
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteraction of vapor-phase and liquid organophosphonates with inorganic surfaces
Swapnil
Das
,
Mirko
Schoenitz
and
Edward L.
Dreizin
 *
*
New Jersey Institute of Technology, Newark, NJ, USA. E-mail: dreizin@njit.edu
First published on 12th February 2025
Abstract
Chemical warfare agents (CWAs) pose a threat to humanity, which motivates research focused on their destruction. Often, research deals with non-toxic simulants of CWAs, such as dimethyl methyl phosphonate (DMMP) and diisopropyl methyl phosphonate (DIMP). These compounds, like CWAs, are liquids at room temperature and boil just below 200 °C. In different scenarios, their interactions with inorganic solids may initially involve either liquid or vapor phases. This paper reviews published experimental data describing how the initial phase (vapor or liquid) of DMMP or DIMP influences the properties of their residues adsorbed to different inorganic surfaces. To facilitate comparisons between different sets of experiments, the focus is on the commonly reported shift and possible split of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak assigned to the phosphoryl group, sensitive to molecular interactions in organophosphorus liquids and detected by Fourier Transform Infrared (FTIR) spectroscopy. Data sets for multiple metal oxides and salts are compared to one another. Systematic and distinct trends are found for the PO peak behavior for residues of evaporated and liquid DMMP and DIMP left on different surfaces. The literature data offer compelling evidence that the properties of residues left by organophosphonates on inorganic surfaces vary depending on the initial phase of the organophosphonate.
O peak assigned to the phosphoryl group, sensitive to molecular interactions in organophosphorus liquids and detected by Fourier Transform Infrared (FTIR) spectroscopy. Data sets for multiple metal oxides and salts are compared to one another. Systematic and distinct trends are found for the PO peak behavior for residues of evaporated and liquid DMMP and DIMP left on different surfaces. The literature data offer compelling evidence that the properties of residues left by organophosphonates on inorganic surfaces vary depending on the initial phase of the organophosphonate.
1. Introduction
Chemical warfare agents (CWAs), which are illegal under the Chemical Weapons Convention,1,2 are extremely toxic chemical compounds, many of which belong to the class of organophosphorus liquids,2,3 Given the significant threat that CWAs pose to humanity, it is imperative to understand how they evolve and decompose when interacting with different solids, e.g., soil components, construction materials, and combustion products of energetic formulations. Such interactions may form condensed products and/or surface residues of CWAs, which can be toxic even if they are altered chemically from the initial compounds. Contaminants in the form of surface residues on various inorganic solids may be transported away from the release site, e.g., as aerosolized dust particles. Their modified chemical compositions may affect the subsequent fate of both evaporated and surface-adsorbed chemical complexes.4Many published experimental studies deal with non-toxic simulants, which mimic the relevant chemical and physical characteristics of CWAs.3,5 In particular, dimethyl methylphosphonate (DMMP) and diisopropyl methylphosphonate (DIMP) are organophosphorus liquids commonly serving as such simulants.6 The structures and some properties of DMMP and DIMP are presented in Table 1.
| DMMP | DIMP | |
|---|---|---|
| Molecular weight, g mol−1 | 124 | 180 |
| Boiling point, °C | 181 | 195 |
| Acute oral toxicity LD50, mg kg−1 | 8210 | 826 |
| Molecular structure |
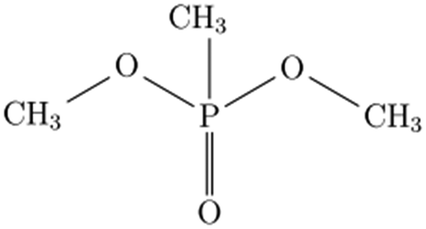
|
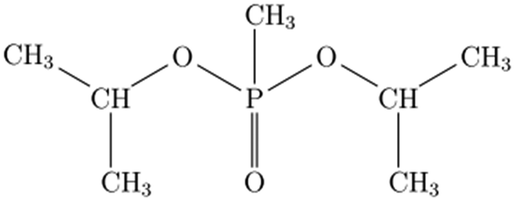
|
Interactions of both DMMP and DIMP with inorganic solids have been studied extensively.4,7–13 The experiments involved both liquid4,11,12,14–18 and vapor-phase13,19–31 organophosphonates leaving residues or forming reaction products on metal oxides,22,24–28,30–32 metal halides12,13,19 and perchlorates.11 Fourier transform infrared spectroscopy (FTIR)8,9,11,12,14–19,22–25,27–29,31,33–38 has been used universally to characterize and identify such residues and products. In many studies, other experimental methods supplemented FTIR, including as, X-ray photoelectron spectroscopy (XPS),26,27,29,31,39–45 thermal analysis,4,18,19,29–31 surface area analysis through Brunauer–Emmett–Teller (BET) measurement,40,46,47 elemental analysis via induction-coupled plasma mass spectroscopy (ICP-MS),11,14,15,23,48,49 and energy-dispersive X-ray spectroscopy (EDX).7,18 Molecular structures and abbreviations for common residues or reaction products identified in the literature for liquid and vapor phase DIMP and DMMP interacting with surfaces between room temperature and ∼200 °C are summarized in Fig. 1. Relevant results, including the characterization methods used in combination with FTIR, are listed in Table 2. Unfortunately, few direct comparisons of the residues left by liquid and vapor-phase organophosphonates can be made.
 | ||
| Fig. 1 Reported residues and products forming by DMMP and DIMP on different surfaces. | ||
| Compound | Initial phase | Surface | Product/residue | Methods in addition to FTIR |
|---|---|---|---|---|
| X = metal. | ||||
| DMMP | Vapor | Al2O3 | MMP, MPA | N/A28,50 |
| CuO | PO3+ or PO2CH4+ | XPS27 | ||
| CeO2 | MMP, MP | XPS,29,31 TPD29,31 | ||
| La2O3 | MMP | N/A28 | ||
| C | MMP, MPA | BET46 | ||
| Y2O3 | MMP | XPS46 | ||
| Fe2O3 | (H3CO)2P(O)-CH3 | XPS28 | ||
| MgO | MMP, | N/A,24 ICP-MS29 | ||
| MoO3 | PO3+ or PO2CH4+ | XPS,30 TPD30 | ||
| V/Al2O3 | MMP, MPA | BET,40 XPS40 | ||
| V/SiO2 | PO43−, MPA | BET,40 XPS40 | ||
| MnO2 | (PO43+) | ICP-MS49 | ||
| Liquid | Al2O3 | MMP | N/A22 | |
| Go/MnO2 | MMP | GC–MS,9 EDX9 | ||
| AlCl3 | X(MMP)3 | ICP-MS51 | ||
| TiCl3 | X(MMP)3 | ICP-MS51 | ||
| CrCl3 | X(MMP)3 | ICP-MS48 | ||
| VCl3 | X(MMP)3 | ICP-MS48 | ||
| DIMP | Vapor | Al2O3 | IMP | N/A22 |
| Liquid | Al2O3 | IMP, MPA | TG,4 MS4 | |
| LiCl3 | X(IMP), X2(PMP) | ICP-MS12 | ||
| NaCl | X(IMP), X2(PMP) | ICP-MS12 | ||
| VoCl3 | X(IMP) | ICP-MS14 | ||
| FeCl3 | X(IMP) | ICP-MS14 | ||
| KCl | X2(PMP) | ICP-MS14 | ||
| CaCl2 | X2(PMP) | ICP-MS14 | ||
| NiCl2 | X2(PMP) | ICP-MS14 | ||
| ZnCl2 | X2(PMP) | ICP-MS14 | ||
Starting with DMMP, its vapors interacting with Al2O3 at 200 °C produced MMP and MPA.28,50 Only MMP was reported as a residue when Al2O3 interacted with liquid DMMP.22 MMP residue was also found after DMMP vapors interacted with CeO2,29,31 La2O3,28 C,46 Y2O3,26,46 MgO,23,28 V/Al2O3 (ref. 40) and V/SiO2.40 The vapors left some surface-bound phosphate complexes, including (H3CO)2P(O)–CH3, PO3+, and PO2CH4, combined with PO43− on Fe2O3,28 MoO3,30 and V/SiO2,40 respectively, as was found using XPS. Temperature-programmed desorption (TPD) analysis was also used to identify MMP, MP, and DMMP fragments such as PO3+ and PO2CH4+ when CeO2 (ref. 29) and MoO3 (ref. 30) were exposed to DMMP vapors. Another commonly reported residue, X(MMP)3, was mainly identified through ICP-MS in the reaction products of liquid DMMP interacting with AlCl3,51 TiCl3,51 CrCl3,48 and VCl3.48 MPA and MMP were proposed as products of the DMMP vapor decomposing on V/Al2O3 (ref. 40) causing a reduction in the specific surface area. Similarly, MPA and MMP products and a reduction in the specific surface were reported for C (carbon)46 after its exposure to DMMP vapor.46
For DIMP, IMP was identified as a residue on Al2O3 interacting with both vapor22 and liquid phases.4 Analysis of the results of thermal analysis and mass spectrometry suggested that IMP and MPA were adsorbed on the Al2O3 surface after its exposure to liquid DIMP.4 Unfortunately, no other data identifying DIMP residues on oxides could be found in the literature. For liquid DIMP interacting with salts, ICP-MS was employed to identify commonly reported products, including X2(PMP) for LiCl3,12 NaCl,12 KCl,14 CaCl2,14 NiCl2 (ref. 14) and ZnCl2 (ref. 14) and X(IMP) for LiCl3,12 NaCl,12 VoCl3 (ref. 14) and FeCl3.14
Although it is clear from the reviewed literature that multiple descriptors can identify compounds and usefully characterize different properties of the reaction products and residues left on solid surfaces interacting with organophosphonates, the available information is fragmentary at best. The available data are inadequate to compare the decomposition products formed by individual liquid or vapor phase organophosphonates interacting with the same solids. Yet, the phase of an organophosphate present as either liquid or vapor may substantially affect its interaction with the solids and determine the compounds and complexes comprising respective products or residues.
In literally all related studies, infrared spectroscopy was used, generating a rather comprehensive and representative data set. Comparing the results of such studies offers a path to begin understanding the effect of the initial phase of the organophosphonate on the residues formed. In most studies, the focus is on three functional groups: PO–C,12,14,15,19,24,33,38,50 O–P–O (ref. 26, 27 and 37) and PO.4,11–19,22,24–28,30–32 Shifts in the PO–C vibrational mode were reported for the vapor-phase DMMP residues on Al2O3,33 ZnO,24 and TiO2.38 Unfortunately, no similar data exists for the residues formed by liquid organophosphonate on the same oxides. Similarly, a limited set of measurements is available characterizing the vibrational O–P–O mode for the residues of vapor phase DMMP on Y2O3,26 CuO (ref. 27) and TiO2.37 At the same time, nearly every study described the phosphoryl group PO. The FTIR data for this specific vibration are available for the broadest range of solids interacting with both liquid and vapor-phase organophosphonates. Respectively, as the first step to understanding the nature of the residues produced on different surfaces from different organophosphonate phases, our focus here is on the spectral characteristics assigned to the PO bond.
These double bonds are particularly sensitive to molecular interactions.16,34,52 For example, it was suggested that both DMMP and DIMP molecules attach to metal oxide surfaces through their phosphoryl oxygen,24,53 as shown schematically in Fig. 2a. The phosphoryl oxygen can attach to either a hydroxy group (Brønsted acid site, Fig. 2a, left) or a metal ion (Lewis acid site, Fig. 2a, right). In Fig. 2, R represents either methyl or isopropyl groups, for DMMP and DIMP, respectively. This is similar to CWAs, for which the PO bond also binds to the surface active sites.33
 | ||
| Fig. 2 Types of bonding sites for (a) vapor and liquid phase DMMP and DIMP on metal oxides22,53 and vapor phase DMMP and DIMP with metal salts19 (b) liquid phase DMMP and DIMP on metal salt.12,14 | ||
Because of its great significance, detecting, characterizing, and eliminating the binding of metal cations to organophosphonates via the phosphoryl group is the focus of many CWA sensing, capture, and destruction technologies. For this reason, the adsorption of organophosphonates via strong binding to metal cations through their phosphoryl oxygen was studied for metal salts, e.g., metal iodides,12 other halides,14 and perchlorates.11 The strength of this interaction (strong chemisorption or weak physisorption) is influenced by types of both anion (e.g., halides, iodides, perchlorates) and cation (e.g., transition metal, rare earth metal) on the salt surface.13,19 Measured shifts in the PO FTIR peak position were interpreted to represent different residual complexes formed on the surface, (cf.Table 2). All complexes include the PO bond, and their identification is based on the bond strength with the interacting solid54 through spectroscopic investigation in combination with other characterization techniques.
For metal salts with no surface hydroxyl groups, both DMMP and DIMP molecules are attached to Lewis acid sites (Fig. 2a).19 It was also proposed that for both liquid DIMP and DMMP on metal salts, the phosphoryl group transforms into phosphate complexes, such as shown in Fig. 2b, producing distinct condensed reaction products, while gaseous products, such as propene, are liberated.12,48
Recent atomistic models also targeted organophosphorus compound/solid interactions27,53,55 where initial interaction is predicted to occur through the PO bond. Density functional Theory (DFT) is now commonly used41,44,56–63 in different studies to predict the reaction mechanism or decomposition pathways but not the reaction products. These models focus on individual organophosphorus molecules interacting with solids and thus do not distinguish whether such molecules come from vapor or liquid phases. Unfortunately, this makes it impossible for the existing models to explain the differences between the residues forming on different surfaces from either vapor or liquid organophosphonates.
Molecular interactions in organophosphorus liquids are complex and involve hydrogen bond acceptors, dipole interactions, steric strain and hindrance, etc. All such interactions are expected to affect how the molecules bind to solid surfaces as compared to identical free molecules approaching the same solid surfaces from the vapor. For CWA and related organophosphonates, these interactions determine the nature, stability, and toxicity of the surface residues, which form at different locations and under various environmental conditions. It is thus important to predict whether residues that form on different solids interacting with liquid differ from those formed by vapor-phase organophosphonates. To explore the possible effects of the molecular interactions in the liquid on the respective reaction products and surface residues, the existing experimental data are reviewed. The goal is to compare to each other products resulting from interactions of various solids with either vapor or liquid DMMP and DIMP phases. The review focuses on the reported changes in characteristics of the phosphoryl group detected in products or residues at low temperatures. The properties of this group are well-known to affect the nature of the reaction products and residues formed by DMMP and DIMP on inorganic solids.
2. Experimental techniques
As noted above, among many experimental methods characterizing organophosphonates and their residues formed on different surfaces, FTIR spectroscopy is commonly used.11–14,19,21,33 In addition to a conventional transmission geometry, surface-sensitive variants such as ATR4 or DRIFTS40 and inelastic electron tunneling spectroscopy22 have been used to obtain vibrational spectra in the relevant wavenumber ranges. Here, the data from vibrational spectra in the near IR range reported by different researchers will be considered systematically, focusing, as noted above, on the characteristics of the phosphoryl group present either in the surface residues or the reaction products of the organophosphonates and solids. Both common and distinctive characteristics for the analyzed products formed after either vapor or liquid DMMP and DIMP interacted with different solids will be elucidated.For liquid organophosphonates interacting with solids, most experiments can be schematically represented by a configuration shown in Fig. 3. Typically, the environment is air2–6 or argon7 at atmospheric or slightly reduced pressure. Different inorganic powders, like halides,14 iodides12 or oxides4,17 are mixed with liquid organophosphonates and then heated in a controlled environment furnace at 2–10 °C min−1,3–7 typically to a temperature well above the boiling point of the organophosphonates. Most of the liquid evaporates during heating. However, powders recovered after heating contain condensed reaction products on the surface or in the bulk, which are then analyzed using infrared spectroscopy.
 | ||
| Fig. 3 Schematic diagram of experiments for studying liquid organophosphonates.4,11,12,14,17 | ||
In some experiments, halides12,14 and perchlorates11 were dissolved with organophosphorus liquids and reaction products precipitated at higher temperatures. The reaction products are metal ions and phosphorus-bearing complexes that typically have a defined composition. They are distinct from the organophosphonate derivatives bound to surfaces.
In one type of experiment exploring vapor phase organophosphonates interacting with inorganic solids, the reactions were carried out at various pressures in a gas cell (Fig. 4a) connected to a high vacuum system (10−1–10−8 torr).13,19,21 The organophosphonates were allowed to react with the solids from seconds to hours. Reaction times were assessed by changes in the physical appearance of the powders. The cell could be operated at room temperature, or it could be heated up to 200 °C. The cell was then removed from the vacuum system and inserted in an FTIR spectrometer for analysis. Since exposure of the solids to gaseous phosphonates produced visible color changes, it should be considered that here, too, the reaction products formed the bulk of the recovered material and did not just reside on the surface.
 | ||
| Fig. 4 Schematic diagram of (a) variable temperature infrared cell13,19,21 (b) flow reactor setup containing powders8,40,49 for studying vapor phase organophosphonates. | ||
In different experiments, organophosphorus gases mixed with a carrier gas (air, helium, or nitrogen) are injected into a flow reactor where they pass over inorganic solids8,40,49 as schematically shown in Fig. 4b. In some experiments the gas may then flow into a gas analyzer (e.g., a gas chromatograph). Initially, the organophosphorus molecules begin adsorbing to the solid and do not reach the gas analyzer. After the solid is saturated, the organophosphonates begin being detected, defining the so-called breakthrough point (BTP).8,40 Temperatures for BTP experiments range from ambient to as high as 700 °C, and residence time can be sub-seconds to several seconds. Pressures are typically around 1 atm. The exposed solids are collected, and the residue is characterized using FTIR.
3. FTIR spectral features of neat DMMP and DIMP and their residues on solids
Adsorption spectra of neat liquid and vapor-phase DMMP and DIMP are available in the literature and serve as useful baselines when interpreting similar spectra produced by their condensed phase reaction products and residues left on solid surfaces. For example, adsorption spectra of neat vapor-phase and liquid DMMP reported in ref. 64 are reproduced in Fig. 5. For DIMP, relevant spectra are reported in.34 Peak assignments for both DMMP65 and DIMP34 spectra are listed in Table 3. The spectral features for liquid and vapor-phase adsorption are similar to each other for both DMMP and DIMP. The most significant difference between the vapor-phase and liquid adsorption spectra occurs for the PO stretching mode peak, appearing within the shaded area between approximately 1200 and 1300 cm−1 in Fig. 5. This is because, as noted earlier, the phosphoryl group is most sensitive to molecular interactions.16,34,52 Indeed, from Table 3, it is noted that the shifts in the position of the PO peak for the vapor and liquid are much greater (2.4% for DMMP and 1.7% for DIMP) than for any other peaks. For both DMMP and DIMP, the PO bond produces peaks in lower wavenumber regions for liquid compared to the vapor phases. This redshift observed for liquids is due to significant molecular interactions, including van der Waals forces, dipole–dipole interactions, and hydrogen bonding.52 Similarly, because of the molecular interactions of the condensed phase residues produced by either DMMP or DIMP with a solid surface, a redshift for the PO peak is always expected.
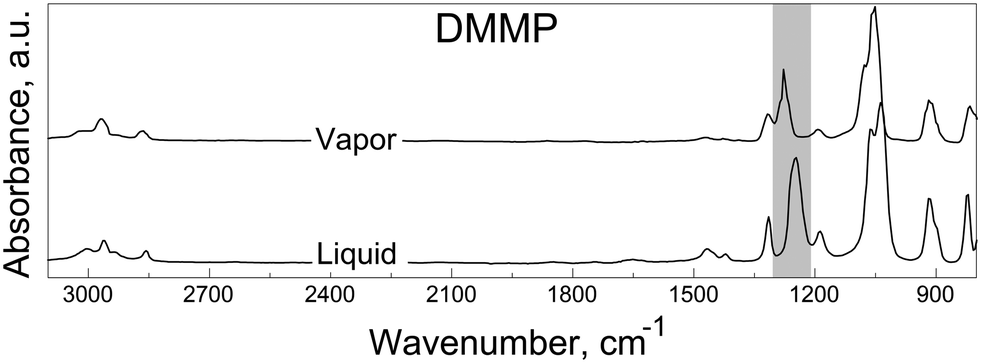 | ||
| Fig. 5 Reference transmission FTIR spectra of vapor and liquid phases of DMMP.64 | ||
| DMMP | DIMP | ||||||
|---|---|---|---|---|---|---|---|
| Assignment | Peak position, cm−1 | Assignment | Peak position, cm−1 | ||||
| Vapor | Liquid | Difference | Vapor | Liquid | Difference | ||
| a: asymmetric; s: symmetric, ν: stretching, δ: bending, ρ: rocking. | |||||||
| ν a(PCH3) | 3014 | 2995 | 19 (0.6%) | ν a(PCH3) | 2985 | 2979 | 6 (0.2%) |
| ν a(CH3O) | 2962 | 2956 | 6 (0.2%) | ||||
| ν s(CCH3) | 2945 | 2934 | 11 (0.4%) | ||||
| ν s(PCH3) | 2921 | 2927 | −6 (−0.2%) | ν(PCH3) | 2886 | 2878 | 8 (0.3%) |
| ν s (CH3O) | 2859 | 2852 | 7 (0.2%) | ||||
| δ a(CCH3) | 1473, 1457 | 1468, 1455 | 5 (0.3%), 2 (0.1%) | ||||
| δ a(CH3O) | 1471 | 1466 | 5 (0.3%) | ||||
| δ s (CH3O) | 1423 | 1421 | 2 (0.1%) | ||||
| δ s(CCH3) | 1387, 1377 | 1386, 1375 | 1 (0.1%), 2 (0.1%) | ||||
| δ s(PCH3) | 1314 | 1312 | 2 (0.2%) | ||||
|
ν(PO) |
1276 | 1245 | 31 (2.4%) |
ν(PO) |
1266 | 1245 | 21 (1.7%) |
| ν a(C–O) | 1075 | 1065 | 10 (0.9%) | ||||
| δ ρ(CCH3) | 1180, 1142, 1112 | 1178, 1142, 1112 | 2 (0.2%), 0, 0 | ||||
| δ s(PCH3) | 1315 | 1314 | 1 (0.1%) | ||||
| ν s(C–O) | 1050 | 1038 | 12 (1.1%) | ||||
| ν(POC) | 1018, 995 | 1011, 986 | 7 (0.7%), 9 (0.9%) | ||||
| δ ρ(PCH3) | 919, 901 | 917, 899 | 2 (0.2%), 2 (0.2%) | ||||
Focusing on the PO peak, spectra of neat DMMP vapor and liquid are compared to those of the residues left by both vapor and liquid DMMP on alumina surfaces in Fig. 6a.17 In both cases, the exposure of the oxide to DMMP occurred at room temperature. The PO peak for the surface residue of the vapor DMMP is shifted to 1230 cm−1, which is a greater shift than for the neat liquid. Thus, the molecular interactions in the residue formed from the DMMP vapor on alumina are stronger than in the neat DMMP liquid. Conversely, for the residue formed on alumina from DMMP liquid, the redshift is smaller than for the neat liquid, suggesting respectively weaker molecular interactions.
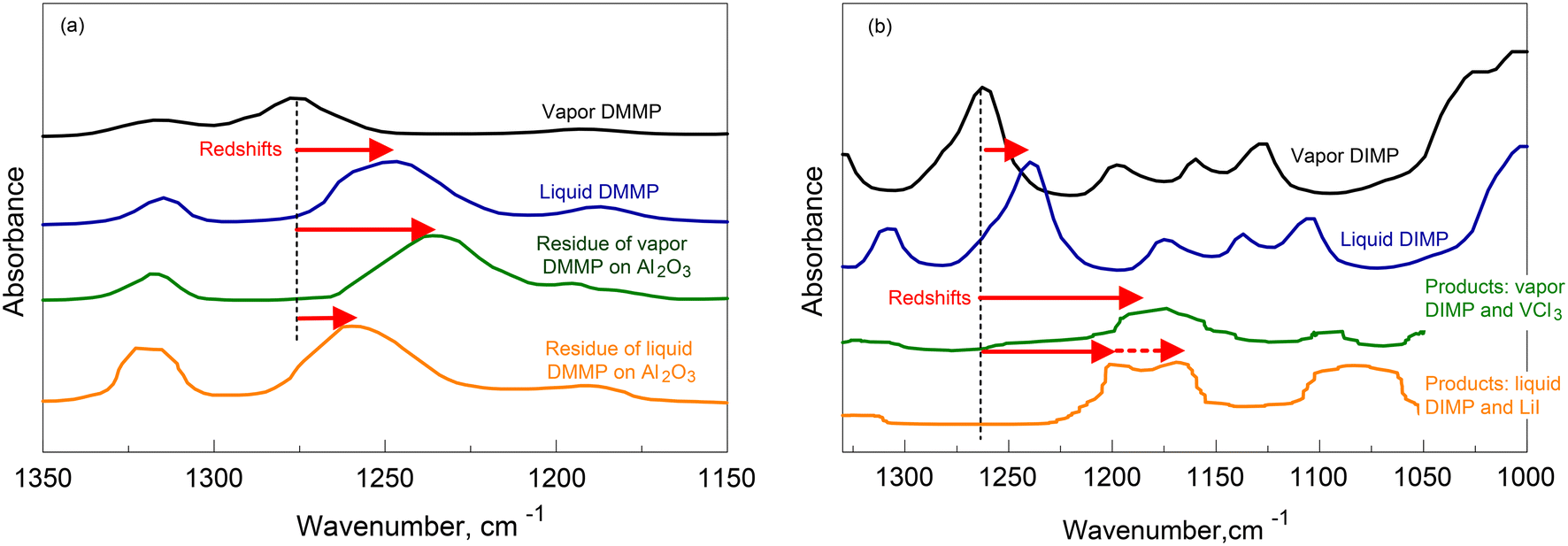 | ||
| Fig. 6 Examples of infrared spectra or organophosphonates interacting with inorganic solids (a) DMMP17 (b) DIMP.12,19 | ||
Different example spectra in Fig. 6b show the shifts of the PO peak for the reaction products of vapor and liquid DIMP with metal halides: LiI (ref. 12) and VCl3.19 In this case, the redshifts observed for the PO peaks are always greater than the shift for the neat liquid. Thus, molecular interactions in both reaction products are stronger than in the liquid DIMP. In addition, for the reaction product with LiI, the PO peak splits. This is common for reaction products of liquid DIMP and metal halides.14,15 The split PO peaks indicate multiple distinct interactions of the PO group with the metal ions in the reaction products.12,14
4. Shifts in the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) O peak positions for different surface residues of DMMP and DIMP
O peak positions for different surface residues of DMMP and DIMP
The examples of FTIR spectra discussed above are part of an extensive dataset reported in the literature for both gas and liquid DMMP and DIMP interacting with different oxides and salt. Bar plots shown in Fig. 7 and 8 compile data from the literature for the PO peak positions for various relevant reaction products. For comparison, the dashed and solid horizontal lines show the positions of the PO peaks for neat vapor and liquid phases, respectively. The data for the PO peak positions formed by interaction with DMMP vapor are shown in Fig. 7a. For all reaction products, a significant redshift is observed. The strongest and weakest shifts are observed, respectively, for FeCl3 and MgO.28 For DIMP, a large set of data is available in the literature for its vapor interacting with various metal salts (chlorides); these are summarized in Fig. 7b and c for rare earth elements13 and transition metal chlorides,13,19 respectively. Only one report characterizing the residue left by the vapor phase DIMP on an oxide, namely, alumina was available,4 as shown in Fig. 7c. For all rare earth element chlorides, the redshift is nearly the same (Fig. 7b). For transition metal chlorides, the shifts vary somewhat; the shift observed for the residue formed on Al2O3 is nearly the same as that for the product with HgCl2 (Fig. 7c).
 | ||
| Fig. 7 PO peak positions sorted based on the redshift; (a) neat vapor-phase DMMP and its residues on different oxide surfaces22,24–32 and reaction product with FeCl3; (b) neat vapor-phase DIMP and its reaction products with rare earth metal salts13 (c) neat vapor-phase DIMP, it residue on alumina and its reaction products with transition metal salts.13,19 | ||
 | ||
| Fig. 8 PO peak positions sorted based on the redshift for (a) neat liquid DMMP and its residues on different oxide surfaces and reaction products with certain halides;9,17,32 (b) neat liquid DIMP and its products with certain metal halides showing the split peak14,15 (c) neat liquid DIMP, its residue on alumina and reaction products with different metal perchlorates.4,11 | ||
A diverse dataset is available for the phosphoryl peak position in surface residues and products formed by liquid DMMP interacting with different solids, as shown in Fig. 8a. For all oxides, there are redshifts in the PO peak position smaller than that for the neat liquid.17 Conversely, for the chlorides, the redshifts are consistently stronger than for the neat liquid.51
For the products of liquid DIMP reacting with all metal halides,12 the PO peak splits into two, as shown in Fig. 8b (cf.Fig. 6b); the peak positions consistently exhibit redshifts compared to the neat liquid. The available data for the products formed by liquid DIMP reacting with metal perchlorates11 and forming a residue on alumina4 are shown in Fig. 8c. As for the liquid DMMP, the PO peak for the residue on alumina shows a smaller redshift compared to the neat liquid. Conversely, stronger than for the neat liquid redshifts (but no splits) are observed for the PO peaks in products of liquid DIMP interacting with all metal perchlorates.
A limited dataset exists in the literature characterizing the residues left on solid surfaces or products of interaction of either liquid or vapor-phase DMMP and DIMP with the same solids. These data are collected in Fig. 9. For comparison, hashed and solid-filled bars show positions of the PO peaks for the respective products and residues formed from the vapor and liquid organophosphonate phases. For DMMP, the data are available for several oxides (Fig. 9a). For all oxides, except MgO, the PO peaks for the residues formed from the vapor phase are observed at substantially lower wavenumbers than those in the residues formed from the liquid. Qualitatively, the same difference in peak positions is observed for the DIMP residues formed on alumina from it vapor and liquid phases (Fig. 9b). Conversely, for products formed with all metal halides, the PO peak positions are at higher wavenumbers for the vapors of DMMP or DIMP compared to similar products formed from the respective liquids. Additionally, peak splits are observed for the products of liquid DIMP with metal halides, which are also shown in Fig. 9b.
 | ||
| Fig. 9 PO peak position for vapor and liquid phase (a) DMMP and (b) DIMP interacting with the same solids.4,12,14,17,19,22,24,25 Hashed and solid-filled bars show the peak positions formed after interaction with the vapor-phase and liquid organophosphates, respectively. | ||
The available experimental data describing the PO peak shifts for residues and products formed by the same (either vapor-phase or liquid) organophosphonates on/with the same solids make it possible to explore correlations between the observed redshifts and characteristics of the respective metal ions. The PO peak positions for the DMMP and DIMP products on different solids as a function of the metal ionization energy (from ref. 66), formation energy (from ref. 67), and Pauling electronegativity (from ref. 68) are plotted in Fig. 10(a and b), (c and d) and (e and f), respectively. Symbols show reported data and lines show tentative implied correlations between the redshifts and respective material parameters. For the products of vapors of both DMMP and DIMP shown by the open symbols, the PO peaks redshift from their respective neat positions in both vapor and neat liquid phases. Relatively large data sets are available for the residues left by vapor phase DMMP and DIMP on oxides and for their reaction products with salts. For ionization energy, both sets form decaying trends in Fig. 10(a and b), showing increasing redshifts for metals with greater ionization energies. This indicates a strong attachment to the surface through the PO bond (cf.Fig. 2a). A similar decaying trend is noted for products formed with salts by liquid DIMP (Fig. 10b). While there is only one data point available for the residue left by liquid DIMP on alumina (Fig. 10b), for liquid DMMP more data points are available for its residues on oxides (Fig. 10a). Surprisingly, these data form an opposite trend, with a decreasing redshift (compared to neat vapor) with the increasing metal ionization energy. The interactions among the residues formed from the liquid-phase interaction of DIMP with the surface are stronger than their interactions with the surface itself. The diverging trends for the PO peak positions observed for residues left on the same oxide surfaces by vapor-phase and liquid DMMP (Fig. 10a and b) are likely pointing at a true difference in respective molecular interactions.
 | ||
| Fig. 10 PO peak position as a function of (a) ionization energy of the metal ions for DMMP interacting with inorganic solid (b) ionization energy of the metal ions for DIMP interacting with inorganic solid (c) heat of formation of the solid per metal atom for DMMP interacting with inorganic solid (d) heat of formation67 of the metal ions for DIMP interacting with inorganic solid (e) electronegativity67 of the metal ions for DMMP interacting with inorganic solid (f) electronegativity of the metal ions for DIMP interacting with inorganic solid. | ||
Similar trends in redshifts, including the divergence of the PO peak positions for residues formed from liquid and vapor phases, are also observed when the data are plotted as a function of the heat of formation of the solids (data for perchlorates were not available) normalized per metal atom in Fig. 10(a and b). An increasing heat of formation increases the redshift, indicating stronger interactions of the metal ions with the oxygen of the PO group. When the positions of the PO peak are plotted as a function of the Pauling electronegativity, they also form a qualitatively similar trend. For both DMMP and DIMP, an increase in electronegativity also increases the peak redshifts from the neat vapor position. For the lanthanide complexes (data in a dashed circle in Fig. 10f), the electronegativities are close to one another; their respective redshifts are also clustered closely together. The remaining open rectangular symbols are for transitional metal complexes. Although they seem to form a trend, they are also close to each other.
The observed correlations between PO peak position, ionization energy, formation energy, or electronegativity were approximately described using simple linear regressions, see eqn (1), where Y [cm−1] is the wavenumber of the PO peak position, X, is the independent variable (ionization energy, formation energy, or electronegativity), m is the slope, and C is the intercept.
| Y = mX + C | (1) |
O peak as functions of ionization energy, formation energy, and electronegativity
| Organophosphates | Phase | Surfaces | R 2 value | Characteristics of the linear fit | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Slope | Intercept, cm−1 | ||||||||||
| Ionization energy | Formation energy | Electronegativity | Ionization energy (cm−1 eV−1) | Formation energy (cm−1 kJ−1 per mole-metal) | Electronegativity (cm−1) | Ionization energy | Formation energy | Electronegativity | |||
| DMMP | Vapor | Oxides | 0.72 | 0.03 | 0.15 | −1.81 | −0.011 | −19.36 | 1252 | 1230 | 1253 |
| Liquid | 0.71 | 0.35 | 0.42 | 0.86 | 0.027 | 18.77 | 1243 | 1242 | 1224 | ||
| DIMP | Vapor | Halides | 0.51 | 0.35 | 0.03 | −2.69 | −0.035 | −19.49 | 1241 | 1208 | 1222 |
| Liquid | 0.79 | 0.15 | 0.12 | −3.11 | −0.044 | −28.58 | 1220 | 1197 | 1190 | ||
| Liquid | Perchlorates | 0.63 | — | 0.02 | −1.88 | — | −8.72 | 1205 | — | 1178 | |
The analysis shown in Fig. 10 suggests that different processes affect how the vapor and liquid organophosphonates adsorb to and interact with different materials. One possible interpretation of such differences may be relevant to the energy with which molecules approach the surface from the vapor and liquid phases. Multiple molecular interactions significant in a liquid, including, for example, steric strain and hindrance, may limit the formation of the bonds with the solid surface, while such limitations do not exist for the vapor phase–surface interactions.
Whatever the reasons for different energies of the PO bond formed by the organophosphonates with surfaces, these differences will be essential to predicting subsequent reactions describing the decomposition of organophosphonates and formation of products, e.g., in respective molecular and atomistic simulations. Both DIMP and DMMP create surface-bound products when they come in contact with the surfaces. For example, in the case of metal oxides, they preferentially attach to acid sites, as shown in Fig. 2a. Subsequent reactions produce phosphorus-bearing complexes that are still bound to the surface;9,22,28,70 some such complexes are illustrated in Fig. 11. Among other factors, the strength of the initially formed PO bond is clearly expected to be important in affecting which complex forms and how it decomposes further. The trends identified here and describing the changes in the PO bond strength should thus be accounted for in future models targeting the respective reactions.
 | ||
| Fig. 11 Possible residue formation paths for DMMP and DIMP on metal oxide surfaces. | ||
The present analysis suggests that more experimental studies addressing the interaction of organophosphonates with solids involving different groups of metals are needed to better understand and quantify the effect of the initial organophosphonate phase (liquid vs. gas) as well as combined effects of ionization energy, electronegativity, and formation energy correlations on the PO shifts (and thus, bond strengths) of the adsorbed moieties. Batch experiments8,13,19,21,40,49 (cf.Fig. 4) combining infrared spectroscopy with elemental analysis, e.g., ICP-MS and others focused on specific sets of solids interacting with both liquid and vapor-phase organophosphates will be particularly instructive. Residues recovered from the same surfaces exposed to both vapor and liquid organophosphonates should be analyzed, in particular.
5. Conclusions
Reviewing the available literature shows a significant shift in the PO peak in the residues and products formed when organophosphonates interact with solids, with variations depending on both the initial phase of the organophosphonate and the type of solid. Both products of reaction and surface residues formed by vapor-phases of both DMMP and DIMP reacting with all solids exhibited redshifts in the PO peak positions, indicating molecular interactions stronger than in respective neat liquids.
In contrast, a different pattern emerged for surface residues of liquid organophosphonates interacting with different oxides. Compared to the respective neat liquids, the redshifts of the PO peak were smaller for the surface residues on metal oxides. Thus, molecular interactions are weakened in the surface residues left on oxides as compared to those in the respective neat liquids. For the products of reaction of liquid organophosphonates with metal salts, the redshifts were (similarly to the respective products formed from vapor-phase organophosphonates) stronger than in the respective neat liquids, pointing to the respectively stronger molecular interactions.
The ionization energy of a metal present in the solid interacting with the organophosphonate plays a role in the type of the formed residue or product: higher ionization energies cause stronger shifts of the PO peak from its original position, whether such shifts point to stronger or weaker molecular interactions compared to those in the respective neat liquids. Molecular interactions in products of both DMMP and DIMP reacting with metal salts are stronger than in the surface residues left by the same compounds on metal oxide surfaces, irrespective of the starting phase. The shifts in the PO bond correlating with electronegativity and formation energy of the respective metal bonds support the theory of organophosphonates bonding to solids through the PO group. The difference between redshifts for residues of vapor and liquid phase organophosphates indicates differences in the residue formation. Future studies should focus on systematic variations of temperatures, pressures, and other parameters affecting the interaction of both vapor and liquid phases of organophosphates, such as DIMP and DMMP, with different inorganic solids involving different groups of metals. The results will provide a more in-depth understanding of correlations between the residue formation and the underlying chemical characteristics of the surfaces, such as ionization energy, the heat of formation, or electronegativity, all affecting the PO bond redshifts. Correlations observed among these parameters can guide the development of better models capable of predicting with confidence the residues left on solids by organophosphonates.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are very grateful to Prof. A. Khalizov of NJIT for many useful discussions and suggestions. This work was supported by the US Defense Threat Reduction Agency, Award HDTRA12020001.References
- H. Follows, Organisation for the Prohibition of Chemical Weapons, 2005 Search PubMed.
- S. Chauhan, R. D'cruz, S. Faruqi, K. Singh, S. Varma, M. Singh and V. Karthik, Environ. Toxicol. Pharmacol., 2008, 26, 113–122 CrossRef CAS PubMed.
- M. Schwenk, Toxicol. Lett., 2018, 293, 253–263 CrossRef CAS PubMed.
- A. Vasudevan, E. I. Senyurt, M. Schoenitz and E. L. Dreizin, J. Hazard. Mater., 2023, 443, 130154 CrossRef CAS PubMed.
- R. A. Moyer, F. R. Sidell and H. Salem, in Encyclopedia of Toxicology, ed. P. Wexler, Academic Press, Oxford, 3rd edn, 2014, pp. 483–488, DOI:10.1016/B978-0-12-386454-3.00635–7.
- S. L. Bartelt-Hunt, D. R. U. Knappe and M. A. Barlaz, Crit. Rev. Environ. Sci. Technol., 2008, 38, 112–136 CrossRef CAS.
- W.-C. Hung, J.-C. Wang and K.-H. Wu, Appl. Surf. Sci., 2018, 444, 330–335 CrossRef CAS.
- V. N. Sheinker and M. B. Mitchell, Chem. Mater., 2002, 14, 1257–1268 CrossRef CAS.
- M. Šťastný, J. Tolasz, V. Štengl, J. Henych and D. Žižka, Appl. Surf. Sci., 2017, 412, 19–28 CrossRef.
- C. Tesvara, C. Walenta and P. Sautet, Phys. Chem. Chem. Phys., 2022, 24, 23402–23419 RSC.
- N. M. Karayannis, C. Owens, L. L. Pytlewski and M. M. Labes, J. Inorg. Nucl. Chem., 1969, 31, 2059–2071 CrossRef CAS.
- N. M. Karayannis, C. M. Mikulski, M. J. Strocko, L. L. Pytlewski and M. M. Labes, Inorg. Chim. Acta, 1971, 5, 357–361 CrossRef CAS.
- G. G. Guilbault and E. P. Scheide, J. Inorg. Nucl. Chem., 1970, 32, 2959–2962 CrossRef CAS.
- N. M. Karayannis, C. M. Mikulski, M. J. Strocko, L. L. Pyptlewski and M. M. Labes, Z. Anorg. Allg. Chem., 1971, 384, 267–279 CrossRef CAS.
- M. M. Labes, C. Owens, N. M. Karayannis and L. L. Pytlewski, J. Phys. Chem., 1971, 75, 637–641 CrossRef CAS.
- V. M. Bermudez, Langmuir, 2013, 29, 1483–1489 CrossRef CAS PubMed.
- B. Aurian-Blajeni and M. M. Boucher, Langmuir, 1989, 5, 170–174 CrossRef CAS.
- E. I. Senyurt, S. Das, T. Kenny, L. J. Groven, M. Schoenitz and E. L. Dreizin, Appl. Surf. Sci., 2024, 672, 160853 CrossRef.
- G. G. Guilbault and J. Das, J. Phys. Chem., 1969, 73, 2243–2247 CrossRef CAS.
- J. J. G. M. van Bokhoven, A. E. T. Kuiper and J. Medema, J. Catal., 1976, 43, 168–180 CrossRef CAS.
- G. G. Guilbault, E. Scheide and J. Das, Spectrosc. Lett., 1968, 1, 167–175 CrossRef CAS.
- M. K. Templeton and W. H. Weinberg, J. Am. Chem. Soc., 1985, 107, 774–779 CrossRef CAS.
- Y. X. Li and K. J. Klabunde, Langmuir, 1991, 7, 1388–1393 CrossRef CAS.
- S. Holdren, R. Tsyshevsky, K. Fears, J. Owrutsky, T. Wu, X. Wang, B. W. Eichhorn, M. M. Kuklja and M. R. Zachariah, ACS Catal., 2019, 9, 902–911 CrossRef CAS.
- C. S. Kim, R. J. Lad and C. P. Tripp, Sens. Actuators, B, 2001, 76, 442–448 CrossRef CAS.
- W. O. Gordon, B. M. Tissue and J. R. Morris, J. Phys. Chem. C, 2007, 111, 3233–3240 CrossRef CAS.
- L. Trotochaud, R. Tsyshevsky, S. Holdren, K. Fears, A. R. Head, Y. Yu, O. Karslıoğlu, S. Pletincx, B. Eichhorn, J. Owrutsky, J. Long, M. Zachariah, M. M. Kuklja and H. Bluhm, Chem. Mater., 2017, 29, 7483–7496 CrossRef CAS.
- M. B. Mitchell, V. N. Sheinker and E. A. Mintz, J. Phys. Chem. B, 1997, 101, 11192–11203 CrossRef CAS.
- D. A. Chen, J. S. Ratliff, X. Hu, W. O. Gordon, S. D. Senanayake and D. R. Mullins, Surf. Sci., 2010, 604, 574–587 CrossRef CAS.
- A. R. Head, X. Tang, Z. Hicks, L. Wang, H. Bleuel, S. Holdren, L. Trotochaud, Y. Yu, L. Kyhl, O. Karslıoǧlu, K. Fears, J. Owrutsky, M. Zachariah, K. H. Bowen and H. Bluhm, Catal., Struct. React., 2017, 3, 112–118 CAS.
- E. D. Davis, W. O. Gordon, A. R. Wilmsmeyer, D. Troya and J. R. Morris, J. Phys. Chem. Lett., 2014, 5, 1393–1399 CrossRef CAS PubMed.
- J. Henych, P. Janoš, M. Kormunda, J. Tolasz and V. Štengl, Arabian J. Chem., 2019, 12, 4258–4269 CrossRef CAS.
- A. E. T. Kuiper, J. J. G. M. van Bokhoven and J. Medema, J. Catal., 1976, 43, 154–167 CrossRef CAS.
- R. M. Crooks, H. C. Yang, L. J. McEllistrem and R. C. Thomas, Faraday Discuss., 1997, 107, 285–305 RSC.
- A. Kiselev, A. Mattson, M. Andersson, A. E. C. Palmqvist and L. Österlund, J. Photochem. Photobiol., A, 2006, 184, 125–134 CrossRef CAS.
- D. A. Panayotov and J. R. Morris, Langmuir, 2009, 25, 3652–3658 CrossRef CAS PubMed.
- D. A. Panayotov and J. R. Morris, J. Phys. Chem. C, 2009, 113, 15684–15691 CrossRef CAS.
- C. Kim, R. Lad and C. Tripp, Sens. Actuators, B, 2001, 76, 442–448 CrossRef CAS.
- A. R. Head, R. Tsyshevsky, L. Trotochaud, B. Eichhorn, M. M. Kuklja and H. Bluhm, J. Phys. Chem. A, 2016, 120, 1985–1991 CrossRef CAS PubMed.
- L. Cao, S. R. Segal, S. L. Suib, X. Tang and S. Satyapal, J. Catal., 2000, 194, 61–70 CrossRef CAS.
- X. Tang, Z. Hicks, L. Wang, G. Ganteför, K. H. Bowen, R. Tsyshevsky, J. Sun and M. M. Kuklja, Phys. Chem. Chem. Phys., 2018, 20, 4840–4850 RSC.
- S. Ma, J. Zhou, Y. C. Kang, J. E. Reddic and D. A. Chen, Langmuir, 2004, 20, 9686–9694 CrossRef CAS PubMed.
- L. Trotochaud, A. R. Head, C. Büchner, Y. Yu, O. Karslıoğlu, R. Tsyshevsky, S. Holdren, B. Eichhorn, M. M. Kuklja and H. Bluhm, Surf. Sci., 2019, 680, 75–87 CrossRef CAS.
- A. R. Head, R. Tsyshevsky, L. Trotochaud, Y. Yu, L. Kyhl, O. Karslıoǧlu, M. M. Kuklja and H. Bluhm, J. Phys. Chem. C, 2016, 120, 29077–29088 CrossRef CAS.
- J. Zhou, K. Varazo, J. Reddic, M. Myrick and D. Chen, Anal. Chim. Acta, 2003, 496, 289–300 CrossRef CAS.
- L. Cao, S. L. Suib, X. Tang and S. Satyapal, J. Catal., 2001, 197, 236–243 CrossRef CAS.
- W. Graven, S. Weller and D. Peters, Ind. Eng. Chem. Process Des. Dev., 1966, 5, 183–189 CrossRef CAS.
- N. M. Karayannis, C. M. Mikulski and L. L. Pytleweski, Inorg. Chim. Acta, Rev., 1971, 5, 69–105 CrossRef CAS.
- S. R. Segal, L. Cao, S. L. Suib, X. Tang and S. Satyapal, J. Catal., 2001, 198, 66–76 CrossRef CAS.
- M. K. Templeton and W. H. Weinberg, J. Am. Chem. Soc., 1985, 107, 97–108 CrossRef CAS.
- C. Mikulski, N. Karayannis and L. Pytlewski, J. Inorg. Nucl. Chem., 1974, 36, 971–981 CrossRef CAS.
- R. J. Piffath, Infrared Spectroscopic Observations on the Fate of Organophosphorus Compounds Exposed to Atmospheric Moisture Part I. G-Agents and Related Compounds, Defense Technical Information Center, Report ECBC-TR-323, 2003 Search PubMed.
- S. Biswas and B. M. Wong, J. Phys. Chem. C, 2021, 125, 21922–21932 CrossRef CAS.
- V. Kumar, N. Upadhyay, A. B. Wasit, S. Singh and P. Kaur, Curr. World Environ., 2013, 8(2), 313–318 CrossRef.
- R. Tsyshevsky, S. Holdren, B. W. Eichhorn, M. R. Zachariah and M. M. Kuklja, J. Phys. Chem. C, 2019, 123, 26432–26441 CrossRef CAS.
- A. R. Head, R. Tsyshevsky, L. Trotochaud, Y. Yu, O. Karslıoǧlu, B. Eichhorn, M. M. Kuklja and H. Bluhm, J. Phys.:Condens. Matter, 2018, 30, 134005 CrossRef PubMed.
- C. A. Walenta, F. Xu, C. Tesvara, C. R. O'Connor, P. Sautet and C. M. Friend, J. Phys. Chem. C, 2020, 124, 12432–12441 CrossRef CAS.
- A. Michalkova, M. Ilchenko, L. Gorb and J. Leszczynski, J. Phys. Chem. B, 2004, 108, 5294–5303 CrossRef CAS.
- V. Bermudez, J. Phys. Chem. C, 2007, 111, 3719–3728 CrossRef CAS.
- V. Bermudez, J. Phys. Chem. C, 2009, 113, 1917–1930 CrossRef CAS.
- N. Sharma and R. Kakkar, J. Comput. Sci., 2015, 10, 225–236 CrossRef.
- T. Ash, T. Debnath, A. Ghosh and A. K. Das, Chem. Res. Toxicol., 2017, 30, 1177–1187 Search PubMed.
- L. Yang, D. Tunega, L. Xu, N. Govind, R. Sun, R. Taylor, H. Lischka, W. A. DeJong and W. L. Hase, J. Phys. Chem. C, 2013, 117, 17613–17622 CrossRef CAS.
- P. M. Chu, F. R. Guenther, G. C. Rhoderick and W. J. Lafferty, J. Res. Natl. Inst. Stand. Technol., 1999, 104, 59 CrossRef CAS.
- L. Bertilsson, I. Engquist and B. Liedberg, J. Phys. Chem. B, 1997, 101, 6021–6027 CrossRef CAS.
- D. R. Lide and W. Haynes, J. Am. Chem. Soc., 2009, 131, 12862–12862 Search PubMed.
- D. D. Wagman, W. H. Evans, V. B. Parker, R. H. Schumm, I. Halow, S. M. Bailey, K. L. Churney and R. L. Nuttall, J. Phys. Chem. Ref. Data, 1982, 11(Supplement 2), 392 Search PubMed.
- J. E. Huheey, Principles of structure and reactivity, Harper & Row, 1983 Search PubMed.
- X. Su, X. Yan and C.-L. Tsai, WIREs Comput. Stat., 2012, 4, 275–294 CrossRef.
- B. Yuan and H. Eilers, Combust. Flame, 2019, 199, 69–84 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |