
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel polymer/halloysite composites with high halloysite content and remarkable mechanical strength
Mingxuan
Zhang
a,
Camila
Sabatini
 b,
Kaiwen
Chen
a,
Steven
Makowka
b,
Ruijia
Hu
c,
Mark
Swihart
a and
Chong
Cheng
*a
b,
Kaiwen
Chen
a,
Steven
Makowka
b,
Ruijia
Hu
c,
Mark
Swihart
a and
Chong
Cheng
*a
aDepartment of Chemical and Biological Engineering, The State University of New York, University at Buffalo, Buffalo, NY 14260, USA. E-mail: ccheng8@buffalo.edu
bDepartment of Restorative Dentistry, The State University of New York, University at Buffalo, Buffalo, NY 14214, USA
cDepartment of Biology, Duke University, Durham, NC 27708, USA
First published on 20th November 2024
Abstract
Halloysite nanotubes (HNTs) are of interest for use in nanocomposites due to their unique cylindrical structure and resulting properties. Various polymer/HNT composites have been studied, but generally these composites have HNTs as a minor component. Here, we report novel polymer/HNT composites with high HNT content facilitated by strong hydrogen-bonding interactions between the polymer and HNTs. These composites with 50–75 wt% HNTs were prepared by in situ polymerization reactions of mixtures comprising HNTs, acrylic acid, triethylene glycol dimethacrylate, potassium persulfate, and water, followed by drying. The chemical structure of composites was verified by Fourier-transform infrared spectroscopy (FTIR). The high dispersity of HNTs in the poly(acrylic acid)-based matrix was demonstrated by scanning electron microscopy (SEM). Studies of mechanical properties illustrated greatly enhanced mechanical strength of the composites relative to the pure polymer matrix, with the highest flexural strength, microhardness, and ultimate tensile strength achieved for the composite with 66.7 wt% HNTs. Thus, the incorporation of high mass fractions of HNTs in a polymer matrix can offer potential benefits for applications requiring superior mechanical properties, in addition to other functions endowed by either HNTs or the polymer matrix. Differential scanning calorimetry (DSC) characterization did not show evidence of a glass transition in the polymer matrices of these composites, and thermogravimetric analysis (TGA) revealed increased thermal stability of the composites relative to the matrices. Swelling tests indicated that the swelling capacity is primarily determined by the amount of polymer present in the composite, and the presence of HNTs may facilitate mass transfer of water within the polymer matrix.
Introduction
Halloysite nanotubes (HNTs), with a chemical composition of Al2Si2O5(OH)4·nH2O (identical with kaolinite), are a class of two-layered aluminosilicate clay mineral, consisting of one alumina octahedron sheet and one silica tetrahedron sheet in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio. Their multilayer tubular structure arise from rolling of aluminosilicate sheets under favorable geothermal conditions.1,2 Such a tubular structure is easily observed in electron microscopy. The specific dimensions, morphologies, and contents of HNTs depend heavily on their origins of excavation.3,4 Generally, commercially available HNTs have an average external diameter of 50–70 nm, an average inner diameter of 10–15 nm, and a length range of about 0.5–1.5 μm. HNTs have received considerable attention due to their unique nanostructure, environmental friendliness, biocompatibility, and natural abundance.5–8
:1 stoichiometric ratio. Their multilayer tubular structure arise from rolling of aluminosilicate sheets under favorable geothermal conditions.1,2 Such a tubular structure is easily observed in electron microscopy. The specific dimensions, morphologies, and contents of HNTs depend heavily on their origins of excavation.3,4 Generally, commercially available HNTs have an average external diameter of 50–70 nm, an average inner diameter of 10–15 nm, and a length range of about 0.5–1.5 μm. HNTs have received considerable attention due to their unique nanostructure, environmental friendliness, biocompatibility, and natural abundance.5–8
HNTs have great potential as nanocarriers for encapsulation and controlled release due to their tunable surface chemistry and inner cavity capable of hosting active guest molecules.9,10 Surface-modified HNTs containing various drug molecules like aspirin, metronidazole, and chlorhexidine have been previously reported.11,12 HNTs can also be used as a reinforcement filler material due to their high mechanical strength and high aspect ratio.13 Many types of polymer-based composites incorporating small amounts of HNTs have been reported, using various polymeric matrices such as epoxy, nylon-6, and polyethylene.14–18 They exhibit enhanced stiffness, impact resistance, compressive strength, tensile strength and flexural strength relative to the pure polymer. Furthermore, the presence of HNTs in composites can enhance thermal stability of polymer matrices due to a barrier effect.19 However, in most polymer/HNT composites, considerable aggregation of HNTs can only be avoided at low HNT contents, because the interactions between HNTs and typical polymer matrices are generally weak, leading to quite limited dispersibility of HNTs in these matrices.9 Enhanced filler-matrix interactions can increase HNT contents in composites. For example, the composites of HNTs and sulphated galactan reported by Cavallaro and co-workers can possess up to 30 wt% HNTs.20
Poly(acrylic acid) (PAA) is a polyelectrolyte capable of absorbing a large amount of water. PAA-based polymers are typically prepared from acrylic acid (AA) or AA with other acrylic monomers via free radical polymerization, and have been used in a range of applications, including diapers, cosmetics, ion-exchange resins, paints and adhesives, and pharmaceuticals.21 However, PAA-based materials typically have poor overall mechanical strength, which severely restricts their use in many fields.22 Several methods have been investigated to enhance the mechanical strength of PAA-based materials, including chemical and/or ionic crosslinking,23–27 and the formation of polymer blends or interpenetrating polymer networks.28,29 HNT-containing PAA-based copolymer composites have been previously reported. Zhang and co-workers studied a poly(acrylic acid-co-acrylamide)/HNT superabsorbent composite with 10 wt% HNTs, which exhibited enhanced mechanical strength and thermal stability relative to the polymer matrix.30 Li et al. investigated an ionic crosslinked poly(acrylamide-co-acrylic acid)/chitosan hydrogel reinforced by a low wt% of HNTs.31 Notably, PAA has been combined with other kinds of nanoscopic fillers, such as graphene, carbon nanotubes, silica nanopsheres, or metal and metal oxide nanoparticles, to form a variety of composites with advanced functional properties.32,33
To the best of our knowledge, polymer/HNT composites with high mass fractions (≥50 wt%) of well-dispersed HNTs have not been reported. Polymer/inorganic composites with high mass fractions of inorganic fillers can possess remarkable properties, and therefore may have a much wider range of applications. For instance, glass ionomer cements utilize PAA and calcined glass powder to form composite cements used as a temporary dental filling material.34 While glass ionomer cements are highly desirable for their ability to provide chemical bonding to the teeth's mineral content, their relatively weak mechanical properties impose limitations that restrict the potential for broader dental applications such as their use as a permanent dental filling material. Therefore, we are keenly interested in developing a general strategy to prepare PAA-based polymer/HNT composites with high wt% of well-dispersed HNTs. Based on the chemical structure of HNTs with a sheet comprising silica tetrahedra on the outer surface, we hypothesize that polymer matrices capable of serving as hydrogen donors may strongly interact with HNTs via their surface oxygen atoms through hydrogen bonding, thereby stabilizing dispersion of HNTs in the matrices and favoring the formation of polymer/HNT composites with high wt% of well-dispersed HNTs (Fig. 1). Accordingly, this hypothesis was tested by choosing PAA-based matrices for the preparation of HNT-containing composites since PAA possesses hydrogen-donating carboxylic acid groups. Subsequently, systematic studies were performed to interpret the structure and properties of the resulting composites, as well as their structure–property relationship. Collectively, our studies demonstrate a novel strategy to achieve polymer/HNT composites with high HNT content, which are new materials with promising properties and application potential.
 | ||
| Fig. 1 Illustration of PAA interaction with HNT surface. | ||
Materials and methods
Materials
AA (99%) was purchased from TCI Chemicals. Potassium persulfate (99%) was obtained from Thermo Scientific. Triethylene glycol dimethacrylate (TEGDMA, 95%) and halloysite nanoclay (i.e., HNTs) were supplied by Sigma Aldrich. All materials were used as received.Preparation of polymer/HNT composites
The composites with 50–75 wt% HNTs were prepared as follows. Five vials of mixtures were prepared. In each vial, AA (2.25 g, 31.2 mmol) as monomer, TEGDMA (0.25 g, 0.87 mmol) as crosslinker, and potassium persulfate (0.010 g, 0.037 mmol) as thermal initiator were mixed with deionized water (2.50 g); the resulting mixture was purged with nitrogen gas for 30 min. Next, 2.50, 3.75, 5.00, 6.25, and 7.50 g of HNTs were added to these vials, respectively. The samples were further mixed by vortexing and ultrasonication to obtain well-mixed dispersions. Ice baths were applied periodically to prevent overheating during ultrasonication. The samples were equilibrated to 25 °C, and then slowly heated to 65 °C for curing over a period of 12 h. The cured samples were processed into the desired shapes, based on the specifications of the individual experiments, and then dried in a vacuum oven at 100 °C for 12 h.Fourier transfer infrared (FTIR) spectroscopy
Attenuated total reflectance (ATR) FTIR analysis was carried out on a Bruker vertex 70 spectrometer fitted with a universal ATR sampling accessory. All data were recorded at room temperature in the spectral range of 600–4000 cm−1. Subsequent data analysis was performed using Origin software.Scanning electron microscopy (SEM)
SEM imaging was performed using a Carl Zeiss AURIGA CrossBeam FIB-SEM instrument. Test samples were sputtered with gold using an SPI-Module sputter coater system for 20 s under argon before testing. The accelerating voltage utilized was 2.0 kV for raw HNTs and 1.5 kV for composites.Transmission electron microscopy (TEM)
TEM imaging was performed using a JEOL JEM 2010 high-resolution electron microscope instrument. Test samples were prepared by drop casting of a dispersion of HNTs in methanol on a standard TEM grid, and followed by drying under nitrogen. The accelerating voltage utilized was 200 kV.Flexural strength
A standard three-point flexural test (ASTM D 793) with a span of 20 mm was used for the testing of cured beam-shaped specimens of polymer/HNT composite (2 mm × 2 mm × 25 mm) at a crosshead speed of 1.0 mm min−1 using a computer-controlled Dillon Quantrol universal testing machine with a 100 N load cell. The cured specimens were hand polished with silicon carbide (SiC) sandpapers of sequentially higher grits (240, 320 and 600) in a longitudinal direction. Ten replicates were prepared for each group.Ultimate tensile strength
Testing of ultimate tensile strength was conducted per the ASTM D-638 testing standard. For each group, ten hourglass-shaped specimens (10 mm long × 2 mm neck wide × 1 mm thick) were fabricated for evaluation of specimens using a universal testing machine (Micro-tester) at a crosshead speed of 1.0 mm min−1.Young's modulus
Testing of Young's modulus was conducted per the ASTM D-638 testing standard. For each group, ten dumbbell-shaped specimens (20 mm long × 2 mm neck wide × 1 mm thick) were fabricated for evaluation of specimens using a computer-controlled Dillon Quantrol universal testing machine with a 500 N load cell at a crosshead speed of 1.0 mm min−1.Compressive strength and modulus
Testing of compressive strength and modulus was conducted per the ASTM D-695 standard. For each group, six cylindrical specimens (3 mm diameter × 2 mm height) were fabricated with a die cutter for evaluation of specimens using a computer-controlled Dillon Quantrol universal testing machine with 2500 N load cell at a crosshead speed of 1.0 mm min−1.Microhardness
Microhardness of the composite materials was evaluated with a Buehler Micromet micro indentation hardness tester using a 300 gram-force (gf) load with a 15 s dwell time. Cured beam-shaped specimens of polymer/HNT composite (2 mm × 2 mm × 25 mm) were embedded in acrylic resin and hand polished with SiC sandpapers of 240, 320 and 600 grit. Six indentations, spaced at least 0.5 mm apart from one another (ASTM E384-11), were made on each specimen with six replicates. The two diagonal lengths of each indentation were measured by a built-in microscope and converted into a microhardness value using the following equation: HV = 0.1891 F/d2, where HV is the microhardness value, F is the load in gf and d is the average length of the diagonals in mm. For each specimen, six hardness values were averaged and reported as a single value.Differential scanning calorimetry (DSC)
DSC analysis was carried out using a DSC Q2000 instrument with a nitrogen flow rate of 50 mL min−1. Test specimens (each with a mass between 9 and 13 mg) were weighed on an analytical scale, capable of being read to four decimal places. Samples were crimped into non-perforated aluminum pans, with an empty aluminum pan as the reference. For each measurement, the sample was repeatedly heated from 25 °C to 150 °C at a heating rate of 10 °C min−1, cooled back to 25 °C and then held isothermal at 25 °C for 10 min. The DSC curves of the second heating cycles were used to analyze the thermal transitions of samples.Thermogravimetric analysis (TGA)
TGA was carried out using a DSC SDT Q600 Thermogravimetric Analyzer with a nitrogen flow rate of 100 mL min−1. Test specimens (each of ∼10–20 mg) were crimped into non-perforated aluminum pans, with an empty aluminum pan as the reference. The samples were held isothermal at 25 °C for 5 min, then heated from 25 to 600 °C at a heating rate of 20 °C min−1.Swelling test
A completely dry, pre-weighed disc-shaped sample was immersed in deionized water for 24 h. Then the sample was removed from the water and weighed after the surface of the sample was blotted dry. Swelling ratio (Q) was calculated for each sample according to the following eqn (1):| Q = (Ms − Md)/Md | (1) |
Statistical analysis
Statistical analysis was conducted using t-test and P-test, with P > 0.05 as not significant (ns).Results and discussion
Design and preparation of composites
PAA is known to be brittle under normal conditions, with poor mechanical performance. PAA is also water-soluble unless it is crosslinked. To address these issues and to achieve PAA-based composites with enhanced mechanical properties, HNTs were used in high mass fractions as the inorganic filler, and a crosslinked PAA-based network was employed as the continuous phase of composites in this work (Fig. 2). | ||
| Fig. 2 Synthesis of polymer/HNT composites. | ||
Incorporating HNTs into the composites can offer several advantages. First, HNTs have a (Si–O–Si) tetrahedral external surface structure that resembles silica, which can enable strong hydrogen bonding interactions with PAA.35,36 Second, HNTs exhibit a high specific surface area of up to 184.8 m2 g−1,37 promoting surface interactions. Third, the significant amount of HNTs in these composites limits volumetric shrinkage of the polymer matrix during drying. Fourth, the tubular morphology of HNTs reduces aggregation, which helps in attaining well-dispersed fillers within the polymer matrix. We hypothesized that the numerous carboxylic groups in PAA could form hydrogen bonds with the outer surface of HNTs, establishing robust surface interactions. This, in turn, enhances the load-bearing capacity of the composites due to the highly adhesive PAA-HNT interface, resulting in composites with remarkable mechanical properties.
To obtain a crosslinked PAA-based network as the continuous phase of composites, TEGDMA was selected as the crosslinker for the free radical polymerization of AA. TEGDMA possesses two methacrylate groups which can effectively crosslink PAA chains. Moreover, its oxyethylene units can form hydrogen bonds with PAA,38 which can further strengthen the cross-linkages in the network. In addition, potassium persulfate (KPS), a water-soluble thermal initiator, was selected for the free radical preparation of the network.
The composites were prepared by free radical polymerization of the reaction mixtures of AA, TEGDMA, KPS, water, and HNTs at 65 °C for 12 h (AA/TEGDMA/KPS/water = 9/1/0.04/10, by mass; HNTs/(AA + TEGDMA) = 1.0, 1.5, 2.0, 2.5, 3.0, by mass), followed by drying under vacuum at 100 °C for 12 h to remove water. The resulting composites contained 50–75 wt% HNTs.
Structural analysis
The chemical structures of the composites were analyzed using FTIR spectroscopy, with AA, TEGDMA, P(AA-co-TEGDMA), and pristine HNTs as controls. As shown in Fig. 3, FTIR spectra of all composites exhibited characteristic absorbance peaks from both the P(AA-co-TEGDMA) matrix and HNTs. Specifically, peaks at ∼1695 cm−1 and ∼2400–3600 cm−1 (very broad) were assigned to (meth)acrylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations and carboxylic O–H stretches of the P(AA-co-TEGDMA) network, respectively; these absorbance positions agree well with those of AA, the major precursor of the network. Moreover, absorbance peaks at 2920 cm−1 and 2860 cm−1 were absent in the FTIR spectra of AA and TEGDMA, but appeared in the FTIR spectra of the composites; they were assigned to the C–H stretches of the polymer backbones, which were formed by free radical polymerization. Notably, FTIR spectra of both AA and TEGDMA showed absorbance at ∼1635 cm−1 for CC vinyl stretch and at ∼815 cm−1 for (meth)acrylic C–H bending; the disappearance of these absorbance peaks in the FTIR spectra of composites indicates the full conversion of vinyl groups of AA and TEGDMA.39 In the FTIR spectra of composites, the absorbance at 1006 cm−1 was assigned to an Si–O–Si stretching mode, and the absorbance features at 3690 cm−1 and 3616 cm−1 were attributed to the O–H stretch of the hydroxyl groups on the HNT inner surface.40 Increasing the HNT mass fraction of the composites led to increased intensities of HNT-related absorbance peaks relative to P(AA-co-TEGDMA)-related features.
O vibrations and carboxylic O–H stretches of the P(AA-co-TEGDMA) network, respectively; these absorbance positions agree well with those of AA, the major precursor of the network. Moreover, absorbance peaks at 2920 cm−1 and 2860 cm−1 were absent in the FTIR spectra of AA and TEGDMA, but appeared in the FTIR spectra of the composites; they were assigned to the C–H stretches of the polymer backbones, which were formed by free radical polymerization. Notably, FTIR spectra of both AA and TEGDMA showed absorbance at ∼1635 cm−1 for CC vinyl stretch and at ∼815 cm−1 for (meth)acrylic C–H bending; the disappearance of these absorbance peaks in the FTIR spectra of composites indicates the full conversion of vinyl groups of AA and TEGDMA.39 In the FTIR spectra of composites, the absorbance at 1006 cm−1 was assigned to an Si–O–Si stretching mode, and the absorbance features at 3690 cm−1 and 3616 cm−1 were attributed to the O–H stretch of the hydroxyl groups on the HNT inner surface.40 Increasing the HNT mass fraction of the composites led to increased intensities of HNT-related absorbance peaks relative to P(AA-co-TEGDMA)-related features.
 | ||
| Fig. 3 FTIR spectra from a) AA, b) TEGDMA, c) P(AA-co-TEGDMA) resin, d) pristine HNTs, and PAA/HNT composites with HNT loadings of e) 50.0 wt%, f) 60.0 wt%, g) 66.7 wt%, h) 71.4 wt% and i) 75.0 wt%. | ||
Electron microscopy methods were used to examine the morphologies of both pristine HNTs and polymer/HNT composites. The SEM image of pristine HNTs showed their rod-like shape with an average outer diameter of ∼60 nm and length up to ∼1 μm (Fig. 4a). TEM imaging further revealed their tubular structure, with an average inner diameter of ∼20 nm (Fig. 4b). Because HNTs can be effectively etched by a strong acid (such as hydrochloric acid) to form porous structures,41 a concern was whether the structure of HNT would be altered in the presence of AA. TEM images of HNTs after AA treatment (65 °C, 12 h, in the presence of 2,2-diphenyl-1-picrylhydrazyl to inhibit radical polymerization) revealed no considerable structural change (Fig. 4c), suggesting that there was no critical structural difference between pristine HNTs and the HNTs in the PAA-based composites. Of note, AA treatment of HNTs for extended time period (e.g., 72 h) may result in a noticeable increase in lumen diameter,42 but the time required to convert AA (together with TEGDMA) to polymer matrix was much shorter in this work.
 | ||
| Fig. 4 a) SEM image of pristine HNTs; b) TEM image of pristine HNTs, c) TEM image of HNTs after treating the pristine HNTs with AA without polymerization. | ||
The morphologies of polymer/HNT composites were investigated by SEM imaging (Fig. 5). Achieving high dispersibility of HNTs is crucial for obtaining consistent properties in the polymer/HNT composites. However, it has proven challenging to disperse even small amounts of HNTs in polymeric matrices.43,44 SEM images of the fracture surfaces of PAA-based polymer/HNT composites provided critical information about the distribution of HNTs within the polymeric matrix. Relative to pristine HNTs (Fig. 5a), the HNTs at the fracture surfaces generally showed reduced width because the surface nanotubes were partly “buried” within the polymer. For the composite with 50.0 wt% HNTs (Fig. 5b), there were significant HNT-free regions (marked as “α”) with dimensions of 1 μm or larger, and only about half of the fractured surface was covered with well-distributed HNTs. With increasing HNT wt%, the surface coverage of HNT-free regions decreased, and composites with 60.0, 66.7, and 71.4 wt% HNTs exhibited predominant surface coverage with well-distributed HNTs (Fig. 5c–e). However, for the composite with the highest HNT fraction of 75.0 wt% (Fig. 5f), while the surface coverage of HNT-free regions essentially disappeared, a region with very crowded presence of HNTs (marked as “β”) was observed, indicating significant aggregation of HNTs in the composite.
 | ||
| Fig. 5 SEM images of a) pristine HNTs, and PAA-based polymer/HNT composites with various HNT loading: b) 50.0 wt%, c) 60.0 wt% d) 66.7 wt%, e) 71.4 wt%, and f) 75.0 wt%. | ||
Studies of mechanical properties
A series of tests were performed for evaluation of the mechanical properties of polymer/HNT composites. The effect of HNT incorporation on flexural strength, microhardness, ultimate tensile strength, compressive strength, ultimate elongation, tensile modulus, and compressive modulus of the polymer/HNT composites was assessed (Fig. 6). Pure P(AA-co-TEGDMA), the matrix material, was also prepared; however, it was too fragile to serve as a valid control for the mechanical testing of the composites. | ||
| Fig. 6 a) Flexural strength, b) microhardness, c) ultimate tensile strength, d) compressive strength, e) ultimate elongation, f) Young's modulus, and g) compressive modulus of PAA-based polymer/HNT composites. | ||
Flexural strength, microhardness, and ultimate tensile strength are commonly used parameters for evaluation of the mechanical properties of brittle materials. The flexural strength values (Fig. 6a) were 66.45 ± 6.35, 106.03 ± 5.66, 114.28 ± 4.67, 100.3 ± 7.66, and 98.48 ± 9.25 MPa for the polymer/HNT composites with HNT contents of 50.0, 60.0, 66.7, 71.4, and 75.0 wt%, respectively. With increasing HNT content, flexural strength first increased and reached a maximum value at 66.7 wt% HNTs, and then decreased gradually.
The microhardness testing of the polymer/HNT composites followed a similar trend (Fig. 6b). The microhardness values for the composites with 50.0, 60.0, and 66.7 wt% HNTs were 37.13 ± 4.04, 43.54 ± 3.53, and 52.51 ± 3.19 Hv, respectively, showing an increase in hardness value as the HNT content increased. The samples with 71.4 and 75.0 wt% HNTs showed microhardness values of 43.91 ± 3.18, and 43.94 ± 2.67 Hv, respectively; these values were considerably lower than the value for the composite with 66.7 wt% HNTs.
The ultimate tensile strength values also showed a similar trend to the microhardness values, since microhardness is internally related to tensile strength (Fig. 6c). The ultimate tensile strength values for the composites with 50.0, 60.0, and 66.7 wt% HNTs were 50.64 ± 3.27, 60.11 ± 4.74, and 73.33 ± 3.72 MPa, respectively. After reaching a peak value at 66.7 wt% HNTs, a decrease in ultimate tensile strength was observed with further increasing HNT content. Composites with 71.4 and 75.0 wt% HNTs had ultimate tensile strength values of 53.54 ± 7.27 and 47.51 ± 7.05 MPa, respectively. Noteworthily, the ultimate tensile strength is lower than the flexural strength for each sample set. This is common for brittle materials as fewer defects have a critical influence in the bending flexural test relative to the tensile test.45
Compressive strength of these samples was also analyzed. The values were 253.1 ± 16.1, 252.3 ± 33.0, 277.8 ± 25.5, 267.0 ± 17.1 and 187.14 ± 68.0 MPa for HNT contents of 50.0, 60.0, 66.7, 71.4, and 75.0 wt%, respectively (Fig. 6d). The differences of the first four data points with the last one were statistically significant. For each sample set, its compressive strength was much larger than tensile strength; such behavior is typical for brittle materials,46 since defects tend to close under compression, while the resolved shear stress (maximizing at ∼45° to compression direction) and induced tensile stress (perpendicular to compression direction) have less magnitude than compressive stress.
The observed trends of the above mechanical properties with respect to HNT content can be explained through the distribution of the filler, as well as the interactions between the filler and the continuous phase. As revealed by SEM imaging (Fig. 5b), the composite with 50.0 wt% HNTs included numerous polymer microscale domains. This resulted in relatively poor mechanical strength of the composite because the polymer microscale domains have limited ability to bear mechanical loads. With increasing HNT content, the domains of well-dispersed HNTs became predominant, enhancing the mechanical strength up to a point where the dispersibility of HNT became an issue. The relatively poor mechanical strength of the composite with 75.0 wt% HNTs can be attributed to aggregation of HNTs, as shown by SEM (Fig. 5f). The interactions between HNTs and the polymeric continuous phase are weak in these domains, due to the small amount of polymer present. Thus, these domains serve as defect points where mechanical failure originates. Conversely, the highest mechanical strength for the composite containing 66.7 wt% HNTs can be attributed to the well-dispersed HNTs in the polymeric matrix, with little to no aggregation.
Ductility of the polymer/HNT composites was measured through their ultimate elongation. These composites with HNT contents of 50.0, 60.0, 66.7, 71.4, and 75.0 wt% gave ultimate elongation values of 9.28 ± 2.53, 9.21 ± 3.25, 8.45 ± 1.21, 5.46 ± 0.94 and 4.91 ± 0.97%, respectively (Fig. 6e). The differences of the first three data points with the last two were statistically significant. Such results suggested notable presence of structural defects (as voids and aggregation defects) in specimens with 71.4 and 75.0 wt% HNTs, causing the material to become much more brittle. Of note, the fracture surfaces of all specimens were quite smooth, as a typical feature of brittle fracture. Meanwhile, the fractured specimens exhibited reduced cross-sectional area as compared to the original ones, indicating the occurrence of plastic deformation before fracture.
Minimal elastic deformation was observed when the composites were tested. Accordingly, their tensile and compressive moduli were measured. Tensile modulus of the composites with 50.0, 60.0, 66.7, 71.4, and 75.0 wt% HNTs were 388.7 ± 117.6, 399.7 ± 60.5, 452.0 ± 69.2, 602.5 ± 216.7 and 1029.0 ± 300.1 MPa, respectively (Fig. 6f). Compressive modulus of the composites with HNT contents with 50.0, 60.0, 66.7, 71.4, and 75.0 wt% were 1164.4 ± 127.3, 1288.1 ± 90.1, 1293.8 ± 26.7, 1079.3 ± 68.4 and 824.9 ± 142.3, respectively (Fig. 6g). The two sets of modulus values exhibited different trends, with tensile modulus increasing significantly, but compressive modulus decreasing notably after HNT contents exceeding 66.7 wt%. The ratio of compressive modulus to tensile modulus decreased dramatically from ∼3 for composites with 50.0, 60.0, 66.7 wt% HNTs, to 1.79 for composite with 71.4 wt% HNTs, and then to 0.80 for the composite with 75.0 wt% HNTs.
The modulus results of the composites can also be explained through their structural features. It is well-documented that solid continuum materials with well-defined homogeneous structures have the same tensile and compressive moduli, but the PAA-based polymer/HNT composites are anisotropic materials, which often show different resistance to elastic tensile and compressive deformation.47 Because HNTs are discrete nano-fillers dispersed in the polymer matrices, interfacial interactions between polymer and HNTs are responsible for holding everything together to resist elastic tensile deformation of the samples as integrated materials. Because of the high intrinsic moduli of HNTs, polymer-bound HNT-rich microscale domains have higher resistance to elastic tensile deformation than the HNT-free polymer microscale domains. The exceptionally high tensile modulus of the composite with 75.0 wt% HNTs can be attributed to the fact that a HNT network bound by polymer binder was formed throughout the material, but HNT networks of composites of lower contents of HNTs were compromised by the presence of HNT-free polymer microscale domains (Fig. 5). Of note, structural defects unfavorably affecting mechanical strengths would not influence tensile modulus so critically because tensile modulus is measured as the initial slope of tensile stress versus tensile strain. On the other hand, compressive modulus shows material's resistance to elastic compressive deformation associated with reduced volume, and therefore, it is sensitive to void volume in the material.48 The notably lower compressive modulus of the composite with 75.0 wt% HNTs may be ascribed to the presence of void volume between HNTs in the regions with very crowded presence of HNTs in which HNTs were not fully covered by polymer.
Overall, relative to the matrix material, which was too brittle to give meaningful data on mechanical properties, the significantly enhanced mechanical properties of the composites are critically related to the strong interfacial interactions between PAA and HNTs. As the addition of HNTs resulted in significant changes in the morphology of the PAA-based polymer matrix, with restricted polymer chain mobility,49 the interactions between the alumina outer layer of HNTs and the carboxylic groups of PAA segments promote surface adhesion. Of note, other approaches have also been reported to improve surface interaction and achieve dispersion of HNTs in a polymer matrix. Raji et al. reported vinyltrimethoxysilane-modified HNTs in polypropylene with enhanced dispersibility.50 Zhu et al. also reported HNTs modified with 3-aminopropyltriethoxysilane to dramatically improve the dispersion of HNTs in a polyimide composite.51 Thus, incorporating surface modification of HNTs may further improve the mechanical properties of composites containing high weight fractions of HNTs.
Studies of other properties
DSC analysis was conducted to examine the thermal properties of polymer/HNT composites, and the corresponding thermograms are shown in Fig. 7. PAA has a glass transition temperature (Tg) value around 102 °C.52 In principle, at low crosslink density, crosslinking may result in an increase of Tg. However, P(AA-co-TEGDMA) did not show a distinguishable glass transition in its DSC curve. This may be ascribed to the greatly reduced mobility of PAA chains due to the presence of TEGDMA-based cross-links. Moreover, the incorporation of HNTs further restricts polymer chain mobility, resulting in the absence of glass transition in all the DSC curves of the polymer/HNT composites. | ||
| Fig. 7 DSC curves of PAA-based polymer/HNT composites and P(AA-co-TEGDMA). | ||
The TGA and differential TGA curves of polymer/HNT composites, with P(AA-co-TEGDMA) and pristine HNTs as controls, are shown in Fig. 8. P(AA-co-TEGDMA), with 90 wt% AA monomer units, showed three stages of weight loss. The first stage with the temperature of maximum weight loss rate (Td,max) at 317.1 °C, the second phase at ∼350–435 °C and the third phase with the Td,max at 443.6 °C were ascribed mainly to anhydride formation with the elimination of water, anhydride degradation, and massive polymer degradation, respectively.53 Relative to linear PAAs,54 P(AA-co-TEGDMA) showed high thermal stability with significantly retarded weight losses because of the presence of network structures. HNTs exhibited a noticeable weight loss of 9.05 wt% at 500–600 °C due to dehydroxylation of their AlOH groups, with Td,max of 584.4 °C.55 The gradual slight weight loss (5.42 wt%) of HNTs below 500 °C can be attributed to the removal of physically bound water and the desorption of water intercalated between HNT planes. The thermogravimetric behavior of polymer/HNT composites were largely based on their compositions, but showed critical differences with that of P(AA-co-TEGDMA) and HNTs. With increasing HNT content, Td,max values increased gradually for the phase of anhydride formation but deceased notably for the phase of anhydride degradation; moreover, the massive polymer degradation, which gave a sharp peak in the differential TGA curve of P(AA-co-TEGDMA), became quite slow in polymer/HNT composites. These results suggested that water released from HNTs retarded anhydride formation of the polymer matrices,56 and the presence of HNTs might significantly stabilize the residues resulted from the phase of anhydride degradation via surface adsorption. Moreover, the Td,max values for dehydroxylation of AlOH groups of HNTs increased with the increase of HNT contents, suggesting that the presence of residues of polymer degradation on surface of HNTs may promote the occurrence of dehydroxylation reaction.
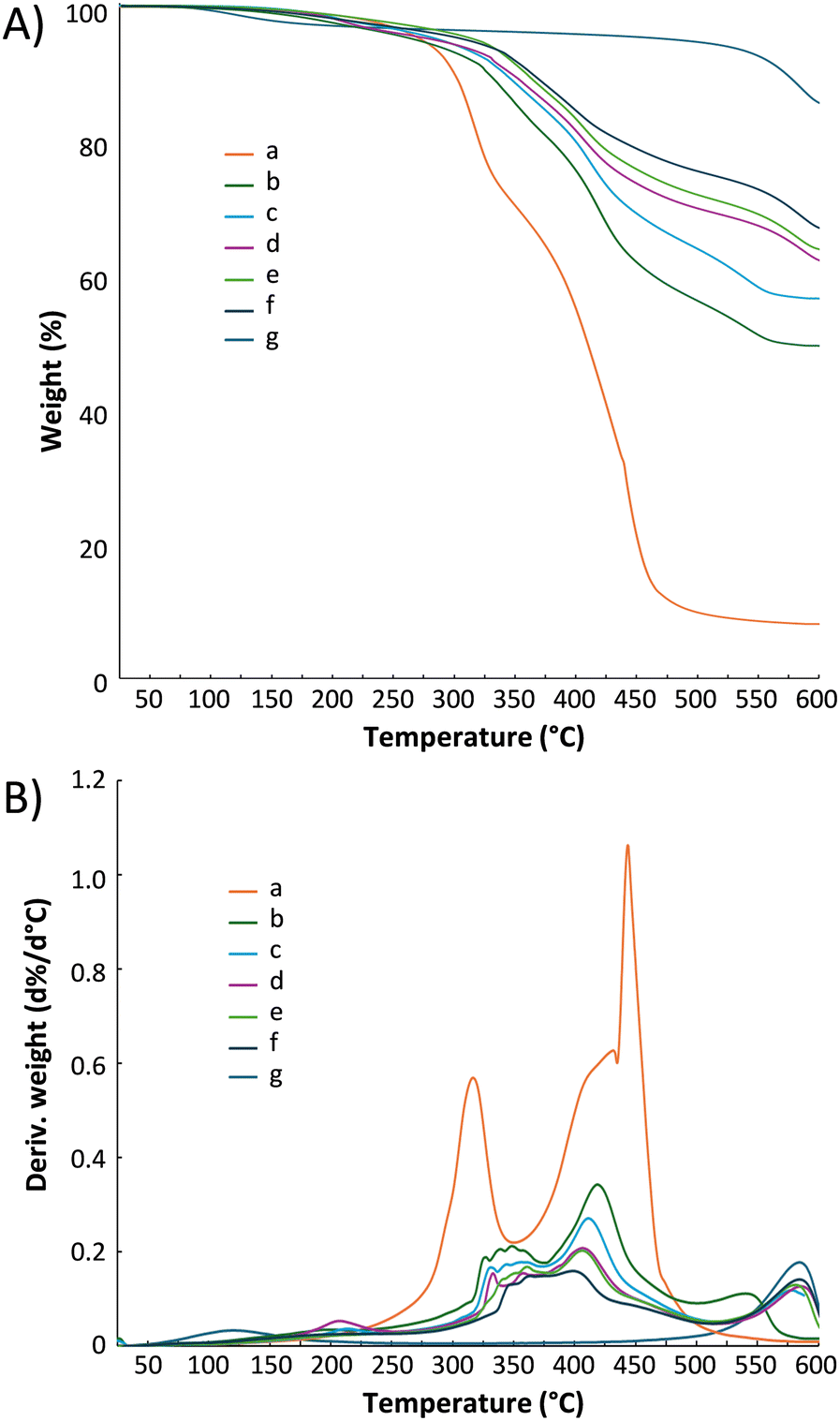 | ||
| Fig. 8 A) TGA curves and B) differential TGA curves for a) P(AA-co-TEGDMA), PAA-based polymer/HNT composites with HNT contents of b) 50.0 wt%, c) 60.0 wt%, d) 66.7 wt%, e) 71.4 wt%, f) 75.0 wt%, and g) pristine HNTs. | ||
The compositions of such composites have major effect on the residual wt% at 600 °C. The polymer matrix, P(AA-co-TEGDMA), gave only 8.11 wt% residue at 600 °C. On the other hand, HNT had 84.6 wt% residue at 600 °C. For the composites, their residue wt% increases with their HNT wt%. For instance, composites with 50.0 and 75.0 wt% HNTs gave 49.45 and 66.26 w% of residue, respectively. For all composites, their residues were mainly derived from HNTs. Overall, the incorporation of high amounts of HNTs into polymer matrices enhances composite thermal stability. Such results may be ascribed to the high thermal stability of HNTs and the barrier properties of HNTs to maximize heat insulation.57 Of note, because of water release during dihydroxylation of HNTs at elevated temperatures,58 the composites may potentially have anti-inflammatory properties.
Swelling tests were conducted for the polymer/HNT composites to assess the impact of HNT incorporation on the swelling properties (Fig. 9), with P(AA-co-TEGDMA) as a control. P(AA-co-TEGDMA) exhibited a swelling ratio of 2.16. The swelling ratios of composites were almost proportional to their wt% of P(AA-co-TEGDMA) matrix, and the composite with 75.0 wt% HNTs and 25.0 wt% P(AA-co-TEGDMA) displayed a swelling ratio of 0.49. The results suggest that the swelling capacity is primarily influenced by the amount of polymer present in the composite, and the incorporation of high HNT contents does not significantly affect the swelling capacity of polymer. In this work, because a high concentration of crosslinker (TEGDMA, 10 wt% relative to AA) was applied, the restriction of swelling by the presence of HNT becomes negligible despite its high weight fractions. PAA-based polymer/HNT composites can not only be quickly hydrated, but also effectively dehydrated as indicated by the absence of free water absorbance in the FTIR spectra, because the presence of HNTs facilitates mass transfer. Of note, the effects of HNTs on the restriction of swelling through physical crosslinking or surface adsorption of polymer chains may become more evident if the polymer matrix was less densely chemically crosslinked.59
 | ||
| Fig. 9 Swelling ratios of PAA-based polymer/HNT composites and P(AA-co-TEGDMA). | ||
Conclusions
This study focused on the effects of high HNT content on morphology, mechanical and thermal properties of TEGDMA-crosslinked PAA-based composites. Polymer/HNT composites with 50.0–75.0 wt% HNTs were fabricated by in situ free radical polymerization. The incorporation of HNTs into the PAA-based matrix resulted in improved thermal stability and greatly enhanced mechanical strength, while not affecting the intrinsic swelling ability of PAA-based matrix. SEM imaging confirmed high compatibility and remarkable dispersion of HNTs in the PAA-based matrix. The composite containing 66.7 wt% HNTs demonstrated the highest flexural strength, microhardness, ultimate tensile strength and compressive strength amongst all polymer/HNT composite materials. These polymer/HNT composites may potentially have other useful properties, such as separation and encapsulation properties, which merit further investigation.Data availability
The data supporting the findings of this work are available from the corresponding author, C. Cheng, upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Jiangsu Qunxin Powder Technology Co., Ltd.References
- P. Yuan, D. Tan and F. Annabi-Bergaya, Appl. Clay Sci., 2015, 112–113, 75–93 CrossRef CAS.
- E. Joussein, S. Petit, J. Churchman, B. Theng, D. Righi and B. Delvaux, Clay Miner., 2005, 40, 383–426 CrossRef CAS.
- I. Wilson and J. Keeling, Clay Miner., 2016, 51, 309–324 CrossRef CAS.
- M. M. Calvino, G. Cavallaro, P. Pasbakhsh, G. Lazzara and S. Milioto, J. Mol. Liq., 2024, 394, 123721 CrossRef.
- S. Zhao, Y. Yuan, Q. Yu, B. Niu, J. Liao, Z. Guo and N. Wang, Angew. Chem., Int. Ed., 2019, 58, 14979–14985 CrossRef CAS PubMed.
- A. Kausar, Polym.-Plast. Technol. Eng., 2018, 57, 548–564 CrossRef CAS.
- M. Du, B. Guo and D. Jia, Polym. Int., 2010, 59, 574–582 CrossRef CAS.
- Y. Zhang, A. Tang, H. Yang and J. Ouyang, Appl. Clay Sci., 2016, 119, 8–17 CrossRef CAS.
- Y. M. Lvov, M. M. DeVilliers and R. F. Fakhrullin, Expert Opin. Drug Delivery, 2016, 13, 977–986 CrossRef CAS PubMed.
- Y. M. Lvov, D. G. Shchukin, H. Mohwald and R. R. Price, ACS Nano, 2008, 2, 814–820 CrossRef CAS.
- T. Barot, D. Rawtani and P. Kulkarni, J. Compos. Sci., 2020, 4, 81 CrossRef CAS.
- H. Lun, J. Ouyang and H. Yang, RSC Adv., 2014, 4, 44197–44202 RSC.
- L. Guimarães, A. N. Enyashin, G. Seifert and H. A. Duarte, J. Phys. Chem. C, 2010, 114, 11358–11363 CrossRef.
- M. Liu, Z. Jia, D. Jia and C. Zhou, Prog. Polym. Sci., 2014, 39, 1498–1525 CrossRef CAS.
- M. Liu, B. Guo, M. Du, X. Cai and D. Jia, Nanotechnology, 2007, 18, 455703 CrossRef.
- Y. Ye, H. Chen, J. Wu and L. Ye, Polymer, 2007, 48, 6426–6433 CrossRef CAS.
- K. Prashantha, H. Schmitt, M.-F. Lacrampe and P. Krawczak, Compos. Sci. Technol., 2011, 71, 1859–1866 CrossRef CAS.
- D. P. A. Pegoretti, R. Thomann, J. Kristof and J. Karger-Kocsis, Polym. Compos., 2015, 36, 869–883 CrossRef.
- S. Srivastava and A. Pandey, Compos. Commun., 2019, 11, 39–44 CrossRef.
- G. Cavallaro, G. Lazzara and S. Milioto, Int. J. Biol. Macromol., 2023, 234, 123645 CrossRef CAS.
- G. Dalei and S. Das, J. Drug Delivery Sci. Technol., 2022, 78, 103988 CrossRef CAS.
- S. O. Bolanta, S. Malijauskaite, K. McGourty and E. J. O'Reilly, ACS Omega, 2022, 7, 9108–9117 CrossRef CAS.
- M. Zhong, Y.-T. Liu, X.-Y. Liu, F.-K. Shi, L.-Q. Zhang, M.-F. Zhu and X.-M. Xie, Soft Matter, 2016, 12, 5420–5428 RSC.
- S. Anjum, P. Gurave, M. V. Badiger, A. Torris, N. Tiwari and B. Gupta, Polymer, 2017, 126, 196–205 CrossRef CAS.
- X. Li, Y. Zhao, D. Li, G. Zhang, S. Long and H. Wang, Polymer, 2017, 121, 55–63 CrossRef CAS.
- G. Miquelard-Garnier, D. Hourdet and C. Creton, Polymer, 2009, 50, 481–490 CrossRef CAS.
- U. Gulyuz and O. Okay, Macromolecules, 2014, 47, 6889–6899 CrossRef CAS.
- S. Y. Kim, H. S. Shin, Y. M. Lee and C. N. Jeong, J. Appl. Polym. Sci., 1999, 73, 1675–1683 CrossRef CAS.
- Y. Huang, J. Lu and C. Xiao, Polym. Degrad. Stab., 2007, 92, 1072–1081 CrossRef CAS.
- Y. Wang, X. Zhang, H. Wei, B. Zhang, X. Xiang and R. Chen, Polym. Compos., 2015, 36, 229–236 CrossRef CAS.
- S.-N. Li, B. Li, Z.-R. Yu, Y. Li, K.-Y. Guo, L.-X. Gong, Y. Feng, D. Jia, Y. Zhou and L.-C. Tang, Composites, Part B, 2020, 194, 108046 CrossRef CAS.
- A. Kausar, J. Plast. Film Sheeting, 2021, 37, 409–428 CrossRef CAS.
- H. Wang, T. Shi and L. Zhai, J. Appl. Polym. Sci., 2006, 102, 1729–1733 CrossRef CAS.
- S. K. Sidhu and J. W. Nicholson, J. Funct. Biomater., 2016, 7, 16 CrossRef.
- X. Shi, S. Xu, J. Lin, S. Feng and J. Wang, Mater. Lett., 2009, 63, 527–529 CrossRef CAS.
- K. Kurihara and M. Mizukami, Proc. Jpn. Acad., Ser. B, 2001, 77, 115–120 CrossRef.
- X. Sun, Y. Zhang, H. Shen and N. Jia, Electrochim. Acta, 2010, 56, 700–705 CrossRef CAS.
- Y. Torii, N. Sugimura, H. Mitomo, K. Niikura and K. Ijiro, Langmuir, 2017, 33, 5537–5544 CrossRef CAS PubMed.
- N. Rekha and S. K. Asha, J. Appl. Polym. Sci., 2008, 109, 2781–2790 CrossRef CAS.
- A. Pumchan, O. Cheycharoen, S. Unajak and C. Prasittichai, New J. Chem., 2021, 45, 9130–9136 RSC.
- Z. Shu, Y. Chen, J. Zhou, T. Li, D. Yu and Y. Wang, Appl. Clay Sci., 2015, 112, 17–24 CrossRef.
- D. Garcia-Garcia, J. M. Ferri, L. Ripoll, M. Hidalgo, J. Lopez-Martinez and R. Balart, Appl. Surf. Sci., 2017, 422, 616–625 CrossRef CAS.
- Y. Chen, L. M. Geever, J. A. Killion, J. G. Lyons, C. L. Higginbotham and D. M. Devine, Polym. Compos., 2017, 38, 2166–2173 CrossRef CAS.
- Q. Chen, Y. Zhao, W. Wu, T. Xu and H. Fong, Dent. Mater., 2012, 28, 1071–1079 CrossRef CAS PubMed.
- D. Leguillon, É. Martin and M.-C. Lafarie-Frenot, C. R. Mec., 2015, 343, 275–281 CrossRef.
- Y. Wang, J. Ding, Z. Fan, L. Tian, M. Li, H. Lu, Y. Zhang, E. Ma, J. Li and Z. Shan, Nat. Mater., 2021, 20, 1371–1377 CrossRef CAS PubMed.
- M. S. Dayal and J. M. Catchmark, Carbohydr. Polym., 2016, 144, 447–453 CrossRef CAS PubMed.
- L. Li, Y. Zhu and J. Yang, Mater. Lett., 2018, 210, 136–138 CrossRef CAS.
- S. Kadanyo, C. N. Matindi, N. N. Gumbi, D. S. Dlamini, Y. Hu, Z. Cui, B. He, B. B. Mamba and J. Li, npj Clean Water, 2023, 6, 19 CrossRef CAS.
- M. Raji, M. E. M. Mekhzoum, D. Rodrigue, A. E. K. Qaiss and R. Bouhfid, Composites, Part B, 2018, 146, 106–115 CrossRef CAS.
- T. Zhu, C. Qian, W. Zheng, R. Bei, S. Liu, Z. Chi, X. Chen, Y. Zhang and J. Xu, RSC Adv., 2018, 8, 10522–10531 RSC.
- C.-K. Chan and I.-M. Chu, Polymer, 2001, 42, 6089–6093 CrossRef CAS.
- C. A. Fyfe and M. S. McKinnon, Macromolecules, 1986, 19, 1909–1912 CrossRef CAS.
- J. J. Maurer, D. J. Eustace and C. T. Ratcliffe, Macromolecules, 1987, 20, 196–202 CrossRef CAS.
- S. A. Hashemifard, A. F. Ismail and T. Matsuura, J. Colloid Interface Sci., 2011, 359, 359–370 CrossRef CAS PubMed.
- A. R. Greenberg and I. Kamel, J. Polym. Sci., Polym. Chem. Ed., 1977, 15, 2137–2149 CrossRef CAS.
- N. Eser, M. Önal, M. Çelik, A. D. Pekdemir and Y. Sarıkaya, Polym. Compos., 2020, 41, 893–899 CrossRef CAS.
- P. Yuan, D. Tan, F. Annabi-Bergaya, W. Yan, M. Fan, D. Liu and H. He, Clays Clay Miner., 2012, 60, 561–573 CrossRef CAS.
- Y. Luo and D. K. Mills, Gels, 2019, 5, 40 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |