded to the textiles' adsorption sites, and/or converted into other reaction products. The efficiency of extraction also demonstrates a good correlation with surface area (R2 = 0.919), as shown in Fig. S9,† highlighting the important role of the surface area in adsorption of CEES. | ||
| Fig. 6 (a) Normalized CEES content and (b) normalized chromatographic peak areas of the compounds detected in the headspace. (c) Normalized CEES content and (d) normalized chromatographic peak areas of the compounds detected in the extracts of spent textiles after exposure to CEES liquid droplets. | ||
To further investigate the surface reactivity and identify the products formed via reactive adsorption, the headspaces and extracts from the spent modified textiles used in the droplet tests were analyzed by GCMS. The normalized chromatographic peak areas are presented in Fig. 6b and d, respectively. Both CT-O and CT-O-UiO-d show similar reactivity, as the same reaction products HEES, BETE, and EVS were detected at similar amounts. HEES is the product of CEES hydrolysis (Fig. 4), possibly due to the hydroxyl groups present on the oxygen-rich surface of CT-O (Fig. 3d), which act as nucleophiles replacing the chlorine atom.61 The effect of hydroxyl groups on CEES hydrolysis has been previously reported.43 BETE is formed through the recombination of intermediates cyclic cations generated from the C–S bond cleavage (Fig. 4).62 The high activity of CT-O-UiO-i in CEES decomposition is seen by a significant increase in the amount of EVS formed, particularly detected in the headspace, but also in the extract. EVS is a product of the CEES dehydrohalogenation/elimination reaction (Fig. 4), and UiO-66 appears to play a crucial role in facilitating this reaction. Its Lewis acid sites of the zirconium clusters can eliminate the chlorine ion, promoting the EVS formation.63,64 Interestingly, CT-O-UiO-d does not show much EVS formation and its main detoxification pathway is through hydroxylation, similarly to pure CT-O. The amount of HEES detected for CT-O-UiO-i was lower than that for CT-O-UiO-d. The differences in the reaction products suggest that the higher loadings and smaller particles of UiO-66 significantly alter the reaction pathways, shifting the mechanism toward elimination rather than hydrolysis. This is attributed to the use of the CT-O hydroxyl groups as seeds for the growth of the small UiO-66 particles in CT-O-UiO-i. The unavailability of these sites makes the dehydrohalogenation pathway more probable than the hydrolysis one (Fig. 4).43,61 Hydrolysis has been reported as one of the main decomposition pathways of CEES by the UiO-66 materials rich in nucleophile and basic sites, in the presence of water.65 Besides, an increase of DEDS was also observed. Interestingly the formation of DEDS was not detected when liquid CEES interacted with the composite textile surfaces, but it was detected in the headspace when the vapors interacted with the composites. This suggests that this decomposition pathway was favored only in the vapor-phase conditions/interactions, indicating that the rate of the S–C cleavage and recombination becomes competitive with the other detoxification pathways when the diffusion is slow.
4. Conclusions
Using two different routes of porous carbon textiles modifications, by the in situ synthesis of the UiO-66 phase and by the deposition of pre-synthesized UiO-66 nanoparticles in the dip-and-dry method, resulted in marked differences in textile surface properties. It was found that oxygen-containing functional groups on the carbon surface played an important role in affecting not only the amount but also the dispersion of the active phase. The combination of oxidation of carbon followed by the in situ approach, led to more UiO loaded (6.7% per weight), with smaller particles compared to that deposited with the dip-and-dry method (1.6% per weight), due to oxygen groups of the carbon surface acting as seeds for the growth of the UiO-66 crystals.The modified textiles showed a high activity in adsorption of mustard gas surrogate, 2-chloroethyl ethyl sulfide (CEES). Maximum weight uptakes were 372, 396, and 361 mg g−1 for CT-O, CT-O-UiO-d, and CT-O-UiO-i, respectively. Despite a 44% decrease in the surface area of CT-O-UiO-i (430 m2 g−1) compared to that of CT-O (772 m2 g−1), the maximum weight uptake upon CEES exposure was only ∼5% lower than that of CT-O. Besides, the deposition of UiO-66 had a positive impact on the strength of adsorption, since fewer species desorbed from the MOF-modified carbon textiles compared to the oxidized one. An important aspect is the ability of the relatively small amount of UiO-66 to bring a marked reactive detoxification capability. The highest reactive detoxification efficiency was found for CT-O-UiO-i, with the conversion of CEES mainly through dehydrohalogenation to EVS. This best activity of CT-O-UiO-i was attributed not only to the amount of UiO-66 phase deposited on the surface but also to the high dispersion of small MOF particles synthesized in the in situ approach. Another interesting finding was the formation of DEDS only when CEES interacted with the textile in the vapor phase. This detoxification path was not favored in the case of liquid CEES interactions with the surface of modified textiles. Therefore, it might be affected by the rate of CEES adsorption from the vapor phase and the kinetics of this reaction.
As shown here, the CEES decomposition can follow different pathways and small differences in the composite preparation can favor one pathway over another. Finding the causes of this is important to design better detoxifying materials. Our study suggests that the availability of the hydroxyl groups in the composite can facilitate hydrolysis and conversely their occupancy can favor dehydrohalogenation. Also, a slow diffusion can limit a reaction rate and make another pathway more predominant, which seems to be the case for the formation of DEDS vs. EVS/HEES. This highlights the complexity of a problem involving different factors and synergies and more studies are needed to give exact directions towards specific CEES decomposition products.
Data availability
All data will be available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research was sponsored by the Army Research Office and was accomplished under Grant Number W911NF-23-1-0204. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the Army Research Office or the U.S. Government. The U.S. Government is authorized to reproduce and distribute reprints for Government purposes notwithstanding any copyright notation herein.References
- M. Balali-Mood, S. Mousavi and B. Balali-Mood, Chronic Health Effects of Sulphur Mustard Exposure with Special Reference to Iranian Veterans, Emerg. Health Threats J., 2008, 1(1), 7068, DOI:10.3402/ehtj.v1i0.7068.
- X. Cheng, C. Liu, Y. Yang, L. Liang, B. Chen, H. Yu, J. Xia, S. Liu and Y. Li, Advances in Sulfur Mustard-Induced DNA Adducts: Characterization and Detection, Toxicol. Lett., 2021, 344, 46–57, DOI:10.1016/j.toxlet.2021.03.004.
- D. A. Giannakoudakis and T. J. Bandosz, Detoxification of Chemical Warfare Agents, Springer International Publishing, Cham, 1st edn, 2018, DOI:10.1007/978-3-319-70760-0.
- M. A. R. Bhuiyan, L. Wang, A. Shaid, R. A. Shanks and J. Ding, Advances and Applications of Chemical Protective Clothing System, J. Ind. Text., 2019, 49(1), 97–138, DOI:10.1177/1528083718779426.
- N. Couzon, J. Dhainaut, C. Campagne, S. Royer, T. Loiseau and C. Volkringer, Porous Textile Composites (PTCs) for the Removal and the Decomposition of Chemical Warfare Agents (CWAs) – A Review, Coord. Chem. Rev., 2022, 467, 214598, DOI:10.1016/j.ccr.2022.214598.
- G. K. Prasad, J. Praveen Kumar, P. V. R. K. Ramacharyulu and B. Singh, Breakthrough Behaviour of Activated Charcoal Cloth Samples against Oxygen Analogue of Sulphur Mustard, Carbon Lett., 2015, 16(1), 19–24, DOI:10.5714/CL.2015.16.1.019.
- K. Ganesan, S. K. Raza and R. Vijayaraghavan, Chemical Warfare Agents, J. Pharm. BioAllied Sci., 2010, 2(3), 166–178, DOI:10.4103/0975-7406.68498.
- D. T. Lee, J. D. Jamir, G. W. Peterson and G. N. Parsons, Protective Fabrics: Metal-Organic Framework Textiles for Rapid Photocatalytic Sulfur Mustard Simulant Detoxification, Matter, 2020, 2(2), 404–415, DOI:10.1016/j.matt.2019.11.005.
- M. Shufeng, D. Man, S. Ao, H. Xiaohui, L. Mengwen, Y. Jiaxin, D. Xiaoyu, Z. Lifeng and Y. Yunxu, The Selective Identification of Nerve Agent and Mustard Gas Simulants Based on the Multi-Functionalized Luminescent Platform of Tb3+@UiO-66-DPA, Microporous Mesoporous Mater., 2022, 339, 112006, DOI:10.1016/j.micromeso.2022.112006.
- Z. Cheng, X. Li, L. Zhang, Z. Yuan, H. Zheng, H. Guo, X. Zhao, J. Chen, Y. Pan, X. Chen, Y. Feng, Z. Luo, G. Tang, X. Jin and J. He, Flexible and MOF-808 Uniformly Assembled Polyimide Nanofiber Membranes with Excellent Mechanical Properties for Rapid Degradation of Chemical Warfare Agents, Chem. Eng. J., 2023, 475, 145912, DOI:10.1016/j.cej.2023.145912.
- J. Zhou, X. Li, C. Chu and J. Cao, One-Pot Synthesis of a Porphyrin Functionalized Metal-Organic Frameworks as a Recyclable Visible-Light-Driven Photosensitizer for Efficient Detoxification of a Sulfur Mustard Simulant in Air, Microporous Mesoporous Mater., 2024, 375, 113163, DOI:10.1016/j.micromeso.2024.113163.
- M. C. Oliver and L. Huang, Advances in Metal–Organic Frameworks for the Removal of Chemical Warfare Agents: Insights into Hydrolysis and Oxidation Reaction Mechanisms, Nanomaterials, 2023, 13(15), 2178, DOI:10.3390/nano13152178.
- M. Kim, S. H. Kim, M. Park and S. G. Ryu, Degradation of Chemical Warfare Agents over Cotton Fabric Functionalized with UiO-66-NH2, RSC Adv., 2018, 8, 41633–41638, 10.1039/c8ra06805d.
- D. Bůžek, J. Demel and K. Lang, Zirconium Metal–Organic Framework UiO-66: Stability in an Aqueous Environment and Its Relevance for Organophosphate Degradation, Inorg. Chem., 2018, 57, 14290–14297, DOI:10.1021/acs.inorgchem.8b02360.
- H.-H. Kim, J. Y. Seo, H. Kim, S. Jeong, K.-Y. Baek, J. Kim, S. Min, S. H. Kim and K. Jeong, Decomposition of the Simulant 2-Chloroethyl Ethyl Sulfide Blister Agent under Ambient Conditions Using Metal–Organic Frameworks, ACS Appl. Mater. Interfaces, 2021, 13(3), 3782–3792, DOI:10.1021/acsami.0c17022.
- D. A. Giannakoudakis and T. J. Bandosz, Defectous UiO-66 MOF Nanocomposites as Reactive Media of Superior Protection against Toxic Vapors, ACS Appl. Mater. Interfaces, 2020, 12(13), 14678–14689, DOI:10.1021/acsami.9b17314.
- X. Zhang, Y. Sun, Y. Liu, Z. Zhai, S. Guo, L. Peng, Y. Qin and C. Li, UiO-66-NH 2 Fabrics: Role of Trifluoroacetic Acid as a Modulator on MOF Uniform Coating on Electrospun Nanofibers and Efficient Decontamination of Chemical Warfare Agent Simulants, ACS Appl. Mater. Interfaces, 2021, 13(33), 39976–39984, DOI:10.1021/acsami.1c12751.
- C. Zhou, B. Yuan, S. Zhang, G. Yang, L. Lu, H. Li and C. Tao, Ultrafast Degradation and High Adsorption Capability of a Sulfur Mustard Simulant under Ambient Conditions Using Granular UiO-66-NH 2 Metal–Organic Gels, ACS Appl. Mater. Interfaces, 2022, 14(20), 23383–23391, DOI:10.1021/acsami.2c02401.
- J. Zhao, D. T. Lee, R. W. Yaga, M. G. Hall, H. F. Barton, I. R. Woodward, C. J. Oldham, H. J. Walls, G. W. Peterson and G. N. Parsons, Ultra-Fast Degradation of Chemical Warfare Agents Using MOF-Nanofiber Kebabs, Angew. Chem., Int. Ed., 2016, 55(42), 13224–13228, DOI:10.1002/anie.201606656.
- K. Ma, T. Islamoglu, Z. Chen, P. Li, M. C. Wasson, Y. Chen, Y. Wang, G. W. Peterson, J. H. Xin and O. K. Farha, Scalable and Template-Free Aqueous Synthesis of Zirconium-Based Metal–Organic Framework Coating on Textile Fiber, J. Am. Chem. Soc., 2019, 141(39), 15626–15633, DOI:10.1021/jacs.9b07301.
- S. Zhang, C. Dong, Y. Wang, J. Huang, S. Gu, H. Yang, X. Liu, W. Xu and D. Ye, Construction of MOF-Loaded Polypropylene Nonwoven Fabrics for Fast Catalytic Hydrolysis of Chemical Warfare Agent Simulants, Mater. Lett., 2022, 326, 132916, DOI:10.1016/j.matlet.2022.132916.
- D. T. Lee, J. Zhao, G. W. Peterson and G. N. Parsons, Catalytic “MOF-Cloth” Formed via Directed Supramolecular Assembly of UiO-66-NH 2 Crystals on Atomic Layer Deposition-Coated Textiles for Rapid Degradation of Chemical Warfare Agent Simulants, Chem. Mater., 2017, 29(11), 4894–4903, DOI:10.1021/acs.chemmater.7b00949.
- D. A. Giannakoudakis, Y. Hu, M. Florent and T. J. Bandosz, Smart Textiles of MOF/g-C 3 N 4 Nanospheres for the Rapid Detection/Detoxification of Chemical Warfare Agents, Nanoscale Horiz., 2017, 2(6), 356–364, 10.1039/C7NH00081B.
- R. Gil-San-Millan, P. Delgado, E. Lopez-Maya, J. D. Martin-Romera, E. Barea and J. A. R. Navarro, Layer-by-Layer Integration of Zirconium Metal–Organic Frameworks onto Activated Carbon Spheres and Fabrics with Model Nerve Agent Detoxification Properties, ACS Appl. Mater. Interfaces, 2021, 13(42), 50491–50496, DOI:10.1021/acsami.1c12095.
- R. Wallace, D. A. Giannakoudakis, M. Florent, C. Karwacki and T. J. Bandosz, Ferrihydrite Deposited on Cotton Textiles As Protection Media Against Chemical Warfare Agent Surrogate (2-Chloroethyl Ethyl Sulfide), J. Mater. Chem. A, 2017, 5, 4972–4981, 10.1039/C6TA09548H.
- P. S. Pauletto, D. A. Giannakoudakis, M. Florent and T. J. Bandosz, The Role of Carbon Textile Surface Functionalities in Reactive Adsorption of Vapor and Liquid 2-Chloroethyl Ethyl Sulfide: Evaluating Interactions at the Interface, Carbon, 2024, 230, 119673, DOI:10.1016/j.carbon.2024.119673.
- D. A. Giannakoudakis and T. J. Bandosz, Building MOF Nanocomposites with Oxidized Graphitic Carbon Nitride Nanospheres: The Effect of Framework Geometry on the Structural Heterogeneity, Molecules, 2019, 24(24), 4529, DOI:10.3390/molecules24244529.
- J. Jagiello and J. P. Olivier, 2D-NLDFT Adsorption Models for Carbon Slit-Shaped Pores with Surface Energetical Heterogeneity and Geometrical Corrugation, Carbon, 2013, 55(2), 70–80, DOI:10.1016/j.carbon.2012.12.011.
- C. Petit and T. J. Bandosz, MOF-Graphite Oxide Composites: Combining the Uniqueness of Graphene Layers and Metal-Organic Frameworks, Adv. Mater., 2009, 21(46), 4753–4757, DOI:10.1002/adma.200901581.
- R. Wu, X. Qian, K. Zhou, H. Liu, B. Yadian, J. Wei, H. Zhu and Y. Huang, Highly Dispersed Au Nanoparticles Immobilized on Zr-Based Metal–Organic Frameworks as Heterostructured Catalyst for CO Oxidation, J. Mater. Chem. A, 2013, 1(45), 14294, 10.1039/c3ta13114a.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability, J. Am. Chem. Soc., 2008, 130(42), 13850–13851, DOI:10.1021/ja8057953.
- A. Policicchio, M. Florent, A. Celzard, V. Fierro, J. Jagiello and T. J. Bandosz, Enhancing the Gas Adsorption Capacities of UiO-66 by Nanographite Addition, Microporous Mesoporous Mater., 2020, 309, 110571, DOI:10.1016/j.micromeso.2020.110571.
- S. Dai, C. Simms, G. Patriarche, M. Daturi, A. Tissot, T. N. Parac-Vogt and C. Serre, Highly Defective Ultra-Small Tetravalent MOF Nanocrystals, Nat. Commun., 2024, 15(1), 3434, DOI:10.1038/s41467-024-47426-x.
- U. Holzwarth and N. Gibson, The Scherrer Equation versus the “Debye-Scherrer Equation”, Nat. Nanotechnol., 2011, 6(9), 534–534, DOI:10.1038/nnano.2011.145.
- Z. Jiang, Y. Liu, X. Sun, F. Tian, F. Sun, C. Liang, W. You, C. Han and C. Li, Activated Carbons Chemically Modified by Concentrated H2SO4 for the Adsorption of the Pollutants from Wastewater and the Dibenzothiophene from Fuel Oils, Langmuir, 2003, 19(3), 731–736, DOI:10.1021/la020670d.
- V. Gomez-Serrano, J. Pastor-Villegas, A. Perez-Florindo, C. Duran-Valle and C. Valenzuela-Calahorro, FT-IR Study of Rockrose and of Char and Activated Carbon, J. Anal. Appl. Pyrolysis, 1996, 36(1), 71–80, DOI:10.1016/0165-2370(95)00921-3.
- R. Ali, Z. Aslam, R. A. Shawabkeh, A. Asghar and I. A. Hussein, BET, FTIR, and RAMAN Characterizations of Activated Carbon from Wasteoil Fly Ash, Turk. J. Chem., 2020, 44(2), 279–295, DOI:10.3906/kim-1909-20.
- M. Florent, D. A. Giannakoudakis, R. Wallace and T. J. Bandosz, Carbon Textiles Modified with Copper-Based Reactive Adsorbents as Efficient Media for Detoxification of Chemical Warfare Agents, ACS Appl. Mater. Interfaces, 2017, 9(32), 26965, DOI:10.1021/acsami.7b10682.
- M. Florent, D. A. Giannakoudakis and T. J. Bandosz, Mustard Gas Surrogate Interactions with Modified Porous Carbon Fabrics: Effect of Oxidative Treatment, Langmuir, 2017, 33(42), 11475–11483, DOI:10.1021/acs.langmuir.7b02047.
- J. Cazaux, Mechanisms of Charging in Electron Spectroscopy, J. Electron Spectrosc. Relat. Phenom., 1999, 105(2–3), 155–185, DOI:10.1016/S0368-2048(99)00068-7.
- A. J. Pertsin and Yu. M. Pashunin, Differential Charging in XPS Studies of Polymer/Metal Interfaces, Appl. Surf. Sci., 1990, 44(3), 171–178, DOI:10.1016/0169-4332(90)90047-4.
- G. Greczynski and L. Hultman, X-Ray Photoelectron Spectroscopy: Towards Reliable Binding Energy Referencing, Prog. Mater. Sci., 2020, 107, 100591, DOI:10.1016/j.pmatsci.2019.100591.
- D. A. Giannakoudakis, P. S. Pauletto, M. Florent and T. J. Bandosz, Impact of Humidity on the Adsorption and Decomposition of Mustard Gas Simulant on Heteroatom-Modified Nanoporous Carbon Textiles, J. Hazard. Mater., 2025, 487, 137155, DOI:10.1016/j.jhazmat.2025.137155.
- S. Y. Bae and M. D. Winemiller, Mechanistic Insights into the Hydrolysis of 2-Chloroethyl Ethyl Sulfide: The Expanded Roles of Sulfonium Salts, J. Org. Chem., 2013, 78(13), 6457–6470, DOI:10.1021/jo400392b.
- G. W. Wagner, O. B. Koper, E. Lucas, S. Decker and K. J. Klabunde, Reactions of VX, GD, and HD with Nanosize CaO: Autocatalytic Dehydrohalogenation of HD, J. Phys. Chem. B, 2000, 104(21), 5118–5123, DOI:10.1021/jp000101j.
- D. A. Giannakoudakis, F. Pearsall, M. Florent, J. Lombardi, S. O'Brien and T. J. Bandosz, Barium Titanate Perovskite Nanoparticles as a Photoreactive Medium for Chemical Warfare Agent Detoxification, J. Colloid Interface Sci., 2018, 531, 233–244, DOI:10.1016/j.jcis.2018.07.053.
- D. A. Giannakoudakis, J. K. Mitchell and T. J. Bandosz, Reactive Adsorption of Mustard Gas Surrogate on Zirconium (Hydr)Oxide/Graphite Oxide Composites: The Role of Surface and Chemical Features, J. Mater. Chem. A, 2016, 4(3), 1008–1019, 10.1039/C5TA09234E.
- K. Vellingiri, L. Philip and K. H. Kim, Metal–Organic Frameworks as Media for the Catalytic Degradation of Chemical Warfare Agents, Coord. Chem. Rev., 2017, 353, 159–179, DOI:10.1016/j.ccr.2017.10.010.
- K. Y. Cho, J. Y. Seo, H. J. Kim, S. J. Pai, X. H. Do, H. G. Yoon, S. S. Hwang, S. S. Han and K. Y. Baek, Facile Control of Defect Site Density and Particle Size of UiO-66 for Enhanced Hydrolysis Rates: Insights into Feasibility of Zr(IV)-Based Metal-Organic Framework (MOF) Catalysts, Appl. Catal., B, 2019, 245, 635–647, DOI:10.1016/j.apcatb.2019.01.033.
- J. E. Mondloch, M. J. Katz, W. C. Isley III, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. DeCoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Destruction of Chemical Warfare Agents Using Metal–Organic Frameworks, Nat. Mater., 2015, 14(5), 512–516, DOI:10.1038/nmat4238.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Metal-Organic Framework Materials as Catalysts, Chem. Soc. Rev., 2009, 38(5), 1450–1459, 10.1039/b807080f.
- Y. Liu, A. J. Howarth, N. A. Vermeulen, S.-Y. Moon, J. T. Hupp and O. K. Farha, Catalytic Degradation of Chemical Warfare Agents and Their Simulants by Metal-Organic Frameworks, Coord. Chem. Rev., 2017, 346, 101–111, DOI:10.1016/j.ccr.2016.11.008.
- Y. Liu, C. T. Buru, A. J. Howarth, J. J. Mahle, J. H. Buchanan, J. B. DeCoste, J. T. Hupp and O. K. Farha, Efficient and Selective Oxidation of Sulfur Mustard Using Singlet Oxygen Generated by a Pyrene-Based Metal–Organic Framework, J. Mater. Chem. A, 2016, 4(36), 13809–13813, 10.1039/C6TA05903A.
- J. Yin, C. Huang, Y. Zhou, L. Zhang, N. Li and R. Sun, Selective Oxidation of 2-Chloroethyl Ethyl Sulfide in Aqueous Media Catalyzed by {Mo72 M30} Nano-Polyoxometalate Clusters Differentiating the Catalytic Activity of Nodal Metals, Ind. Eng. Chem. Res., 2022, 61(23), 7699–7708, DOI:10.1021/acs.iecr.2c00479.
- S. C. Stout, S. C. Larsen and V. H. Grassian, Adsorption, Desorption and Thermal Oxidation of 2-CEES on Nanocrystalline Zeolites, Microporous Mesoporous Mater., 2007, 100(1–3), 77–86, DOI:10.1016/j.micromeso.2006.10.010.
- S. Popiel, J. Nawała, D. Dziedzic, M. Söderström and P. Vanninen, Determination of Mustard Gas Hydrolysis Products Thiodiglycol and Thiodiglycol Sulfoxide by Gas Chromatography-Tandem Mass Spectrometry after Trifluoroacetylation, Anal. Chem., 2014, 86(12), 5865–5872, DOI:10.1021/ac500656g.
- Y. Liu, A. J. Howarth, J. T. Hupp and O. K. Farha, Selective Photooxidation of a Mustard-Gas Simulant Catalyzed by a Porphyrinic Metal-Organic Framework, Angew. Chem., Int. Ed., 2015, 54(31), 9001–9005, DOI:10.1002/anie.201503741.
- C. T. Buru, M. B. Majewski, A. J. Howarth, R. H. Lavroff, C. W. Kung, A. W. Peters, S. Goswami and O. K. Farha, Improving the Efficiency of Mustard Gas Simulant Detoxification by Tuning the Singlet Oxygen Quantum Yield in Metal-Organic Frameworks and Their Corresponding Thin Films, ACS Appl. Mater. Interfaces, 2018, 10(28), 23802–23806, DOI:10.1021/acsami.8b05792.
- M. Athar, P. Rzepka, D. Thoeny, M. Ranocchiari and J. Anton van Bokhoven, Thermal Degradation of Defective High-Surface-Area UiO-66 in Different Gaseous Environments, RSC Adv., 2021, 11(61), 38849–38855, 10.1039/D1RA05411B.
- D. A. Giannakoudakis, J. A. Arcibar-Orozco and T. J. Bandosz, Effect of GO Phase in Zn(OH)2/GO Composite on the Extent of Photocatalytic Reactive Adsorption of Mustard Gas Surrogate, Appl. Catal., B, 2016, 183, 37–46, DOI:10.1016/j.apcatb.2015.10.014.
- A. Saxena, A. K. Srivastava, A. Sharma and B. Singh, Kinetics of Adsorption of 2-Chloroethylethylsulphide on Al2O3 Nanoparticles with and without Impregnants, J. Hazard. Mater., 2009, 169(1–3), 419–427, DOI:10.1016/j.jhazmat.2009.03.112.
- S. Fichtner, J. Hofmann, A. Möller, C. Schrage, J. M. Giebelhausen, B. Böhringer and R. Gläser, Decomposition of 2-Chloroethylethylsulfide on Copper Oxides to Detoxify Polymer-Based Spherical Activated Carbons from Chemical Warfare Agents, J. Hazard. Mater., 2013, 262, 789–795, DOI:10.1016/j.jhazmat.2013.09.052.
- J. K. Mitchell, J. A. Arcibar-Orozco and T. J. Bandosz, Reactive Removal of 2-Chloroethyl Ethyl Sulfide Vapors under Visible Light Irradiation by Cerium Oxide Modified Highly Porous Zirconium (Hydr) Oxide, Appl. Surf. Sci., 2016, 390, 735–743, DOI:10.1016/j.apsusc.2016.08.118.
- J. A. Arcibar-Orozco, S. Panettieri and T. J. Bandosz, Reactive Adsorption of CEES on Iron Oxyhydroxide/(N-)Graphite Oxide Composites under Visible Light Exposure, J. Mater. Chem. A, 2015, 3(33), 17080–17090, 10.1039/C5TA04223B.
- R. Gil-San-Millan, E. López-Maya, M. Hall, N. M. Padial, G. W. Peterson, J. B. DeCoste, L. M. Rodríguez-Albelo, J. E. Oltra, E. Barea and J. A. R. Navarro, Chemical Warfare Agents Detoxification Properties of Zirconium Metal-Organic Frameworks by Synergistic Incorporation of Nucleophilic and Basic Sites, ACS Appl. Mater. Interfaces, 2017, 9(28), 23967–23973, DOI:10.1021/acsami.7b06341.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5lf00142k |
| This journal is © The Royal Society of Chemistry 2025 |