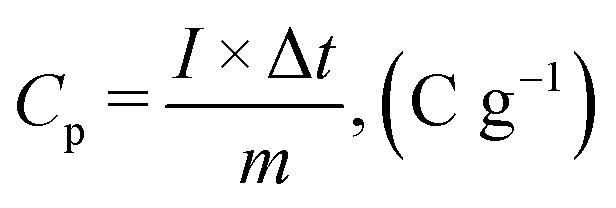Iron oxide@CoFe-LDH nanocomposites for highly stable aqueous hybrid supercapacitors†
Received
12th January 2025
, Accepted 13th March 2025
First published on 18th March 2025
Abstract
CoFe-LDH (layered double hydroxide) nanomaterials are widely explored as battery-type electrode materials owing to their excellent redox activity, layered structure, and fast ion diffusion. However, their practical application is often hindered by poor cyclic stability. The nanocomposite of CoFe-LDH with iron oxide has great potential to overcome this limitation. The layered structure of CoFe-LDH facilitates a fast ion diffusion and realizes synergistic activities of multiple metal elements, while iron oxide prevents the self-restacking and aggregation of CoFe-LDH layers, which ultimately enhance their structural stability and electrochemical performance. In this work, we prepared an Fe16O20/CoFe-LDH (FO@CoFe-LDH) nanocomposite via a single-step hydrothermal method. As composition tuning was a major concern to regulate the electrochemical performance, two samples with different compositions were prepared by tuning the mole ratios of Co and Fe. Electrochemical investigations of FO@CoFe-LDH1 (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Co:Fe) demonstrated a specific capacity of 84 C g−1 at 1 A g−1, while FO@CoFe-LDH2 (3:2 ratio of Co:Fe) was limited to 25 C g−1 at 1 A g−1 in a 6 M KOH electrolyte solution. Furthermore, an aqueous hybrid supercapacitor (AHS) fabricated using FO@CoFe-LDH1 as the positive electrode and activated carbon (AC) as the negative electrode exhibited remarkable cyclic stability, retaining 99.9% after 4000 cycles. This study demonstrates the potential of FO@CoFe-LDH1 nanocomposites as battery-type electrodes for AHS devices, paving the way for durable energy storage devices.
:1 ratio of Co:Fe) demonstrated a specific capacity of 84 C g−1 at 1 A g−1, while FO@CoFe-LDH2 (3:2 ratio of Co:Fe) was limited to 25 C g−1 at 1 A g−1 in a 6 M KOH electrolyte solution. Furthermore, an aqueous hybrid supercapacitor (AHS) fabricated using FO@CoFe-LDH1 as the positive electrode and activated carbon (AC) as the negative electrode exhibited remarkable cyclic stability, retaining 99.9% after 4000 cycles. This study demonstrates the potential of FO@CoFe-LDH1 nanocomposites as battery-type electrodes for AHS devices, paving the way for durable energy storage devices.
Introduction
The intensifying demand for portable electronic devices, electric vehicles, the internet of things (IoT), and advancements in industrialization 5.0 has led to a substantial increase in the global need for energy storage solutions.1–3 Electrochemical supercapacitors are a highly promising technology to fulfil the demand for rapid power delivery in various applications, including quick power devices, electronic gadgets, and electric vehicles.4–6 Unlike traditional batteries, supercapacitors excel in delivering quick bursts of energy and exhibit high power density, making them ideal for applications requiring rapid charge and discharge cycles.
Among the various types of electrochemical supercapacitors, hybrid supercapacitors are well suited to meet the demand for higher energy densities, which is crucial for applications requiring both rapid power delivery and extended energy storage.7,8 Hybrid supercapacitors combine the features of electric double-layer capacitors (EDLCs) and pseudocapacitors or battery types, leveraging the advantages of both to achieve superior performance. However, traditional EDLCs are known for their high-power density and low energy density. Additionally, hybrid supercapacitors incorporate additional materials or mechanisms that enhance the energy storage capabilities.9 By integrating elements, such as battery-type materials or utilizing different charge storage mechanisms, hybrid supercapacitors offer a significantly higher capacity. Although hybrid supercapacitors combine high energy density with fast charge–discharge capabilities, they have some drawbacks.10 They generally show low power density and less cycle stability compared with conventional EDLCs and pseudocapacitors.11,12 To date, various battery-type nanomaterials have been extensively studied for their potential to enhance the performance of hybrid supercapacitors, such as NiOH,13 Co3O4,14 V2O5,15 and layered double hydroxide (LDH).16 Among these, nickel hydroxide (NiOH) stands out owing to its high capacitance and good conductivity, which contribute to its excellent redox properties and stability. Cobalt oxide (Co3O4) is another notable material, offering high energy density and robust pseudocapacitive behaviour for high capacity and stability, which improve energy density. Similarly, vanadium oxide (V2O5) is renowned for its high energy density and strong electrochemical performance, making it a promising candidate for battery-type electrodes. These electrode materials offer several advantages over other materials; however, they suffer from low power density and poor cycle stability.
Therefore, strategically designing effective electrode materials is essential.17 Recently, there has been significant interest in battery-type electrode materials for hybrid supercapacitors. Layered double hydroxides (LDHs) stand out as a promising choice owing to their flexible chemical composition, layered structure, bimetallic characteristics, facile anion exchange capability, high theoretical specific capacitance, and excellent redox activity.18–20 LDHs consist of a Brucite-like host layer with tuneable oxidized metal components and interlayer water molecules or anions, represented by the formula [(M2+)1−x(M3+)x(OH)2]x + (An−)x/n·yH2O,7,21 where M2+ denotes divalent metal cations (e.g., Ni2+, Co2+, Cu2+, and Mg2+), M3+ represents trivalent metal cations (e.g., Fe3+, Al3+, V3+, and Cr3+), and x denotes the ratio of M3+ to (M2+ + M3+). ‘An−’ refers to exchangeable anions such as Cl−, NO3−, CO32−, SO42−, or [CH3COO]−. Despite these advantages, LDHs face several challenges, including intrinsic poor conductivity, limited lifespan, and relatively low rate capability. Among the various LDH materials, cobalt–iron layered double hydroxides (CoFe-LDHs) are highly favoured for supercapacitor applications owing to their exceptional electrochemical properties, such as higher redox reactions involving cobalt and iron ions and layered structure facilities ionic insertion/desertion. Nevertheless, they struggle with poor cycle stability owing to the lower conductivity, structural degradation and layer staking of CoFe-LDH. To overcome this problem, heterostructure LDH or composite materials with other metal oxides are the ultimate solution because of the improved electrical conductivity achieved through the integration of metal oxides. However, the addition of metal oxides to CoFe-LDH enhances electrical conductivity, preserves structural stability, and prevents the stacking of CoFe-LDH layers.22,23
Several efforts have been made to enhance the electrochemical performance of CoFe-LDH, Fang et al. prepared CoFe-LDH by applying the reflux method and achieved a specific capacitance of ∼2358 F g−1 at 0.5 A g−1 and 83% capacitance retention at 1400 cycle.24 Patil et al. developed CoFe-LDH nanosheets on Ni foam using a one-pot hydrothermal method and accomplished the specific areal capacity of 2263 mC cm2 at 5 mV s−1 and achieved 92.6% capacity retention after 5000 charge–discharge cycles.25 The Ni3S2/CoFe-LDH/NF was prepared by applying hydrothermal and electrodeposition methods that demonstrated the cycle stability of 93.4% after 5000 cycles.26 Liu et al. synthesized hierarchical MgCo2O4@CoFe by applying the hydrothermal method and achieved a specific capacitance of 2007 F g−1 at 1 A g−1 and achieved 80.2% cycle stability after 5000 cycles.27
In this work, we developed Fe16O20 (FO) functionalized CoFe-LDH (FO@CoFe-LDH) using a one-step hydrothermal method. This synthesis approach provided better structural, morphological and compositional control in designing materials than previous studies. This further results in the effective integration of iron oxide with CoFe-LDH, which considerably enhances the performance of the hybrid supercapacitor. FO@CoFe-LDH offers excellent redox behaviour and conductivity. Further, a substantial improvement in cycle stability is achieved, addressing one of the key challenges in hybrid supercapacitors. Therefore, the heterostructure of FO@CoFe-LDH offers excellent redox behaviour and conductivity that not only boosts energy density but also extends the operational lifespan of the hybrid supercapacitor, making it a promising electrode for advanced energy storage applications.
Experimental
Materials
Cobaltous nitrate hexahydrate AR (Co (NO3)2·6H2O), ferric chloride (FeCl3), urea (NH2CONH2), sodium hydroxide AR (NaOH), potassium hydroxide LR (KOH), and ethanol (C2H6O) were obtained from SD Fine-Chem Ltd., India. Whatman-42 filter paper was procured from Molychem, India. Activated carbon (AC) with a surface area of 2000 m2 g−1 and an ash content of 0.21% and carbon paper were purchased from Global Nanotech, India.
Preparation of CoFe-LDH
FO@CoFe-LDH1 was synthesized using a one-step hydrothermal method. To prepare FO@CoFe-LDH1 (3:1), 6 mmol of urea was dissolved in 50 ml of deionized (DI) water. Subsequently, 3 mmol of cobalt nitrate hexahydrate AR and 1 mmol of ferric chloride were added to the urea solution. The pH was adjusted to 8–9 by the drop-wise addition of 1 M NaOH and stirred for 30 minutes at room temperature. The prepared solution was placed into a Teflon-lined autoclave and subjected to heating at 180 °C for 12 hours (Fig. 1). After cooling naturally, the product was washed with ethanol and DI water until a neutral pH was obtained. Further, it was dried at 70 °C for 12 hours in an electric oven. Similarly, FO@CoFe-LDH2 samples were prepared with a mole ratio of 3:2 (Co–Fe). The major advantage of selecting the hydrothermal method.
 |
| | Fig. 1 Schematic representation of preparation of FO@CoFe-LDH1 and FO@CoFe-LDH2. | |
Material characterization
The crystal structure properties of FO@CoFe-LDH1 and FO@CoFe-LDH2 were examined by XRD using CuKα radiation with a Rigaku MiniFlex, Japan in the 2θ range from 10° to 70°. Morphological investigations were performed using field emission scanning electron microscopy (FE-SEM) and elemental analysis with FE-SEM-EDS (energy dispersive spectroscopy) equipped with S-4800, Hitachi, Ibaraki, Japan, high-resolution transmission electron microscopy (HR-TEM) instrument with selected area electron diffraction (SAED) using a TECNAI G2-20 TEM-300 kV instrument. Elemental analysis was performed via XPS using a Versaprobe II, ULVAC-PHI Inc., Chigasaki, Kanagawa, Japan. The functional groups were examined using FT-IR (Bruker make IR apha2, Germany) in the range of 500–4000 cm−1, and Raman spectroscopy (Xplora, Horiba, Japan with a 532 nm) was measured in the range of 100 cm−1–800 cm−1. Additionally, the optical band gap estimation was carried out through UV-visible spectroscopy (Thermo Scientific spectrometer, Multiscan SkyHigh) in the range of 200–800 nm. Furthermore, the specific surface area and porosity of both samples were determined using a Quantachrome Instruments system, version 5.21.
Working electrode preparation
The active nanomaterials of FO@CoFe-LDH1 and FO@CoFe-LDH2 were applied onto working electrodes using a simple drop-casting technique for a three-electrode measurement setup. Initially, 5 mg of each active material was dispersed in 2 mL of a solution comprising equal parts of ethanol and water, along with 10 μL of Nafion binder. The mixture was then sonicated to ensure uniform dispersion. Subsequently, 10 μL of this prepared solution was drop cast onto a glassy carbon (GC) electrode and dried for 30 minutes under an IR lamp (200 W).
Electrochemical measurements
The electrochemical properties of FO@CoFe-LDH1 and FO@CoFe-LDH2 were investigated using a three-electrode setup with a PGSTAT302N Autolab system. The reference, working, and counter electrodes were silver/silver chloride (Ag/AgCl3), glassy carbon, and platinum wire, respectively. All measurements were conducted in a 6 M KOH electrolyte solution. The electrochemical charge kinetics were analysed using frequency response analysis (FRA) at frequencies ranging from 0.01 Hz to 100 kHz.
Charge balancing for device fabrication
For the fabrication of an asymmetric supercapacitor, charge balancing can be done by utilizing the following equations:| |  | (1) |
where Cs+ and Cs− represent the specific capacitance values and ΔV+ and ΔV− represent potential windows of the positive and negative electrodes, respectively. However, the specific capacitance, energy density, and power density were determined using the following equations:
For specific capacity,
| |  | (2) |
where
Cp represent the specific capacity,
I is the current (A), m denotes mass loading on electrode, and Δ
t indicates discharge time (s);
for specific energy density,
| |  | (3) |
where
Eg – specific energy densities,
Cs – specific capacitance (F g
−1), and
V – potential window (V);
for specific power density,
| |  | (4) |
where
Eg is the energy density (W h kg
−1) and Δ
t is the charge time.
Hybrid aqueous supercapacitor (HAS) fabrication
The positive electrode for the hybrid aqueous supercapacitor (HAS) was prepared as follows: 5 mg of FO@CoFe-LDH1 and 5 μL of Nafion binder were added to 0.5 mL of DI water and sonicated for 5 minutes to form a homogeneous solution. From the prepared solution, 0.3 mL was drop-cast onto carbon paper and left to dry under an infrared (IR) lamp for 10 minutes. Similarly, the negative electrode was prepared by mixing 5 mg of activated carbon (AC) with 0.5 mL of deionised water and 5 μL of Nafion binder. After sonication for 5 minutes, 0.5 mL of the prepared solution was drop-cast onto carbon paper and dried under an IR lamp for 10 minutes. Both electrodes were then sandwiched with Whatman-42 filter paper as a separator and assembled in a Swagelok cell containing a 6 M KOH aqueous electrolyte.
Results and discussion
Morphological analysis
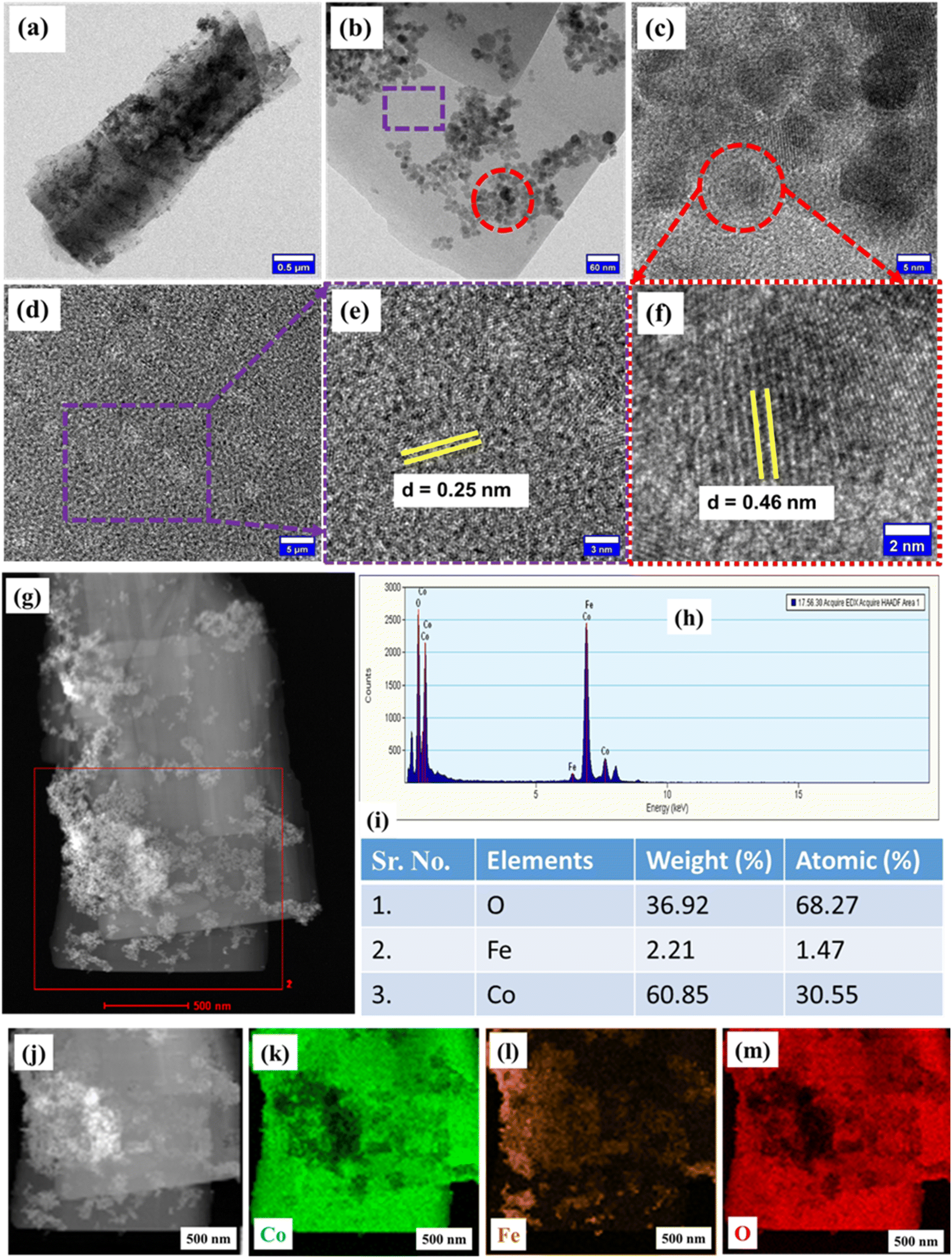
Morphological analysis conducted using FE-SEM at various scales is depicted in Fig. 2(a–f). Fig. 2(a–d) shows the FE-SEM images of FO@CoFe-LDH1 at 5 μm, 3 μm, 2 and 1 μm, emphasizing a flake-like morphology of FO@CoFe-LDH1. However, Fig. 2(e and f) show iron oxide nanoparticles spread on the surface of these CoFe-LDH flakes. Fig. 2(g) shows the FE-SEM image of FO@CoFe-LDH1 at a 3 μm scale, and Fig. 2(h) shows the EDS spectra of FO@CoFe-LDH1, confirming the presence of carbon (C), oxygen (O), cobalt (Co), and iron (Fe). Furthermore, the details morphological investigation was performed with TEM imaging and HR-TEM imaging of FO@CoFe-LDH1 samples, as depicted in Fig. 3. Fig. 3(a) displays the TEM image of FO@CoFe-LDH1 at a scale of 0.5 μm, revealing a flake-like morphology with black spots that indicate the presence of Fe16O20 nanoparticles on the surface of flakes. Fig. 3(b) shows a TEM image at 60 nm, displaying the Fe16O20 nanoparticles distribution across the CoFe-LDH flakes. Fig. 3(c) presents an HR-TEM image of a selected region (red circle) from Fig. 3(b), highlighting the Fe16O20 nanoparticles. Fig. 3(d) displays an HR-TEM image from a region (violet box) in Fig. 3(b), while Fig. 3(e) illustrates a closer view from Fig. 3(d). The HR-TEM image in Fig. 3(e) reveals a d-spacing of 0.25 nm corresponding to the (012) plane of the CoFe-LDH flakes. Moreover, Fig. 3(f) shows an HR-TEM image of the region marked by the red dotted circle in Fig. 3(c), confirming a d-spacing of 0.46 nm attributed to the (006) planes of Fe16O20 nanoparticles. Additionally, Fig. S1(a and b)† display a bar graph of the length and width distribution of the flakes, respectively, highlighting the average length of 3.61 μm and the width of 1.39 μm. Fig. S1(c)† illustrates the bar graph of the area distribution for Fe16O20 nanoparticles, revealing an average area of 46.10 nm2. Meanwhile, Fig. S1(d)† shows a bar graph of the diameter distribution of Fe16O20 nanoparticles, with an average diameter of 6.77 nm. Additionally, the elemental analysis of FO@CoFe-LDH1 was studied at 500 nm with TEM-EDS spectra, as portrayed in Fig. 3(g). Fig. 3(h) shows the EDS spectra of FO@CoFe-LDH1, confirming the presence of oxygen (O), cobalt (Co), and iron (Fe) along with estimated element weight (%) and atomic (%), as shown in table (Fig. 3i). Individual elemental mapping was carried out for detailed visualization, as shown in Fig. 3(j–m). Fig. 3(j) presents the HRTEM image of FO@CoFe-LDH1 at 500 nm, while Fig. 3(k–m) depicts the individual elemental mappings of Co, Fe, and O, respectively, confirming the uniform distribution of these elements.
 |
| | Fig. 2 FE-SEM images of FO@CoFe-LDH1 at 5 μm (a), 3 μm (b), 2 μm (c) 1 μm (d), 0.3 μm (e), 200 nm (f), (g) SEM image of FO@CoFe-LDH1 at 3 μm and (h) EDS spectra of FE-SEM image of Fig. 2(g). | |
 |
| | Fig. 3 TEM images of FO@CoFe-LDH1 at 0.5 μm (a) and 60 nm (b), (c) HR-TEM image of the selected red circle of Fig. 3b, (d) HR-TEM image of the selected region (violet dash rectangle) of Fig. 3b, (e) magnified HR-TEM image at 3 nm, (f) HR-TEM image of the selected region of Fig. 3c, (g) TEM image of FO@CoFe-LDH1 at 500 nm, (h) EDS spectra of FO@CoFe-LDH1, (i) EDS elemental composition table, (j) TEM image of FO@CoFe-LDH1 at 500 nm, and (k–m) individual elemental mapping of Co, Fe, and O, respectively. | |
Crystal structural analysis
The crystal structures of FO@CoFe-LDH1 and FO@CoFe-LDH2 were investigated using XRD, as displayed in Fig. 4(a). The XRD spectra of FO@CoFe-LDH1 and FO@CoFe-LDH2 match with standard JCPDS file number 50-235 and correspond to the Brucite phase of Co (OH)2 and Fe (OH)2 in CoFe-LDH, respectively. Additionally, some extra peaks align with the standard JCPDS file number 96-901-4244, indicating the presence of an orthorhombic crystal structure of Fe16O20. No additional peaks were observed, but a decrease in peak intensity of FO@CoFe-LDH2 compared to FO@CoFe-LDH1 was noted, which was attributed to reduced crystallinity via structural disorder.
 |
| | Fig. 4 Combine (a) XRD spectra and (b) Raman spectra of FO@CoFe-LDH1 and FO@CoFe-LDH2. | |
Chemical property analysis
Raman spectroscopy was utilized to determine the vibrational modes of FO@CoFe-LDH1 and FO@CoFe-LDH2, as depicted in Fig. 4(b). Fig. 4(b) shows the Raman spectra of FO@CoFe-LDH1 (brown spectra) and FO@CoFe-LDH2 (violet spectra). The Raman shift peak at 115.21 cm−1 is assigned to the lattice vibration mode of the brucite-like layers in CoFe-LDH. The peaks at 186 cm−1 and 253 cm−1 correspond to the symmetric vibration of metal hydroxide (M–OH) in Co–(OH)2 and Fe–(OH)2, respectively.28 However, the peak at 465 cm−1 is attributed to the stretching vibration OH–O of CoFe-LDH.29 The peak at 508 cm−1 is connected with metal oxygen (M–O) symmetric stretching vibration.30,31 Furthermore, the Raman shift peak at 598 cm−1 is associated with the Fe–O linkage bond of Fe16O20. Hence, the peak positions in both samples are identical except for peak intensities, signifying the minor change in chemical bonding and stretching vibrations.
Furthermore, the inorganic functional groups of FO@CoFe-LDH1 and FO@CoFe-LDH2 were studied via FT-IR spectroscopy, as represented in Fig. S2(a).† The peak at 564.21 cm−1 corresponds to the metal-hydroxide bond (M–OH) within the layered double hydroxide structure, while the peaks at 661.21 cm−1 and 806.13 cm−1 are associated with metal–oxygen (M–O) bonds.7,32 A distinct peak at 978.53 cm−1 is attributed to the deformation vibrations of water molecules (H2O) or hydroxyl groups (OH−).33,34 Additionally, a peak at 1322.12 cm−1 associated with carbonate anions (CO32−) present in two brucite-like hydroxide layers and the deformation vibrations of water (H–O–H) are observed at 1546.72 cm−1.32,33 Moreover, peaks at 3374.62 cm−1 and 3505.46 cm−1 correspond to the O–H stretching vibrations of water molecules in the interlayered structure of CoFe-LDH.35 The optical band gap of FO@CoFe-LDH1 and FO@CoFe-LDH2 was calculated by Tauc plot using the following equation:
where ‘
α’ is the absorption coefficient, ‘
hν’ is the incident photon energy, ‘
K’ is the energy independent constant, and ‘
Eg’ is the optical bandgap of materials. In this equation, the exponent ‘
n’ represents the nature of the transition. For the direct bandgap,
n = 2, and for the indirect bandgap,
n = 1/2. Furthermore, FO@CoFe-LDH1 and FO@CoFe-LDH2 show a direct bandgap. Fig. S3(a) and (b)
† show the absorbance spectra of FO@CoFe-LDH1 and FO@CoFe-LDH2, respectively. For FO@CoFe-LDH1, the band gap was found to be 1.94 eV (Fig. S3(b)
†), while it increased to 2.0 eV (Fig. S3(c)
†) for FO@CoFe-LDH2, which has a higher iron mole ratio concentration. The larger band gap of FO@CoFe-LDH2 can be attributed to quantum confinement effects, enhanced interactions between Fe
16O
20 and CoFe-LDH, and compositional changes that influence the material's electronic and structural properties. Therefore, FO@CoFe-LDH1, with its lower band gap, is more suitable for supercapacitor applications.
Surface area analysis
The specific surface areas of FO@CoFe-LDH1 and FO@CoFe-LDH2 were determined using BET analysis, as illustrated in Fig. 5. Fig. 5(a) and (b) show the nitrogen adsorption–desorption isotherms of FO@CoFe-LDH1 and FO@CoFe-LDH2, respectively, both exhibiting H3 hysteresis, indicating a mesoporous nature with slit-like or plate-like pores. The specific surface areas of FO@CoFe-LDH1 and FO@CoFe-LDH2 are 52 m2 g−1 and 45 m2 g−1, respectively. These mesoporous features arise from the LDH structure of CoFe-LDH, which is integrated with Fe16O20 nanoparticles. Fig. 5(c and d) display the pore diameter vs. pore volume plot of FO@CoFe-LDH1 and FO@CoFe-LDH2, respectively. The average pore diameters of FO@CoFe-LDH1 and FO@CoFe-LDH2 are 8.7 nm and 8.6 nm, respectively, while their average pore volumes are 3.03 × 10−2 cm3 g−1 and 2.60 × 10−2 cm3 g−1, respectively. Furthermore, Fig. 5(e and f) present the pore diameter versus pore area plots, revealing average pore surface areas of 21 m2 g−1 and 18 m2 g−1 for FO@CoFe-LDH1 and FO@CoFe-LDH2, respectively. Therefore, the reduction in specific surface area, pore volume and pore surface area is observed in FO@CoFe-LDH2 because of the incorporation of excess iron oxide nanoparticles blocking the pores, leading to a reduction in the porosity. Consequently, the changes in particle size and structure reduce the available surface area for adsorption or reaction while decreasing the pore sizes, leading to a decline in both pore volume and specific surface area in the FO@CoFe-LDH2.
 |
| | Fig. 5 (a and b) Nitrogen adsorption–desorption isotherm, (c and d) pore diameter vs. pore volume, and (e and f) pore diameter vs. pore surface area of FO@CoFe-LDH1 and FO@CoFe-LDH2, respectively. | |
Elemental analysis
The elemental analysis of FO@CoFe-LDH1 was studied with XPS spectroscopy, as depicted in Fig. S4† and 6. Fig. S3† shows the XPS survey spectra of FO@CoFe-LDH1 validating the presence of Co, Fe, C and O. Fig. 6(a) shows the deconvoluted XPS spectra of Co 2p; peak at binding energy 782.55 eV and 796.59 eV belongs to Co 2p3/2 and Co 2p1/2, respectively. Peaks at binding energies at 780.95 eV and 796.59 eV are associated with Co2+.28 However, peaks at binding energies of 786.20 eV, 780.45 eV and 802.58 eV are ascribed to satellite peaks of Co2+. Fig. 6(b) shows the deconvoluted XPS spectra of Fe 2p, and peaks at binding energies of 712.74 eV and 724.64 eV belong to Fe 2p3/2 and Fe 2p1/2 of Fe3+, respectively.28,36 Furthermore, peaks at binding energies of 710.12 eV and 717.64 eV are assigned to the satellite peak of Fe3+.37Fig. 6(c) shows the deconvoluted XPS spectra of O 1s of FO@CoFe-LDH1. The peak at a binding energy of 529.48 is assigned to the presence of metal oxygen (M–O) and the peak at binding energies of 530.63 eV and 531.1 eV corresponds to hydroxide groups (OH−) or adsorbed water molecules on the surface.7,28 For CoFe-LDH, these peaks indicate the presence of hydroxyl groups that is part of the layered structure or water molecules adsorbed onto the surface or within the interlayers of the hydroxide. Moreover, the peak at 532.16 eV is attributed to the metal hydroxide (M–OH) groups in CoFe-LDH, reflecting the chemical state of oxygen in these hydroxide environments within the layered structure.16Fig. 6(d) shows the deconvoluted XPS spectra of C 1s of FO@CoFe-LDH1, and the peak at 284.58 eV corresponds to carbon atoms in C–C often originating from hydrocarbons. However, peaks at 285.92 eV and 289.18 eV are associated with the C–O–C and O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O of CoFe-LDH, respectively.7,28
C–O of CoFe-LDH, respectively.7,28
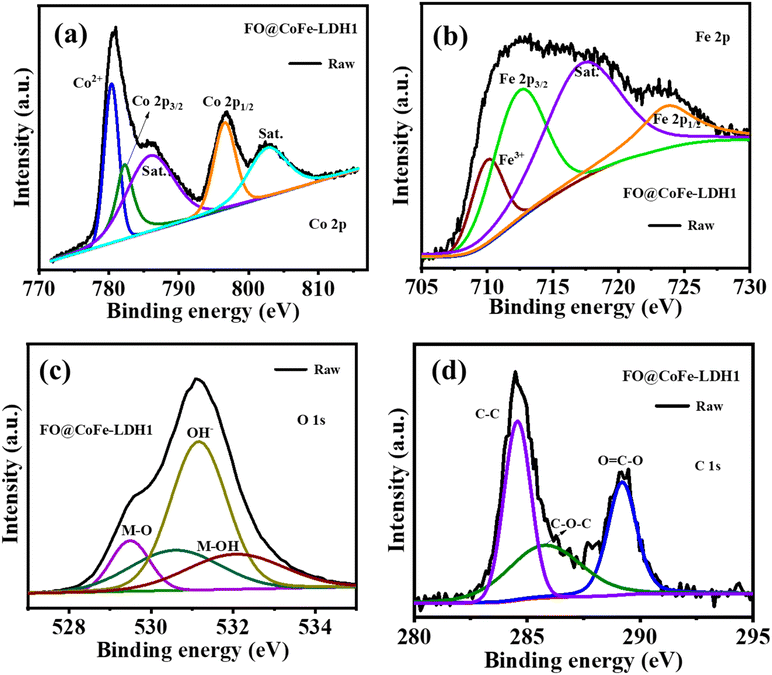 |
| | Fig. 6 Deconvoluted XPS spectra for FO@CoFe-LDH1 of Co 2p (a), Fe 2p (b), O 1s (c) and C 1s (d), respectively. | |
Electrochemical investigations
The electrochemical performance of FO@CoFe-LDH1 and FO@CoFe-LDH2 was investigated using a three-electrode system with a 6 M KOH aqueous electrolyte. Fig. 7(a) and (b) show the CV curves of FO@CoFe-LDH1 and FO@CoFe-LDH2 with respect to different voltages at 30 mV s−1, respectively. This confirms that the maximum working potential (0.8 V) ranges from −0.2 V to 0.6 V. Further, this was confirmed by performing the GCD curve at different voltages on 1 A g−1, as depicted in Fig. 7(c and d). Moreover, Fig. 7(e) displays the combined CV curves of FO@CoFe-LDH1 and FO@CoFe-LDH2 at 5 mV s−1 and reveals the better performance of FO@CoFe-LDH1 (blue colour curve) as the larger area under CV curves. Nevertheless, FO@CoFe-LDH1 shows a couple of oxidation peaks at 0.25 V (a peak 1) and 0.58 V (a peak 2), while two reduction peaks at 0.17 V (c peak 1) and 0.54 V (c peak 2) are due to the redox reactions of Co and Fe species in the layered double hydroxide (LDH) structure as per eqn (6).38,39 Additionally, FO@CoFe-LDH2 shows a decrease in the peak current of the oxidation and reduction of 0.25 V and 0.17 V, respectively. The peak current decreases owing to an excessive amount of Fe, which is attributed to the saturation of active sites, reducing the efficiency of electrochemical reactions. In addition, at a higher mole ratio of Fe concentrations, more defects or impurities are introduced into the material, which can act as recombination centres for charge carriers, thereby decreasing the efficiency of charge storage and transfer. Fig. S5(a) and (b)† show the CV curves of FO@CoFe-LDH1 and FO@CoFe-LDH2 at various scan rates. At lower scan rates, the CV curve exhibits battery-like behaviour, while at higher scan rates, it transitions to a semi-rectangular shape, indicating an increased pseudocapacitive contribution due to rapid surface redox reactions.
 |
| | Fig. 7 (a–d) CV and GCD curves at different voltages of FO@CoFe-LDH1 and FO@CoFe-LDH2, (e and f) combined CV and GCD curves of FO@CoFe-LDH1 and FO@CoFe-LDH2, (g) Combined specific capacity at various scan rates of FO@CoFe-LDH1 and FO@CoFe-LDH2, and (h) combined Nyquist plots of FO@CoFe-LDH1 and FO@CoFe-LDH2; inset is equivalence circuit diagram, (i) Nyquist plot of selected high frequency region (black doted circle) of Fig. 7h. | |
| | |
Co(OH)2 + OH− ↔ CoOOH + H2O + e−
| (6) |
Fig. 7(f) illustrates the combined GCD curves of FO@CoFe-LDH1 and FO@CoFe-LDH2 at 0.5 A g
−1. Both prepared materials display the charge/discharge plateaus between the potential windows of 0.1 V and 0.6 V in the GCD curve, confirming the battery type materials.
40 However, FO@CoFe-LDH1 shows a higher chare-discharge time compared to FO@CoFe-LDH2. The decrease in the GCD time with increasing Fe concentration is evident in the CV data. Additionally, Fig. S5(c and d)
† shows the GCD curves of FO@CoFe-LDH1 and FO@CoFe-LDH2 at different current densities. Compared to both GCD curves, the charge/discharge plateaus diminish at higher current densities. At lower current densities (0.5 A g
−1 and 1 A g
−1), the system has sufficient time to achieve equilibrium, allowing for well-defined plateaus that correspond to redox reactions. However, at higher current densities (from 2 A g
−1 to 5 A g
−1), the electrochemical reactions are forced to occur more rapidly, leading to kinetic constraints and increased EDLC contribution.
41,42
Furthermore, the specific capacity of FO@CoFe-LDH1 and FO@CoFe-LDH2 was calculated from the discharge curve according to eqn (2). Fig. 7(g) shows the specific capacity at various current densities, and FO@CoFe-LDH1 achieves a specific capacity of 84 C g−1 at 1 A g−1, while FO@CoFe-LDH2 is limited to 25 C g−1 at 1 A g−1.
Charge storage kinematics
The electrochemical charge kinetics were examined using frequency response analysis (FRA) at frequencies ranging from 0.01 Hz to 100 kHz in a 6 M KOH, as shown in Fig. 7(h and i). Fig. 7(h) shows the combined Nyquist plots of FO@CoFe-LDH1 and FO@CoFe-LDH2, and the inset shows an equivalent circuit diagram featuring the series resistance (Rs), charge transfer resistance (Rct), and Warburg resistance (Zw). Fig. 7(i) shows the Nyquist plot of the selected high frequency region (black dotted circle) of Fig. 7(h). The Rs values for FO@CoFe-LDH1 and FO@CoFe-LDH2 are 5.07 Ω and 6.08 Ω, respectively, and a lower Rs value of FO@CoFe-LDH1 indicates lower internal resistance and higher conductivity within the electrode.43 The Rct values are 66.69 Ω for FO@CoFe-LDH1 and 866.95 Ω for FO@CoFe-LDH2. The significantly lower Rct value for FO@CoFe-LDH1 suggests a better interface between the electrode and the electrolyte compared to FO@CoFe-LDH2. Furthermore, the Zw values for FO@CoFe-LDH1 and FO@CoFe-LDH2 are 66.76 Ω and 876 Ω, respectively. The lower Zw value for FO@CoFe-LDH1 indicates faster OH− diffusion within the layered structure of CoFe-LDH.7,44 Further, the details of electrochemical output are summarised in Table S1.†
However, the Trasatti method was utilized to investigate the inner and outer charges of electrodes by cyclic voltammetry at different scan rates, as per eqn (7):45
As the scan rate increases, it becomes difficult for ions to diffuse into the inner surfaces of the electrode owing to limited interaction time. However, at very low scan rates, all the inner surfaces are accessible, and the charge (
Q) tends to approach the total charge (
QT) of the electrode. To determine
QT, a plot of 1/
Q against the square root of the scan rate (
ν1/2) is created, as shown in
Fig. 8(a). Meanwhile, the outer charge (
Qo) is obtained by plotting
Qo against
v−1/2, as depicted in
Fig. 8(b). The
QT values are 86 C g
−1 and 84 C g
−1, while the
Qo values are 72 C g
−1 and 12 C g
−1 for FO@CoFe-LDH1 and FO@CoFe-LDH2, respectively. However, the electrochemical charge storage kinetics is also explored with a correlation of current with respect to the scan rate of cyclic voltammetry as per
eqn (8):
46where
i is the current, ‘
a’ and ‘
b’ are adjustable parameters, and
ν represents scan rates. Furthermore, when the ‘
b’ value is less than 0.5, it suggests a diffusion-controlled reaction, and when the ‘
b’ value is 1, it suggests a surface-dominant mechanism of the electrochemical cell. However, the ‘
b’ value can be obtained by obtaining the slope of log (scan rate)
versus log (current) plot of different scan rates.
Fig. 8(c) illustrates ‘
b’ values of 0.9 and 1 estimated from the cathode current peak 1 and peak 2 for FO@CoFe-LDH1, while
Fig. 8(d) illustrates that anode current peak 1 and peak 2 are 0.85 and 1, respectively. The ‘
b’ values from cathode current peak 1 and peak 2 are 0.94 and 0.97 and anode current peak 1 and peak 2 are 0.72 and 0.95 of FO@CoFe-LDH2, respectively (
Fig. 8(e and f)). This suggests that FO@CoFe-LDH1 has a more capacitive contribution.
 |
| | Fig. 8 (a and b) Trasatti methods for FO@CoFe-LDH1 and FO@CoFe-LDH2; (c and d) log (scan rates, mV s−1) vs. log (cathode current, mA) and log (scan rates, mV s−1) vs. log (anode current, mA) of FO@CoFe-LDH1, and (e and f) log (scan rates, mV s−1) vs. log (cathode current, mA) and log (scan rates, mV s−1) vs. log (anode current, mA) of FO@CoFe-LDH2. | |
However, the diffusion coefficient is represented by D, which demonstrates how fast ions can move through a solution by a diffusion process. The diffusion coefficient achieved from cyclic voltammetry helps in understanding the transport of electroactive species in solution and their interaction with electrode surfaces. Nevertheless, the diffusion coefficient (D) was estimated by employing the Randles–Servcik eqn (9):47
| | | ip = (2.69 × 105)n3/2AD1/2Co1/2, | (9) |
where the diffusion coefficient (
D) is in square centimetres per second (cm
2 s
−1), the concentration of the electrolyte (
Co) is in moles per cubic centimetre (mol cm
−3), the scan rate (
ν) is in volts per second (V s
−1), the number of involved electrons (
n) is 1, and the peak current density per square centimetre (mA cm
−2). Fig. S6(a and b)
† illustrate the scan rate
vs. anode peak current of peak 1 and peak 2 of FO@CoFe-LDH1 and FO@CoFe-LDH2, respectively. The diffusion coefficients of peak 1 and peak 2 of FO@CoFe-LDH1 are 1.96 × 10
−4 cm
2 s
−1 and 2.62 × 10
−4 cm
2 s
−1, respectively. Furthermore, the diffusion coefficients of peak 1 and peak 2 of FO@CoFe-LDH2 are 1.95 × 10
−4 cm
2 s
−1 and 1.75 × 10
−4 cm
2 s
−1, respectively. The diffusion coefficient of FO@CoFe-LDH2 decreases owing to the excessive presence of Fe, which results in the blockage of active sites due to the decrease in specific surface area, pore volume, and pore surface area, thereby hindering the effective transport of ions and reducing overall electrochemical performance.
Aqueous hybrid supercapacitor (AHS)
The three-electrode electrochemical analysis confirmed better performance of FO@CoFe-LDH1 than FO@CoFe-LDH2. Therefore, it is selected for the fabrication of an aqueous hybrid supercapacitor (AHS) using activated carbon (AC) as the negative electrode and FO@CoFe-LDH1 as the positive electrode. Fig. 9(a) shows the CV curves of AC and FO@CoFe-LDH1 at 10 mV s−1 and confirms the working potential window of 1.6 V. Fig. 9(b) shows the CV curves of AC//FO@CoFe-LDH1 at various scan rates. At lower scan rates (10–30 mV s−1), the CV curves exhibit more pronounced redox behaviour. Conversely, at higher scan rates (from 50 mV s−1 to 10 mV s−1), the redox peaks tend to fade owing to the rapid scanning rate that limits the time constraints for the redox reactions.48,49 Consequently, the system shifts towards surface charge storage mechanisms rather than relying on deep, reversible redox processes. The observed charge storage primarily reflects capacitance from the electrode surface rather than the faradaic redox activity at higher scan rates. Fig. 9(c) shows the GCD curves of AC//FO@CoFe-LDH1 at various current densities. At lower current densities (0.5 and 1 A g−1), the charge and discharge plateaus on the GCD curves are well defined, reflecting stable electrochemical reactions and efficient energy storage mechanisms. However, at higher current densities, such as 2 to 5 A g−1, the charge and discharge plateaus tend to fade. This occurs because the rapid rates of charge and discharge at these higher currents cause kinetic limitations and increased resistive effects within the cell.50 Furthermore, the specific capacitance, specific energy density, and specific power density were estimated by utilising eqn (3) and (4). Fig. 9(d) illustrates the specific capacitance of AC//FO@CoFe-LDH1 at various current densities, highlighting a maximum specific capacitance of 25 F g−1 at 1 A g−1. Fig. 9(e) presents the Ragone plot for AC//FO@CoFe-LDH1, showing a maximum specific energy density of 9.2 W h kg−1 and a maximum power density of 3857 W kg−1. The charge kinematic of AC//FO@CoFe-LDH1 was studied with EIS, as depicted in Fig. 9(f and g). Fig. 9(f) shows the Nyquist plot of AC//FO@CoFe-LDH1, and the inset is the equivalence circuit diagram. Fig. 9(g) shows the Nyquist plot of the selected high frequency region (red dotted circle) of Fig. 9(f). The Rs value is 0.055 Ω, indicating that the supercapacitor has minimal internal resistance. However, the Rct value of 0.036 Ω signifies an excellent interface between the electrode and electrolyte. Additionally, the Warbag resistance (Zw) value of 0.091 Ω implies fast OH− diffusion in the layer structure. Fig. 9(h) illustrates the capacitance retention percentage of AC//FO@CoFe-LDH1 up to 4000 cycles, demonstrating a retention rate of 99.9%. In contrast, Fig. 9(i) presents the GCD curves of AC//FO@CoFe-LDH1, comparing the 1st cycle and the 4000th cycle at 3 A g−1, demonstrating no significant change in charge/discharge time and maintaining high stability even at 4000 cycles.
 |
| | Fig. 9 (a) CV curves of AC and of FO@CoFe-LDH1 at 10 m V s−1, AHS characterization: (b and c) CV and GCD curves at various scan rates and current densities, (d) specific capacitance at various current densities, (e) Ragone plot, (f) Nyquist plot and inset is the equivalence circuit diagram, (g) Nyquist plot of selected high frequency region (red doted circle) of Fig. 9f, (h) capacitance retention percentage at various cycle numbers, and (i) GCD curves of 1st cycle and 4000th cycle at 3 A g−1. | |
Conclusions
In conclusion, we successfully prepared iron oxide/CoFe-LDH (FO@CoFe-LDH) by applying one-step hydrothermal methods. The synergistic combination of iron oxide with CoFe-LDH's layered structure facilitates fast ion diffusion and redox activity. Further, iron oxide plays a significant role in preventing the self-restacking and aggregation of CoFe-LDH layers, enhancing the structural stability and electrochemical performance of the composite. The electrochemical investigation exhibits a specific capacity of 84 C g−1 at 1 A g−1 (FO@CoFe-LDH1), while that of FO@CoFe-LDH2 is limited to 25 C g−1 at 1 A g−1. The AHS device using FO@CoFe-LDH1 as the positive electrode and activated carbon (AC) as the negative electrode demonstrated remarkable cyclic stability, retaining 99.9% capacitance after 4000 cycles. This electrode showed comparatively better stability (Table S2†). These findings highlight the effectiveness of the iron oxide/CoFe-LDH nanocomposite in addressing the cyclic stability challenges of CoFe-LDH materials, paving the way for the development of durable and efficient energy storage devices.
Data availability
Data generated and analysed in this study are included in the article and its ESI.†
Author contributions
Harishchandra S. Nishad: conceptualization, formal analysis and investigation, writing – original draft. Jaewong Lee: manuscript modifications and analysis. Sagar Mane: TEM, Raman, XPS data curation. Pravin S. Walke: corresponding author, review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Thanks to the Director of National Centre for Nanoscience and Nanotechnology, University of Mumbai, Mumbai, India-400098. Special thanks to Prof. Pradip Sarawade for providing BET measurements at Department of Physics, University of Mumbai.
References
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CAS.
-
M. G. Petrescu, A. Neacşa, E. Laudacescu and M. Tănase, Ind. 5.0 Creat. Innov. Organ., 2023, pp. 71–90 Search PubMed.
- B. H. Xiao, K. Xiao, J. X. Li, C. F. Xiao, S. Cao and Z. Q. Liu, Chem. Sci., 2024, 15, 11229–11266 CAS.
- A. Inman, T. Parker, Y. Zhang, M. Saraf and Y. Gogotsi, Adv. Energy Mater., 2024, 14, 2402367 CAS.
- S. A. Siddiqui, S. Das, S. Rani, M. Afshan, M. Pahuja, A. Jain, D. Rani, N. Chaudhary, R. Jyoti, S. Ghosh, C. B. Riyajuddin and K. Ghosh, Small, 2024, 20, 2306756 CAS.
- O. A. AlKawak, J. R. R. Kumar, S. S. Daniel and C. V. K. Reddy, J. Energy Storage, 2024, 77, 109835 Search PubMed.
- H. S. Nishad, V. Kotha, P. Sarawade, A. C. Chaskar, S. Mane, J. Lee and P. S. Walke, J. Mater. Chem. A, 2024, 12, 9494–9507 CAS.
- F. Liao, G. Yang, Q. Cheng, L. Mao, X. Zhao and L. Chen, Electrochim. Acta, 2022, 428, 140939 CrossRef CAS.
- Y. Jiang and J. Liu, Energy Environ. Mater., 2019, 2, 30–37 Search PubMed.
- S. S. Shah, M. A. Aziz, P. I. Rasool, N. Z. K. Mohmand, A. J. Khan, H. Ullah, X. Feng and M. Oyama, Sustainable Mater. Technol., 2024, 39, e00814 Search PubMed.
- W. Liu, H. Li and R. Y. Tay, Nanoscale, 2024, 16, 4542–4562 RSC.
- H. Aydın, Ü. Kurtan, B. Üstün, S. N. Koç, E. Akgül and M. Demir, Mater. Res. Bull., 2024, 180, 113062 Search PubMed.
- M. Mohanraj, S. P. Shaji, K. S. Rajni and M. Ulaganathan, Electrochim. Acta, 2024, 482, 143972 Search PubMed.
- S. K. Meher and G. R. Rao, J. Phys. Chem. C, 2011, 115, 15646–15654 CAS.
- Y. Xu, X. Yang, X. Li, Y. Gao, L. Wang and W. Lü, J. Power Sources, 2024, 623, 235399 Search PubMed.
- M. Prajapati, C. Ravi Kant, A. Hussain and M. V. Jacob, New J. Chem., 2024, 48, 18376–18391 CAS.
- H. H. Hegazy, J. Khan, E. A. Noshaba Shakeel, M. I. Alabdullkarem, H. A. Saleem and I. S. Yahia, RSC Adv., 2024, 14, 32958–32977 CAS.
- H. Sun, S. Liu, S. Liu and S. Wang, Appl. Catal., B, 2014, 146, 162–168 CAS.
- H. Xing, X. Deng and X. Wang, J. Colloid Interface Sci., 2025, 679, 43–53 CAS.
- P. K. Ray and K. Parida, Mater. Adv., 2025, 6, 84–116 CAS.
- Z. Wang, Y. Song, R. Li, R. Li, R. Jia, K. Nie, H. Xie, X. Xu and L. Lin, Dalton Trans., 2025, 54, 821–831 CAS.
- Y. Liu, C. Wang, H. Sun, L. Duan, Z. Yang, X. Wang and J. Liu, J. Colloid Interface Sci., 2025, 682, 1051–1061 CAS.
- Q. He, W. Jia, X. Wu and J. Liu, CrystEngComm, 2024, 27, 55–63 Search PubMed.
- X. Fang, S. Han, D. Liu and Y. Zhu, Chem. Phys. Lett., 2020, 746, 137282 CAS.
- D. S. Patil, S. A. Pawar, S. H. Lee and J. C. Shin, J. Electroanal. Chem., 2020, 862, 114012 CAS.
- Y. Wang, W. Zhang, X. Guo, Y. Liu, Y. Zheng, M. Zhang, R. Li, Z. Peng and Y. Zhao, Appl. Surf. Sci., 2021, 561, 150049 CAS.
- Z. Liu, Y. Liu, Y. Zhong, L. Cui, W. Yang, J. M. Razal, C. J. Barrow and J. Liu, J. Power Sources, 2021, 484, 229288 CAS.
- Y. Zhang, M. Yang, X. Jiang, W. Lu and Y. Xing, J. Alloys Compd., 2020, 818, 153345 CAS.
- J. Guo, Z. Wei, K. Wang and H. Zhang, Int. J. Hydrogen Energy, 2021, 46, 27529–27542 CAS.
- S. Jo, N. Jayababu and D. Kim, J. Alloys Compd., 2022, 904, 164082 CAS.
- R. Li, J. Xu, Q. Pan, J. Ba, T. Tang and W. Luo, ChemistryOpen, 2019, 8, 1027–1032 CAS.
- N. F. Shahid, A. Jamal, G. U. Haq, M. Javed, M. Saifullah and M. A. R. Anjum, Energy Adv., 2023, 2, 2109–2118 CAS.
- Q. Li, P. Man, L. Yuan, P. Zhang, Y. Li and S. Ai, Mol. Catal., 2017, 431, 32–38 CAS.
- G. Abellán, J. A. Carrasco, E. Coronado, J. Romero and M. Varela, J. Mater. Chem. C, 2014, 2, 3723–3731 RSC.
- S. O. Ganiyu, T. X. Huong Le, M. Bechelany, G. Esposito, E. D. Van Hullebusch, M. A. Oturan and M. Cretin, J. Mater. Chem. A, 2017, 5, 3655–3666 RSC.
- C. Hao, Y. Wu, Y. An, B. Cui, J. Lin, X. Li, D. Wang, M. Jiang, Z. Cheng and S. Hu, Mater. Today Energy, 2019, 12, 453–462 CrossRef.
- C. Huang, C. Ni, L. Yang, T. Zhou, C. Hao, X. Wang, C. Ge and L. Zhu, J. Alloys Compd., 2020, 829, 154536 CrossRef CAS.
- J. Xu, K. Liao, K. Song, J. Wu, X. Hu, H. Gao, F. Hu and J. P. Cheng, J. Solid State Electrochem., 2018, 22, 1037–1045 CrossRef CAS.
- J. Xi, M. S. Javed, S. Asim, M. Idrees, S. S. A. Shah, M. A. Assiri, W. Mai and C. Hu, Mater. Chem. Front., 2021, 5, 3436–3447 RSC.
- M. P. Harikrishnan and A. Chandra Bose, Energy Fuels, 2023, 37, 10799–10826 CrossRef.
- J. He, Y. Zhou, S. Wu, L. Jin, J. Cao, M. Demir and P. Ma, Inorg. Chem., 2024, 63, 13755–13765 CrossRef CAS PubMed.
- X. Zhou, X. Li, D. Chen, D. Zhao and X. Huang, J. Mater. Chem. A, 2018, 6, 24603–24613 RSC.
- M. Dinari, H. Allami and M. M. Momeni, Energy Fuels, 2021, 35, 1831–1841 CrossRef CAS.
- Y. Zhang, J. Zhang, H. Li, H. Yao and X. Wang, Int. J. Electrochem. Sci., 2024, 19, 100590 CrossRef CAS.
- H. Vogt, Electrochim. Acta, 1994, 39, 1981–1983 CrossRef CAS.
- V. Tripathi, S. P. Gupta, K. Kumar and L. S. Panchakarla, J. Energy Storage, 2023, 70, 107974 CrossRef.
- H. Lv, H. Rao, Z. Liu, Z. Zhou, Y. Zhao, H. Wei and Z. Chen, J. Energy Storage, 2022, 52, 104940 CrossRef.
- Y. Qiao, G. Liu, R. Xu, R. Hu, L. Liu, G. Jiang, M. Demir and P. Ma, Electrochim. Acta, 2023, 437, 141527 CrossRef CAS.
- T. M. Masikhwa, M. J. Madito, D. Y. Momodu, J. K. Dangbegnon, O. Guellati, A. Harat, M. Guerioune, F. Barzegar and N. Manyala, RSC Adv., 2016, 6, 46723–46732 RSC.
- S. P. Gupta, S. W. Gosavi, D. J. Late, Q. Qiao and P. S. Walke, Electrochim. Acta, 2020, 354, 136626 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sagar M.
Mane
a,
Sagar M.
Mane