
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradation of chemical warfare simulants over CeO2 and Gd-doped CeO2 aerogels: divergent results of DMMP and DFP†
Travis G.
Novak
 *a,
Janna
Domenico
b,
Alex
Balboa
c,
Wesley O.
Gordon
c,
Austin E.
Herzog‡
d,
Nam Q.
Le
b,
Evan R.
Glaser
a,
Paul A.
DeSario§
a and
Debra R.
Rolison
a
*a,
Janna
Domenico
b,
Alex
Balboa
c,
Wesley O.
Gordon
c,
Austin E.
Herzog‡
d,
Nam Q.
Le
b,
Evan R.
Glaser
a,
Paul A.
DeSario§
a and
Debra R.
Rolison
a
aU.S. Naval Research Laboratory, Washington, D.C., USA. E-mail: travis.g.novak.civ@us.navy.mil
bJohns Hopkins University Applied Physics Laboratory, Laurel, MD, USA
cU.S. Army Combat Capabilities Development Command Chemical Biological Center, Aberdeen Proving Ground, MD, USA
dU.S. Naval Research Laboratory, Washington, D.C., USA
First published on 7th February 2025
Abstract
Ceria (CeO2) is a promising material for binding and degrading organophosphorus chemical warfare agents (CWAs). This oxide boasts reactive surface hydroxyls, offers a photo-excitable band gap state, and exhibits a propensity to form reactive oxygen species (ROS). Given the challenges of working with live CWAs, researchers generally evaluate CeO2 and other reactive sorbents against a simulant molecule, but these simulants may differ in degradative pathways and fail to accurately predict performance of the sorbent against CWAs. Here, we report key divergence in the properties of Gd-doped CeO2 (GCO) aerogels against two simulants, finding that Gd3+ substitution for Ce4+ impedes degradation of dimethyl methylphosphonate (DMMP) but improves degradation of diisopropyl fluorophosphonate (DFP). We attribute the difference to enhanced ROS stabilization on the GCO surface. Computational analysis of the two simulants as well as the CWA sarin (GB) reveals ROS improve the binding of fluorophosphorus molecules but not fluorine-free DMMP. Our findings identify GCO as a potentially effective material against CWAs and highlight the limits of DMMP as an arbiter for evaluating materials' efficacy.
Introduction
Toxic organophosphorus (OP) compounds, such as chemical warfare agents (sarin, soman) and common pesticides (parathion, malathion) pose serious health threats to both military and civilian populations. The traditional filtration media employed for protection against OP molecules consist of activated charcoal or other high surface-area carbon materials, which typically bind the intact OP molecule with minimal degradation,1,2 making the exposed material a hazard in itself, creating additional disposal burdens, and eliminating any possibility of catalytic turnover.Recent research focuses on high surface-area oxides3 or metal–organic frameworks (MOFs)4 to unlock destructive adsorption of OP threats. Ceria (CeO2), in particular, offers many properties favorable for OP adsorption and degradation.5 CeO2 strongly bonds phosphoryl groups (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at Lewis acid sites6 and attacks OP compounds through surface OH groups.7 Under UV illumination, CeO2 can photogenerate reactive oxygen species (ROS), notably superoxide (O2−), that effectively catalyze various reactions.8 Our group recently described alkaline-treated CeO2 aerogels as a potent material for destructive adsorption of dimethyl methylphosphonate (DMMP),9 a commonly studied simulant for chemical warfare agents. The hydroxylated surface of CeO2 aerogels generate hydrolysis products in the dark and more fully mineralized oxidation products under UV illumination, outperforming both TiO2 and commercial CeO2 nanoparticles under the same conditions.
O) at Lewis acid sites6 and attacks OP compounds through surface OH groups.7 Under UV illumination, CeO2 can photogenerate reactive oxygen species (ROS), notably superoxide (O2−), that effectively catalyze various reactions.8 Our group recently described alkaline-treated CeO2 aerogels as a potent material for destructive adsorption of dimethyl methylphosphonate (DMMP),9 a commonly studied simulant for chemical warfare agents. The hydroxylated surface of CeO2 aerogels generate hydrolysis products in the dark and more fully mineralized oxidation products under UV illumination, outperforming both TiO2 and commercial CeO2 nanoparticles under the same conditions.
Although the reactivity of CeO2 nanostructures against OP compounds is widely acknowledged, it remains to be seen how well this efficacy translates to reactivity against chemical warfare agents. Common OP molecules studied, such as DMMP, diisopropyl methylphosphonate (DIMP),10,11 or p-nitrophenylphosphate (DMNP),12 do not contain fluorine, but the P–F bond in sarin is highly reactive and could impact degradation pathways. Over other reactive oxides, such as CuO (ref. 13) and TiO2,14 degradation of sarin proceeds through scission of the P–F bond. To date, the only study evaluating CeO2 against a live agent (sarin) did so in highly idealized conditions (O2-free), and did not evaluate any potential photodegradation pathways.15
Here, we evaluate CeO2 and Gd3+-doped CeO2 (GCO) aerogels for destructive sorption of the fluorine-containing chemical warfare simulant diisopropyl fluorophosphate (DFP), contrasting the results against the more commonly-studied DMMP. We find that while Gd3+ doping slightly impedes reactivity against DMMP, it improves DFP degradation, showing key divergence between the simulant molecules. Computational analysis and electron paramagnetic resonance (EPR) spectroscopy reveal that the defective surface of GCO, with its population of stabilized ROS, more strongly binds DFP, but not necessarily DMMP.
Results and discussion
Synthesis and characterization of CeO2 and GCO aerogels
We synthesize CeO2 and Gd3+-doped CeO2 (GCO) aerogels using previously reported sol–gel routes utilizing chloride precursors followed by supercritical drying in CO2.9,16,17 Our previous study of DMMP degradation over CeO2 aerogels reveals that residual Cl from the synthesis blocks Lewis acid sites and terminal hydroxyl formation, leading to minimal degradation of DMMP.9 To remove residual Cl and promote surface –OH groups, we soak the aerogels in a mildly alkaline solution (pH 9.5) for 48 h. An illustration of the GCO surface (Fig. 1a) highlights the features predicted to be beneficial for CWA degradation (terminal –OH groups, reactive oxygen species, surface oxygen vacancies, and Lewis acid sites). | ||
| Fig. 1 (a) Schematic of GCO showing features for OP binding/degradation. (b) XPS Cl 2p region and (c) XPS O1s region for CeO2 and GCO aerogels before and after the alkaline soak. (d) Raman spectroscopy with inset showing the shift and broadening of the F2g peak. | ||
Brunauer–Emmett–Teller (BET) surface area analysis (Fig. S1†) reveals that the alkaline treatment does not significantly affect aerogel morphology, with the post-treatment surface area calculated to be within 5% of the starting value. Gd3+ substitution into Ce sites slows crystallite growth in the aerogels, resulting in substantially higher surface area (142.4 m2 g−1) for GCO compared to CeO2 (80.2 m2 g−1).17 Average Barrett–Joyner–Halenda (BJH) pore sizes of CeO2 and GCO aerogels are 7.7 and 6.8 nm, respectively, which is twice that of template-based approaches to CeO2 (3.6 nm)6 used for OP degradation.15 DFT pore-size distribution plots (Fig. S2†) show a greater incremental volume of 2–10 nm pores for GCO, while CeO2 has a higher fraction of 10–20 nm pores.
X-ray photoelectron spectroscopy (XPS) establishes that the alkaline treatment changes the surface chemistry. GCO has more residual Cl than CeO2, perhaps a product of its higher surface area or strong Cl binding at oxygen vacancies. The calculated Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ce ratio (or Cl:Ce + Gd in the case of GCO) is 0.25 for CeO2 and 0.38 for GCO. Residual Cl falls to trace levels in both aerogels after the alkaline treatment (Fig. 1b).
:Ce ratio (or Cl:Ce + Gd in the case of GCO) is 0.25 for CeO2 and 0.38 for GCO. Residual Cl falls to trace levels in both aerogels after the alkaline treatment (Fig. 1b).
The two broad peaks in the O 1s region (Fig. 1c) correspond to lattice oxygen in CeO2 (Oa, 528.5 eV),18,19 while the higher binding energy peak at 530.7 eV (Ob) convolves O bound to Ce3+ (ref. 19) and various Ce hydroxides.20,21 After Cl removal, the Ob peak increases in intensity, indicative of additional hydroxyl groups on the surface. The 2.2 eV gap between the binding energies of Oa and Ob speciation matches previously reported values for terminal hydroxyls;22 bridging hydroxyls typically have a gap of <1.5 eV.23,24 This Ob increase is more pronounced for GCO compared to CeO2, indicating the GCO surface is richer in OH groups than CeO2. A comparison of commercial CeO2 nanoparticles (COM-CeO2) titrated to pH 3.0 or pH 5.0 using aqueous HCl prior to the alkaline treatment (Fig. S3†) yields minimal differences in the O 1s region, showing that the higher OH content in GCO is likely a result of its higher surface area and Gd3+-fixed concentration of oxygen vacancies rather than the higher Cl content. The Raman spectra (Fig. 1d) show a shift of the main F2g peak, likely related to smaller crystallites in GCO, as well as the appearance of α and β peaks in the GCO (Fig. 1d), indicating abundant oxygen-vacancies not present in CeO2.25
IR analysis of simulant degradation
We compare the performance of aerogels through in situ diffuse-reflectance infrared Fourier transform spectroscopy (DRIFTS), where the powdered sample is exposed to DFP for 1 h (dose step), then DFP vapor flow is stopped while continuing the flow of zero air (purge step). Fig. 2 shows a comparison of four aerogel samples during the dose step under dark conditions: CeO2, GCO, and the alkaline-soaked variants of each (designated CeO2-s and GCO-s) during the dose step under dark conditions. Unlike our previous report on DMMP degradation with CeO2 and CeO2-s aerogels, where the pristine CeO2 bound but did not degrade DMMP,9 we observe abundant products of DFP degradation over all aerogel variants. A previous study asserts that DFP is more easily hydrolyzed than DMMP due to the presence of F in the molecule,26 which may explain the higher apparent reactivity against DFP in the dark. A schematic of possible degradation pathways of DFP, DMMP, and sarin (GB) is shown in Fig. S4.† | ||
| Fig. 2 DRIFTS spectra under DFP dosing over four aerogels variants. Spectra are recorded at 1 min intervals and progress from blue to red. | ||
Although all four aerogels degrade DFP, analysis of the product distribution suggests different pathways are present. The degradation pathway is determined by two factors i) the presence of oxygen vacancies, which increases for GCO compounds and ii) whether the surface is rich with terminal hydroxyls from the soak step or residual chloride from aerogel synthesis. Signals in both CeO2-s and GCO-s are obscured due to large OH/H2O losses. All aerogels show a loss of hydroxyl stretching groups at 3660 cm−1 and 3690 cm−1, as well as a bending –OH mode at 1330 cm−1 and 1550 cm−1 for soaked samples. However, the losses are more severe for the soaked samples corresponding to the higher amount of surface hydroxyl groups bound, with roughly equal losses for both CeO2 and GCO. Peaks at 2975 cm−1 and from 940–1155 cm−1 are attributable to POC–H and P–O–C stretches, respectively, and are only present in the non-soaked samples, but whether these occur from intact or partially degraded DFP is unclear. A PO peak at 1220 cm−1 could correspond to either intact DFP or phosphonic acid, but its value is closer to the latter.27–29
Starting with CeO2, dosing with DFP generates primarily alcohol products, as evidenced by an OH stretch at 3150 cm−1, and an OH bend at 1415 cm−1. The C–OH stretch at 1095 cm−1 arises from hydrolysis of the P–OCR bond. A large peak at 1690 cm−1 corresponds to a CO stretch, likely from the oxidation of the hydrolysis product into acetone by an oxygen vacancy. A small peak from 1620–1570 cm−1 is identified as a metal-alkoxide C–O stretch (MC–O) from surface-bound degradation product. The C–H bend of these moieties occurs between 1100–1200 cm−1 and are mixed with the other identified peaks.30,31
The CeO2-s aerogel vibrational spectrum is largely the same as that of the CeO2 aerogel except for the greater decrease in the intensity of OH/H2O modes. However, in this sample the metal-alkoxide peak shifts to 1610 cm−1 and is more prominent than in the unsoaked aerogel, due to interacting with the abundant hydroxyls on the surface. The C–O stretch of the alcohol also shifts from 1095 cm−1 to 1125 cm−1 and broadens compared to CeO2, possibly from this same surface interaction with the hydroxyl groups. The GCO-s aerogel is nearly identical to the CeO2-s aerogel, although the intensity assigned to the CO oxidation product increases compared to the alcohol hydrolysis product. This increase in the oxidized product is likely encouraged by the additional oxygen vacancies present when Gd3+ is incorporated. GCO has the largest deviation in observed products, as the presence of alcohol products is nearly undetectable, instead yielding almost entirely CO ketone product, with a trace of the metal-alkoxide C–O stretch mentioned for CeO2 and the P containing peaks.
During the purge step after DFP exposure (Fig. 3), we see a large increase in the O–H stretches of the hydrolyzed alcohol product as well as the mineralized PO stretch in the CeO2 aerogel. A peak appears at 1045 cm−1, which most likely corresponds to the P–O–C stretch of partially or undegraded DFP. We also see a strong increase in the C–O stretch of an alcohol for CeO2-s, although it shifts to 1065–1005 cm−1 without a corresponding increase in the O–H alcohol stretch. This result is likely caused by the deprotonation of the alcohol from the surface hydroxyl groups. The GCO sample shows a large increase in mineralized PO stretch, as well as a broad shoulder at 1250 cm−1 that may arise from a PO–H interaction. As with CeO2, the 1045 cm−1 peak of partially or undegraded DFP is prominent, as well as a more noticeable POC–H stretch at 2975 cm−1 in the absence of the alcohol O–H peaks. This P–O–C stretch may shift due to H-bonding interactions with surface hydroxyl groups, but assignment is obscured by the presence of other peaks. GCO-s also shows an increase in deprotonated alcohol peaks as well as mineralized PO stretches, but with a noticeable loss of CO peaks as the acetone is purged from the surface during this step.
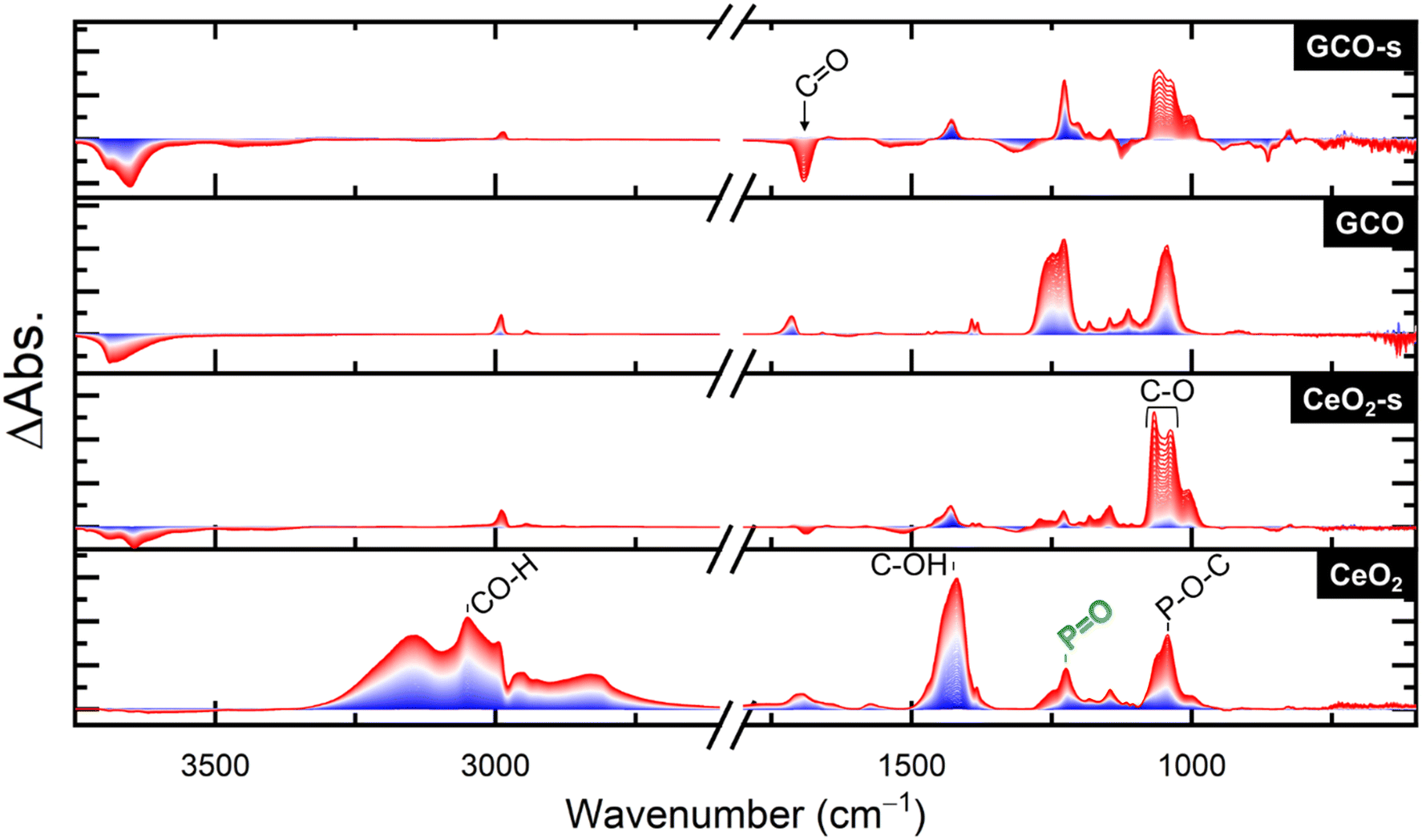 | ||
| Fig. 3 DRIFTS spectra under a N2 + O2 purge after DFP exposure. Spectra are recorded at 1 min intervals and progress from blue to red. | ||
When exposed to broadband (BB) illumination (Fig. S5†), a variety of peaks appear corresponding to C–F IR modes (1300–900 cm−1).32 Due to the numerous new peaks identified, the photo-fluorination seems to show no clear site specificity. The spectra for CeO2-s and GCO-s are nearly identical from 1300–950 cm−1, with peaks corresponding to C–F stretches in hydrolysis products. The only difference between the two spectra is a shoulder at 1010 cm−1 assigned to halogenated acetone, present in every sample except for CeO2-s. The CeO2 aerogel matches the soaked samples up to 1200 cm−1, but at higher wavenumbers more closely matches the GCO sample. The peaks from 1230–1270 and 1370–1400 cm−1 are indicative of C–Cl stretches;33,34 their absence in CeO2-s and GCO-s further substantiates removal of surface Cl. GCO differs from the other aerogels by the absence of a 1030 cm−1 peak where the C–F peaks in the alcohol species are present. The identification of this peak as a ketone C–F stretch, which we noted as a shoulder for CeO2 and GCO-s, informs us that little or no oxidized hydrolysis product forms over CeO2-s. Surface hydroxyls in CeO2-s may effectively fill or cover most surface oxygen vacancies, and so this catalyst forms mostly alcohol hydrolysis products with acetone lost in the purge step.
From these DRIFTS data, we conclude that both the soaked aerogels show faster appearance of the mineralized PO peak corresponding to faster hydrolysis of DFP. While hydrolysis to alcohol is present for both soaked aerogels, the extra oxygen vacancies in GCO contribute to further oxidation of these alcohols to acetone, most prevalently seen in unsoaked GCO, which solely makes this oxidized product. The acetone purges more easily from the surface than the alcohol product. Upon BB illumination, remaining organic products are photo-halogenated, inserting either DFP-derived F or residual surface Cl into these compounds.
Given the differences in product formation over the four aerogels, it is difficult to determine the degree of DFP degradation based on DRIFTS analysis alone. The amount of intact DFP remaining is determined using dose extraction and analysis using a GC-MS, which identifies products that remain bound to the surface after DFP exposure. Differences in the reactivity of pristine and alkaline-treated aerogels become apparent here, with CeO2-s and GCO-s showing far less intact simulant than with CeO2 and GCO (Fig. S6†). GCO-s also slightly outperforms CeO2-s in terms of intact DFP remaining, despite its higher surface area likely allowing for greater uptake. CeO2-s and GCO-s both outperform commercial CeO2 nanoparticles (COM-CeO2), which do not suffer from residual chlorine but have much lower surface area. Very few other products were unambiguously identifiable in the GC-MS, with the exception of methyl phosphonic acid (MPA) in the case of CeO2 aerogel. We note that phosphonic acid, a product of highly degraded DFP, would not be detectable using this protocol.35 Phosphonic acid is a likely product given the lack of other detectable products as well as the prominent PO peaks observed in DRIFTS. Similar to our previous results with DMMP9 and other recent work with oxides against DFP,29 we find that terminal hydroxyls are effective in promoting more complete degradation of OP simulants. The abundant terminal hydroxyls present on alkaline-treated CeO2-s and GCO-s aerogels drive hydrolysis of DFP, while residual Cl content in pristine CeO2 and GCO prevents terminal hydroxyl formation.
Although the effect of the alkaline treatment is consistent between simulants, the improved performance against DFP with GCO-s vs. CeO2-s contrasts with our findings against DMMP, where Gd3+-doping slightly impeded degradation. While both aerogel variants show product formation, the relative intensity of the O–P–O modes, both during dose (Fig. S7†) and purge (Fig. S8†) steps, indicates a greater fraction of product forms over CeO2-s. A previous study concludes that doping CeO2 with another trivalent cation (Y3+) similarly impedes performance against DMMP.36
Computational insights into OP adsorption on pristine and defective CeO2
To further elucidate the differences observed experimentally between CeO2 and GCO and between simulants, we modeled the interactions of DMMP, GB (sarin), and DFP with CeO2 surfaces using density functional theory (DFT). Two surface models were evaluated in terms of adsorption by simulants and agents: a pristine CeO2 (111) surface and a defective CeO2 (111) surface with surface oxygen vacancies, the latter being a model for the GCO material. To simulate the O2-rich reaction conditions in this investigation, we model the defective surfaces in the presence of an O2 molecule (Fig. S9†).The formation of an oxygen vacancy (Ovac) creates excess donor electrons at the surface in Ce 4f states, creating a corresponding electronic defect state within the bandgap. The formation of the surface Ovac is unfavorable by 1.8 eV per defect at CeO2 (111) based on differences in electronic internal energy, in good agreement with other literature reports.37 Upon binding of neutral O2, these excess donor electrons partially localize near the bound species, and the resulting equilibrium O–O distance of 1.44 Å is consistent with a peroxide O22− ion. At the CeO2 (111) surface, O22− binding to an Ovac is thermodynamically favorable by 1.6 eV per molecule with respect to the internal electronic energy.
Models built from the optimized surface and OP geometries were used to predict stable geometries of DMMP, GB, and DFP on pristine CeO2 and defective CeO2 (Fig. 4). Energies corresponding to these structures were used to compute binding energies, which are listed in Table 1. We note two trends in the binding energies. First, the binding of each OP molecule is predicted to be stronger on the defective surface than on the pristine surface. Second, on both surfaces, DMMP is predicted to bind more weakly (−19.8 to −20.9 kcal mol−1) than GB and DFP (−22.5 to −27.5 kcal mol−1). DFP may therefore be a better simulant for GB in terms of adsorption characteristics on CeO2 surfaces. Interestingly, these trends are not correlated with trends in the length of the dative (PO)–Ce bond, which is relatively consistent: 2.48 to 2.54 Å. We therefore infer that, while the dative bond is still likely the strongest single interaction, as in other OP-oxide systems, its strength is relatively insensitive, and trends in overall binding energy are instead attributed to differences in other side group interactions with the surface.
 | ||
| Fig. 4 Bound geometries of organophosphorus (OP) compounds on two variations of CeO2 surfaces computed using DFT. (a) The pristine CeO2 (111) surface with no adsorbate and then with bound (b) DMMP, (c) GB, and (d) DFP. (e) The defective CeO2 surface with surface Ovac and bound O22− and then (f–h) bound with the same three OP adsorbates. Key interatomic distances are labeled in each system, and distances are highlighted in bold between the OP phosphorus center and co-adsorbed O22−. Atom colors are C: gray, H: white, O: red (purple in O22−), P: orange, F: green, Ce: off-white. | ||
On the pristine surface, these other interactions are dominated by hydrogen bonding between the OP compound and surface oxygen sites, consistent with recent reports by Li et al.6,15 On the defective surface, a significant interaction may also be possible between the bound OP and co-adsorbed ROS. We observe a strong correlation in the defective CeO2 systems between the binding energies and the phosphorus–ROS distances, which are annotated in boldface in Fig. 4. The weakest binding by DMMP is correlated with the longest distance (−20.9 kcal mol−1 and 3.72 Å), and the strongest binding by DFP is correlated with the shortest (−27.5 kcal mol−1 and 3.18 Å). This trend could correlate with strength of hydrogen bonding between the OP and ROS, as well as electrostatic interaction directly between the phosphorus center and ROS. The latter interaction would also be consistent with well-established GB decomposition mechanisms by nucleophilic attack on the phosphorus center.
To help discern the origin of the differences among the surface–adsorbate interactions, Bader partial charge analysis was performed on the six systems of interest as described in the Materials and methods section. The resulting partial charges, summed over different portions of interest in each system, are shown in Table 2. Tables describing atomic partial charges for key individual atoms are shown in Tables S1 and S2 in the ESI.† In all six systems, there is a slight net positive charge on the adsorbate and negative charge on the surface, consistent with the expected formation of a dative bond via donation of the (PO) lone pair to a surface Ce site. In the defective systems, a large negative charge is localized on the ROS, consistent with the peroxide ion. In all cases, the local charge on the OP-binding Ce site is more positive in the adsorbed state, and the magnitude of this effect is also larger in the presence of ROS for all adsorbates.
| System | Sum of atomic partial charges | ||||
|---|---|---|---|---|---|
| Adsorbate | Surface | Adsorbate | Surface | CeO2 + Ovac | ROS |
| DMMP | CeO2 + Ovac + ROS | 0.053 | −0.056 | 1.13 | −1.19 |
| GB | CeO2 + Ovac + ROS | 0.031 | −0.034 | 1.19 | −1.22 |
| DFP | CeO2 + Ovac + ROS | 0.013 | −0.016 | 1.20 | −1.22 |
| DMMP | CeO2 | 0.038 | −0.041 | ||
| GB | CeO2 | 0.003 | −0.005 | ||
| DFP | CeO2 | 0.014 | −0.017 | ||
Volumes of the basins computed as part of the Bader analysis can also complement the partial charges and geometries in correlations with adsorption strength. The Bader volumes of the ROS in the DMMP and GB systems, taken as the union of the volumes of the two oxygen atoms, are shown in Fig. 5. The Bader volume of the ROS is significantly smaller when co-adsorbed with GB than DMMP, consistent with the shorter P–ROS distance and with stronger total binding energy. In contrast, the partial charges on the P and F atoms, ROS, and surface Ce atom (Tables S1 and S2†), and corresponding bond lengths are very similar between the systems with bound DMMP, GB, and DFP.
 | ||
| Fig. 5 Structures of (a) DMMP and (b) GB on the (111) CeO2 surface, co-adsorbed with Ovac-stabilized ROS (purple). In each subfigure, the Bader basin computed for the ROS is shown in yellow and annotated with corresponding volume and partial charge. The Bader volume of the ROS, and likewise the ROS–P distance, is significantly larger for DMMP than GB. | ||
Observation of ROS through EPR spectroscopy
To confirm computational results regarding the role of ROS in binding and degradation of OP molecules, we perform electron paramagnetic resonance (EPR) spectroscopy for the CeO2, GCO, La-doped CeO2 (LCO), and Y-doped CeO2 (YCO) aerogels (Fig. 6). EPR is a powerful tool for directly observing unpaired electrons present in ROS, such as superoxide (O2˙−) or hydroxyl radicals (OH·).39 CeO2 shows prominent EPR signals at g = 2.003, characteristic of trapped electrons at Ovac in CeO2.40 A small peak corresponding to Ce3+ may be present as well, but we did not observe any ROS features for the CeO2 aerogel. We attempted EPR for the GCO aerogels, but the presence of Gd broadens the EPR signal,41,42 making identification of ROS impossible.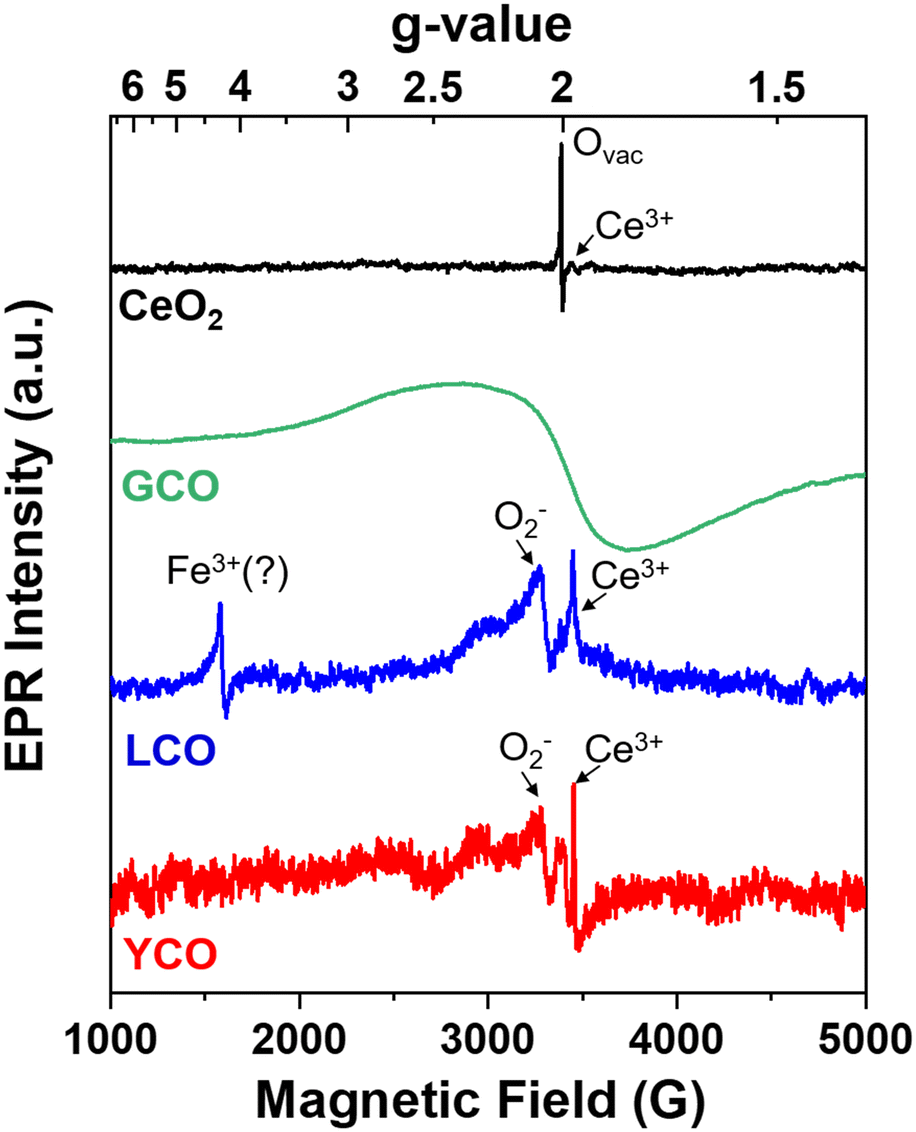 | ||
| Fig. 6 Normalized EPR spectra of CeO2, GCO, YCO, and LCO aerogels. LCO and YCO show significant ROS (O2˙−) signals, likely obscured in GCO due to the broad, wavy EPR lines. | ||
For LCO and YCO aerogels, EPR signals at g = 2.055 appear, corresponding to superoxide (O2˙−),43,44 along with a sharp signal at g = 1.964 due to Ce3+. In LCO, another feature at g = 4.27 is prominent and matches a g-value attributed to Fe3+ in Fe-doped oxide materials.45 Iron is a known impurity in La and notoriously difficult to fully remove,46 so we conclude that some residual Fe is likely present in our precursor salt. Given the similar characteristics of Gd, Y, and La in the CeO2 lattice, namely substituting into Ce sites and generating Ovac, these results align with computational results implicating stabilization of ROS when Ovac are present. Alkaline-treated GCO, LCO, and YCO aerogels also perform similarly against DMMP (Fig. S10†), further suggesting that their performance against simulants is a product of 3+ cation substitution and subsequent Ovac formation.
Conclusions
The ability of CeO2 surfaces to not only bind, but also rapidly degrade organophosphorus molecules makes ceria a promising material for use in protective applications against acute toxic threats. Our study of Gd3+-doped CeO2 (GCO) aerogels highlights their excellent properties against chemical warfare simulants and serves to caution researchers against relying solely on non-fluorinated simulants for materials evaluation. When comparing materials with appropriate hydroxyl-rich surface chemistries (CeO2-s and GCO-s), CeO2-s outperforms GCO-s against DMMP, but the opposite is true when evaluated against DFP (GCO-s outperforms CeO2-s). Computational analysis and EPR spectroscopy reveal that the divergent results may stem from the role of reactive oxygen species (ROS) in these systems. ROS on defective CeO2 surfaces (analogous to GCO) improve binding of DFP and sarin (GB), but are less effective against DMMP.Materials and methods
Aerogel synthesis
The CeO2 and Gd3+-doped CeO2 aerogels were prepared by previously established sol–gel synthetic methods.16,17 For CeO2 aerogels, 2.39 g of CeCl3·7H2O (99.9%, Aldrich) was dissolved in 10 g of anhydrous methanol (99.8%, Fisher) before adding 6 g of propylene oxide (99.5%, Aldrich). For Gd3+-substituted CeO2, 10 mol% of GdCl3·6H2O (99.99%, Alfa Aesar) was substituted for an equivalent amount of Ce precursor. The solution was allowed to gel overnight before rinsing several times with isopropanol (ACS grade, Fisher) and then acetone (ACS grade, Fisher). The gels were dried from supercritical CO2 (Leica EM CPD200 auto-dryer) after undergoing 99 CO2 rinsing cycles over ∼4 h to ensure that all acetone was exchanged.The dried, amorphous aerogels were calcined in air at 500 °C for 2 h (5 °C min−1 ramp). The calcined aerogel was soaked in an alkaline solution to remove residual Cl.9 The aerogel was suspended in a 10 vol% aqueous ethanol solution (200 mg aerogel/100 mL) and titrated to pH 9.5 with 1 M and 0.1 M NaOH. The aerogels were left to soak for 48 h, then vacuum filtered over a 0.1 μm polyvinylidene difluoride (PVDF) membrane, rinsed with Milli-Q® water, and dried overnight at 40 °C in air.
DRIFTS IR – simulant trials and dose-extraction
Simulant degradation studies were performed in an in situ DRIFTS environmental accessory coupled to a Thermo Scientific Nicolet 6700 FT-IR spectrometer (MCT/A detector, KBr beamsplitter, 24 bit digitizer, 3.7974 cm s−1 optical velocity, aperture 65, 2.0 sample gain). Approximately 20–30 mg of aerogel was loaded into a 6 mm diameter porous ceramic cup and dried under zero grade air (Airgas, 20% O2, balance N2, and trace impurity) for several days before loading into the DRIFTS accessory. The DRIFTS accessory (Pike Technologies DiffusIR™ Environmental Chamber) was evacuated under vacuum for several days to minimize any residual contamination prior to introduction of sample aerogel. The cell was maintained at 25 °C for the full duration of the dosing experiments.Flow was introduced into the cell in a top-to-bottom configuration wherein the vapor stream flows through the packed aerogel bed and exits through the bottom of the ceramic cup before exiting the cell. Zero grade air (Airgas, 20% O2, balance N2, and trace impurity) was used as the carrier gas and purge gas. First, carrier gas was flowed through the packed bed at a flow rate of 1.45 sccm long enough to allow the DRIFTS baseline signal to stabilize. The carrier was next diverted through a microsaturator cell (BioChemWare, 13 × 30 mm) containing a liquid reservoir of DFP in a temperature-controlled water-circulating chamber maintained at 20 °C. After 1 h of dosing the sample with DFP (Sigma-Aldrich DFP D0879-1G LOT# MKCQ1976/Toronto Research Chemicals CAT# D455300 LOT# 12-YMK-148-4) vapor, the cell was purged for 1 h with carrier gas (bypassing the saturator) to remove excess agent vapor from the system. After purging for 1 h, the carrier gas flow was maintained at a constant 1.45 sccm while the cell was illuminated for 1 h with a 200 W mercury–xenon lamp (Newport Model 67005 housing with 6290 Ozone free lamp) interfaced with fiber optic bundle positioned ∼6 mm above the sample cup. DRIFTS single-beam spectra were recorded at 1-min intervals (128 scans per spectra, 58.85 s per spectra, 2 cm−1 resolution, Happ–Genzel apodization, Mertz phase correction) throughout the duration of the initial purge and subsequent 1 h DFP vapor dosing, 1 h post-dose purge, and 1 h broadband illumination steps. The final spectrum of each step was used as the background for the ensuing step and difference spectra are plotted after background subtraction.
After agent exposure, the spent aerogel was placed into a 20 mL scintillation vial, vortexed for 60 s in acetonitrile and the solution was placed into a 2 mL Luer-slip plastic syringe to be filtered through a 0.45 μm nylon membrane syringe filter into a 2 mL silanized GC vial. Subsequently, five (5) μL of N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) derivatizing agent47 was added into the extract, sealed quickly, vortexed, and placed onto an Agilent GC autosampler contained within an Agilent 6890 N Network GC System coupled to a 5973 Network Mass Selective Detector. Peaks corresponding to DFP and degradation products were integrated and the relative percentage of DFP and products were determined by their respective area ratios in order to qualitatively assess the degradation capacity of various materials.
Characterization
Multipoint Brunauer–Emmett–Teller (BET) surface area was determined using the adsorption leg of the N2 physisorption isotherm (Micromeritics ASAP 2020). Approximately 120 mg of the samples were degassed under vacuum for 10 h at 150 °C prior to measurement. Average pore sizes were calculated using the Barrett–Joyner–Halenda (BJH) model on the desorption isotherm. X-ray photoelectron spectroscopy was performed using a Thermo Scientific Nexsa with Al Kα source operating at 72 W (12 kV × 6 mA) and a flood gun to minimize charging. Spot size, pass energy, and energy step size were 400 μm, 50.0 eV, and 0.100 eV, respectively. Raman spectroscopy was performed using a Renishaw confocal inVia microscope with a 514 nm laser source, imaged through a 50× objective lens, with 1 s exposure time at 10 acquisitions.The electron paramagnetic resonance (EPR) spectra were acquired using a commercial Bruker EMX spectrometer operating at a frequency of 9.51 GHz. A few milligrams of the rare earth-doped CeO2 aerogel powders were put in 4 mm OD low-loss quartz tubes with the tubes inserted in the middle of the cylindrical microwave cavity. The spectrometer was equipped with a liquid helium flow system that allowed for temperature control from 4.2–300 K. Typical microwave powers of 1–2 mW with 1–2 Gauss modulation amplitude and 100 kHz field modulation were employed for these experiments. The g-values were calibrated with use of a DPPH (2,2-diphenyl-1-picrylhydrazyl) standard.
Density functional theory calculations
Electronic structure was computed with density functional theory (DFT) using Quantum Espresso v6.7.0.48 All calculations were periodic and used the PBE functional with the projector augmented wave method49 and Grimme's D3 dispersion model. The Hubbard parameter U50 was incorporated for Ce 4f orbitals and was set to 4.5 eV.51 Convergence studies were performed on bulk ceria with respect to the plane wave energy cutoff of 120 Ry which produced significantly more accurate electronic structures, energies, and final bound geometries, owing primarily to improved resolution of Ce f states compared to default cutoffs recommended for the pseudopotentials. Atomic positions and cell vectors were optimized using the Broyden–Fletcher–Goldfarb–Shannon algorithm within thresholds of 10−4 atomic units (a.u.) in energy and 10−3 a.u. in force. A 4 × 4 × 4 Monkhorst–Pack k-point mesh was used for the bulk ceria optimization, and the resultant optimized lattice vectors were a = 5.438 Å, in agreement with prior experimental and theoretical reports.38 Structures were visualized using Jmol v14, an open-source Java viewer for chemical structures in 3D (http://www.jmol.org/). Full DFT input/output files are included in the supplementary information.The (111) surface was cleaved from the optimized bulk structure and subjected to geometry optimization. The model slab consists of 144 atoms (9 atomic layers) with supercell lattice vectors a = b = 15.46 Å and c = 22.90 Å. The surface model included 15 Å of vacuum along the c direction and the electronic structure was calculated using a dipole correction. For computational efficiency, surface calculations were carried out at a smaller k-point grid of 2 × 2 × 1. The atoms in the bottom CeO2 layer were frozen to bulk positions while all other atomic positions were optimized. In addition, (111) models with a surface oxygen vacancy (Ovac) and with Ovac and bound O2 were prepared. Because the Ovac creates two excess electrons, the defective (111) surface represents a system in a triplet state, therefore, spin-polarized DFT calculations were performed to compute the total energies of these systems. Atomic structures of the OP molecules of interest were optimized using plane-wave DFT in the same calculation domains. Binding enthalpies (ΔE) were computed on surface models using the following equation:
| ΔE = Esurf+ads − Eads − Esurf |
Density of states (DOS) and projected density of states (PDOS) were generated by performing non-self-consistent-field calculations with a denser k-point grid of 12 × 12 × 1. Bader charge analysis52,53 was used to quantify changes in atomic partial charges due to adsorption. From the optimized charge density grid, a code developed by Henkelman et al. was used to partition the grid into Bader volumes.54,55 The charge densities within the resulting Bader volumes are used to approximate the actual charge states of the atoms.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Travis G. Novak: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing. Janna Domenico: formal analysis, investigation, methodology, writing. Alex Balboa: formal analysis, investigation, methodology. Wesley O. Gordon: formal analysis, investigation, methodology. Austin E. Herzog: formal analysis, writing. Nam Q. Le: formal analysis, investigation, methodology, writing. Evan R. Glaser: investigation, formal analysis, methodology. Paul A. DeSario: funding acquisition, conceptualization, investigation. Debra R. Rolison: funding acquisition, validation, conceptualization, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding provided by the Defense Threat Reduction Agency (DTRA, HDTRA CB10992). A. E. H. acknowledges the National Research Council for support through a Naval Research Laboratory/National Research Council Postdoctoral Associateship. This work was supported by the Office of Naval Research.Notes and references
- Q. Han, L. Yang, Q. Liang and M. Ding, Carbon, 2017, 122, 556–563 CrossRef CAS.
- S. A. Bhatti, F. H. Memon, F. Rehman, Z. Bhatti, T. Naqvi and K. H. Thebo, Rev. Inorg. Chem., 2022, 42, 283–295 CrossRef CAS.
- T. G. Novak, P. A. DeSario, J. W. Long and D. R. Rolison, Front. Mater., 2021, 8, 674798 CrossRef.
- Y. Chen, X. Zhang, M. R. Mian, F. A. Son, K. Zhang, R. Cao, Z. Chen, S.-J. Lee, K. B. Idrees, T. A. Goetjen, J. Lyu, P. Li, Q. Xia, Z. Li, J. T. Hupp, T. Islamoglu, A. Napolitano, G. W. Peterson and O. K. Farha, J. Am. Chem. Soc., 2020, 142, 21428–21438 CrossRef CAS PubMed.
- S. Bhasker-Ranganath, C. Zhao and Y. Xu, Surf. Sci., 2021, 705, 121776 CrossRef CAS.
- T. Li, R. Tsyshevsky, L. Algrim, M. McEntee, E. M. Durke, B. Eichhorn, C. Karwacki, M. R. Zachariah, M. M. Kuklja and E. E. Rodriguez, ACS Appl. Mater. Interfaces, 2021, 13, 54597–54609 CrossRef CAS PubMed.
- Q. Z. Li, H. Z. Fan, Z. Z. Wang, J. J. Zheng, K. L. Fan, X. Y. Yan and X. F. Gao, ACS Catal., 2023, 13, 504–514 CrossRef CAS.
- A. E. Herzog, T. J. Michael, A. D. Dunkelberger, M. D. Johannes, D. R. Rolison, P. A. DeSario and T. G. Novak, Nanoscale, 2024, 16, 9659–9679 RSC.
- T. G. Novak, R. B. Balow, M. R. Buck, D. R. Rolison and P. A. DeSario, ACS Appl. Nano Mater., 2023, 6, 3075–3084 CrossRef CAS.
- A. Emelianova, E. A. Basharova, A. L. Kolesnikov, E. V. Arribas, E. V. Ivanova and G. Y. Gor, J. Phys. Chem. B, 2021, 125, 4086–4098 CrossRef CAS PubMed.
- A. Emelianova, A. Reed, E. A. Basharova, A. L. Kolesnikov and G. Y. Gor, ACS Appl. Mater. Interfaces, 2023, 15, 18559–18567 CrossRef CAS PubMed.
- Y. Liao, T. R. Sheridan, J. Liu, Z. Lu, K. Ma, H. Yang, O. K. Farha and J. T. Hupp, ACS Catal., 2024, 14, 437–448 CrossRef CAS.
- M. B. Leonard, E. Bruni, M. Hall, T. Li, E. E. Rodriguez and E. M. Durke, J. Phys. Chem. Lett., 2022, 13, 11663–11668 CrossRef CAS PubMed.
- Y. C. Quintero and R. Nagarajan, Surf. Sci., 2018, 675, 26–35 CrossRef CAS.
- T. Y. Li, M. Leonard, R. Tsyshevsky, M. McEntee, C. Karwacki, E. M. Durke, M. M. Kuklja and E. E. Rodriguez, Mater. Chem. Front., 2023, 7, 1855–1866 RSC.
- T. G. Novak, P. A. DeSario, M. D. Johannes, T. H. Brintlinger, R. H. DeBlock, J. W. Long, C. N. Chervin, R. M. Stroud and D. R. Rolison, Chem. Mater., 2022, 34, 5644–5653 CrossRef CAS.
- T. G. Novak, P. A. DeSario, T. H. Brintlinger, R. H. DeBlock, J. W. Long and D. R. Rolison, ACS Sustainable Chem. Eng., 2023, 11, 2853–2860 CrossRef CAS.
- F. Liang, Y. Yu, W. Zhou, X. Xu and Z. Zhu, J. Mater. Chem. A, 2015, 3, 634–640 RSC.
- H. Sohn, G. Celik, S. Gunduz, D. Dogu, S. Zhang, J. Shan, F. F. Tao and U. S. Ozkan, Catal. Lett., 2017, 147, 2863–2876 CrossRef CAS.
- Z. Liu, T. Duchoň, H. Wang, D. C. Grinter, I. Waluyo, J. Zhou, Q. Liu, B. Jeong, E. J. Crumlin, V. Matolín, D. J. Stacchiola, J. A. Rodriguez and S. D. Senanayake, Phys. Chem. Chem. Phys., 2016, 18, 16621–16628 RSC.
- O. Amadine, Y. Essamlali, A. Fihri, M. Larzek and M. Zahouily, RSC Adv., 2017, 7, 12586–12597 RSC.
- B. Liu, B. Zhang, J. Ji, K. Li, J. Cao, Q. Feng and H. Huang, Appl. Catal., B, 2022, 304, 120952 CrossRef CAS.
- Y. Hong, M. Yu, J. Lin, K. Cheng, W. Weng and H. Wang, Colloids Surf., B, 2014, 123, 68–74 CrossRef CAS PubMed.
- M. Yu, J. Gong, Y. Zhou, L. Dong, Y. Lin, L. Ma, W. Weng, K. Cheng and H. Wang, J. Mater. Chem. B, 2017, 5, 3955–3963 RSC.
- D. N. Durgasri, T. Vinodkumar and B. M. Reddy, J. Chem. Sci., 2014, 126, 429–435 CrossRef CAS.
- A. Kiselev, A. Mattson, M. Andersson, A. E. C. Palmqvist and L. Österlund, J. Photochem. Photobiol., A, 2006, 184, 125–134 CrossRef CAS.
- A. J. Mott and P. Rez, Spectrochim. Acta, Part A, 2012, 91, 256–260 CrossRef CAS PubMed.
- D. J. McGarvey, J. R. Stuff, B. R. Williams and H. D. Durst, Spectrosc. Lett., 2000, 33, 795–819 CrossRef CAS.
- M. B. Leonard, T. Li and E. E. Rodriguez, ACS Appl. Mater. Interfaces, 2024, 16, 38757–38767 CrossRef CAS PubMed.
- D. A. Köse and H. Necefoğlu, J. Therm. Anal. Calorim., 2008, 93, 509–514 CrossRef.
- C. T. Lynch, K. S. Mazdiyasni, J. S. Smith and W. J. Crawford, Anal. Chem., 1964, 36, 2332–2337 CrossRef CAS.
- A. Harsanyi and G. Sandford, Green Chem., 2015, 17, 3000–3009 RSC.
- T. Yamanaka, M. Kawasaki, M. D. Hurley, T. J. Wallington, W. F. Schneider and J. Bruce, Phys. Chem. Chem. Phys., 2007, 9, 4211–4217 RSC.
- S. i. Mizushima, T. Shimanouchi, T. Miyazawa, I. Ichishima, K. Kuratani, I. Nakagawa and N. Shido, J. Chem. Phys., 1953, 21, 815–818 CrossRef CAS.
- Z. Witkiewicz, S. Neffe, E. Sliwka and J. Quagliano, Crit. Rev. Anal. Chem., 2018, 48, 337–371 CrossRef CAS PubMed.
- T. Li, L. Algrim, M. McEntee, R. Tsyshevsky, M. Leonard, E. M. Durke, C. Karwacki, M. M. Kuklja, M. R. Zachariah and E. E. Rodriguez, J. Phys. Chem. C, 2022, 126, 17923–17934 CrossRef CAS.
- J. Paier, C. Penschke and J. Sauer, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS PubMed.
- N. Q. Le, C. E. Ekuma, B. I. Dunlap and D. Gunlycke, J. Phys. Chem. C, 2018, 122, 2832–2839 CrossRef CAS.
- Y. Zhao, B.-T. Teng, X.-D. Wen, Y. Zhao, Q.-P. Chen, L.-H. Zhao and M.-F. Luo, J. Phys. Chem. C, 2012, 116, 15986–15991 CrossRef CAS.
- Z. Xiao, Y. Li, F. Hou, C. Wu, L. Pan, J. Zou, L. Wang, X. Zhang, G. Liu and G. Li, Appl. Catal., B, 2019, 258, 117940 CrossRef CAS.
- C. Oliva, F. Orsini, S. Cappelli, P. Arosio, M. Allieta, M. Coduri and M. Scavini, J. Spectrosc., 2015, 2015, 491840 Search PubMed.
- M. Scavini, M. Coduri, M. Allieta, P. Masala, S. Cappelli, C. Oliva, M. Brunelli, F. Orsini and C. Ferrero, IUCrJ, 2015, 2, 511–522 CrossRef CAS PubMed.
- L. Wang, Y. Yu, H. He, Y. Zhang, X. Qin and B. Wang, Sci. Rep., 2017, 7, 12845 CrossRef PubMed.
- J. Guo, R. Song, Z. Li, D. Pan, H. Xie, Y. Ba, M. Xie, S. Fan, X. Yang, H. Zhang, H. Yu, S. Zhang, J. Du, L. He and L. Wang, Adv. Energy Sustainability Res., 2022, 3, 2200106 CrossRef CAS.
- N. A. Zhuk, V. P. Lutoev, B. A. Makeev, S. V. Nekipelov, A. V. Koroleva, A. V. Fedorova, M. V. Yermolina, D. S. Beznosikov and L. O. Karlova, J. Mater. Res. Technol., 2020, 9, 4173–4182 CrossRef CAS.
- S. Pang, W. Lu, Z. Yang, X. Zhang, D. Chen, D. Wu, L. Zhou and R. Miao, J. Rare Earths, 2021, 39, 875–880 CrossRef CAS.
- T. P. Mawhinney and M. A. Madson, J. Org. Chem., 1982, 47, 3336–3339 CrossRef CAS.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H. Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H. V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- A. Dal Corso, Comput. Mater. Sci., 2014, 95, 337–350 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- M. F. Camellone and S. Fabris, J. Am. Chem. Soc., 2009, 131, 10473–10483 CrossRef PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- M. Yu and D. R. Trinkle, J. Chem. Phys., 2011, 134, 064111 CrossRef PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lf00390j |
| ‡ NRC Postdoctoral Associate at U.S. Naval Research Laboratory, Washington, DC, Dr. Herzog is performing research at NRL but is not employed by NRL. |
| § Current address: Advanced Naval Platforms Division, Office of Naval Research, Arlington, VA, USA. |
| This journal is © The Royal Society of Chemistry 2025 |