
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Charge carrier dynamics in semiconductor–cocatalyst interfaces: influence on photocatalytic activities
Dipendu
Sarkar
ab,
Jishu
Pramanik
ab,
Soumita
Samajdar
ab,
Maitrayee
Biswas
a and
Srabanti
Ghosh
 *ab
*ab
aEnergy Materials & Devices Division (EMDD), CSIR – Central Glass and Ceramic Research Institute, Raja S. C. Mullick Road, Jadavpur, Kolkata, 700032, India. E-mail: ghosh.srabanti@gmail.com; srabanti@cgcri.res.in
bAcademy of Scientific & Innovative Research (AcSIR), Ghaziabad, 201002, India
First published on 3rd April 2025
Abstract
Electron transfer dynamics at semiconductor–cocatalyst interfaces are critical for efficient solar fuel generation, including water splitting, pollutant degradation, CO2 reduction, and N2 fixation. These interfaces facilitate charge separation, suppress recombination, and enable photoexcited charge carriers to transfer to active sites for photocatalytic reactions. The formation of Schottky or ohmic junctions, energy band alignment, and surface properties significantly influence charge transfer efficiency. Advances in theoretical modeling, such as density functional theory (DFT) and several experimental techniques like ultrafast spectroscopy and in situ X-ray photoelectron spectroscopy, have offered profound insights into these processes. Understanding and optimizing these dynamics is essential for developing high-performance photocatalytic systems to harness solar energy and address global energy demands sustainably. This review offers a concise explanation of charge transfer mechanisms at semiconductor–cocatalyst interfaces, explored through various experimental methodologies and theoretical frameworks. Exploring the underlying mechanism will open new avenues for advancing high-performance semiconductor photocatalytic technologies. The conclusion sheds light on the challenges and promising opportunities for enhancing the understanding and investigation of interfacial electron transfer dynamics in semiconductor–cocatalyst systems.
 Dipendu Sarkar | Dipendu Sarkar pursued his B.Sc. in Physics from Midnapore College (Autonomous), Paschim Midnapore, India in 2018, and M.Sc. in Physics from Ramakrishna Mission Vivekananda Educational and Research Institute, Howrah, India in 2020. He is engaged in Ph.D. research under the guidance of Dr. Srabanti Ghosh in the Energy Materials & Devices Division at CSIR-Central Glass & Ceramic Research Institute, Kolkata, India. His research focuses on modifying the band structure of bismuth-based semiconductor heterostructures to enhance photocatalytic solar fuel production, including H2 and CO, CH4 generation. |
 Jishu Pramanik | Jishu Pramanik completed his B.Sc. in Physics from Midnapore College (Autonomous), Paschim Midnapore, India in 2018 and M.Sc. from Vidyasagar University, Paschim Midnapore, India in 2020. He is currently pursuing his Ph.D. under the supervision of Dr. Srabanti Ghosh in the Energy Materials & Devices Division at CSIR-Central Glass & Ceramic Research Institute, Kolkata, India. His research is centered on the fabrication of perovskite-based semiconductor heterostructures for advanced photo-electrocatalytic applications, with a particular focus on nitrogen fixation and ammonia production. |
 Soumita Samajdar | Soumita Samajdar obtained her B.Sc. in Chemistry from Presidency University, Kolkata, India in 2019, followed by an M.Sc. in Chemistry from the same University in 2021. She is currently pursuing her Ph.D. under the guidance of Dr. Srabanti Ghosh in the Energy Materials & Devices Division at CSIR-Central Glass & Ceramic Research Institute, Kolkata, India. Her work primarily involves the design and development of two-dimensional material-based nanocomposite photocatalysts for applications in solar energy conversion, water splitting, hydrogen production, and environmental remediation. |
 Maitrayee Biswas | Maitrayee Biswas is a research scholar in Dr. Srabanti Ghosh's research group in the Energy Materials & Devices Division at CSIR-Central Glass & Ceramic Research Institute, Kolkata, India. She graduated in Chemistry from University of Calcutta, Kolkata, India in 2020 and completed her postgraduation in Chemistry (specialization in Organic chemistry) from the West Bengal State University, Kolkata, India in 2022. Her main research is dedicated to the fabrication of conjugated polymers and its nanocomposites-based photocatalysts for solar energy conversion, water splitting, photocatalytic H2 generation, and CO2 reduction. |
 Srabanti Ghosh | Dr. Srabanti Ghosh is working as a Senior Scientist at CSIR – Central Glass & Ceramic Research Institute, India. She received her PhD degree from UGC-DAE CSR, Kolkata Centre, and Jadavpur University. She is a recipient of the Marie Curie Fellowship at University of Paris SUD, France, and GOT ENERGY TALENT Marie Curie Fellow Fellowship at Universidad De Alcala, Spain. Her main research interests include nano materials, electrochemical devices, photocatalysis, and therapeutics. She co-authored 115 publications, 4 patents, edited 6 books, and h-index: 44, citation: 6573. She mentored master students, Ph. D. – 03 completed, and Ph. D. – 05 (ongoing). She has been conferred with a few prestigious awards such as MRSI Young Scientists award, top 5% of highly cited authors in RSC Materials portfolio, Young Investigator Award in Gordon Radiation Chemistry, USA, Top 2% Most Influential Indian Scientist by Stanford University, 2019–2024. Recently, she received Society of Materials Chemistry (SMC) Bronze Medal-2024, and MRSI Medal for 2024. |
1. Introduction
Semiconductor photocatalysts are essential in addressing high energy demand and other pressing challenges facing society today, including energy sustainability, ecological pollution, atmospheric changes, and rising average global temperatures.1,2 Semiconductor nanostructures possess the remarkable ability to harness sunlight and convert it into a renewable energy source through chemical reactions, establishing them as a foundation of modern clean energy and environmental technologies.3–5 Despite their potential, semiconductor photocatalysts face challenges such as limited absorption of sunlight beyond the UV range, fast recombination of electron–hole (e−–h+) pairs, and stability issues under prolonged use.6–8 Advancements in bandgap engineering, surface modification, heterostructure formation, doping, vacancies, and cocatalyst depositions are crucial for enhancing the efficiency and practicality of the semiconductors.9–11 Among them, cocatalysts play a critical role in photocatalysis by enhancing efficiency through charge carrier extraction and trapping, promoting charge separation and transfer, minimizing e−–h+ pair recombination, and offering active sites for catalytic reactions, including hydrogen evolution, pollutant degradation, CO2 reduction, and N2 fixation.12–14 Conceptually, cocatalysts that trap electrons are referred to as reduction cocatalysts, while those that capture holes are known as oxidation cocatalysts. Most metals (such as Pt, Pd, Ru, Au, Ag, etc.) and metal oxides/sulfides typically capture electrons and have been identified as reduction cocatalysts.15–21 In contrast, materials like RuO2, CoOx, MnO2, FePO4, etc., tend to trap holes, making them oxidation cocatalysts.22–27 So far, most studies have concentrated on noble metal-based cocatalysts due to their ability to form Schottky or ohmic junctions at the interfaces, facilitating efficient, unhindered charge transfer.28–31 As an example, Liu et al.32 developed a novel Ag@Si2O3@TiO2–Au plasmonic semiconductor photocatalyst, where Ag nanoparticles functioned as plasmonic resonators. The Ag–Au coupling amplified the local electric field at the TiO2–Au interface, resulting in improved charge separation and enhanced visible light absorption. In another study, Jeong et al.33 synthesized Ag nanoparticles deposited on BiVO4 for improved photocatalytic water splitting. This improvement was credited to the enhanced charge generation and separation driven by the linear surface plasmon resonance effect. However, noble metal-free, cost-effective, and earth-abundant cocatalysts have also been synthesized for photocatalytic activity. Xiao et al.34 developed ultrathin CoOx cocatalysts on tantalum oxynitride and found that the predominant presence of Co2+ ions created more attractive sites and stronger interactions for photogenerated holes, significantly boosting the photocatalytic water oxidation efficiency.In this review, recent progress in charge transfer at the semiconductor–cocatalysts interfaces has been discussed. Various types of cocatalysts, their synthesis methods, charge transfer dynamics, and their applications in diverse photocatalytic processes, including photodegradation of pollutants, water splitting, conversion of CO2, and N2 fixation, have been focused. We also provided an overview of the interfacial charge transfer mechanisms as elucidated by theoretical studies. Lastly, the challenges and future opportunities for the practical implementation of photocatalysts based on semiconductor–cocatalyst systems have been emphasized.
2. Architecture of cocatalysts for surface modification of semiconductor
Cocatalysts can be integrated with semiconductors in various forms (Fig. 1). Based on the roles of cocatalysts in photocatalytic reactions, four distinct semiconductor–cocatalyst structures have been classified. | ||
| Fig. 1 Schematic presentation of cocatalysts–semiconductor configuration. | ||
2.1. Semiconductor–reduction cocatalyst structure
The efficiency of H2 evolution or CO2 reduction in most semiconductors is often limited by fast recombination of e−–h+ pairs and slow surface reaction kinetics. To efficiently extract electrons to the surface of the photocatalyst, depositing a reduction cocatalyst is a promising strategy.35 It serves as both an electron reservoir and a provider of active sites for proton reduction, significantly enhancing the efficiency of the proton reduction process.The photocatalytic performance of bare g-C3N4 is constrained by its limited visible light absorption range, high photogenerated e−–h+ pairs recombination rate, low electrical conductivity, and restricted number of active surface sites. Hence, Zhao et al.36 synthesized PdCuHx (x = hydrogen content in the compound) nanocrystals deposited on ultrasonically exfoliated C3N4 nanosheets. Initially, PdCu nanocrystals were synthesized on g-C3N4 nanosheets by reducing K2PdCl and CuCl2 in an aqueous solution, followed by a solvothermal treatment in DMF (N,N-dimethylformamide) to produce C3N4–PdCuHx hybrids, as shown in Fig. 2a. The Pd and PdCu nanocrystals exhibited truncated cubic structures and were uniformly distributed across the surface of the g-C3N4 nanosheets as shown in TEM images. However, the corners of the Pd9Cu1Hx nanocubes appeared rounded in C3N4–Pd9Cu1Hx (Fig. 2b), whereas, in the C3N4–PdHx material, adjacent nanocubes merged into irregular nanorods, suggesting significant volume expansion following the hydriding process via solvothermal reaction. Upon adding metal cocatalysts to C3N4, a notable reduction in photoluminescence intensity was observed, indicating the suppression of e−–h+ recombination (Fig. 2c). As reported, doping H atoms into the PdCu cocatalyst lattice significantly enhanced the electron trapping capability of PdCu. Furthermore, the H2 atoms within the Pd lattice are crucial in determining the rate-limiting step of the reduction reactions. Additionally, Cu atoms serve as highly active sites for the reduction processes within the Pd lattice. Therefore, the synergy of these effects collectively enhanced the photocatalytic reduction reaction of C3N4 when PdCuHx was used as the reduction cocatalyst (Fig. 2d). With the hydride treatment, the average CH4 production rate increased by 2.8, 4.0, and 2.6 times for C3N4–PdHx, C3N4–Pd9Cu1Hx, and C3N4–Pd7Cu3Hx, respectively. Remarkably, C3N4–Pd9Cu1Hx demonstrated the best performance by achieving a selectivity of 100% and an average CH4 production rate of 0.018 mmol h−1.
 | ||
| Fig. 2 (a) Schematic representation of the synthesis process for loading reduction cocatalyst onto C3N4-based photocatalysts. (b) Transmission Electron Microscopy (TEM) image of C3N4–Pd9Cu1Hx. Inset: high-Resolution Transmission Electron Microscopy (HRTEM) image of C3N4–Pd9Cu1Hx. (c) Photoluminescence spectra (PL) of C3N4-based photocatalysts (excitation wavelength = 390 nm). (d) The diagrammatic representation shows the photocatalytic process for converting CO2 to CH4 with C3N4–PdCuHx as the photocatalyst. Reproduced from ref. 36 with permission from [Wiley-VCH], copyright [2018]. (e) TEM image of TiO2–PdH0.43 NCs. (f) Schematic illustration of photocatalytic reduction mechanism of TiO2–Pd NCs and TiO2–PdH0.43 NCs. Reproduced from ref. 37 with permission from [Springer Nature], copyright [2017]. | ||
In another study, Zhu et al.37 loaded Pd nanocubes (NCs) as cocatalysts onto TiO2 nanosheets and hydriding Pd into β-phase PdH0.43. This transformation notably enhanced the efficiency of photocatalytic reduction. As observed from the TEM image (Fig. 2e), PdH0.43 nanocrystals preserved their cubic-shaped morphology, demonstrating that the cocatalysts retained their shape effectively after the hydriding process. The photocatalytic mechanism depicted in Fig. 2f indicates that employing pure Pd as a cocatalyst hindered charge transfer at the TiO2–Pd interface. Interestingly, hydridation significantly improved charge separation and transfer at the TiO2–PdH0.43 interface extended the lifespan of photoexcited charge carriers, and decreased the H2 evolution rate. Notably, TiO2–PdH0.43 NCs produced 82.40 μmol g−1 CH4 in 4 h, ∼11 times higher than the CH4 yield from TiO2–Pd NCs. As a result, hydriding the cubic Pd co-catalyst enhanced the CH4 production selectivity increasing it from 3.2% to 63.6%.
2.2. Semiconductor–oxidation cocatalyst structure
In recent years, photocatalytic and photoelectrochemical (PEC) water splitting has gained attention as a potential strategy for producing clean and renewable solar fuels, offering an effective solution to environmental issues and energy scarcity.38 The light-induced oxygen evolution reaction (OER), characterized by a complex four-electron transfer mechanism, O–H bond cleavage, and O–O bond formation is limited by slow charge transfer, sluggish reaction kinetics, and high energy demands. These factors present challenges and opportunities for advancing efficient semiconductor photocatalysts for water oxidation.39 Photon absorption, charge separation, positive charge transport, and surface reactions are essential for designing materials with optimized composition, structure, and morphology, which play key roles in OER. Various methodologies have been applied to enhance photocatalytic processes, among them OER co-catalyst loading being a result-oriented technique to improve charge separation and transport which facilitate photocatalytic water oxidation.40 Especially, d-block metals and their oxides are commonly paired with photocatalysts as cocatalysts to suppress charge recombination and improve surface reactivity. In this context, W. Li and his group demonstrated using Co3O4 and Pt as dual cocatalysts integrated with perylene diimide (PDI) polymer for efficient photocatalytic oxygen production.41 As shown in Fig. 3a the distribution of Co, Pt, and Co3O4 is observed through HRTEM. Under light irradiation, the O2 formation rate increased by 4.1 times compared to pristine PDI, and by incorporating Pt as a cocatalyst, the O2 evolution increased to 24.4 mmol g−1 h−1 (Fig. 3b). To achieve a more comprehensive insight into charge transfer, the contact potential difference (CPD) was measured, revealing that the work function of PDI is lower than that of Co3O4, indicating that electrons are more easily released from the surface. When PDI contacts Co3O4, electron transfer from PDI to Co3O4 equilibrates their Fermi levels, inducing upward band bending that drives photogenerated holes to Co3O4, whereas, at the PDI–Pt interface, Pt's lower work function causes downward band bending, facilitating electron transfer from PDI's conduction band to Pt. (Fig. 3c). From the surface voltage, surface charge density, and transient photocurrent density interfacial electric field was calculated (Fig. 3d). The strength of the interfacial electric field (Fig. 3e), reveals a 15.4 times enhancement for PDI/Co3O4, and a 23.9-fold increase for PDI/Co3O4/Pt compared to pristine PDI, emphasizing the notable benefits of dual cocatalysts in creating internal electric fields, as they facilitate the separation and transport of electrons and holes across the interfaces.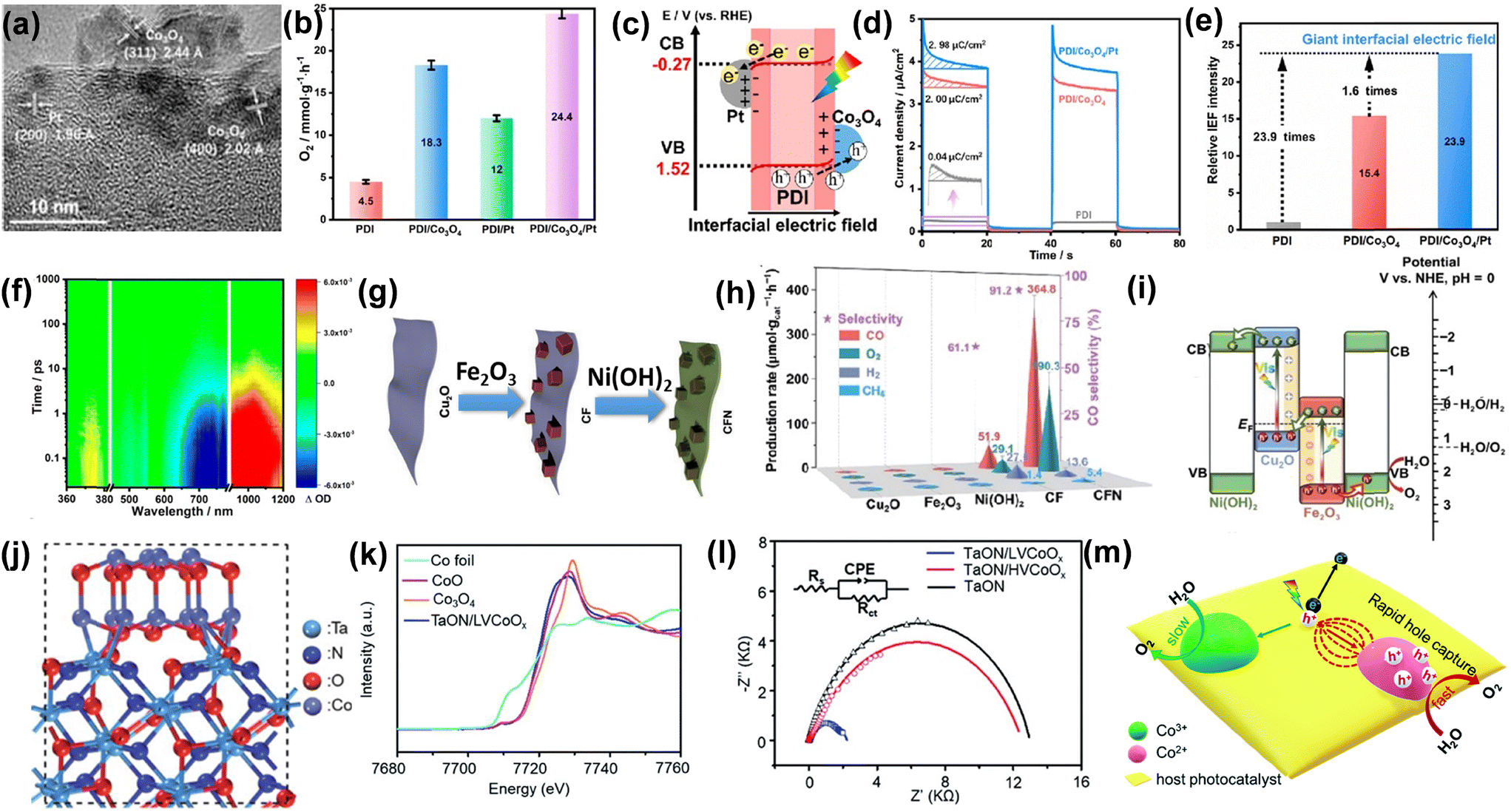 | ||
| Fig. 3 (a) HRTEM image of the PDI/Co3O4/Pt composite. (b) Comparison of photocatalytic O2 evolution rates among PDI, PDI/Co3O4, PDI/Pt, and PDI/Co3O4/Pt. (c) Diagram illustrating the band structure and charge-transfer pathway at the interface of a PDI/Co3O4/Pt heterostructure. (d) Surface charge density determined using an electrochemical approach. (e) Intensity of the relative interfacial electric field. (f) fs-TA of PDI/Co3O4/Pt. Reproduced from ref. 41 with permission from [American Chemical Society], copyright [2023]. (g) Preparation method for Cu2O/Fe2O3@Ni(OH)2 (CFN). (h) Photocatalytic activity and charge kinetics evaluation. (i) Suggested charge transfer pathway of CFN. Reproduced from ref. 42 with permission from [Springer Nature], copyright [2024]. (j) Diagram depicting the crystal structure of TaON/LVCoOx synthesized using the PMOD technique. (k) The normalized Co K-edge XANES spectra of TaON/LVCoOx and the control samples reveal the predominance of Co2+ species in the bulk. (l) EIS Nyquist plots. (m) The suggested mechanism for the photocatalytic water oxidation process on CoOx-decorated TaON highlights the enhanced hole transport and reaction on the Co2+ species. Reproduced from ref. 43 with permission from [The Royal Society of Chemistry], copyright [2021]. | ||
Fig. 3f shows the femtosecond transient absorption (fs-TA) data of PDI/Co3O4/Pt under a 420 nm laser flash. Positive absorption bands at 360–380, 550–590, and 900–1200 nm correspond to electron transitions in PDI, while negative ground-state bleach signals at 450–780 nm indicate charge recombination. Thus, the dual cocatalysts generate interfacial electric fields, directing holes to Co3O4 and electrons to Pt, enhancing charge separation for water oxidation. In another study, Zhao et al.42 developed a ternary Cu2O/Fe2O3@Ni(OH)2 (CFN) system by coating a Cu2O/Fe2O3 (CF) Z-scheme heterojunction with Ni(OH)2, serving as a bifunctional, noble-metal-free cocatalyst (Fig. 3g). It can be seen from (Fig. 3h) that ternary catalyst CFN demonstrated an improved rate of O2 yield rate 190.3 μmol g−1 h−1. The transient photocurrent response reveals that the current density increase in CFN surpasses that of Fe2O3, Cu2O, and CF, suggesting that charge recombination is suppressed through the hybridization of Fe2O3 and Cu2O, and this effect is further enhanced by the Ni(OH)2 coating. A Z-scheme charge transfer mechanism is proposed in CF and CFN. As shown in Fig. 3i, when Fe2O3 comes into contact with Cu2O, driven by their Fermi level difference, free electrons transfer from Cu2O to Fe2O3, leading to an internal electric field creation at the Cu2O–Fe2O3 interface, accompanied by upward band bending in Cu2O and downward band bending in Fe2O3. Y. Xiao et al.43 demonstrated that CoOx, as a cocatalyst, can be effectively embedded in tantalum oxynitride (TaON) for OER. Fig. 3j illustrates the schematic configuration of TaON/LVCoOx. Co K-edge X-ray absorption near edge structure (XANES) analysis was used to examine the bulk average oxidation states of CoOx co-catalysts, and as shown in Fig. 3k, the Co K-edge XANES spectrum of TaON/LVCoOx aligns with that of the reference sample CoO, suggesting the predominant presence of Co2+ species in the co-catalyst bulk. Higher photocurrent response and lower charge transfer resistance (Fig. 3l) indicate that low valent Co2+ (TaON/LVCoOx) is superior to high valent Co2+ (TaON/HVCoOx) and pristine TaON towards photocatalytic performance. Depending on the above result a mechanism is illustrated in Fig. 3m. The high proportion of Co2+ species in the CoOx co-catalyst of TaON/LVCoOx acts as an effective hole trap, generating strong attraction forces that significantly enhance charge separation and transport during the photocatalytic water oxidation process. Interestingly, TaON/LVCoOx modified with 5 mg Co(acac)2 on TaON using PMOD method for 30 min exhibited the highest O2 evolution rate up to 6.10 ± 0.17 mmol g−1 h−1, which is nearly 4 times greater than the value of pristine TaON (1.27 ± 0.10 mmol g−1 h−1). In conclusion, modifying semiconductor photocatalysts with co-catalysts significantly enhances their photocatalytic performance, particularly in oxidation reactions. The introduction of co-catalysts facilitates efficient charge separation, improving the overall catalytic efficiency by reducing e−–h+ recombination. These co-catalysts, typically consisting of noble or non-noble metals, form active sites that facilitate both the adsorption and activation of reactants, improving charge transport to the semiconductor surface. Additionally, co-catalysts can alter the light absorption characteristics of the photocatalyst, leading to improved photocatalytic activity under visible light. By optimizing the combination of semiconductor and co-catalyst, a photocatalytic system can achieve a balance between increased efficiency, enhanced selectivity, and sustainable energy use.
2.3. Reduction cocatalyst–semiconductor–oxidation cocatalyst (R–S–O) structure
The R–S–O configuration is superior to the semiconductor–reduction cocatalyst and semiconductor–oxidation cocatalyst structure due to its unique ability to enhance charge separation, improve reaction kinetics, and boost photocatalytic efficiency.44 Upon illumination, e−–h+ pairs are generated and separated, with electrons from the CB and holes from the VB transferred to the reduction and oxidation cocatalyst, respectively, through the interfaces. For example, He et al.44 deposited Ag2S and NiS dual cocatalysts on CdS nanorods that showed an efficient minimized charge carrier recombination. Here, Ag2S and NiS acted as reduction and oxidation cocatalysts, respectively.The HRTEM image showed distinct lattice fringes of Ag2S, NiS, and CdS as presented in Fig. 4a. The CdS nanorods exhibited high crystallinity with exposed (001) facets, while the (012) planes of Ag2S and the (101) planes of hexagonal NiS were reported. Fig. 4b represented the Kelvin probe force microscopy (KPFM) image of CdS nanorods coated simultaneously with Ag2S and NiS dual cocatalysts under 420 nm illumination where A1, B1, and B2 represented the position of Ag2S, NiS, and CdS nanorods, respectively (Fig. 4c). The change in surface potential of the material was evidenced by the colour change upon light irradiation. The surface energy of p-type NiS showed an increasing trend after light irradiation, indicating holes transfer from CdS to the NiS cocatalyst. A decrease in surface potential upon illumination for Ag2S implies CdS donating electrons to NiS. The schematic illustration of charge transfer through the interfaces is shown in Fig. 4d. The photoexcited electrons were transferred to Ag2S through the Schottky junction and holes to NiS through the p–n junction formed at the cocatalyst and CdS nanorod interface. Therefore, the electrons in Ag2S were involved in the reduction reaction, while the holes in NiS facilitated the oxidation reaction, establishing an R–S–O configuration. The H2 evolution rates of CdS/Ag2S and CdS/NiS attained 7.24 and 6.43 mmol g−1 h−1, respectively. Astonishingly, CdS/Ag2S/NiS demonstrated the highest H2 production rate of 48.28 mmol g−1 h−1, almost 45-fold higher than CdS. Additionally, the H2 evolution rate and calculated apparent quantum yield (AQY) of 49.5% of CdS/Ag2S/NiS surpassed those of other CdS-based photocatalysts. In a similar work, Di et al.45 adopted a two-step photodeposition technique for synthesized CoOx and MoS2 dual cocatalysts onto CdS nanorods. Fig. 5a displayed the synthesis steps of the CdS–CoOx–MoS2 nanocomposite.
 | ||
| Fig. 4 (a) HRTEM image of CdS/Ag2S/NiS. (b) KPFM image of CdS/Ag2S/NiS under light irradiation of 420 nm. (c) Surface potential profiles under 420 nm light irradiation. (d) Pictorial representation of electron and hole migration process in CdS/Ag2S/NiS under illumination. Reproduced from ref. 44 with permission from [Elsevier], copyright [2021]. | ||
 | ||
| Fig. 5 (a) Schematic diagram of CdS–CoOx–MoS2 nanocomposite preparation. (b) and (c) HRTEM image of CdS–CoOx–MoS2 nanocomposite. (d) Proposed charge transfer mechanisms at CdS–CoOx and CdS–MoS2 interfaces. Reproduced from ref. 45 with permission from [Elsevier], copyright [2022]. | ||
Firstly, CoOx was deposited on CdS nanorods using Co(NO3)2 as a precursor, followed by the deposition of MoS2 onto CdS–MoS2 using (NH4)2MoS4 as the precursor. As shown in Fig. 5b, the HRTEM image of the CdS–CoOx–MoS2 nanocomposite displays (100) and (101) lattice fringes of CdS nanorods and the (002) fringe of MoS2. An amorphous particle on the CdS nanorod surface is identified as CoOx (Fig. 5c). Due to the low loading of MoS2 and CoOx, only a few reduction and oxidation sites on the CdS nanorods were occupied by the cocatalysts. Upon illumination, photoexcited electrons were transferred from CdS to MoS2, while holes migrated from CdS to CoOx through the formation of a p–n junction, as shown in Fig. 5d. As a result, MoS2 and CoOx functioned as reduction and oxidation cocatalysts, respectively, effectively enhancing the hydrogen production efficiency of CdS nanorods. The CdS–MoS2–CoOx composite delivered an impressive photocatalytic H2 production rate of 7.4 mmol g−1 h−1, coupled with an apparent quantum efficiency (QE) of 7.6% at 420 nm. A unique covalent connection of α-Fe2O3/TpPa-1-COF/FeP-PC (FCF-3) was synthesized by Xu and co-workers to promote an overall water splitting reaction.46 α-Fe2O3 and FeP-PC served as the oxidation and reduction cocatalysts, respectively, effectively preventing the agglomeration of TpPa-1-COF and enhancing the migration and parting of light-induced charge carriers. The as-prepared spindle-like α-Fe2O3 and FeP-PC were progressively functionalized with organic groups using 3-aminopropyltriethoxysilane (APTES), yielding APTES-α-Fe2O3 and APTES-FeP-PC. Subsequently, the covalently linked FCF-3 photocatalyst was synthesized by incorporating APTES-α-Fe2O3 and APTES-FeP-PC into the reaction process of a disordered and ordered yarn-like morphology TpPa-1-COF as shown in Fig. 6a. The UV-vis DRS spectra demonstrated that α-Fe2O3/TpPa-1-COF, TpPa-1-COF/FeP-PC, and FCF-3 exhibited a similar reflectance pattern to TpPa-1-COF, with a single reflection edge. In contrast, FCF-3 and the physically mixed α-Fe2O3 + TpPa-1-COF + FeP-PC displayed two reflection edges, suggesting the creation of a novel hybrid material via an in situ chemical bonding process, as shown in Fig. 6b.
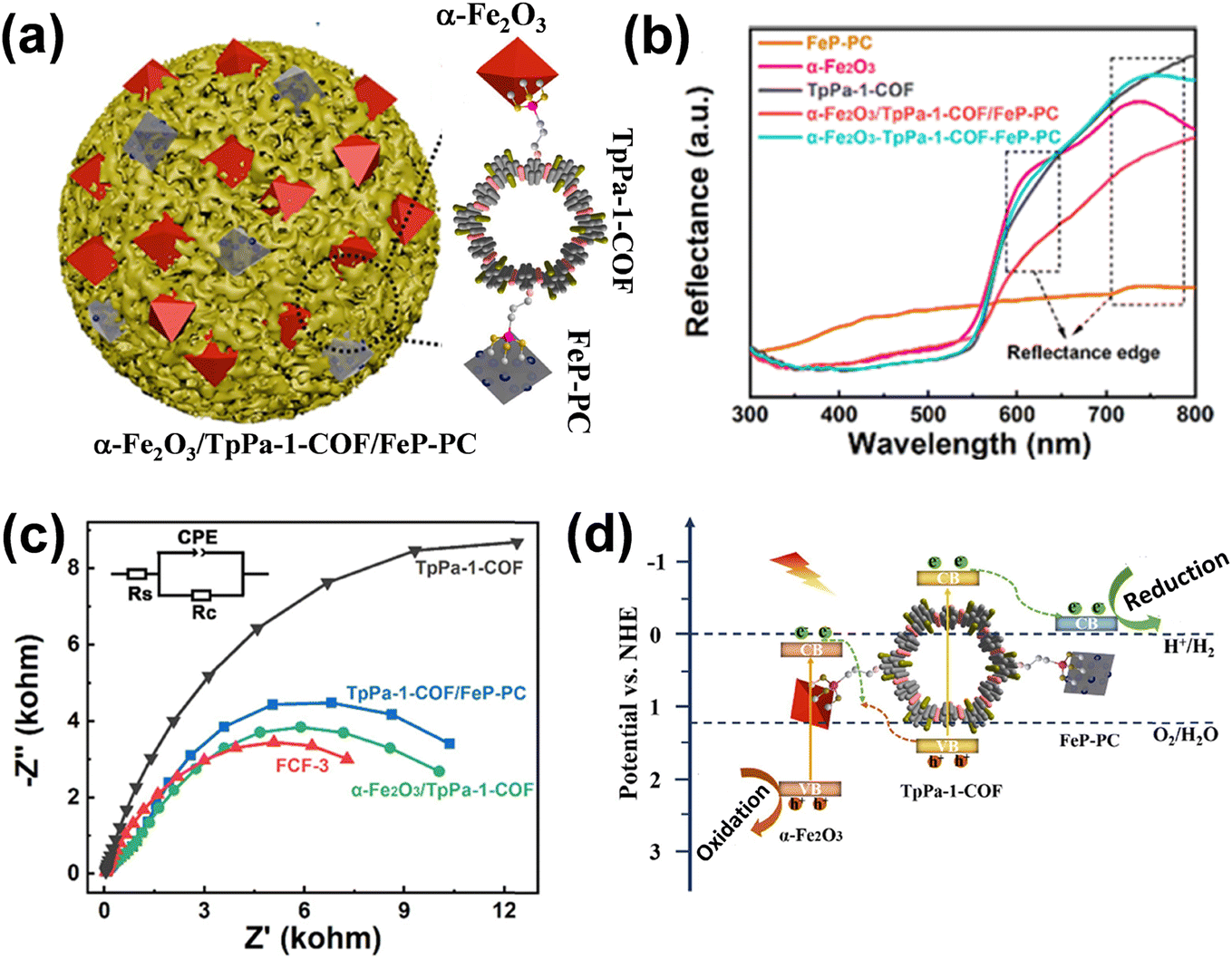 | ||
| Fig. 6 (a) Schematic illustration of the α-Fe2O3/TpPa-1-COF/FeP-PC heterojunction synthesis. (b) UV-vis Diffuse Reflectance Spectroscopy (DRS) of α-Fe2O3, FeP-PC, TpPa-1-COF, α-Fe2O3/TpPa-1-COF/FeP-PC, and α-Fe2O3-TpPa-1-COF-FeP-PC. (c) EIS Nyquist plots of TpPa-1-COF, α-Fe2O3/TpPa-1-COF, TpPa-1-COF/FeP-PC and FCF-3. (d) Mechanistic illustration of α-Fe2O3/TpPa-1-COF/FeP-PC. Reproduced from ref. 46 with permission from [Elsevier], copyright [2022]. | ||
The EIS Nyquist plots (Fig. 6c) displayed the lowest charge transfer resistance of FCF-3, indicating that the covalent bonding between FeP-PC and α-Fe2O3 as cocatalysts effectively enhanced charge transfer efficiency. Therefore, upon illumination, electrons from the CB of α-Fe2O3 transferred to the valence band of TpPa-1-COF via the establishment of a direct Z-scheme heterojunction at the interface of α-Fe2O3 cocatalyst and TpPa-1-COF (Fig. 6d). Next, electrons from the CB of TpPa-1-COF moved toward the CB of FeP-PC cocatalyst. Thus, α-Fe2O3 and FeP-PC acted as oxidation and reduction cocatalysts, respectively, enhancing the H2 and O2 generation of α-Fe2O3/TpPa-1-COF/FeP-PC. Due to the reduced charge by electron transfer from α-Fe2O3 to TpPa-1-COF, the α-Fe2O3/TpPa-1-COF showed a little enhancement of H2 and O2 release activities (16.87 and 8.37 μmol g−1 h−1). Noted that the volcano-type trend is observed between the overall water splitting rate of α-Fe2O3/TpPa-1-COF/FeP-PC and the loading of FeP-PC. After additional deposition of FeP-PC, the optimal FCF-3 reached a HER rate of 97.45 μmol g−1 h−1 and a stoichiometric OER rate of 48.68 μmol g−1 h−1.
2.4. Plasmonic metal/sensitizer–semiconductor–cocatalyst structure
The broad-spectrum light absorption of plasmonic nanoparticles using plasmon leads to generating a significant number of e− and h+ for light-driven catalytic reactions. Plasmon nanoparticles with minimal defects and high structural crystallinity can prevent charge carrier recombination, promoting photocatalytic efficiency.47,48 The plasmonic photocatalysts mainly work on the excitation and relaxation principle of plasmons. Light absorption and scattering, local electromagnetic field enhancement (LEFM), improved hot carriers (HCs) injection, and enhanced thermal effect are the main pathways of plasmon-assisted semiconductor photocatalysis.49,50 The catalytic process in plasmonic nanoparticles is furnished by the surface interactions on semiconductor photocatalysts and the direct transformation of reactants adsorbed on their surface.1,51 In this review, the charge transfer (HET, HHT, PICTT) and plasmon-induced resonant energy transfer (PIRET) process in plasmonic metal nanostructures have been discussed as shown in Fig. 7. | ||
| Fig. 7 Directional LSPR decay routes at the metal/semiconductor interface, including (a) HET, (b) HHT, (c) PICTT, and (d) PIRET. | ||
Single-atom catalysts (SACs) have emerged as highly promising photocatalysts due to superior atomic utilization efficiency and exceptional catalytic performance.69 Fundamentally, SACs achieve complete metal dispersion on the surface, ensuring maximum metal utilization. This unique and ideal characteristic makes them highly effective for developing efficient and cost-effective heterogeneous catalysts, particularly for optimizing noble metals such as Pt, Pd, Ru, etc.70 Moreover, the vast flexibility in integrating single-atom metal centers with various host materials, along with the precise tuning of their local coordination environment, offers significant potential for designing highly efficient SACs.71 From these benefits, SACs have provided an opportunity to develop efficient photocatalysis with higher activity and selectivity.72 Furthermore, heterojunctions formed between semiconductors and cocatalysts generate built-in electric fields that facilitate charge separation. This leads to the formation of type-II, Z-scheme, and S-scheme heterojunctions, which regulate the directionality and efficiency of electron transfer, effectively minimizing charge recombination and enhancing redox reactions.
In 2019, Bera et al.73 reported enhanced hydrogen production under visible light of BiFeO3 (BFO) perovskite by sensitizing it with gold nanoparticles (Au NPs) using a simple hydrothermal method, followed by radiolysis. TEM image represented the homogeneous distribution pattern of Au NPs on the surface of BFO-Ns after gamma radiation (Fig. 8a).
 | ||
| Fig. 8 (a) TEM image of Au/BFO-Ns heterostructure, (b) UV-visible DRS of pure BFO-Ns and Au/BFO-Ns heterostructures, (c) Hot electron transfer from the surface of the Au NPs to the CB of BiFeO3 in Au/BFO heterostructures. Reproduced from ref. 73 with permission from [Elsevier], copyright [2019]. (d) Spatial plasmonic distribution of electric field as a function of the monochromic incident light, (e) Mechanistic study of the photocatalytic H2 evolution. Reproduced from ref. 74 with permission from [American Chemical Society], copyright [2023]. (f) Site-selective growth of Cu2O on Au bipyramids, (g) TEM image of Cu2O nanoparticles decorated on Au bipyramids, (h) Moiré patterns of Au nanoplatelets decorated with Cu2O nanostructures. Reproduced from ref. 75 with permission from [American Chemical Society], copyright [2022]. | ||
Fig. 8b shows the DRS spectra of BFO-Ns and Au/BFO-Ns. Due to the strong electronic interaction of Au NPs with BFO-Ns, the increment of existing peaks of BFO-Ns at 350 nm and 450 nm have been observed. Additionally, a typical absorption peak at ∼600 nm was attributed to the plasmonic absorption of Au NPs. Fig. 8c revealed the band edge positions of Au/BFO heterostructures and the likely mechanism of photocatalytic H2 generation when visible light is illuminated on the Au/BFO heterostructures. This shows that the incident photons and electrons participate in resonance, which results in electronic oscillation. The energy states of BFO move upward, which is greater than the CB of the neighbouring semiconductor. On the other hand, the hot electrons (excited electrons) of Au may transfer to the CB of the BFO. The maximum photocatalytic H2 generation (∼2.1 mmol h−1 g−1) was achieved with Au/BFO-Ns as a photocatalyst, using methanol as a sacrificial agent.
In another study, Liu et al.74 nano-encapsulated CuOx with carbon supported on TiO2 (TC@C) and observed the in-operando photoreduction of CuOx to generate Cu nanoparticles under light irradiation. Fig. 8d shows the plasmon spatial distribution of the electric fields as a function of monochromatic light irradiation. The presence of Cu nanoparticles leads to a 5.8 times enhancement in the electric field intensity due to the LSPR effect of the Cu nanoparticles, resulting in an improvement in photocatalytic efficiency. Fig. 8e provides a mechanistic interpretation of the photocatalytic reactions occurring over the TC@C composites. The photocatalytic performance of TC@C composites was enhanced due to hot electron transfer into the CB of TiO2, which promotes the reduction of H+ ions to generate H2. In another investigation, Jia and co-workers monitored the differential site-selective growth behaviour of Cu2O on gold nanocrystals by controlling the surfactant concentration.75 The different morphologies of the Au nanocrystals direct the preferential growth of Cu2O nanostructures on the edges and ends of the Au nanorods and nanoplates which in turn influences their photocatalytic performance (Fig. 8f). The TEM image (Fig. 8g) reveals that the preferential growth of Cu2O at the two terminating positions of gold nanorods leads to the development of dumbbell-shaped nanocrystals. Fig. 8h depicts the Moire patterns of the growth of Cu2O on the three edges of Au nanoplates generated due to the superposition of the lattice structures of Au and Cu2O. The preferential growth of Cu2O on the three edges of gold nanocrystals has been observed from the Moire pattern. The dumbbell-shaped nanostructures of Cu2O loaded at the tips of the Au bipyramids result in superior photocatalytic performance towards CO2 reduction due to the hot hole injection process.
3. Electron transfer at the interfaces by various techniques
The electron transfer mechanism at the semiconductor–co-catalyst interface plays a crucial role in determining the efficiency of photocatalytic reactions. Understanding this mechanism is essential for designing more efficient photocatalysts. Theoretical approaches provide valuable insights into the charge transfer dynamics and help guide the development of better semiconductor–co-catalyst systems.X. Zhang et al.76 demonstrated that the incorporation of gold (Au) nanoparticles as co-catalysts, through modifications to the electronic structure, can enhance the efficiency of photocatalytic systems. Fig. 9a shows the synthetic strategy of TiO2/MoSx–Au by the initial lactic acid-induced MoSx deposition on the TiO2 surface. For a depth understanding of the cocatalyst mechanism different technique is used like ISI-XPS (Intensity-Dependent Surface Imaging X-ray Photoelectron Spectroscopy) spectra and fs-TA spectra. The principle of TA spectroscopy in photocatalytic applications involves measuring changes in the absorption spectrum of a material after it is photoexcited. When the photocatalyst is exposed to light, it generates photoexcited charge carriers (electrons and holes). The TA spectra track the dynamics of these carriers by observing light absorption over time, which reflects their relaxation, recombination, and migration behaviours. In photocatalytic studies, TA spectroscopy is employed to analyse the lifetime, migration, and recombination rates of the photogenerated carriers, offering valuable insights into the efficiency of charge separation and their role in catalytic reactions. And, the principle of ISI-XPS in photocatalytic applications is based on monitoring the surface chemical states and electronic properties of a photocatalyst under different light exposure conditions. ISI-XPS combines conventional XPS with light irradiation, enabling the analysis of changes in the chemical composition, binding energy, and oxidation states of elements on the surface of the photocatalyst during photoexcitation. ISI-XPS is particularly valuable for understanding the catalytic mechanism, surface dynamics, and stability of photocatalysts under illumination. By examining how the surface electronic structure evolves with light exposure, it helps in optimizing photocatalytic materials for reactions like water splitting, CO2 reduction, and pollutant degradation. In this research study, after loading MoSx–Au, the TiO2/MoSx–Au composite shows a significant increase in the contact potential difference (CPD), rising by approximately 185.4 mV (from −51.3 to 134.1 mV) under photonic irradiation, accompanied by a colour shift from blue to red. This change is attributed to the increased hole buildup on the surface of TiO2 (Fig. 9b and c), providing strong evidence that photogenerated electrons are rapidly moved from TiO2 to the MoSx–Au co-catalyst. To further confirm the photogenerated electrons transfer and their accumulation on the Au active sites of TiO2/MoSx–Au, in situ XPS analysis was conducted (Fig. 9d). The Au 4f7/2 and Au 4f5/2 peaks in TiO2/MoSx–Au shift Significantly in the direction of lower binding energies (from 83.6 eV to 83.5 eV) upon exposure, indicating that the electrons are transferred from TiO2 to MoSx–Au and predominantly accumulate on the electron-deficient Auδ+ sites, thereby enhancing the photocatalytic H2O2 production rate. To gain deeper insights into the electron transfer dynamics in TiO2/MoSx–Au, fs-TA spectra were performed with careful attention (Fig. 9e and f).
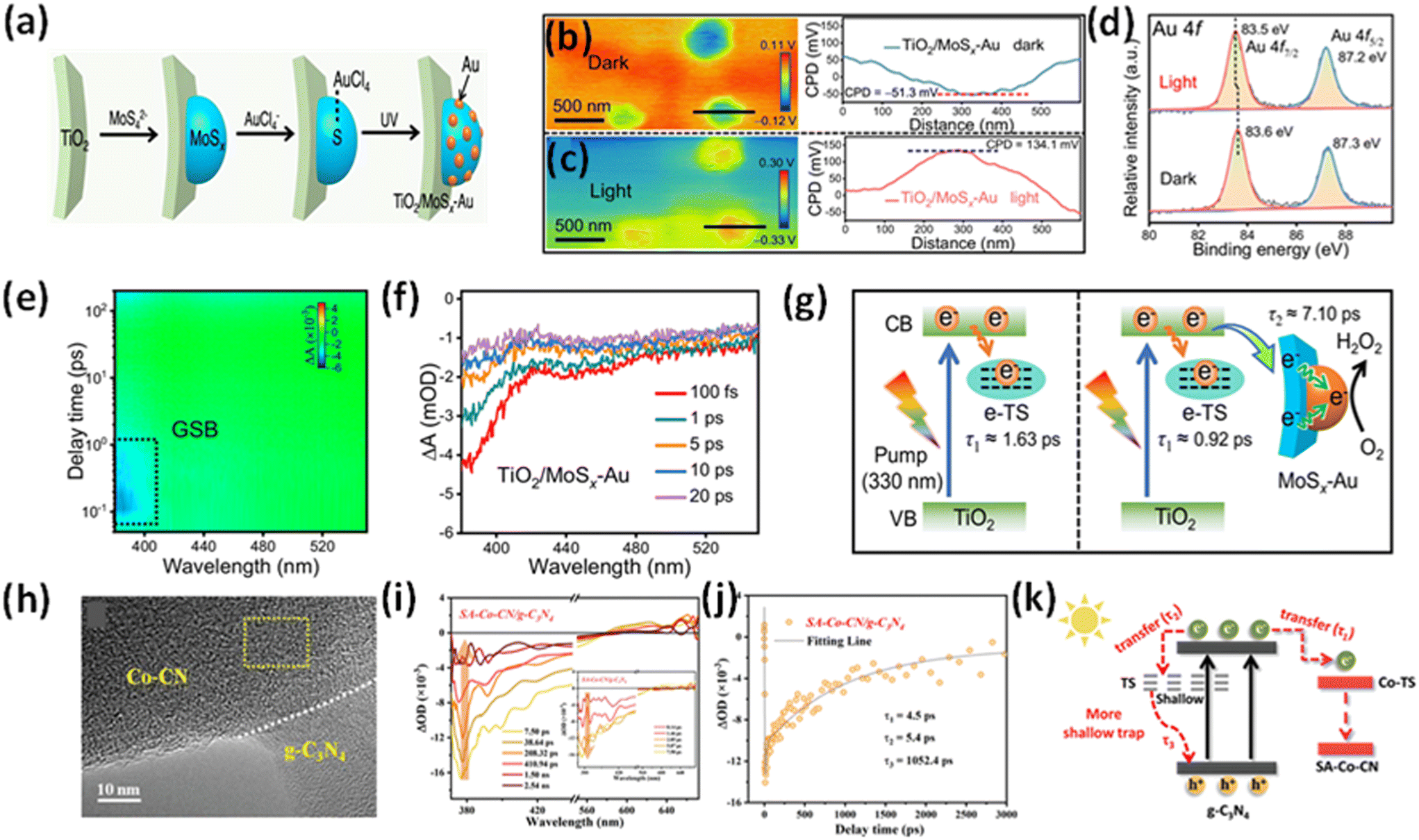 | ||
| Fig. 9 (a) A schematic illustration depicts the synthesis of TiO2/MoSx–Au, involving lactic acid-induced MoSx deposition on the TiO2 surface, followed by S-induced selective photodeposition of the Au cocatalyst onto the MoSx layer. (b) and (c) The KPFM image and corresponding surface potential profiles of TiO2/MoSx–Au were analyzed under dark conditions and 365 nm LED light illumination. (d) The ISI-XPS spectra of Au 4f for TiO2/MoSx–Au were recorded before and after light illumination. (e) Pseudo-color plots of TiO2/MoSx–Au, with GSB representing ground-state bleaching. (f) The fs-TA spectra of TiO2/MoSx–Au were recorded within a timescale of 20 ps. A schematic illustration depicts the decay pathways of photogenerated electrons in (g) TiO2 and TiO2/MoSx–Au. Reproduced from ref. 76 with permission from [Springer Nature], copyright [2024]. (h) HRTEM image of SA-Co-CN/g-C3N4 (i) fs-TA spectra of SA-Co-CN/g-C3N4. (j) The corresponding transient absorption kinetics for SA-Co-CN/g-C3N4 are shown. (k) A schematic representation illustrates the suggested charge localization model in SA-Co-CN/g-C3N4. Reproduced from ref. 77 with permission from [Wiley-VCH], copyright [2023]. | ||
The pseudo colour plots for TiO2, TiO2/Au, and TiO2/MoSx–Au reveal a typical photobleaching peak (∼380 nm), which corresponds to ground-state bleaching (GSB) and reflects the relaxation of the excited state. Monitoring the GSB signal at 380 nm within 20 ps shows stronger intensities in TiO2/Au and TiO2/MoSx–Au compared to TiO2, indicating enhanced electron accumulation in the former two systems. Decay kinetics were analyzed using biexponential equations for the 380 nm signal within 25 ps to further investigate the interfacial electron transfer. The short-lived τ1 (1.63 ps) represents electron trapping at e-TS, while the long-lived τ2 corresponds to interfacial electron transfer from TiO2 to the cocatalyst. A1 and A2 denote the decay fractions during electron trapping and transfer, respectively. In the TiO2 system, the dominant process within 25 ps is electron trapping at e-TS (Fig. 9g). Interestingly, the τ1 values for TiO2/Au and TiO2/MoSx–Au decrease significantly to 0.36 and 0.92 ps, respectively, indicating rapid electron transfer from TiO2 to the Au (τ2 = 5.88 ps) and MoSx–Au (τ2 = 7.10 ps) cocatalysts. Notably, TiO2/MoSx–Au exhibits a higher A2 value (A2 = 39.6%) than TiO2/Au (A2 = 32.3%), suggesting more efficient electron transfer from TiO2 to Au, facilitated by the MoSx mediator. This improved electron transfer in TiO2/MoSx–Au aligns well with results from photoelectrochemical and transient-state photoluminescence (TRPL) measurements. Collectively, these findings provide strong evidence that the MoSx–Au cocatalyst acts as an effective platform for rapid electron transfer, enabling the subsequent H2O2 production reaction at the electron-deficient Auδ+ sites, ultimately leading to high photocatalytic H2O2 yields. M. Qian et al.77 designed a heterojunction between of Co-CN single-atom catalysts (SACs) and g-C3N4 for heterogeneous photo-Fenton-like reactions. HRTEM image (Fig. 9h) of SA-Co-CN/g-C3N4 shows the formation of heterojunctions between Co-CN and g-C3N4. Transient absorption spectroscopy demonstrates the modulated charge transfer and trapping in the SA-Co-CN/g-C3N4 heterostructure, leading to significantly enhanced reactive oxygen species generation under light irradiation. The femtosecond-TA spectra of SA-Co-CN/g-C3N4 (Fig. 9i) revealed a negative absorption signal between 370 and 570 nm. A positive absorption band (570–670 nm) observed in the fs-TA spectra of SA-Co-CN/g-C3N4, attributed to excited-state absorption, indicates the presence of abundant shallowly trapped electrons in the SA-Co-CN/g-C3N4. The negative signals exhibit a rapid increase in peak intensity at early time scales following photoexcitation (inset in Fig. 9i), attributed to the immediate generation of charge carriers and their direct excitation from the ground state to the emissive state. The generation of shallowly trapped excitations in SA-Co-CN/g-C3N4 aids in retaining photoinduced charges, allowing them to contribute to the surface photocatalytic reaction. These results highlight the controlled trapping behaviour of photoinduced electrons by the island-like single-atom Co catalyst, which enhances photo-redox reactions by increasing the concentration of charge carriers. A tri-exponential decay function was used to model the kinetic traces (Fig. 9j). Interestingly, SA-Co-CN/g-C3N4 exhibits two rapid decay components, corresponding to electron transfer to the SA-Co-CN trapping sites (τ1 = 4.5 ps) and shallow ST (τ1 = 5.4 ps), respectively. The fs-TA decay behaviour of SA-Co-CN/g-C3N4 differs from that of pure g-C3N4, confirming the enhanced charge trapping and transport facilitated by the island-like Co-CN SACs. Considering the above result, a charge trapping model is proposed (Fig. 9k), where it is shown that by the formation of an electric field, an electron transfer has occurred between g-C3N4 and SA-Co-CN. Also, SA-Co-CN/g-C3N4 forms a shallower trap than pristine g-C3N4, which also confirms the effective segregation and rapid movement of charge carriers, which increase the catalytic activity.
4. Applications of semiconductor–cocatalyst electron transfer in photocatalysis
The semiconductor–cocatalyst interface is vital for photocatalysis, as it governs charge transfer, enhances e−–h+ separation, and reduces recombination. Cocatalysts provide active sites for reactions like water splitting and CO2 reduction.78 Optimizing interface properties, such as band alignment and bonding, improves charge flow and overall photocatalytic performance. Engineering this interaction is key to advancing solar energy conversion efficiency.4.1. Pollutant degradation
The continuous discharge of persistent non-biodegradable effluents from industries into the water bodies has resulted in the deterioration of the aquatic ecosystems and provided a detrimental effect on the quality of human life.79,80 Therefore, wastewater remediation via advanced redox processes has become the global consensus in recent years to minimize water pollution and safeguard our environment. Photocatalytic degradation of these harmful contaminants using semiconductors offers a desirable and eco-friendly approach towards the removal of these toxic dyes, heavy metals and antibiotics.81,82 In this regard, special emphasis has been laid on the rational fabrication of visible-light active photocatalysts with favourable band positions suitable for redox reactions.83,84 However, the limitations of a single semiconductor, like inefficient visible light absorption, sluggish charge transfer dynamics and rapid charge recombination, trigger the combination of the semiconductors with co-catalysts to improve the photocatalytic performance.For example, Guo and coworkers explored the co-catalytic effect of the cobalt phosphide nanoparticles (CoP) deposited on g-C3N4 nanosheets via a solvothermal method.85 The TEM image reveals the formation of the CoP nanoparticles with an average size of 5–7 nm bound to the surface of g-C3N4 nanosheets as observed in Fig. 10a. The photocatalytic performance of the g-C3N4/CoP composites was examined towards tetracycline (TC) degradation and the composites with an optimized loading of CoP nanoparticles exhibited a remarkable rate of photodegradation (0.0275 min−1) with an efficiency of 96.7% in 120 minutes which is 10.2 times higher than that of pristine g-C3N4 nanosheets (0.0027 min−1) as depicted in Fig. 10b and c. To elucidate the role of active species involved in the redox reaction, various scavengers like ethylene diamine tetra-acetic acid disodium salt, isopropanol, and benzoquinone were added to capture the holes, ˙OH and ˙O2− radicals respectively. It was observed that the introduction of isopropanol and benzoquinone significantly reduced the degradation efficiency to 31.4% and 22.1% respectively, which signifies that ˙O2− and ˙OH radicals play a major role in the photocatalytic degradation of tetracycline (Fig. 10d).
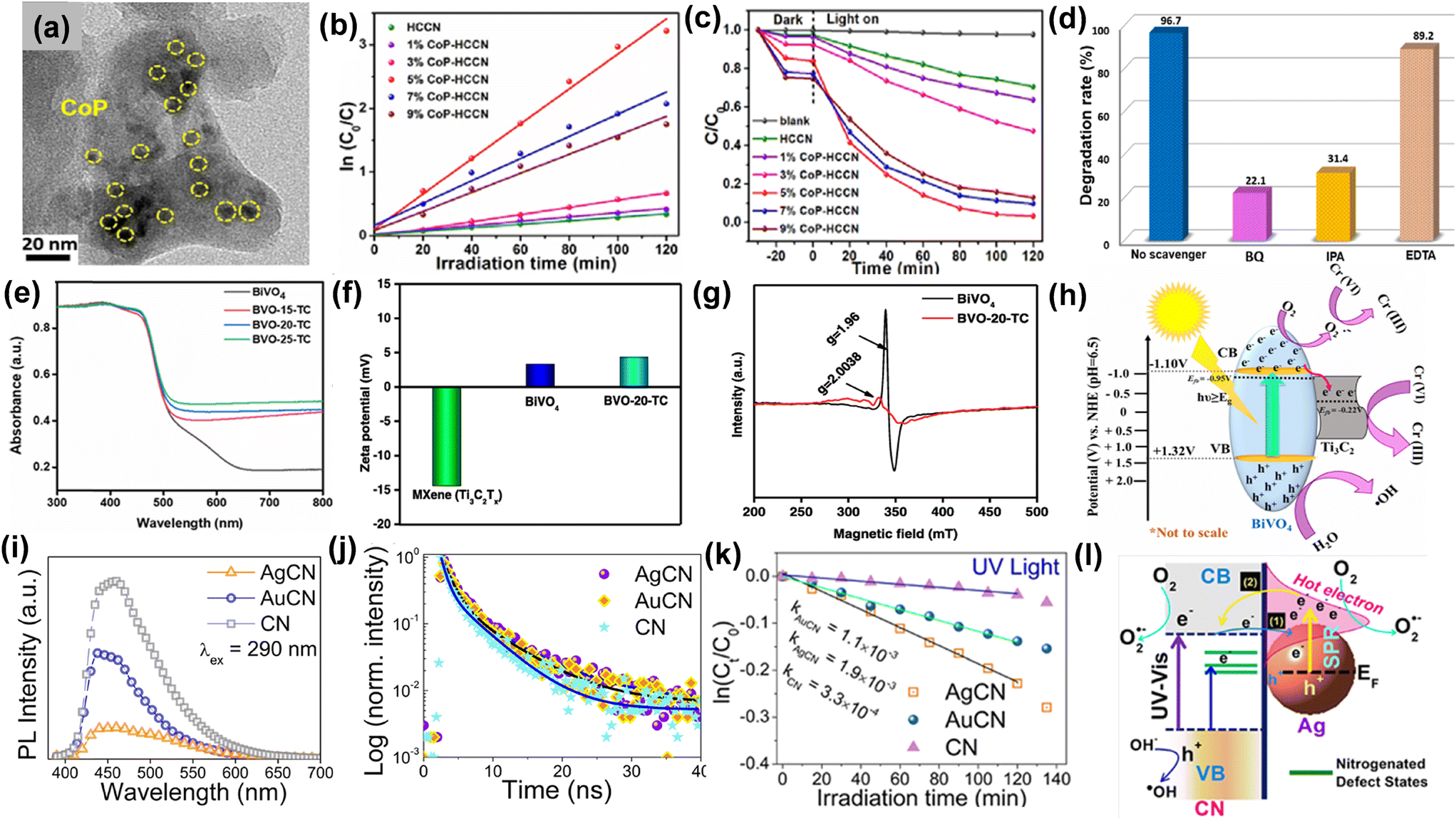 | ||
| Fig. 10 (a) TEM image of CoP cocatalyst loaded carbon nitride. (b) Photodegradation of tetracycline in the presence of visible illumination. (c) Pseudo-first-order kinetics. (d) Influence of different scavengers on the photodegradation of TC. Reproduced from ref. 85 with permission from [Elsevier], copyright [2020]. (e) DRS of photocatalysts. (f) Mean zeta-potentials of bare BiVO4, MXene, and BVO-20-TC in the presence of light. (g) Electron paramagnetic resonance (EPR) spectra of pristine BiVO4 and BiVO4/MXene composites. (h) Charge transfer mechanism occurring at the BiVO4/MXene interface. Reproduced from ref. 86 with permission from [American Chemical Society], copyright [2023]. (i) PL spectra, (j) TRPL spectra, (k) rate constants for photocatalytic RhB degradation, (l) electron transfer mechanism occurring at the semiconductor–plasmonic metal interface. Reproduced from ref. 87 with permission from [American Chemical Society], copyright [2019]. | ||
Razafintsalama et al.86 coupled MXene as a co-catalyst with BiVO4 nanoparticles and observed that the MXene-based BiVO4 heterostructures exhibited superior activity towards photocatalytic Cr(VI) reduction up to 96.4% within 30 minutes. The DRS reveals that the MXene cocatalyst-loaded BiVO4 heterostructure exhibits enhanced visible light absorption compared to pristine BiVO4 (Fig. 10e). The net surface charge density of pure MXene and the heterostructures was determined by the zeta-potential measurements. It was observed that BiVO4 exhibited positive potential, which signifies that BiVO4 possesses superior electrical stability whereas the Ti3C2Tx MXene exhibits a negative zeta-potential of −14.42 mV. The optimized Ti3C2Tx-based BiVO4 heterostructure exhibits a mean zeta potential of 4.29 mV on account of the stronger electrostatic interaction and extensive electroactive coupling between MXene and BiVO4 as observed from Fig. 10f. The EPR spectra indicate that the EPR signal intensity reduces after heterostructure formation in comparison with pristine BiVO4, which implies that the Ti3+ and V4+ act as trapping centres for the photogenerated charge carriers and lowers the rate of e−–h+ recombination as depicted in Fig. 10g. The enhanced photocatalytic performance may be attributed to the development of Schottky junction between BiVO4 and MXene (Fig. 10h), where MXene acts as an electron reservoir and co-catalyst thereby facilitating the reduction process. The combined role of Au and Ag nanoparticles as co-catalysts and plasmonic metals was explored by Kashyap and coworkers, who examined the rate of photocatalytic rhodamine B degradation with Ag and Au-supported g-C3N4 nanosheets.87 The steady-state photoluminescence spectra reveal that the PL intensity decreases considerably for the metal nanoparticles supported g-C3N4 (CN) nanosheets compared to pristine nanosheets which susuggestsower e−–h+ recombination after decoration with Au and Ag nanoparticles (Fig. 10i). The TRPL measurements reveal that the average lifetime of the photogenerated electrons in the CN nanosheets is 1.41 ns which increases to 2.47 and 3.06 ns after incorporation of Au and Ag nanoparticles to CN, respectively. The photoexcited electrons get transferred from CN to Au and Ag and remain trapped in the metallic nanoparticles which prolongs the lifetime of the charge carriers as shown in Fig. 10j. It was observed (Fig. 10k) that there was three-fold and five-fold augmentation in the rate constants for the photocatalytic RhB degradation in case of AuCN and AgCN composites respectively compared to pristine CN in the presence of UV radiation. The mechanism of electron transfer occurring at the metal–semiconductor interface depicted in Fig. 10l reveals that three processes bandgap, sub-bandgap, and plasmonic excitation processes occur simultaneously in the wavelength range of 250–600 nm. In the wavelength range from 400–600 nm, plasmonic excitation occurs in Ag due to the surface plasmon resonance absorption peak at 476 nm for Ag, and the plasmon-induced hot electrons get transferred from the Ag nanoparticles to CN. Bandgap excitation mostly occurs in the UV region (below 420 nm) for carbon nitride and electrons get transferred from the CB of CN to the plasmonic metals, thereby accelerating the photocatalytic redox reactions.
4.2. Water splitting
The interfacial charge transfer between semiconductors and cocatalysts enhances water-splitting efficiency.88–90 Efficient charge transfer minimizes e−–h+ recombination, ensuring photogenerated carriers participate in the desired redox reactions. The semiconductor absorbs light to generate e−–h+ pairs, while the cocatalyst facilitates the hydrogen evolution reaction (HER) or oxygen evolution reaction (OER) by acting as an active site. The nature of the interface—band alignment, surface states, and chemical interactions—directly influences the transfer dynamics. Optimizing this interface is crucial for achieving high-performance photocatalysts for sustainable hydrogen production. For example, CoPi cocatalyst and oxygen vacancy (OV) induced WO3/BiVO4 photoanode was designed for overall water splitting by Liu et al.91 As illustrated in Fig. 11a, the (WO3/BiVO4)-OV exhibits a significantly higher enhancement in photocurrent densities across the applied potential range than WO3-OV/BiVO4. This indicated that oxygen vacancies confined exclusively to WO3 are inadequate for effectively mitigating surface charge recombination. Furthermore, the deposition of the CoPi cocatalyst led to a substantial increase in photocurrent density, reaching 2.9 mA cm−2 at 1.6 V vs. RHE. This enhancement highlights the strong interfacial contact in the (WO3/BiVO4)-OV/CoPi photoanode. As shown in Fig. 11b, WO3 photoanode exhibits a peak applied bias photon to current conversion efficiency (ABPE) of 0.07% at 1.01 V vs. RHE, while WO3/BiVO4 achieves 0.18% at 0.95 V vs. RHE. Notably, the (WO3/BiVO4)-OV/CoPi photoanode reaches a maximum ABPE of 0.38% at 0.88 V vs. RHE, 5-fold higher than bare WO3, highlighting the synergistic effect of OVs and CoPi integration. The charge separation efficiency (Fig. 11c) of (WO3/BiVO4)-OV/CoPi is approximately four times higher than that of bare WO3. This suggests enhanced charge separation at the WO3/BiVO4 interface, facilitated by integrating oxygen vacancies and the CoPi cocatalyst. A type-II heterojunction forms at the WO3/BiVO4 interface, while oxygen vacancies on WO3 and BiVO4 create a stepwise band structure that facilitates charge migration (Fig. 11d). Additionally, CoPi cocatalyst captures holes, enhancing water oxidation efficiency. In another study, Boppella and coworkers explored the role of black phosphorus (BP) supported Ni2P which acted as the reduction co-catalyst for achieving enhanced photocatalytic performance towards water splitting.92 The TEM image shows that the Ni2P cocatalyst-loaded BP is formed on the CN nanosheets, which indicates the development of the Ni2P@BP/CN heterostructure (Fig. 11e). The HRTEM image reveals that the lattice fringes correspond to the diffraction planes of BP, Ni2P and carbon nitride (CN), which indicate stronger interfacial contact between Ni2P cocatalyst loaded BP and CN as depicted in Fig. 11f. The PL spectra indicate a substantial reduction in PL intensity for hybridized material as compared to pristine CN, suggesting enhanced electron transfer across interfaces following cocatalyst deposition (Fig. 11g). The rate of photocatalytic H2 generation using this hybrid (858.2 μmol g−1 h−1) was 50 times higher as compared to pure CN (17.2 μmol g−1 h−1) as (Fig. 11h). The 2D–2D interfacial coupling between the co-catalyst and CN facilitates electron migration, charge separation, lowers the activation energy barrier and promotes water reduction.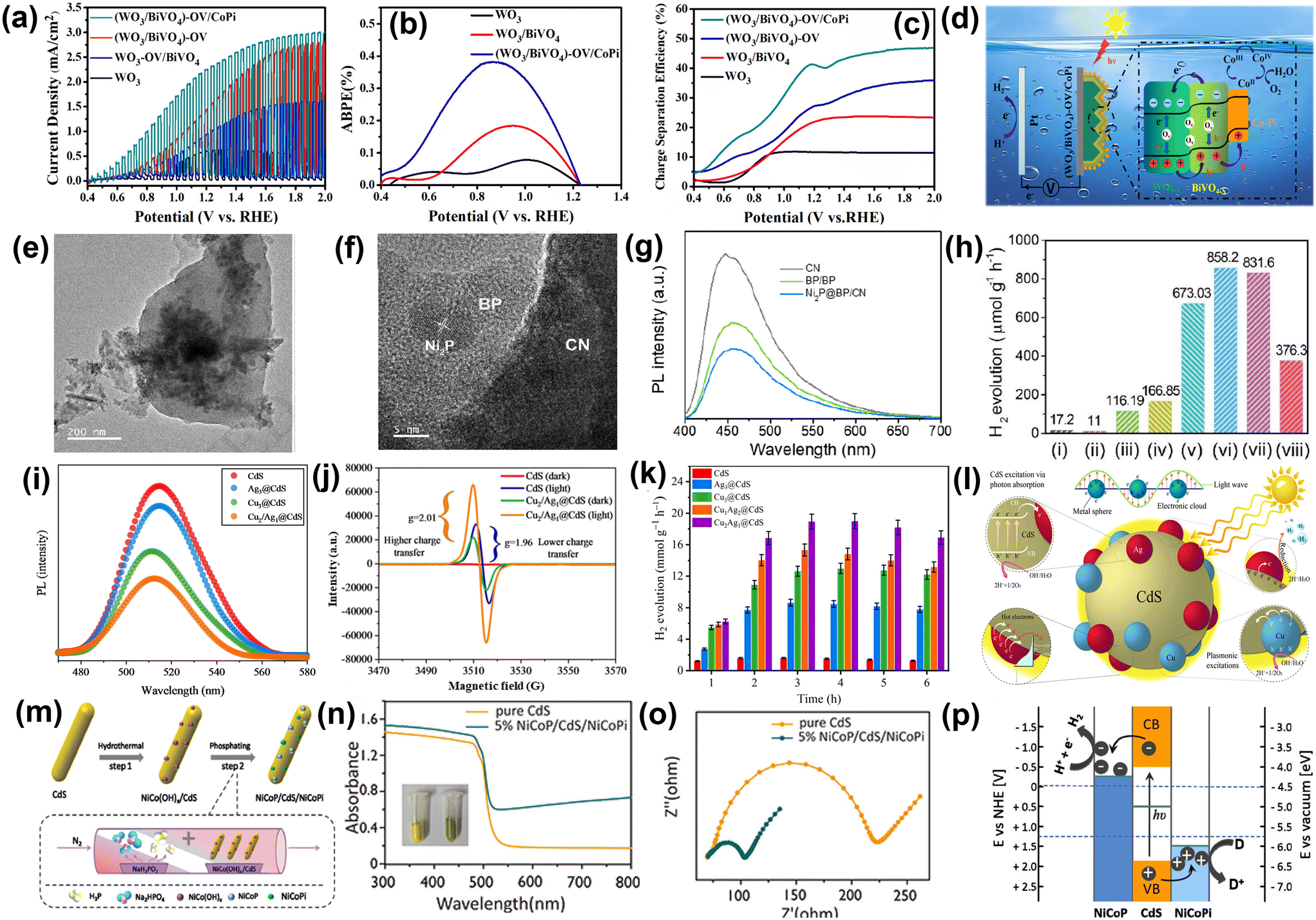 | ||
| Fig. 11 (a) Chopped linear sweep photocurrent–potential curves. (b) ABPE curves. (c) Charge separation efficiency of photoanodes. (d) Schematic description of the PEC water oxidation process in the (WO3/BiVO4)-OV/CoPi Photoanode. Reproduced from ref. 91 with permission from [American Chemical Society], copyright [2021]. (e) TEM image (f) HRTEM image of Ni2P@BP/CN. (g) PL of pristine CN, binary BP/CN, and Ni2P@BP/CN. (h) Rate of photocatalytic H2 evolution of (i) CN; (ii) Ni2P@BP; (iii) BP/CN (2 wt%); (iv) Pt/CN (2 wt%); (v) Ni2P@BP/CN (1 wt%); (vi) Ni2P@BP/CN (2 wt%); (vii) Ni2P@BP/CN (3 wt%); and (viii) Ni2P/CN. Reproduced from ref. 92 with permission from [Elsevier], copyright [2019]. (i) PL results of CdS, Ag3@CdS, Cu3@CdS, and Cu2/Ag1@CdS; (j) EPR of photocatalysts; (k) hydrogen production rates of different catalysts; (l) mechanism of photocatalytic H2 production over Cu2/Ag1@CdS catalysts. Reproduced from ref. 93 with permission from [American Chemical Society], copyright [2024]. (m) Schematic representation of dual-cocatalyst-loaded CdS photocatalyst. (n) DRS of pristine CdS and dual-cocatalyst-loaded CdS. Inset: colour change of the samples from yellow to green. (o) Nyquist plot of bare CdS and NiCoP/CdS/NiCoPi. (p) Charge transfer mechanism occurring at the interface of dual co-catalyst loaded CdS. Reproduced from ref. 94 with permission from [American Chemical Society], copyright [2020]. | ||
E. Hussain et al.93 deposited copper (Cu) and silver (Ag) metals as cocatalysts over the surface of CdS using chemical reduction via hydrothermal method. The results indicate that catalysts containing 2% Cu and 1% Ag on CdS demonstrate the highest activity, achieving 18.93 mmol g−1 h−1 with a quantum efficiency of 45.04%. Fig. 11i represents the photoluminescence (PL) spectra of pristine CdS and different ratios of Cu/Ag on CdS at 370 nm. Here, Ag cocatalysts promote the quenching of electrons to be involved in proton (H+) reduction, and Cu metal enhances its LSPR hot electrons to the surfaces of CdS. Therefore, the PL intensity of Cu2/Ag1@CdS (2% Cu and 1% Ag on CdS) decreases, indicating a higher charge transfer to the active sites for the water reduction reaction. From electron paramagnetic resonance (EPR) spectroscopy (Fig. 11j), it is observed that in dark conditions, Cu2/Ag1@CdS catalysts exhibited low EPR intensities due to unpaired electrons in Cu and Ag. Under visible light, the electrons promote from the VB to the CB and experience a stronger magnetic field, which leads to higher g values (2.01) than pristine CdS (1.96). This increasing EPR intensity result implies that Cu2/Ag1@CdS have more reacting electrons compared to bare CdS catalysts. Cu2/Ag1@CdS delivers 18.93 mmol g−1 h−1 of hydrogen with 45.04% QE at 420 nm and is found to be the most active catalyst. The increased hydrogen activity of Cu2/Ag1@CdS is due to the enhanced SPR conduction of hot electrons to the CdS surface, promoting efficient charge transfer to active sites, as shown in Fig. 11k. When exposed to sunlight, CdS photocatalysts absorb photons, generating e−–h+ pairs, with electrons moving to the CB and holes remaining in the VB. The Cu and Ag cocatalysts enhance photocatalytic activity by generating SPR electrons and forming Schottky barriers, with silver creating stronger barriers. At the same time, copper provides consistent SPR electron generation on the CdS surface during photoreactions. Silver forms more stable junctions with CdS due to its larger size compared to copper, which creates fewer effective junctions. Silver reacts with dissolved oxygen to form oxides, which are reduced back to metallic silver during photoreaction, creating electron pools that enhance H+ ion reduction. The synergy between SPR electrons from copper and Schottky barriers from silver effectively prevents recombination, resulting in higher photocatalytic H2 generation (Fig. 11l). The effect of redox dual co-catalyst nickel cobalt phosphide (NiCoP) and phosphate (NiCoPi) on the light-driven catalytic performance of CdS nanorods was explored by Zhao and co-workers which was synthesized via hydrothermal method followed by one-step phosphating using sodium hypophosphate as the phosphorus source (Fig. 11m).94 It was observed that the CdS nanorods modified with redox-active dual co-catalysts exhibit lower charge transfer resistance at the electrode–electrolyte interface owing to the enhancement in charge carrier separation after co-catalyst incorporation in comparison with bare CdS nanorods as observed from Fig. 11n. The presence of dual co-catalysts increases the rate of photocatalytic H2 evolution (80 mmol g−1 h−1) by 202 times compared to pristine CdS (0.4 mmol g−1 h−1) under visible light illumination as shown in Fig. 11o. Fig. 11p represents the plausible charge transfer occurring at the semiconductor–dual co-catalyst interface. The electrons migrate from the CdS nanorods to the co-catalyst NiCoP, and reduction occurs from the surface of NiCoP, whereas holes transfer from the VB of CdS to the co-catalyst and promote the oxidation process. Therefore, the NiCoP and NiCoPi co-catalysts act as electron and hole acceptors, respectively, and boost the photocatalytic performance of CdS nanorods.
4.3. CO2 reduction
The combustion of fossil fuels leads to substantial CO2 emissions, exacerbating the global energy crisis and accelerating climate change.95–97 Thus, the photocatalytic conversion of CO2 into HCOOH, HCHO, CH3OH, CO, and CH4 has garnered significant attention in recent years.98,99 However, enhancing the effectiveness and selectivity of photocatalytic CO2 reduction remains insufficiently explored. Cocatalysts are crucial in enhancing charge separation and transfer efficiency, boosting the activity and selectivity of CO2 reduction, improving the stability of photocatalysts, and minimizing reverse reactions.100,101For example, Di et al.102 loaded Co2N cocatalyst on BiOBr ultrathin nanosheets to prepare Co2N/BiOBr semiconductors for boosting photocatalytic CO2 reduction. Nanosheets with BiOBr structures adorned with Co2N nanoparticles exhibited uniform distribution and strong interfacial contact, as illustrated in Fig. 12a. The Co2N/BiOBr hybrid materials demonstrated enhanced photocatalytic CO2 reduction to CO, with Co2N/BiOBr-1 achieving the highest CO production yield. Co2N/BiOBr-1 hybrid material displayed 67.8 μmol g−1 h−1 CO production rate which was ∼6 times higher than that of pristine BiOBr (Fig. 12b). The reduced activity of Co2N/BiOBr materials with higher Co2N content could be attributed to the excessive Co2N overshadowing BiOBr, thereby hindering its light absorption. The reaction mechanism of CO2 conversion to CO is described as follows (eqn (1)–(6)) (Fig. 12c):
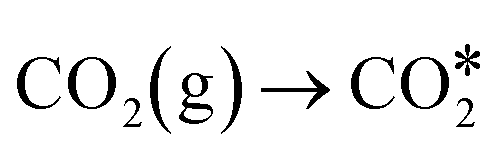 | (1) |
| H2O → H+ + OH− | (2) |
 | (3) |
 | (4) |
| COOH* + H+ + e− → CO* + H2O | (5) |
| CO* → CO | (6) |
 | ||
| Fig. 12 (a) TEM image of Co2N/BiOBr-1. (b) CO2 photoreduction activity over Co2N/BiOBr. (c) Schematic representation of photocatalytic CO2 reduction over Co2N. (d) Ultrafast TA spectroscopy over Co2N/BiOBr-1. Reproduced from ref. 102 with permission from [Elsevier], copyright [2021]. (e) PL spectra and (f) impedance spectra. (g) Photocatalytic H2, CO, and CH4 generation rates. (h) The most stable configurations of CO2 adsorption on Pt (730) and PtCu (730) facets, along with their adsorption energies (dark blue spheres represent Pt atoms; brown, dark, and red spheres represent Cu, C, and O atoms, respectively). Reproduced from ref. 103 with permission from [The Royal Society of Chemistry], copyright [2017]. (i) Raman spectra, (j) EIS Nyquist plots, (k) product yields and CO selectivity. (l) Reaction mechanism for the photothermocatalytic CO2 reduction reaction. Reproduced from ref. 104 with permission from [American Chemical Society], copyright [2021]. | ||
Lang and co-workers designed for the first time a high-index facet cocatalyst for enhanced photocatalytic CO2 reduction.103 They have loaded highly (730) facets exposed concave nanocubes of PtCu alloy cocatalyst on C3N4 nanosheets for efficient CH4 production. As shown in Fig. 12e, the PL intensity significantly decreased upon PtCu loading onto C3N4, indicating that PtCu nanocrystals efficiently inhibited e−–h+ recombination in C3N4 by facilitating effective electron transfer across the PtCu/C3N4 interface. Furthermore, C3N4–PtCu NCs (PtCu nanocubes loaded on C3N4 nanosheets) and C3N4–PtCu CNCs (PtCu concave nanocubes loaded on C3N4 nanosheets) materials displayed comparable PL intensities, indicating both materials exhibited similar charge separation capabilities. Under visible light irradiation, the C3N4–PtCu materials exhibit a significantly higher photocurrent response than C3N4, with both showing comparable performance, indicating that PtCu nanocubes and concave nanocubes have similar electron-trapping and charge-separation capabilities (Fig. 12f). Besides, the CO and CH4 production efficiencies of C3N4–PtCu CNCs were 2 and 3-fold greater, respectively, than those of C3N4–PtCu NCs. Additionally, the addition of PtCu onto the surface of C3N4 effectively suppressed the side reaction of H2 generation from H2O, as shown in Fig. 12g. Density functional theory (DFT) simulations were conducted to analyse the photocatalytic mechanism by determining the CO2 adsorption energies on Pt (730) and PtCu (730) (Fig. 12h). It was suggested that the high-index facet engineering, attributed to the low coordination number of Pt atoms on the concave (730) surface, enhances CO2 adsorption energy and boosts photocatalytic performance.
The photothermal effect accelerates charge migration and optimizes reactant adsorption and desorption, boosting the efficiency of photocatalytic CO2 reduction. For example, Cai et al.104 decorated Au nanoparticles on mesoporous TiO2 nanospheres (mTiO2) with surface oxygen defects. The plentiful surface oxygen vacancies (OVs) facilitated the even distribution and formation of smaller Au nanoparticles, thereby increasing the availability of active sites. Consequently, the combined effects of the photothermal properties, oxygen vacancies (OVs), and Au cocatalysts synergistically improved charge separation and migration, as well as CO2 adsorption and activation. The characteristic Raman peak at 143 cm−1 exhibited a slight shift to a higher wavenumber in Au-loaded materials, highlighting a robust interaction between Au and oxygen in TiO2, as shown in Fig. 12i. The smallest semicircular arc for 0.5Au/mT-H6 demonstrated enhanced charge carrier mobility, suggesting that the combination of OVs and the Au cocatalyst synergistically promotes the separation and movement of photoexcited charge carriers through the Au/mTiO2 interface (Fig. 12j). The yield rates for CO and CH4 in 0.5Au/mT-H6 were 0.14 μmol m−2 and 0.17 μmol m−2, respectively, while in 0.5Au/mTiO2, the yield rates for CO and CH4 were 0.04 μmol m−2 and 0.11 μmol m−2, respectively, as shown in Fig. 12k. The presence of Au NPs cocatalyst not only enhances the yields of CO and CH4 but also boosts H2 production. However, the coexistence of OVs and Au NPs cocatalysts effectively inhibits H2 generation. As shown in Fig. 12l, under solar light irradiation, photoexcited electrons transferred from TiO2 to the OVs and Au nanoparticles, resulting in Schottky junction formation at the interfaces. Additionally, the intense photothermal effect supplied substantial thermal energy, promoting the migration of electrons from TiO2 to the surface OVs and Au NPs. This facilitated effectual charge separation, allowing increased CO generation as well as CH4 production.
In recent years, the photocatalytic conversion of single-carbon compounds into high-value multi-carbon (C2+) products, such as C2H4, C3H6, C2H5OH, HOCH2CH2OH, etc., has emerged as a highly promising research frontier due to its potential for combustion fuel, chemical solvent, medical use, etc., yet remains challenging for complex reaction pathways and selectivity issues. Furthermore, C2+ products possess a wider range of applications and exhibit greater added value compared to C1 compounds. Ji et al.105 engineered Au–CeO2 nanocomposites featuring Au–O–Ce active sites at the microinterface, enabling highly selective CO2 conversion into C2H6. These nanocomposites demonstrated a C2H6 production rate of 11.07 μmol g−1 h−1 and an impressive selectivity of 65.3%, positioning them among the most advanced photocatalysts available. Compared to other cocatalysts, Cu species have attracted considerable interest due to their ability to enhance multi-electron transfer, effectively utilize weakly bound d-band electrons, and benefit from a narrow band gap along with strong CO2 adsorption and activation capabilities. In particular, the valence state and coordination environment of Cu species plays a pivotal role in governing the selectivity of C2+ products. For example, Shi et al.106 atomically dispersed In and Cu onto the polymeric carbonitride (PCN) that showed excellent photoreduction of CO2 to ethanol (production rate of 28.5 μmol g−1 h−1) and a high selectivity of 92%. The In–Cu interaction on the surface of PCN enhanced the charge separation by accelerating charge transfer from PCN to the metal sites. Additionally, intermetallic charge transfer from In to Cu via In–N–Cu bridges enhances the electron density at Cu active sites, thereby reducing the energy barrier for C–C coupling. Zhang et al.107 effectively enhanced the photocatalytic performance of defective titania (TiO2-SBO) catalysts by incorporating diverse active sites through the co-modification of Cu and thiocyanate anion (SCN−). In this study, SCN-Cu/TiO2-SBO-3 achieved a C2H4 production rate of 4.7 μmol g−1 h−1 with a selectivity of 40% for CO2 conversion.
In summary, cocatalysts play a crucial role in improving e−–h+ pair separation and transfer, effectively mitigating photocorrosion by rapidly extracting charge carriers. Additionally, they facilitate CO2 activation, enhance the selectivity of CO2 reduction, and suppress undesired reverse reactions, leading to improved photocatalytic efficiency. However, the majority of studies focus on noble metal-based cocatalysts, alloys, and Ru-complex systems that enhance photocatalytic CO2 reduction reactions. Therefore, noble metal-free cocatalysts such as Cu-, Ni-based cocatalysts, graphene, carbon nanotubes, etc., need to be explored more in CO2 reduction. Additionally, the development of novel cocatalysts with high activity, selectivity, stability, and cost-effectiveness is essential for advancing semiconductor-based photocatalytic CO2 reduction.
4.4. Ammonia (NH3) production
Photocatalytic nitrogen fixation (PNF) offers an environmentally friendly alternative to the energy-intensive Haber–Bosch process, operating under mild conditions without the need for high temperature and pressure.108 PNF can use water and nitrogen to produce ammonia, utilizing the oxidative power of photogenerated holes. Co-catalysts play a crucial role in enhancing the efficiency and performance of the photocatalytic nitrogen reduction reaction by improving charge separation, enhancing electron transfer, and lowering the activation energy. In this regard Zhang et al.109 synthesized Ni3B/VN-CN by electrostatic self-assembly method, where the exact amount of Ni3B and VN-CN was mixed in water and sonicated for few hours (Fig. 13a). As reported, Schottky junction photocatalyst, which is free of noble metals, is composed of g-C3N4 nanosheets with nitrogen vacancies (VN-CN) combined with metallic Ni3B nanoparticles (Ni3B/VN-CN), designed for the reduction of N2 to ammonia. Notably, the 15% Ni3B/VN-CN photocatalyst demonstrated the smallest arc radii in the Nyquist plots (Fig. 13b), indicating the lowest charge transfer resistance. This suggests the most efficient charge transfer across the interface between VN-CN and Ni3B among the tested photocatalysts. The ammonia production rate for the optimized Ni3B/VN-CN photocatalyst is 7.68 mM g−1 h−1, which is 6.7 times greater than that of the pristine CN, with a rate of 1.15 mM g−1 h−1 (Fig. 13c). Based on the analysis, the proposed mechanism for the Ni3B/VN-CN composite photocatalyst in nitrogen reduction reaction (NRR) is illustrated in Fig. 13d. Upon light irradiation, excited electrons are transferred to the surface of VN-CN and then flow to Ni3B due to its lower Fermi level. The electron-rich Ni3B then serves as a co-catalyst, facilitating the adsorption and activation of N2 molecules, which leads to the photoreduction of nitrogen. The enhanced photocatalytic NRR performance of Ni3B/VN-CN can be attributed to three key factors. First, the nitrogen vacancies in CN not only narrow the bandgap and enhance light absorption but also improve electrical conductivity, aiding charge transfer. Second, the Schottky junction formed at the interface of Ni3B and VN-CN significantly promotes charge transfer while suppressing recombination. Third, the metallic Ni3B, with its lower work function, acts as a co-catalyst, providing additional active sites that accelerate the photocatalytic NRR. Together, these factors contribute to the exceptional photocatalytic performance of Ni3B/VN-CN compared to pure CN and VN-CN. Shen and his group demonstrated black phosphorus (BP) as an active cocatalyst modified with CdS as a photocatalyst.110Fig. 13e, HRTEM shows the lattice stripes of CdS and BP, which confirm the formation of BPNS/CdS. As depicted in Fig. 13f, the yellow and blue isosurfaces represent areas of electron accumulation and depletion, respectively, resulting from intercalation. Notably, significant charge rearrangement is observed at the interfaces between BPNSs and CdS. As shown in Fig. 13g, both BPNS/CdS and CdS electrodes exhibit quick and consistent photocurrent responses observed in each illumination cycle. The photocurrent density of the 1.5% BPNSs/CdS electrode was approximately 13.58 μA cm−2, while the bare CdS electrode showed a photocurrent density of around 6.79 μA cm−2, which shows that cocatalyst-modified CdS is more efficient than bare CdS. After 6 hours of irradiation, the average NH3 production rates for BPNS/CdS samples with 0.5, 1.0, 1.5, 2.0, and 2.5% BPNSs were determined to be 28.79, 34.85, 57.64, 45.83, and 32.99 μmol L−1 h−1, respectively (Fig. 13h). Based on photocatalytic tests and theoretical simulations presented above, a photocatalytic mechanism is proposed to explain the effective N2 reduction capability of the BPNS/CdS photocatalyst. As illustrated in Fig. 13i, under light illumination, electrons in the VB of CdS are excited to the CB, generating photogenerated charge carriers. In the absence of BPNSs, these electrons tend to recombine quickly with the holes, resulting in low charge separation efficiency, which enhances the photocatalytic activity. | ||
| Fig. 13 (a) A schematic illustration depicts the formation process of the Ni3B/VN-CN photocatalyst. (b) Impedance plots of CN, VN-CN, and 15% Ni3B/VN-CN. (c) The ammonia yield of the synthesized samples is reported. (d) A possible mechanism for the photocatalytic NRR on the Ni3B/VN-CN photocatalyst is proposed. Reproduced from ref. 109 with permission from [American Chemical Society], copyright [2023]. (e) HRTEM images of 1.5% BPNS/CdS photocatalyst. (f) The charge density difference at the BPNS/CdS heterojunction interfaces are shown, with the yellow and blue isosurfaces representing electron gain and loss, respectively. (g) The time-dependent photocurrent response of different BPNS/CdS samples and bare CdS is examined. (h) The NH3 formation rates of different samples are compared. (i) A proposed photocatalytic mechanism for efficient nitrogen fixation on BPNS/CdS photocatalysts is presented. Reproduced from ref. 110 with permission from [American Chemical Society], copyright [2020]. (j) HRTEM images of BiOBr/Au-CP. (k) PL spectra and (l) NH3 yield rates of BiOBr, BiOBr/Au-C, BiOBr/Au-CP, and BiOBr/Au-T. (m) The mechanism of internal charge transfer and photocatalytic nitrogen fixation in BiOBr/Au-CP. Reproduced from ref. 111 with permission from [Elsevier], copyright [2023]. | ||
In different work, C. Wang et al.111 demonstrated the enhancement of photocatalytic NH3 production BiOBr nanosheets modified with Au clusters (Au-CP) and Au nanoparticles (Au-C) as cocatalysts. The formation of a heterogeneous structure between the interface of BiOBr and Au was distinctly observed through TEM imaging (Fig. 13j). The PL spectra of BiOBr and BiOBr/Au are shown in Fig. 13k, excited at 470 nm. The PL intensity of BiOBr/Au is reduced compared to pure BiOBr. Notably, BiOBr/Au-CP exhibits a lower e−–h+ recombination rate than BiOBr/Au-C. The photocatalytic performance of the catalysts was evaluated and it was clear from Fig. 13l that the BiOBr/Au-CP catalyst achieved the highest nitrogen fixation rate, reaching 80.52 μmol g−1 h−1, which is nearly four-fold higher than that of pure BiOBr (22.32 μmol g−1 h−1). Furthermore, the BiOBr/Au-CP sample demonstrates significantly superior photocatalytic activity compared to BiOBr/Au-C. The mechanism behind photocatalytic nitrogen fixation of BiOBr/Au-CP, as derived from the results, is depicted in Fig. 13m. Unlike traditional methods that focus on small-sized co-catalysts, BiOBr serves as the model carrier, and the BiOBr/Au composite photocatalytic system is created by combining Au nanoclusters and Au nanoparticles of two different sizes. Initially, the noble metal Au creates a Schottky barrier at the interface between the semiconductor and the co-catalyst, facilitating the separation and transfer of charge carriers generated by light while significantly reducing the recombination of electrons and holes. In the BiOBr/Au-CP sample, the smaller Au nanoclusters provide numerous active sites for N2 reduction, while the larger Au nanoparticles contribute to an enhanced plasmonic absorption effect, further improving the photocatalytic nitrogen fixation properties. As a result, the BiOBr/Au-CP system achieves an optimal balance between catalytic activity and light absorption efficiency, exhibiting the most effective photocatalytic nitrogen fixation performance when exposed to visible light.
In conclusion, modifying photocatalysts with co-catalysts significantly enhances the efficiency of photocatalytic nitrogen fixation for ammonia production. By incorporating co-catalysts, such as noble metals or metal nanoparticles, the separation of photo-generated charge carriers is improved, which reduces recombination rates and promotes efficient charge transfer. This results in increased catalytic activity for nitrogen reduction. Co-catalysts also provide additional sites that facilitate nitrogen adsorption and activation, further boosting the reaction rate. Moreover, the synergy between the co-catalyst and photocatalyst can optimize the light absorption properties and enhance overall photocatalytic performance. Thus, the design of photocatalytic systems with appropriately chosen co-catalysts holds great promise for sustainable and efficient ammonia production, offering an eco-friendly alternative to traditional Haber–Bosch processes.
A comprehensive table (Table 1) has been compiled to summarize recent advancements in semiconductor–cocatalyst interfaces and their diverse applications in photocatalysis.
| Photocatalyst | Cocatalyst | Loading method | Application in photocatalysis | Performance | Ref. |
|---|---|---|---|---|---|
| SrTiO3 | Pt (reduction) & CoOx (oxidation) | Photo-deposition | H2 and O2 production | H2 production rate = 15.9 μmol gcat−1 h−1 | 112 |
| O2 evolution rates = 7.6 μmol gcat−1 h− 1 | |||||
| TiO2 | CuRu | Photo-reduction | H2 production | H2 production rate = 5.316 mmol g−1 h−1 | 113 |
| CdS | Ni | — | H2 production and pollutant degradation | H2 production rate = 13.267 mmol g−1 h−1 | 114 |
| Degradation of reactive red = 0.125 min−1 and tetracycline hydrochloride = 0.069 min−1 | |||||
| TiO2 | ReOSx | Photosynthesis | H2 production | H2 production rate = 7.168 mmol g−1 h−1 | 115 |
| SiC | Fe | Incipient-wetness impregnation and chemical reduction | CH4 production | CH4 yield = 30.0 μmol g−1 h−1 | 116 |
| Selectivity = 94.3% | |||||
| TiO2 | WB5−x | — | CH4 production | CH4 yield = 13.0 μmol gcat−1 h−1 | 117 |
| Selectivity = 95.9% | |||||
| rGO/La2Ti2O7 | NiFe-LDH | Ultrasonication | H2 production | H2 production rate = 532 μmol g−1 h−1 | 118 |
| TiO2/CdS | Pt | Chemical reduction | H2 production | H2 production rate = 3.074 μmol g−1 h−1 | 119 |
| NiS/CdZnS | Ni@NiO | Hydrothermal method | H2 production | H2 production rate = 34.456 mmol g−1 h−1 | 120 |
| TiO2 | Cu2O@Cu-MOF core–shell | — | CH4 production | CH4 yield = 366.0 μmol g−1 h−1 | 121 |
| Selectivity = 95.5% | |||||
| CdS nanorods | NiS | Hydrothermal method | NH3 production | NH3 production rate = 2.8 mg L−1 | 122 |
| g-C3N4 | WP nanoparticle | — | CO production | CO production rate = 376 μmol g−1 h−1 | 123 |
| TiO2 | Pt | Photodeposition | CH4 production | CH4 yield = 60.1 μmol gcat−1 h−1 | 124 |
| ZnxCd1−xS | Ti3C2 nanoparticles | — | H2 production | H2 production rate = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 342 μmol g−1 h−1 342 μmol g−1 h−1 |
125 |
| TiO2 | NiSe2 | Supercritical fluid process | H2 production | H2 production = 219.2 mmol gcat−1 h−1 | 126 |
| g-C3N4 | Pt@Ni(OH)2 | Photodeposition | H2 production | H2 production = 298 μmol g−1 h−1 | 127 |
| CdS | Ni(OH)2 | Electrostatic self-assembly process | H2 production | H2 production = 40.18 μmol g−1 h−1 | 128 |
| g-C3N4 | CoxNiyP | Impregnation + phosphorization | H2 production | H2 production rate = 239.3 μmol g−1 h−1 | 129 |
| Y2Ti2O5S2 | Co3O4 | Impregnation method | O2 evolution | O2 production = 298 μmol h−1 | 130 |
| WO3 | B2O3−xNx | Direct thermal oxidation | O2 evolution | O2 production = 74 μmol h−1 | 131 |
5. Theoretical approach on electron transfer in semiconductor–cocatalysts interfaces
While experimental studies confirm that cocatalysts significantly enhance charge separation and boost photocatalytic activity, several key questions regarding their mechanisms and optimization remain unresolved. For instance, the precise positioning of cocatalysts on semiconductor surfaces remains unclear. Additionally, the impact of cocatalyst adsorption on the electronic properties of photocatalysts and the underlying mechanisms by which cocatalyst loading enhances charge separation require further investigation. Thus, theoretical studies are essential for understanding charge transfer mechanisms at the semiconductor–cocatalyst interface, as they offer a comprehensive quantum mechanical perspective of the electronic structure. For instance, Li and colleagues analyzed the band structure of n-NiS2 and n-NiS employing DFT calculation from the Γ point in the Brillouin zone to gain insight into the progressive electron transfer at the interfaces of n-NiS/n-NiS2 heterojunction cocatalysts and the p-Si substrate.132 The semi-metallic nature of NiS2 was observed at the Γ point in the Brillouin zone, as depicted in Fig. 14a, with the CB of n-NiS2 primarily attributed to contributions from Ni and S, as observed from the partial density of state (DOS). However, the CB of NiS is mainly dominated by Ni (Fig. 14b). | ||
| Fig. 14 Band structure and DOS of (a) NiS2 and (b) NiS. (c) Schematic representation of charge transfer on NNH/Si for enhanced H2 generation. Reproduced from ref. 132 with permission from [Wiley-VCH], copyright [2021]. The calculated work functions of (d) TiO2(101) surface (e) Au(001) surface. (f) The band structure alignment of Au catalysts with TiO2, both before and following contact. Reproduced from ref. 133 with permission from [Elsevier], copyright [2019]. | ||
These findings suggest that n-NiS2 is capable of transferring electrons to both Ni and S, whereas n-NiS is limited to accepting electrons exclusively from Ni. The charge transfer mechanism at the interface of the NiS2/NiS heterostructure (NNH) and p-Si, based on their band structure, has been analyzed and illustrated in Fig. 14c. As depicted in the upper half of Fig. 14c, n-NiS functions as a progressive medium in the p-Si/n-NiS/n-NiS2 junction, facilitating improved electron transfer from the p-Si substrate to n-NiS2 under light irradiation, thereby enhancing the H2 evolution reaction. Conversely, in the p-Si/n-NiS2/n-NiS configuration, electron transfer from n-NiS2 to n-NiS is impeded due to the higher CB edge potential of n-NiS, which adversely affects H2 generation performance (lower half of Fig. 14c). In another study, the charge transfer mechanism between TiO2 photocatalyst and Au metal cocatalyst has been investigated using DFT by Wang et al.133 TiO2 nanoparticles exposing the (101) facet and Au nanoparticles exposing the (001) facet was found to have work functions of 5.55 eV (Fig. 14d) and 5.27 eV (Fig. 14e), respectively. Based on these values, the CB and VB edge potentials of TiO2 were calculated, revealing two possible mechanisms for the interaction between TiO2 and the Au cocatalyst: a Schottky junction and an ohmic junction (Fig. 14f). Since the Au cocatalyst has a lower work function than TiO2, electrons were transferred from Au to TiO2. Upon illumination, the LSPR of Au nanoparticles significantly enhanced light absorption by generating additional hot electrons. Furthermore, the formation of an ohmic contact between Au nanoparticles and TiO2 facilitated the transfer of electrons from the CB of TiO2 to Au, contributing to the photocatalytic activity. In this process, the Au cocatalyst was an electron-capturing centre and provided active sites for photocatalytic reactions.
In another study, Liu et al.134 conducted a DFT analysis to evaluate the comparative stabilities of the low-index surfaces of β-Ga2O3 and identify the energetically favourable adsorption sites for Nin (n = 1–4) and (NiO)n (n = 1–4) clusters cocatalysts on these surfaces. It was reported that the binding energy is lowest for Ni4/Ga2O3(100), indicating improved stability of the Ni4 cluster on the β-Ga2O3 surface (Fig. 15a). As shown in Fig. 15b, an unoccupied state of Ni4 was observed at ∼2 eV above the Fermi level. Upon photoexcitation, electrons transitioned from the occupied states of O to the unoccupied states of Ga. Since the unoccupied state of Ni4 overlapped with that of Ga, electrons were further transferred to the Ni4 cluster, where they contributed to the hydrogen generation process. For Ni4O4/Ga2O3(100), as depicted in Fig. 15c, four Ni–O bonds were identified, with two Ni atoms bonded to O(I) and the other two Ni atoms bonded to O(III) at equal distances. Additionally, Ga–O bonds were also observed with bond lengths of 2.038 Å and 2.042 Å. The projected density of states (PDOS) and total density of states (TDOS) of Ni 3d and O 2p states for Ni4O4 indicated the significant overlap between O and Ni states. Under illumination, electrons from the Ni4O4 cluster are inclined to transfer to the VB of Ga2O3, as the occupied states of the Ni4O4 cluster are at a higher energy level than the VB maximum of Ga2O3 (Fig. 15d). Therefore, available holes in the VB of the Ni4O4 cluster took part in the oxygen evolution reaction. The photocatalytic roles of edge and surface atoms differ due to their distinct coordination environments, which alter the atomic ratio between edge and surface atoms. Wu et al.135 solved this issue by theoretically studying the N2 fixation at the edge and surface of the Pd cocatalyst deposited on TiO2 semiconductor. As shown in Fig. 15e, the adsorption energies of N2 on the edges and (100) surfaces of Pd nanocubes were −0.72 eV and −0.27 eV, respectively, indicating a significantly stronger interaction between N2 and the edges. Additionally, electrons accumulated at the edges of the Pd nanocubes can back-donate to the 2π* orbital of N2 during their interaction, as observed in Fig. 15f. Moreover, the d-center of Pd atoms at the edges moves closer to the Fermi energy level, positioned at −1.51 eV, which is notably higher than the −1.80 eV for surface atoms and −2.36 eV for bulk atoms (Fig. 15g). This study demonstrated that the edges of the Pd cocatalyst exhibit superior N2 fixation, attributed to their low-coordination and electron-rich sites.
 | ||
| Fig. 15 (a) Structural parameters of the most stable configurations of Ni4/Ga2O3(100). (b) DOSs for the stable Ni4 configuration on the Ga2O3(100) surface, with the vertical dashed line marking the Fermi level. (c) Structural parameters of the most energetically favourable configurations of Ni4O4/Ga2O3(100). (d) DOSs for the stable configuration of Ni4O4 adsorbed on Ga2O3(100) surface. The vertical dashed line represents the Fermi level. Reproduced from ref. 134 with permission from [The Royal Society of Chemistry], copyright [2015]. (e) The potential energy surface for N2 hydrogenation on a Pd nanocubes. (f) Charge density difference of Pd nanocubes. Pd atoms are highlighted in green, while the yellow colour indicates an increase in electron density. (g) The partial density of states of Pd atoms at the edge, bulk, and surface of the Pd nanocubes. Reproduced from ref. 135 with permission from [The Royal Society of Chemistry], copyright [2021]. | ||
In summary, several important parameters, for example, band alignment, charge density distribution, and semiconductor–cocatalyst interfacial properties have been explored through DFT calculations which is useful for understanding the charge transfer dynamics. Simulating the interactions between the semiconductor and cocatalyst, DFT provides a detailed analysis of band bending, adsorption characteristics, and reaction intermediates, thereby providing the pathways of photocatalytic mechanism with high precision.
6. Summary & future prospects
Understanding the electron transfer dynamics at the semiconductor–cocatalysts interfaces is pivotal for advancing the field of solar fuel generation. This review has highlighted the significant progress in decoding the photocatalytic mechanisms through advanced experimental strategies and theoretical approaches. Herein, we have discussed the key role of cocatalysts in enhancing the performance of photocatalytic systems by promoting efficient charge separation, reducing recombination losses, and providing active sites for redox reactions. These findings have been key to making processes like pollutant removal, water splitting, CO2 reduction, and nitrogen fixation more efficient. We reviewed various semiconductor–cocatalyst architectures (semiconductor–oxidation cocatalysts, semiconductor–reduction cocatalysts, R–S–O cocatalysts, and plasmonic metal–semiconductor cocatalysts) based on their applications. Mainly noble metal-based cocatalysts and alloys have been widely reported as cocatalysts to improve photocatalytic activities. Recently, noble metal-free cocatalysts such as Cu-, Ni- and Co-based cocatalysts, MXene, MOFs, and COFs have been developed and utilized for photocatalytic applications. Additionally, we have examined the role of electron and hole transfer across semiconductor–cocatalyst interfaces in various photocatalytic applications, such as pollutant removal, water splitting, CO2 reduction, and N2 fixation. Our findings highlight that efficient charge transfer through these interfaces significantly enhances photocatalytic performance. Although significant progress has been made, numerous key challenges must be addressed. One of the primary challenges lies in achieving precise control over the interfacial properties of semiconductor–cocatalyst systems. Tailoring the electronic and structural interactions at the interface is essential to optimize charge transfer and reaction kinetics. Furthermore, the long-term stability of photocatalysts under operational conditions remains a significant concern, particularly when subjected to harsh reaction environments. The high cost and limited availability of noble metal-based cocatalysts also pose challenges for large-scale deployment, necessitating the exploration of earth-abundant and cost-effective alternatives. To overcome these challenges, future research should prioritize the development of advanced characterization techniques capable of probing interfacial charge dynamics at the atomic and molecular levels in real time. Techniques such as in situ spectroscopy, and high-resolution microscopy could offer unparalleled insights into the transient processes at the interface. Additionally, incorporating machine learning and high-throughput computational modeling could accelerate the discovery of novel semiconductor–cocatalyst combinations with superior properties. Another exciting avenue for future exploration lies in expanding the scope of photocatalytic applications. While water splitting and CO2 reduction have been extensively studied, more attention should be directed toward N2 fixation, which holds immense potential for sustainable fuel and chemical production. Developing multi-functional cocatalysts capable of driving multiple reactions simultaneously could further enhance the versatility of photocatalytic systems. Lastly, the integration of photocatalytic systems into practical applications requires addressing scalability and system-level optimization. Combining photocatalysts with emerging technologies, such as tandem systems and hybrid photoelectrochemical devices, may bridge the gap between laboratory-scale studies and industrial deployment.Data availability
This review does not include any primary research findings, software, or code, nor does it involve the generation or analysis of new data.Author contributions
Dipendu Sarkar: conceptualization, investigation, writing – original draft, review & editing. Jishu Pramanik: description of methodology, writing – original draft, review & editing. Soumita Samajdar: description of methodology, writing – original draft, review & editing. Maitrayee Biswas: description of methodology, writing – original draft, review & editing. Srabanti Ghosh: conceptualization, writing – original draft, review & editing, visualization, description of methodology, investigation, formatting and supervision.Conflicts of interest
The authors confirm that they have no conflicts of interest to disclose.Acknowledgements
Dr. Srabanti Ghosh acknowledges the Science and Engineering Research Board (SERB) through “SERB-POWER Grant” (project no. SPG/2020/000720) for financial support. The authors, SS and DS, express their gratitude to CSIR, India, for granting the Senior Research Fellowship award. Additionally, JP acknowledges the Department of Science & Technology (DST), India, for providing the INSPIRE Fellowship award.References
- S. Li, P. Miao, Y. Zhang, J. Wu, B. Zhang, Y. Du, X. Han, J. Sun and P. Xu, Adv. Mater., 2021, 33, 2000086 CrossRef CAS.
- S. Qin, N. Denisov, H. Kim and P. Schmuki, Angew. Chem., Int. Ed., 2024, 63, e202316660 Search PubMed.
- D. Liu and C. Xue, Adv. Mater., 2021, 33, 2005738 Search PubMed.
- A. Meng, L. Zhang, B. Cheng and J. Yu, Adv. Mater., 2019, 31, 1807660 Search PubMed.
- Y. Li, S. Zhang, C. He, H. Yao, C. Guo, W. Wang and Y. Hu, ACS Catal., 2025, 15, 2315–2327 Search PubMed.
- S. Ghosh, D. Sarkar, S. Bastia and Y. S. Chaudhary, Nanoscale, 2023, 15, 10939–10974 Search PubMed.
- X. Wang, J. You, J. Ren, Y. Xue, J. Tian and H. Zhang, Appl. Catal., B, 2024, 345, 123722 Search PubMed.
- Z. Xie, Q. Liu, H. Zhao, H. Chen, G. Jia, E. Lei, C. Wang and Y. Zhou, Catal. Commun., 2024, 186, 106837 Search PubMed.
- S. Ghosh, P. S. Das, S. Bera, D. Sarkar, K. Roy, S. Nath, P. Ghosh, C. K. Ghosh and A. R. Allu, ACS Appl. Energy Mater., 2024, 7, 10906–10920 Search PubMed.
- S.-M. Wu and P. Schmuki, Adv. Mater., 2025, 37, 2414889 Search PubMed.
- L. Liu, M. Zang, L. Li, Y. Zhang, L. Wang, X. Zhou, C. Xin and X. Tai, Sci. Rep., 2025, 15, 448 Search PubMed.
- C. Hu, X. Chen, J. Low, Y.-W. Yang, H. Li, D. Wu, S. Chen, J. Jin, H. Li, H. Ju, C.-H. Wang, Z. Lu, R. Long, L. Song and Y. Xiong, Nat. Commun., 2023, 14, 221 CrossRef CAS PubMed.
- L.-W. Chen, Y.-C. Hao, Y. Guo, Q. Zhang, J. Li, W.-Y. Gao, L. Ren, X. Su, L. Hu, N. Zhang, S. Li, X. Feng, L. Gu, Y.-W. Zhang, A.-X. Yin and B. Wang, J. Am. Chem. Soc., 2021, 143, 5727–5736 Search PubMed.
- S. M. Thabet, H. N. Abdelhamid, S. A. Ibrahim and H. M. El-Bery, Sci. Rep., 2024, 14, 10115 Search PubMed.
- X. Zhang, D. Gao, B. Zhu, B. Cheng, J. Yu and H. Yu, Nat. Commun., 2024, 15, 3212 CAS.
- T. Nakamoto, S. Iguchi, S. Naniwa, T. Tanaka and K. Teramura, Catal. Sci. Technol., 2023, 13, 4534–4541 Search PubMed.
- M. Zhang, X. Wang, X. Qi, H. Guo, L. Liu, Q. Zhao and W. Cui, J. Catal., 2022, 413, 31–47 Search PubMed.
- T. Ma, W. Li, J. Li, W. Duan, F. Gao, G. Liao, J. Li and C. Wang, J. Colloid Interface Sci., 2024, 658, 476–486 CAS.
- W. Li, Y. Dang, J. Li, T. Ma, G. Liao, F. Gao, W. Duan, J. Li, X. Wang and C. Wang, Chem. Eng. J., 2023, 472, 144793 CAS.
- W. Li, G. Liao, W. Duan, F. Gao, Y. Wang, R. Cui, X. Wang and C. Wang, Appl. Catal., B, 2024, 354, 124108 Search PubMed.
- W. Li, W. Duan, G. Liao, F. Gao, Y. Wang, R. Cui, J. Zhao and C. Wang, Nat. Commun., 2024, 15, 6763 Search PubMed.
- H. Suzuki, S. Nitta, O. Tomita, M. Higashi and R. Abe, ACS Catal., 2017, 7, 4336–4343 Search PubMed.
- Z. U. Rahman, N. Wei, M. Feng and D. Wang, Int. J. Hydrogen Energy, 2019, 44, 13221–13231 CAS.
- S. Chen, S. Shen, G. Liu, Y. Qi, F. Zhang and C. Li, Angew. Chem., Int. Ed., 2015, 54, 3047–3051 CrossRef CAS PubMed.
- X.-S. Xing, M. Bao, P. Wang, X. Wang, Y. Wang and J. Du, Appl. Surf. Sci., 2022, 572, 151472 CrossRef CAS.
- C. Xia, Y. Li, M. Je, J. Kim, S. M. Cho, C. H. Choi, H. Choi, T.-H. Kim and J. K. Kim, Nano-Micro Lett., 2022, 14, 209 CrossRef CAS PubMed.
- S. Y. Xiao, Y. Liu, X. F. Wu, L. T. Gan, H. Y. Lin, L. R. Zheng, S. Dai, P. F. Liu and H. G. Yang, J. Mater. Chem. A, 2021, 9, 14786–14792 RSC.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CAS.
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga, H. Murakami, Y. Ohki, N. Yoshida and T. Watanabe, J. Am. Chem. Soc., 2008, 130, 1676–1680 CAS.
- B. Li, T. Gu, T. Ming, J. Wang, P. Wang, J. Wang and J. C. Yu, ACS Nano, 2014, 8, 8152–8162 CAS.
- J. U. Salmón-Gamboa, M. Romero-Gómez, D. J. Roth, A. V. Krasavin, P. Wang, W. Dickson and A. V. Zayats, Nanoscale Adv., 2021, 3, 767–780 Search PubMed.
- M. Liu, X. Jin, S. Li, J.-B. Billeau, T. Peng, H. Li, L. Zhao, Z. Zhang, J. P. Claverie, L. Razzari and J. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 34714–34723 Search PubMed.
- S. Y. Jeong, H.-M. Shin, Y.-R. Jo, Y. J. Kim, S. Kim, W.-J. Lee, G. J. Lee, J. Song, B. J. Moon, S. Seo, H. An, S. H. Lee, Y. M. Song, B.-J. Kim, M.-H. Yoon and S. Lee, J. Phys. Chem. C, 2018, 122, 7088–7093 CAS.
- S. Y. Xiao, Y. Liu, X. F. Wu, L. T. Gan, H. Y. Lin, L. R. Zheng, S. Dai, P. F. Liu and H. G. Yang, J. Mater. Chem. A, 2021, 9, 14786–14792 CAS.
- S. Bai, W. Yin, L. Wang, Z. Li and Y. Xiong, RSC Adv., 2016, 6, 57446–57463 RSC.
- L. Zhao, F. Ye, D. Wang, X. Cai, C. Meng, H. Xie, J. Zhang and S. Bai, ChemSusChem, 2018, 11, 3524–3533 Search PubMed.
- Y. Zhu, C. Gao, S. Bai, S. Chen, R. Long, L. Song, Z. Li and Y. Xiong, Nano Res., 2017, 10, 3396–3406 Search PubMed.
- C. Liu, U. Burghaus, F. Besenbacher and Z. L. Wang, ACS Nano, 2010, 4, 5517–5526 Search PubMed.
- S. Lin, H. Huang, T. Ma and Y. Zhang, Adv. Sci., 2021, 8, 2002458 CrossRef CAS.
- M. Setvín, U. Aschauer, P. Scheiber, Y.-F. Li, W. Hou, M. Schmid, A. Selloni and U. Diebold, Science, 2013, 341, 988–991 CrossRef PubMed.
- W. Li, Z. Wei, Y. Sheng, J. Xu, Y. Ren, J. Jing, J. Yang, J. Li and Y. Zhu, ACS Energy Lett., 2023, 8, 2652–2660 CrossRef CAS.
- W. Zhao, W. Mo, Y. Zhang, L. Hu, Y. Zheng, Z. Chen, X. Niu, Y. Zhao, L. Liu, S. Zhong and S. Bai, Nano Res., 2024, 17, 5022–5030 CrossRef CAS.
- S. Y. Xiao, Y. Liu, X. F. Wu, L. T. Gan, H. Y. Lin, L. R. Zheng, S. Dai, P. F. Liu and H. G. Yang, J. Mater. Chem. A, 2021, 9, 14786–14792 Search PubMed.
- B. He, C. Bie, X. Fei, B. Cheng, J. Yu, W. Ho, A. A. Al-Ghamdi and S. Wageh, Appl. Catal., B, 2021, 288, 119994 CrossRef CAS.
- T. Di, Q. Deng, G. Wang, S. Wang, L. Wang and Y. Ma, J. Mater. Sci. Technol., 2022, 124, 209–216 Search PubMed.
- M.-L. Xu, J.-R. Li, X.-M. Wu, T. Yu, G.-Y. Qin, F.-J. Wang, L.-N. Zhang, K. Li and X. Cheng, Appl. Surf. Sci., 2022, 602, 154371 Search PubMed.
- C. Brissaud, L. V. Besteiro, J.-Y. Piquemal and M. Comesaña-Hermo, Sol. RRL, 2023, 7, 2300195 Search PubMed.
- S. Ghosh, D. Rashmi, S. Bera and R. N. Basu, Int. J. Hydrogen Energy, 2019, 44, 13262–13272 Search PubMed.
- T. Wang, H.-J. Wang, J.-S. Lin, J.-L. Yang, F.-L. Zhang, X.-M. Lin, Y.-J. Zhang, S. Jin and J.-F. Li, Chin. J. Struct. Chem., 2023, 42, 100066 Search PubMed.
- J. Gangareddy, P. Rudra, M. Chirumamilla, S. Ganisetti, S. Kasimuthumaniyan, S. Sahoo, K. Jayanthi, J. Rathod, V. R. Soma, S. Das, N. N. Gosvami, N. M. A. Krishnan, K. Pedersen, S. Mondal, S. Ghosh and A. R. Allu, Small, 2024, 20, 2303688 CrossRef CAS PubMed.
- S. Ghosh, A. K. Mallik and R. N. Basu, Solar Energy, 2018, 159, 548–560 CAS.
- P. Christopher and M. Moskovits, Annu. Rev. Phys. Chem., 2017, 68, 379–398 Search PubMed.
- M. Kim, M. Lin, J. Son, H. Xu and J.-M. Nam, Adv. Opt. Mater., 2017, 5, 1700004 Search PubMed.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 Search PubMed.
- G. Liu, Y. Lou, Y. Zhao and C. Burda, Acc. Chem. Res., 2022, 55, 1845–1856 CrossRef CAS PubMed.
- T. P. Rossi, P. Erhart and M. Kuisma, ACS Nano, 2020, 14, 9963–9971 Search PubMed.
- L. Zhou, Q. Huang and Y. Xia, Chem. Rev., 2024, 124, 8597–8619 Search PubMed.
- M. Gutiérrez, Z. Lian, B. Cohen, M. Sakamoto and A. Douhal, Nanoscale, 2023, 15, 657–666 Search PubMed.
- M. Kim, M. Lin, J. Son, H. Xu and J.-M. Nam, Adv. Opt. Mater., 2017, 5, 1700004 CrossRef.
- J. B. Khurgin, Nanophotonics, 2020, 9, 453–471 CrossRef CAS.
- E. Pensa, J. Gargiulo, A. Lauri, S. Schlücker, E. Cortés and S. A. Maier, Nano Lett., 2019, 19, 1867–1874 CrossRef CAS.
- E. Cortés, Adv. Opt. Mater., 2017, 5, 1700191 CrossRef.
- K. Wu, J. Chen, J. R. McBride and T. Lian, Science, 2015, 349, 632–635 CrossRef CAS PubMed.
- Z. Yang, N. Ghorai, S. Wu, S. He and T. Lian, ACS Nano, 2025, 19, 1547–1556 CAS.
- L.-Y. Hsu, W. Ding and G. C. Schatz, J. Phys. Chem. Lett., 2017, 8, 2357–2367 Search PubMed.
- J. Li, S. K. Cushing, F. Meng, T. R. Senty, A. D. Bristow and N. Wu, Nat. Photonics, 2015, 9, 601–607 CrossRef CAS.
- A. Diwan, P. Yadav, A. S. Shekhawat, C. Akila, M. Dhatchayani, R. Sharma, A. M. Shrivastav, R. Kumar, T. Srivastava and S. K. Saxena, J. Phys. Chem. Lett., 2024, 15, 5171–5176 CAS.
- C. Jia, X. Li, N. Xin, Y. Gong, J. Guan, L. Meng, S. Meng and X. Guo, Adv. Energy Mater., 2016, 6, 1600431 Search PubMed.
- R. T. Hannagan, G. Giannakakis, R. Réocreux, J. Schumann, J. Finzel, Y. Wang, A. Michaelides, P. Deshlahra, P. Christopher, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Science, 2021, 372, 1444–1447 CAS.
- C. S. Vennapoosa, S. Varangane, B. M. Abraham, V. Bhasin, S. Bhattacharyya, X. Wang, U. Pal and D. Chatterjee, J. Phys. Chem. Lett., 2023, 14, 11400–11411 CAS.
- Y. Lu, Z. Zhang, H. Wang and Y. Wang, Appl. Catal., B, 2021, 292, 120162 CrossRef CAS.
- X. Wu, H. Zhang, S. Zuo, J. Dong, Y. Li, J. Zhang and Y. Han, Nano-Micro Lett., 2021, 13, 136 CrossRef CAS.
- S. Bera, S. Ghosh, S. Shyamal, C. Bhattacharya and R. N. Basu, Sol. Energy Mater. Sol. Cells, 2019, 194, 195–206 CrossRef CAS.
- P. Liu, A. Dörfler, A. A. Tabrizi, L. Skokan, D. Rawach, P. Wang, Z. Peng, J. Zhang, A. P. Ruediger and J. P. Claverie, ACS Appl. Mater. Interfaces, 2023, 15, 27832–27844 CrossRef CAS.
- H. Jia, F. Li, T. H. Chow, X. Liu, H. Zhang, Y. Lu, J. Wang and C. Zhang, Nano Lett., 2022, 22, 7268–7274 CrossRef CAS PubMed.
- X. Zhang, D. Gao, B. Zhu, B. Cheng, J. Yu and H. Yu, Nat. Commun., 2024, 15, 3212 CAS.
- M. Qian, X.-L. Wu, M. Lu, L. Huang, W. Li, H. Lin, J. Chen, S. Wang and X. Duan, Adv. Funct. Mater., 2023, 33, 2370072 CrossRef.
- S. Tang, Y. Xia, J. Fan, B. Cheng, J. Yu and W. Ho, Chin. J. Catal., 2021, 42, 743–752 CAS.
- M. Shabil Sha, H. Anwar, F. N. Musthafa, H. Al-Lohedan, S. Alfarwati, J. R. Rajabathar, J. Khalid Alahmad, J.-J. Cabibihan, M. Karnan and K. Kumar Sadasivuni, Sci. Rep., 2024, 14, 3608 CrossRef CAS.
- S. P. Kulkarni, Y. M. Chitare, V. V. Magdum, P. D. Sawant, S. V. Talekar, S. A. Pawar, D. B. Malavekar, S. Ansar, J. H. Kim and J. L. Gunjakar, ACS Appl. Nano Mater., 2024, 7, 11411–11422 CAS.
- Y. Jiang, Z. Wang, W. Yang, P. Yang, X. Feng, P. Qin and F. Huang, Nano Lett., 2024, 24, 12597–12604 CAS.
- E. E. Ghadim, M. Walker and R. I. Walton, Dalton Trans., 2023, 52, 11143–11157 RSC.
- V. Van Pham, T. K. Truong, L. V. Hai, H. P. P. La, H. T. Nguyen, V. Q. Lam, H. D. Tong, T. Q. Nguyen, A. Sabbah, K.-H. Chen, S.-J. You and T. M. Cao, ACS Appl. Nano Mater., 2022, 5, 4506–4514 CrossRef.
- S. Ghosh, H. Remita and R. N. Basu, Appl. Catal., B, 2018, 239, 362–372 CAS.
- F. Guo, X. Huang, Z. Chen, H. Sun and L. Chen, Chem. Eng. J., 2020, 395, 125118 CrossRef CAS.
- A. R. Razafintsalama, R. P. Mishra, M. K. Sahoo, M. Mrinalini, B. Sahoo, P. Ravelonandro and Y. S. Chaudhary, Langmuir, 2023, 39, 12725–12739 CrossRef CAS PubMed.
- T. Kashyap, S. Biswasi, A. R. Pal and B. Choudhury, ACS Sustainable Chem. Eng., 2019, 7, 19295–19302 CrossRef CAS.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- H. Zhao, L. Jian, M. Gong, M. Jing, H. Li, Q. Mao, T. Lu, Y. Guo, R. Ji, W. Chi, Y. Dong and Y. Zhu, Small Struct., 2022, 3, 2100229 CrossRef CAS.
- J. Zhang, T. Bai, H. Huang, M.-H. Yu, X. Fan, Z. Chang and X.-H. Bu, Adv. Mater., 2020, 32, 2004747 CrossRef CAS.
- J. Liu, W. Chen, Q. Sun, Y. Zhang, X. Li, J. Wang, C. Wang, Y. Yu, L. Wang and X. Yu, ACS Appl. Energy Mater., 2021, 4, 2864–2872 CrossRef CAS.
- R. Boppella, W. Yang, J. Tan, H.-C. Kwon, J. Park and J. Moon, Appl. Catal., B, 2019, 242, 422–430 CAS.
- E. Hussain, A. Ishaq, M. Z. Abid, M. Z. Waleed, A. Rauf, R. Jin and K. Rafiq, ACS Appl. Energy Mater., 2024, 7, 1914–1926 Search PubMed.
- Y. Zhao, Y. Lu, L. Chen, X. Wei, J. Zhu and Y. Zheng, ACS Appl. Mater. Interfaces, 2020, 12, 46073–46083 Search PubMed.
- C. Wang, R. L. Thompson, J. Baltrus and C. Matranga, J. Phys. Chem. Lett., 2010, 1, 48–53 Search PubMed.
- L. Liu, D. T. Pitts, H. Zhao, C. Zhao and Y. Li, Appl. Catal., A, 2013, 467, 474–482 Search PubMed.
- S. Krejčíková, L. Matějová, K. Kočí, L. Obalová, Z. Matěj, L. Čapek and O. Šolcová, Appl. Catal., B, 2012, 111–112, 119–125 CrossRef.
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863–20868 CAS.
- T. Takayama, K. Tanabe, K. Saito, A. Iwase and A. Kudo, Phys. Chem. Chem. Phys., 2014, 16, 24417–24422 Search PubMed.
- T. Yui, A. Kan, C. Saitoh, K. Koike, T. Ibusuki and O. Ishitani, ACS Appl. Mater. Interfaces, 2011, 3, 2594–2600 Search PubMed.
- M. Yamamoto, T. Yoshida, N. Yamamoto, T. Nomoto, Y. Yamamoto, S. Yagi and H. Yoshida, J. Mater. Chem. A, 2015, 3, 16810–16816 Search PubMed.
- J. Di, C. Chen, C. Zhu, P. Song, M. Duan, J. Xiong, R. Long, M. Xu, L. Kang, S. Guo, S. Chen, H. Chen, Z. Chi, Y.-X. Weng, H. Li, L. Song, M. Wu, Q. Yan, S. Li and Z. Liu, Nano Energy, 2021, 79, 105429 CAS.
- Q. Lang, Y. Yang, Y. Zhu, W. Hu, W. Jiang, S. Zhong, P. Gong, B. Teng, L. Zhao and S. Bai, J. Mater. Chem. A, 2017, 5, 6686–6694 Search PubMed.
- S. Cai, J. Chen, Q. Li and H. Jia, ACS Appl. Mater. Interfaces, 2021, 13, 14221–14229 CrossRef CAS PubMed.
- J. Ji, R. Li, H. Zhang, Y. Duan, Q. Liu, H. Wang and Z. Shen, Appl. Catal., B, 2023, 321, 122020 CAS.
- H. Shi, H. Wang, Y. Zhou, J. Li, P. Zhai, X. Li, G. G. Gurzadyan, J. Hou, H. Yang and X. Guo, Angew. Chem., Int. Ed., 2022, 61, e202208904 Search PubMed.
- L. Zhang, T. Liu, T. Liu, S. Hussain, Q. Li and J. Yang, Chem. Eng. J., 2023, 463, 142358 Search PubMed.
- T. Kandemir, M. E. Schuster, A. Senyshyn, M. Behrens and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 12723–12726 Search PubMed.
- Q. Zhang, Q. Li, H. Li, X. Shi, Y. Zhou, Q. Ye, R. Yang, D. Li and D. Jiang, Inorg. Chem., 2023, 62, 12138–12147 Search PubMed.
- Z.-K. Shen, Y.-J. Yuan, P. Wang, W. Bai, L. Pei, S. Wu, Z.-T. Yu and Z. Zou, ACS Appl. Mater. Interfaces, 2020, 12, 17343–17352 Search PubMed.
- C. Wang, J. Wang and X. Yu, Colloids Surf., A, 2023, 662, 131055 Search PubMed.
- S. Odabasi Lee, H. S. Moon, I. Hong, S. K. Lakhera and K. Yong, Appl. Surf. Sci., 2024, 665, 160298 Search PubMed.
- M. Wang, P. Wang, X. Wang, F. Chen and H. Yu, J. Mater. Sci. Technol., 2024, 174, 168–175 Search PubMed.
- W. Zhu, L. Yang, F. Liu, Z. Si, M. Huo, Z. Li and Z. Chen, J. Alloys Compd., 2024, 973, 172747 Search PubMed.
- D. Gao, W. Zhong, X. Zhang, P. Wang and H. Yu, Small, 2024, 20, 2309123 Search PubMed.
- Q. Lin, J. Zhao, P. Zhang, S. Wang, Y. Wang, Z. Zhang, N. Wen, Z. Ding, R. Yuan, X. Wang and J. Long, Carbon Energy, 2024, 6, e435 Search PubMed.
- A. Yu. Kurenkova, A. D. Radina, V. S. Baidyshev, P. V. Povalyaev, E. E. Aidakov, E. Yu. Gerasimov, D. D. Mishchenko, A. V. Zhurenok, A. Ya. Pak, E. A. Kozlova and A. G. Kvashnin, Appl. Surf. Sci., 2024, 661, 160095 CrossRef.
- R. Boppella, C. H. Choi, J. Moon and D. Ha Kim, Appl. Catal., B, 2018, 239, 178–186 CrossRef CAS.
- X. Ning, J. Li, B. Yang, W. Zhen, Z. Li, B. Tian and G. Lu, Appl. Catal., B, 2017, 212, 129–139 CrossRef CAS.
- D. Zhang, C. Zhang, K. Chai, Y. Li and Z. Lv, Sep. Purif. Technol., 2024, 337, 126408 CrossRef CAS.
- F. Dai, M. Zhang, J. Han, Z. Li, S. Feng, J. Xing and L. Wang, Nano Res., 2024, 17, 1259–1266 CrossRef CAS.
- X. Gao, L. An, D. Qu, W. Jiang, Y. Chai, S. Sun, X. Liu and Z. Sun, Sci. Bull., 2019, 64, 918–925 CrossRef CAS.
- X. Zhang, J. Yan, F. Zheng, J. Zhao and L. Y. S. Lee, Appl. Catal., B, 2021, 286, 119879 CrossRef CAS.
- Y. Wang, Q. Lai, F. Zhang, X. Shen, M. Fan, Y. He and S. Ren, RSC Adv., 2014, 4, 44442–44451 CAS.
- J. Ran, G. Gao, F.-T. Li, T.-Y. Ma, A. Du and S.-Z. Qiao, Nat. Commun., 2017, 8, 13907 CrossRef CAS.
- S. Jayachitra, D. Mahendiran, P. Ravi, P. Murugan and M. Sathish, Appl. Catal., B, 2022, 307, 121159 CrossRef CAS.
- S. Sun, Y.-C. Zhang, G. Shen, Y. Wang, X. Liu, Z. Duan, L. Pan, X. Zhang and J.-J. Zou, Appl. Catal., B, 2019, 243, 253–261 CrossRef CAS.
- L. Mao, Q. Ba, X. Jia, S. Liu, H. Liu, J. Zhang, X. Li and W. Chen, RSC Adv., 2019, 9, 1260–1269 RSC.
- F. Xue, Y. Si, M. Wang, M. Liu and L. Guo, Nano Energy, 2019, 62, 823–831 CrossRef CAS.
- L. Lin, V. Polliotto, J. J. M. Vequizo, X. Tao, X. Liang, Y. Ma, T. Hisatomi, T. Takata and K. Domen, ChemPhotoChem, 2022, 6, e202200209 CrossRef CAS.
- Y. P. Xie, G. Liu, G. Q. Lu and H.-M. Cheng, Nanoscale, 2012, 4, 1267–1270 RSC.
- S. Li, G. Yang, P. Ge, H. Lin, Q. Wang, X. Ren, S. Luo, D. Philo, K. Chang and J. Ye, Small Methods, 2021, 5, 2001018 CrossRef CAS PubMed.
- R. Wang, J. Shen, K. Sun, H. Tang and Q. Liu, Appl. Surf. Sci., 2019, 493, 1142–1149 CrossRef CAS.
- T. Liu, I. Tranca, J. Yang, X. Zhou and C. Li, J. Mater. Chem. A, 2015, 3, 10309–10319 RSC.
- S. Wu, J. Zhang, W. Chen, P. Xing, X. Liu, B. Teng, L. Zhao and S. Bai, J. Mater. Chem. A, 2021, 9, 26036–26044 RSC.
| This journal is © The Royal Society of Chemistry 2025 |