
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Porous carbons: a class of nanomaterials for efficient adsorption-based hydrogen storage
Lila A. M.
Mahmoud
 a,
Jemma L.
Rowlandson
bc,
David J.
Fermin
a,
Valeska P.
Ting
*de and
Sanjit
Nayak
*d
a,
Jemma L.
Rowlandson
bc,
David J.
Fermin
a,
Valeska P.
Ting
*de and
Sanjit
Nayak
*d
aSchool of Chemistry, Cantocks Close, University of Bristol, Bristol BS8 1TS, UK
bBristol Composites Institute, University of Bristol, Bristol, BS8 1TR, UK
cSchool of Electrical, Electronic and Mechanical Engineering, University of Bristol, Bristol, BS8 1TR, UK
dSchool of Civil, Aerospace and Design Engineering, University of Bristol, Bristol, BS8 1TR, UK. E-mail: s.nayak@bristol.ac.uk
eResearch School of Chemistry, Australian National University, Canberra ACT 2601, Australia. E-mail: valeska.ting@anu.edu.au
First published on 30th October 2024
Abstract
Hydrogen has become a promising clean energy source as governments worldwide aim to reduce their reliance on fossil fuels and achieve net-zero emissions. However, a major barrier for hydrogen economy is the challenges associated with the efficient storage of hydrogen, due to its low density, ultra-low boiling point, and extreme volatility. Current practice of using high-pressure tanks has safety concerns and is costly. As a potential solution, adsorption-based hydrogen storage using porous materials has shown great promise due to fast kinetics and their ability to store a comparable amount of hydrogen at much lower pressure. This approach takes advantage of physisorption of hydrogen in porous materials with high surface areas. A number of different classes of materials have been studied for adsorption-based hydrogen storage. Among these materials, porous carbon has shown great promise due to its high surface area, tunable pore size, versatile surface chemistry, scalability, and high chemical and thermal stability. This review provides a comprehensive overview of porous carbon materials, such as graphene, carbon nanotubes, and activated carbons, for hydrogen storage. We delve into the fundamental principles and mechanisms behind adsorptive hydrogen storage, focusing on the critical roles of surface area, pore size, and surface chemistry in determining hydrogen uptake. Strategies to enhance hydrogen storage capacity through structural and chemical modifications are discussed. Additionally, we examine the life cycle assessment of porous carbons and explore recent advancements in machine learning applications to optimize their performance. Finally, we offer insights into the future outlook of porous carbons as a sustainable hydrogen storage solution.
1. Hydrogen as a sustainable energy source: challenges and opportunities
The development of sustainable and efficient energy systems is crucial for meeting the global demand for clean energy. Our increasing consumption of fossil fuels contributes to the release of greenhouse gases into the atmosphere, leading to global warming and climate change.1 In addition, limited reserves, uneven distribution, and price instability of fossil fuels are increasingly becoming driving forces for many countries and economies to turn to renewable sources.2 Hydrogen is a promising energy vector for replacing fossil fuels as it is potentially renewable and clean,3 producing only water, heat and energy when used in a fuel cell. Additionally, hydrogen has the highest gravimetric energy density (energy stored per unit mass) of any commonly used fuel, at 142 MJ kg−1, compared to 44 MJ kg−1, 53.6 MJ kg−1, and 45.4 MJ kg−1 for gasoline, natural gas, and diesel, respectively,4,5 making it one of the most efficient and sustainable sources of energy.6 Despite the positive aspects of using hydrogen, a successful transition to a hydrogen-powered economy requires the remediation of some of its downsides for practical use (as shown in Scheme 1). For example, hydrogen has a very low critical temperature of 33 K and under ambient conditions it exists as a gas with a very low density of 0.083 kg m−3.7 This combination of low gas density and extremely low critical temperature makes it highly challenging for developing compact high-density hydrogen storage systems. To overcome this problem, it is crucial to find solutions to increase the volumetric energy density (energy storage per unit volume) of hydrogen for practical applications. As a guideline, based on the requirements for light-duty vehicles with an ultimate driving range of more than 500 miles, the US Department of Energy (US DOE) has established goals for gravimetric and volumetric hydrogen storage capacities for on-board vehicle applications while maintaining safety, cost and performance requirements (Table 1).8 | ||
| Scheme 1 Schematic showing the hydrogen cycle as part of hydrogen economy. | ||
| Storage parameter | Units | 2020 | 2025 | Ultimate |
|---|---|---|---|---|
| System gravimetric capacity | ||||
| Usable, specific energy from H2 (net useful energy/max system mass) | kW h kg−1 (kg H2/kg system) | 1.5 (0.045) | 1.8 (0.055) | 2.2 (0.065) |
| System volumetric capacity | ||||
| Usable energy density from H2 (net useful energy/max system volume) | kW h L−1 (kg H2/L system) | 1.0 (0.030) | 1.3 (0.040) | 1.7 (0.050) |
The US DOE identified that maximising hydrogen storage capacity is the one of the essential factors to be taken into consideration for successful utilization of hydrogen for on-board applications. However, other factors like operating cost, durability/operability, and charging and discharging rate, and safety have also been identified with defined targets. In a report published by U.S. DRIVE (Driving Research and Innovation for Vehicle efficiency and Energy sustainability) the ultimate cost for an on-board hydrogen storage system is set at $8 USD per kW h.8 Other goals include operability under ambient conditions in a range of −20 °C and 60 °C and minimum and maximum system delivery pressures of 5 to 20 bar (absolute), an ultimate operational life cycle of 1500 times, quick refill, and a target of more than 90% energy efficiency for hydrogen storage systems.8 Clearly, the development of hydrogen storage materials that meet these goals is a top priority to realize the targets set by the US DOE. In order to achieve these targets, researchers and engineers are exploring a wide range of potential storage materials and methods. Currently, several types of hydrogen storage technologies are being studied,10 such as (i) compression in high-pressure cylinders, (ii) liquification of hydrogen at cryogenic temperatures, (iii) adsorption into porous materials with high surface areas, (iv) interstitial adsorption of hydrogen on metals and clathrates, (v) via liquid organic hydrogen carriers, (vi) using complex hydrides, (vii) precursors with binding of hydrogen through the formation of covalent or ionic bonds, (viii) chemical precursors like ammonia that can decompose to hydrogen and (ix) via the oxidation of reactive metals in the presence of water. Physical storage methods include compression and liquefaction. Compression is the most popular method of hydrogen storage for on-board applications.11 However, despite being a highly developed method of storage, compression can be economically unfavourable as storing hydrogen under high pressures (often up to 70 MPa) can compromise energy equivalent to 13–18% of its lower heating value.6,12 Additionally, safety concerns and practical limitations become critical as a result of limited durability of pressurised tank materials. In addition, any further increase in volumetric capacity for hydrogen would require further increases in pressure, but that would result in compromising gravimetric density, due to the need for thicker tank walls that increase the weight and cost of the storage system.10,13 For liquefaction, hydrogen is usually stored at temperatures below 30 K at ambient pressures and requires well-insulated storage tanks. Both methods have proven to be energy-intensive and costly. Standards established for hydrogen storage tank systems for light-duty, heavy duty and ground storage applications require use of expensive materials like composites of carbon fibres, polymer liners, and steel.14,15 Therefore, material-based approaches for efficient hydrogen storage have attracted much attention during the past two decades, as they offer solutions to the limitations set by the two aforementioned methods.
Storage into solid-state materials depends on the material–hydrogen interactions, and mainly involves physical, chemical and intermediary interactions. Physisorption or physical adsorption is an exothermic non-specific process in which molecular hydrogen is adsorbed on the surface of the material via weak van der Waals forces.16 The overall combination of short-range repulsive forces between H2 molecules and long-range attractive forces results in a relatively shallow and small minimum in the potential energy curve (shown in the Lennard-Jones potential diagram in Fig. 1a). Due to this spontaneous interaction, there is no activation energy barrier to facilitate the adsorption of hydrogen on the surface of the adsorbent, giving rise to the fast kinetics of physisorption.17,18 Physisorption involves low enthalpies of adsorption (<10 kJ mol−1) and it is inversely related to ambient temperature, hence it is necessary to maintain cryogenic temperatures for efficient storage.16,19 Physisorption is most effective in porous materials with high specific surface areas (SSA), including MOFs, porous carbon, covalent organic frameworks (COFs), and organic polymers. Chemisorption, on the other hand, is an activated process that involves the selective adsorption of the adsorbate on the active sites of the adsorbent through the formation of strong chemical bonds.20 The enthalpy of chemisorption on metal surfaces is between 40 and 160 kJ mol−1.21 The potential energy curves for chemisorbed and physiosorbed hydrogen are shown in Fig. 1.
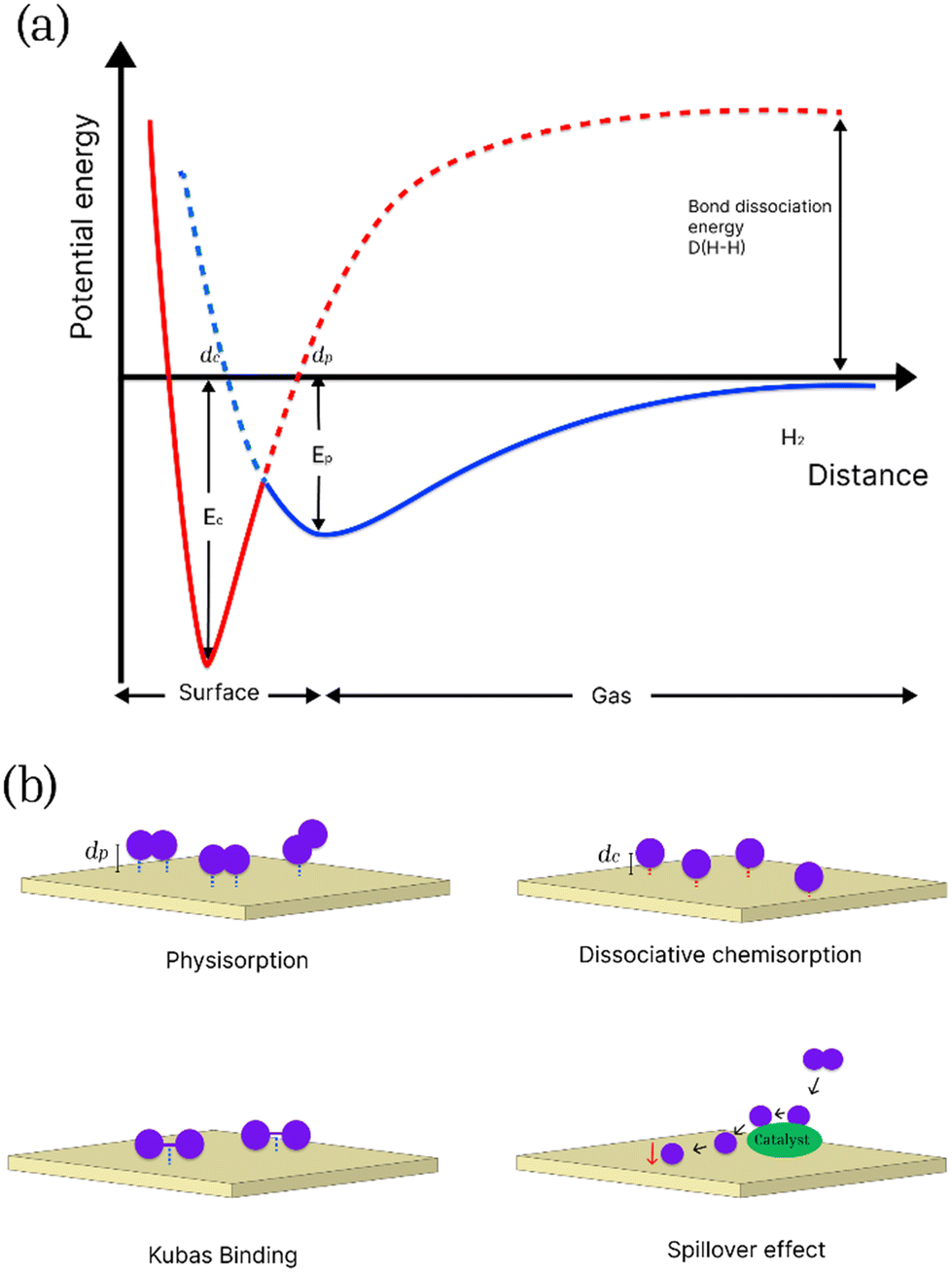 | ||
| Fig. 1 (a) Potential energy curves for physisorbed and chemisorbed hydrogen as a function of distance d from the surface of the adsorbate. Adapted from ref. 17; (b) a schematic showing the different types of mechanisms for hydrogen adsorption on an adsorbate surface. | ||
Two minima are shown at distances dc and dp, corresponding to the equilibrium distance for chemisorption and physisorption, respectively. In chemisorption hydrogen is absorbed into the crystal lattice of the adsorbent, such as metals to create a hydrogenated metallic phase at ambient temperatures and pressures, resulting in dissociation into its atomic form and the formation of metal hydrides or complex hydrides.22 However, despite high storage uptake chemisorption predominantly suffers from slow kinetics and desorption due to the presence of an activation barrier. High temperatures are needed to release the stored hydrogen due to the high binding energy of hydrogen to the substrate. For example, a high adsorption capacity of 8.7 wt% has been achieved for lithium–beryllium hydrides with sorption and desorption temperatures of 300 °C and 320 °C, respectively.23,24 Various types of materials that store hydrogen by chemisorption have been studied for hydrogen storage, including widely studied metal hydrides like MgH2 and LiBH4.25,26
Further modifications of solid-state materials have prompted study into other proposed intermediary adsorption and storage methods via hydrogen spill-over and quasi-molecular interactions (otherwise known as Kubas binding) which are shown in Fig. 1.27 In these proposed processes, the presence of a metal catalyst or a heteroatom on the surface of porous materials is believed to facilitate adsorption by increasing binding enthalpies and therefore, can contribute towards improved adsorption kinetics and hydrogen storage capacity of the materials.28 Kubas binding involves the donation of σ-bonding electrons to an empty d-orbital and π back-donation from a filled d-orbital into the empty σ* anti-bonding orbital of hydrogen, hence causing elongation of an H–H bond.29 For practical applications, the primary advantages of hydrogen storage by physisorption are the rapid adsorption and desorption kinetics and more practical operating conditions when compared to chemisorption.30 Among porous materials that are widely studied, MOFs an COFs can be advantageous in their tuneable structure, highly crystalline nature, and they can be synthesised fairly consistently. The very high surface area makes these materials attractive for hydrogen storage applications. In addition, the presence of metal binding sites and the ability to post-synthetically modify the linkers in MOFs provide additional advantages over other types of porous materials. However, the major caveat to using MOFs, COFs and porous polymers like PIMs is that despite the high surface area, they can be limited by relatively poor chemical and thermal stability, and challenges involved in sustainable large-scale synthesis. Porous carbon on the other hand can be a cost-effective alternative. They can be more easily synthesised in large industrial scales and can be sourced from recyclable carbon-rich waste materials, including bio-based precursors. Porous carbons have attracted much attention recently (Fig. 2), as they offer several advantages over other materials. They also offer a multitude of optimization strategies for improving hydrogen storage capacity due to their high chemical and thermal stability compared to other materials, such as MOFs and porous polymers.31–35 The following table (Table 2) provides a comparative overview of various types of porous materials for hydrogen storage applications, highlighting the benefits and drawbacks of these materials based on different parameters.
 | ||
| Fig. 2 Graph showing the rising trend in the number of publications per year based on a search for “porous carbon” and “hydrogen storage” on Web of Science. Accessed on 26th of September 2024. | ||
| Parameter | MOFs | COFs | PIMs | Porous carbon |
|---|---|---|---|---|
| Porosity | Very high, tuneable pore sizes and shape (e.g. ranging from 2 to more than 50 nm)36 | High porosity. Tuneable pore sizes | Moderate porosity. Tenable porosity | High porosity, though not as tuneable as MOFs. Non-uniform pore sizes |
| Surface area | Extremely high surface area (e.g. experimental highest BET SSA for NU-110, 7140 m2 g−1. Theoretical upper limit of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 m2 g−1)37 600 m2 g−1)37 |
High surface area (surface area can reach 5000 m2g−1)38 | Moderate surface area (around 1000 m2 g−1)39 | High surface area (e.g. highest experimental surface area for activated carbon is 3839 m2 g−1)40 |
| Thermal and chemical stability | Moderate chemical and thermal stability41 | Moderate thermal and chemical stability42 | Moderate thermal stability (e.g. PIM-7 had thermal decomposition temperature of 480 °C;43 PIM-1 retained stability for 400 days with loss of 0.7 wt% at 77 K and 100 bar)44 | Very high chemical and oxidative stability45 |
| Synthesis | Variety of synthesis methods. Involving building blocks, metal salts and organic linkers (e.g. solvothermal, microwave-assisted, mechanical)46 | Variety of synthesis methods. Complex synthesis and processability. Difficulty to synthesize on a larger scale (microwave, solvothermal, mechanical)38 | Moderate synthesis difficulty47 | Variety of synthesis methods. Accessible and cheaper sources of precursor (e.g. biomass). Activation methods can involve chemical or physical methods48,49 |
| Hydrogen storage capacity | High hydrogen storage capacity (highest gravimetric total capacity reported for DUT-32 is 14.21 wt% at 80 bar and 77 K;50 highest gravimetric excess capacity reported for NU-100 is 9.13 wt% at 56 bar and 77 K)51 | High storage capacity (COF-103 has gravimetric hydrogen storage capacity of 6.6 wt% at 35 bar and 77 K)52 | Moderate H2 uptake of 1.7 wt% (77 K, 10 bar)39 | High hydrogen storage capacity (e.g. excess gravimetric storage capacity is 7.0 wt% at 77 K and 20 bar)53 |
| Functionalisation | Easy to functionalise by post-synthetic, or in situ modifications.54 Presence of metal binding sites for adsorption | Lack of H2 binding sites. Potential for modification55,56 | Lack of binding sites, potential for functionalisation by post synthetic modification57–59 | Lack of H2 binding sites. Can be functionalised by heteroatom doping and metal nanoparticles |
| Environmental impact | Presence of toxic starting materials, solvents and waste products. Can be synthesised by mechanochemical methods | Use of solvents and energy in conventional solvothermal methods | Wet chemical methods use solvents like DMSO and toluene. Process can be energy intensive.60 Can be synthesised by mechanochemical methods | Moderate environmental impact. Depends on the synthesis method and choice of precursor. Biomass derived carbon can be sustainable and green |
| Cost | High cost due to use of expensive organic linkers, metal salts, solvents, as well as processes such as heating and drying61 (e.g. synthesis costs could range from 35–71 and 13 to 36 USD per kg for solvent-assisted and solvent-free synthesis, respectively)61 | Solvothermal synthesis can be energy intensive and uses a large amount of solvents, and expensive COF monomers62 therefore costly. However, some environment friendly synthesis methods (e.g., mechanochemical) are available | High cost due to use of large amounts of organic solvents | Can be cost effective by choosing the precursors and activation method |
In this review, we will briefly outline different types of porous carbon materials for hydrogen storage, with an emphasis on their synthesis methods and strategies for improving storage capacity and operating conditions. Lastly, we will outline the future prospects of incorporating carbon-based materials into a sustainable hydrogen economy, by presenting the challenges and potential for their use as future hydrogen storage materials.
2. Properties of porous materials influencing H2 storage by physisorption
The physical characteristics of porous carbon play a critical role in determining hydrogen uptake capacity (shown in Fig. 3). Firstly, as physisorption is a key factor in H–adsorbate interactions on porous carbon, the SSA is an important parameter for improving the hydrogen uptake capacity of porous materials. Typically, a higher SSA means more adsorption sites are available for hydrogen to interact with the surface. As a rule of thumb, materials exhibit a 1 wt% increase in gravimetric H2 uptake per 500 m2 g−1 SSA (measured at 77 K). This correlation is well-known in the community as Chahine's rule.9 However, porous materials frequently surpass this due to another important factor, the pore size, as we will discuss next. Secondly, although there is a relationship between the surface area and hydrogen uptake, it is important to also consider the interrelation between the surface area and pore volume in controlling hydrogen storage capacity. The size and shape of the pores greatly affect hydrogen interactions, by influencing hydrogen's accessibility to adsorption sites of the material and the strength of the interactions. Based on pore diameter IUPAC has defined three types of pores: macropores (pore diameter ≥ 50 nm), mesopores (pore diameters between 2 nm and 50 nm), and micropores (pore diameter ≤ 2 nm).63 Typically, micropores provide the optimal size for better hydrogen adherence to the adsorbent walls. The pore sizes are small enough to result in the overlap of potential fields of opposing pore walls, resulting in the increase of its isosteric enthalpy of adsorption, hence adsorbing hydrogen at much higher densities than its gaseous form,64,65 with a reported density of up to 100 kg m−3.66 It has been found that the pore diameter for maximal hydrogen uptake ranges from 0.6–0.7 nm at elevated pressure and 77 K.67 A smaller pore size than this would not allow dynamic hydrogen atoms to be contained within the pores, and a larger one will decrease the interaction between the molecular hydrogen and pore walls. A number of studies have led to this conclusion, by demonstrating optimal hydrogen uptake within the narrow range of pore size distribution (PSD) below 1 nm.68 Hence, it is essential to consider the importance of the PSD when developing porous materials for hydrogen storage applications. Another important parameter for hydrogen storage capacity is the micropore volume. It is a measure of the extent of the material's microporosity which is determined from the cumulative volume of micropores. There is a positive direct relationship between micropore volume and hydrogen storage capacity, with higher micropore volumes in porous materials correlating to increased hydrogen uptake capacity.69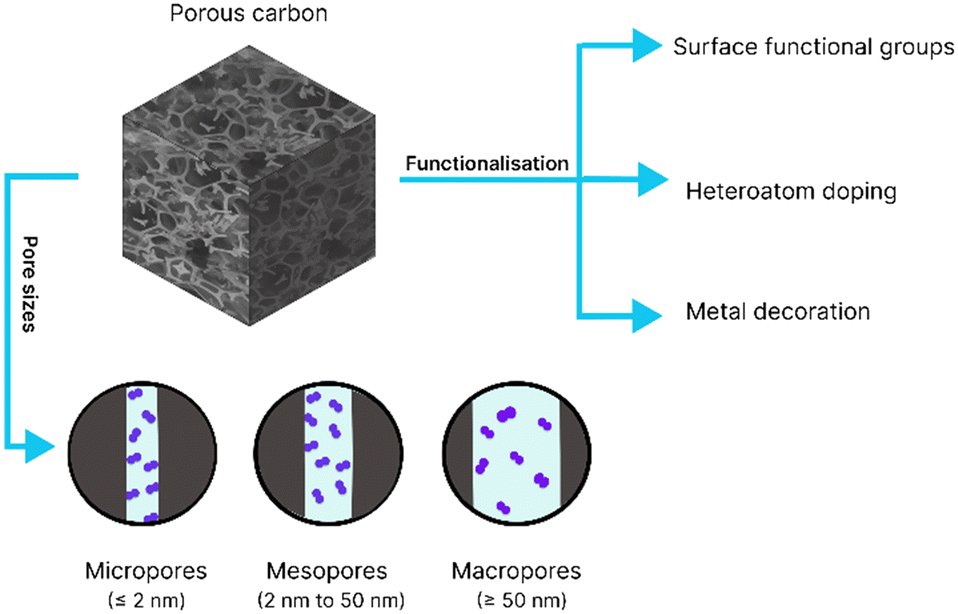 | ||
| Fig. 3 Schematic showing the structure property relationship of porous carbon, the pore size and types of functionalization, which are important factors in enhancing hydrogen uptake in carbon-based sorbents. | ||
Another important parameter that directly influences the H2 storage capacity is the isosteric enthalpy of adsorption. It is defined as the amount of heat released when an adsorbate binds to an adsorbent. It is also referred to as the isosteric heat of adsorption.70,71 However, many researchers have recommended the use of enthalpy of adsorption over the heat of adsorption, as enthalpy represents the thermodynamic change. A higher magnitude of enthalpy corresponds to stronger adsorbent–adsorbate interactions, and hence, more hydrogen will be adsorbed at a given temperature and time.72 In porous carbon materials, the enthalpy of adsorption of hydrogen is relatively low, ranging from −4 to −6 kJ mol−1, as it relies on physisorption of hydrogen to the surface via weak van der Waals interactions.73 The optimal enthalpy of adsorption for hydrogen storage is between −15 and −20 kJ mol−1 under ambient conditions.74 Such isosteric heat of enthalpies can be achieved by optimizing the conditions for adsorption, chemical, and physical characteristics. Surface functionalization plays an important part in enhancing hydrogen uptake in porous carbon materials. Modifying the surface chemistry with heteroatoms, metal nanoparticles, and other functional groups has been proven to increase the interactions of hydrogen with the materials. For example, porous carbons doped with Ni, Fe, or Mn have shown an increased magnitude of enthalpy of adsorption to 6.52, 6.31 and 6.14 kJ mol−1, respectively, as opposed to 6.07 kJ mol−1 for undoped porous carbon. This indicates increased H2–adsorbate interactions.75 These functional groups and metal nanoparticles facilitate stronger adsorption by providing additional binding sites for hydrogen atoms and by creating localized regions of polarity through dipole–dipole interactions.76 Surface functionalization with metal catalysts like Pt and Pd modifies the electronic structure of the carbon material, resulting in charge redistribution and transfer, hence giving rise to a catalytic phenomenon called the spillover effect.9,77 First reported in 1964, the spillover effect is defined as the migration of hydrogen atoms catalysed by a metal nanoparticle to an H+ accepting site on the catalyst support while simultaneously reducing the metal sites.77,78 Temperature programmed reduction (TPR) experiments conducted by Briggs et al. have postulated the mechanism and its effect in enhancing hydrogen storage capacities using Pd and CuO catalysts on a carbon nanotube support.79 Further experiments as well as DFT studies have supported the spillover effect as a potential mechanism for enhanced hydrogen storage.80 Although the exact mechanism for the spillover effect is still debated, it has been proposed as a primary mechanism for enhanced hydrogen uptake capacity of porous materials at room temperature in multiple studies.81–84
2.1. Practical hydrogen storage
In order to successfully incorporate porous materials as a suitable and practical method for hydrogen storage, a number of parameters need to be considered. The term practical hydrogen capacity assesses the potential of materials for widespread applications. This includes factors like stability, cost, ease of synthesis/fabrication, and safety. Additionally, it also includes factors relating to its performance, like reversibility (kinetics), recyclability, adsorption capacity, and thermodynamic properties, which govern the operating conditions for efficient storage and delivery.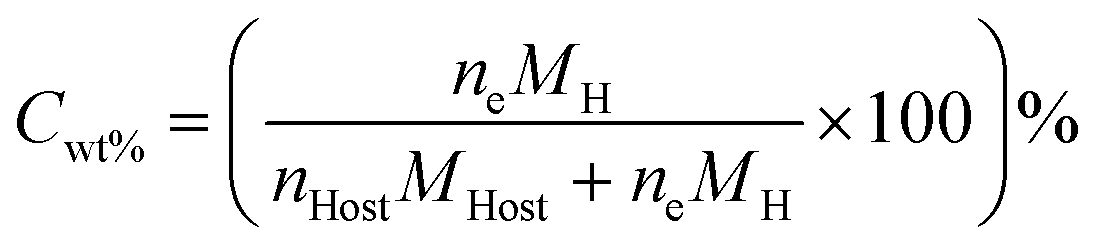 | (1) |
 | (2) |
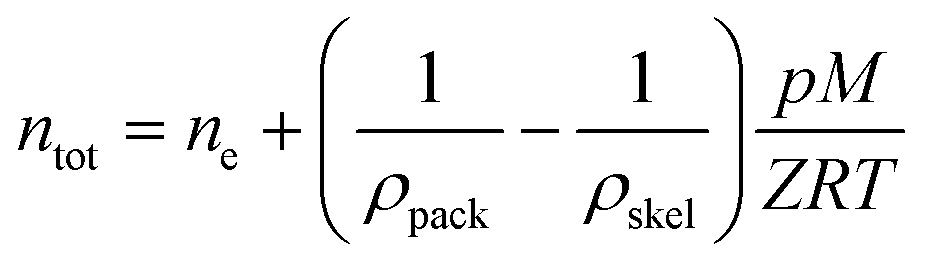 | (3) |
 | ||
| Fig. 4 Scheme illustrating (a) excess adsorption and (b) net adsorption. Grey dots represent the free hydrogen molecules and coloured dots represent the adsorbed hydrogen molecules. Blue dots in (a) are hydrogen molecules of the bulk phase that would be present without interaction between the molecules and adsorbent. Red dots represent the hydrogen molecules adsorbed on the surface due to interaction. Blue dots in (b) represent the amount of hydrogen in the bulk phase that would occupy the skeletal volume under the given experimental conditions. | ||
Volumetric capacity is defined as the amount adsorbed per unit volume, measured in units of H2 kg m−3. Although it gives useful insight on the amount of hydrogen adsorbed, it assumes that the volume doesn't change with adsorption of hydrogen. As with gravimetric storage, it is difficult to determine the exact amount of adsorbed hydrogen, and hence, an assumption is made that the adsorbed hydrogen occupies all the pores in the material. This is called the total pore approximation.91
Both methods of quantifying the adsorbed hydrogen can provide useful insight into the material's characteristics and its ability to store hydrogen. At low temperatures, the trend in improving the gravimetric storage capacity of porous materials seems to be correlated with the BET surface area, as highlighted previously in Chahine's rule. Based on this correlation, research has been focused on microporous materials to develop materials with ultrahigh surface areas to achieve high hydrogen storage capacity. MOFs like NU-100 with a reported BET surface area of around 6143 m2 g−1 showed an excess gravimetric capacity of 9.95 wt% at a temperature of 77 K and pressure of 5.6 MPa.51 However, efforts to improve gravimetric capacities alone might not be the optimal strategy for practical hydrogen storage.92 Other metrices, such as the volumetric hydrogen storage capacity, also need to be taken into consideration. Volumetric measurements require knowledge of the material's bulk density or packing volume, and hence, are correlated with the surface area occupied per unit volume, known as the volumetric surface area. This factor can be optimised by either reducing the bulk volume, or enhancing the surface area, using various approaches including powder compression, incorporation of porous materials into monoliths or polymer composites,44,93 and enhancing interpenetration in the case of open framework materials like MOFs.94
| nuc(Pi) = nabs(Pi) − nabs(Pbk) | (4) |
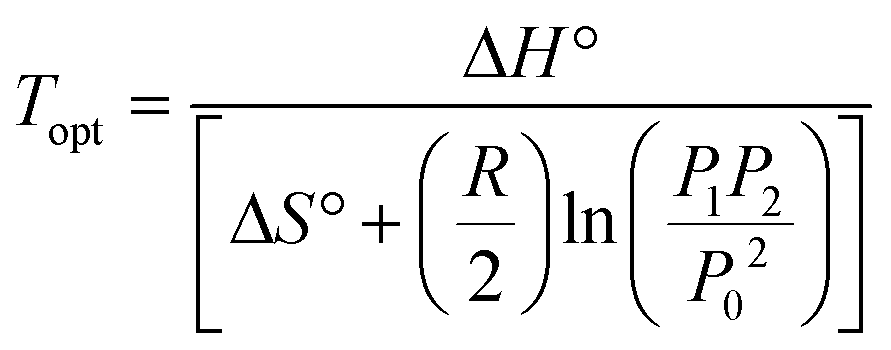 | (5) |
Based on the IUPAC classification, microporous carbon materials show type I (a) and type I (b) adsorption behaviour at 77 K,97 meaning that the equilibrium uptake of hydrogen initially increases with increasing pressure, until a plateau or a saturation point is reached at higher pressures. The adsorption isotherm of a material relates to its potential for reversible hydrogen storage. Usable storage capacity is the amount of hydrogen that can be adsorbed and released within lower and upper limits of operating pressure. As shown in Fig. 5, the usable capacity can be visualised as isobars plotted by calculating the difference between the upper and lower pressure limits of pressure at a given temperature. In Fig. 3(c), the usable capacity is maximum at the optimum operating temperature, and as it is shown earlier in eqn (5), its value is dependent on the upper and lower pressure limits.
 | ||
| Fig. 5 Plots of absolute uptake and usable capacity of hydrogen against various temperatures and pressures showing: (a) typical type I adsorption isotherms plotted at various temperatures, T1, T2 and T3. (b) Usable capacity when plotted at isobars for various pressures, P0, P1, P2, P3, and P4 at different temperatures. (c) Isobars with the usable capacity as the change in absolute adsorption at difference in pressure ΔP. Reproduced with permission from ref. 96. | ||
Another important quality of the material is its cycling stability. It is the ability of the adsorbate material to recover its hydrogen storage capacity along hydrogen charging and discharging cycles.98 Recyclability depends on the chemical and thermal stability of the material at the temperatures of hydrogen cycling. However, it can be negatively impacted by impurities present in the gas as well; adsorption of contaminants is more likely to reduce the hydrogen storage capacity, due to favoured adsorption of gaseous contaminants including water and nitrogen onto the adsorption sites.99,100
3. Porous carbon
As mentioned earlier, porous carbons are a promising class of materials for hydrogen storage due to their high surface area, functionalization, high stability, and tailorable pore size. Additionally, porous carbons are a group of materials with various structural diversity, starting from the simplest 2-D sheet-like structure of graphene to the complex and interconnected network of activated carbons. These groups vary greatly in their structural morphology, including the surface area and PSD, in addition to their synthesis methods and precursors. For example, it has been shown that activated carbon can be sourced from biomass waste materials which makes it an economically favorable material to synthesize. Template-derived carbons made from carbide and zeolite precursors, and activated carbons have shown the highest hydrogen storage capacity to date, showing a maximum hydrogen storage capacity of 6.9 wt% and 8.1 wt%, respectively, at 2 MPa and 77 K.101,102 This is primarily due to their high micropore volume and SSA, as a result of their tuneable synthesis and activation methods. Moreover, other types of carbon have been explored, including graphene and graphene-based materials like graphene and nanotubes, with an estimated theoretical hydrogen storage capacity of 7 wt% at 77 K and 1 MPa, and with computer simulations, respectively. The high volume of research conducted on different classes of porous carbon for hydrogen storage is reflected in the number of papers published (shown in Table 3). Additionally, Fig. 6 presents a 3D-scatter plot of different types of carbon materials. As can be observed, most studies are carried out at 77 K and 20 bar and ambient temperatures. From this comparative plot it can be seen that graphene, activated carbons, and template derived carbons occupy high ranges of uptake capacity under these operating conditions. The comparison also indicates a general trend of much lower hydrogen uptake of carbon nanotubes than the other carbon materials. Additionally, dispersion of the uptake capacity can reflect on the reproducibility of the results, as activated carbons can reach high surface areas and a bigger range of pore size distribution. This can make observations like Chahine's rule more prominent. However, in order to observe a more reliable trend, a systematic comparison of a much larger amount of data is required.| Type of carbon | Modification | BET surface area/m2 g−1 | Pore volumea (0.95–0.99) (micropore volume)/cm3 g−1 | Excess hydrogen capacity/wt% (temperature, pressure) | Comments | Reference, year |
|---|---|---|---|---|---|---|
| a Pore volume at P/P0 of ranges 0.95 to 0.99. | ||||||
| Graphene | Perforated graphene | ∼2900 | 1.4 | 5.5 (77 K, 30 bar) | Perforated graphene by KOH activation | 103 (2015) |
| Graphene nanosheets | 640 | — | 0.72 (298 K and 100 bar) | — | 104 (2010) | |
| Hierarchical graphene | 1305 | — | 4.01 (77 K, 1 bar) | — | 105 (2013) | |
| Pd-Doped graphene sheet catalyst/activated carbon composite (Pd-GS/AC) | — | — | 0.82 (298 K, 8 MPa) | — | 82 (2013) | |
| 1% Pd-doped graphene | — | — | 8.67 (298 K, 60 bar) | — | 106 (2016) | |
| Pt-Doped graphene | 478.3 | 1.81 | 0.15 (303 K and 57 bar) | Hydrogen capacity increased by a factor of 2.23 compared to undoped graphene | 107 (2011) | |
| Graphene oxide | — | — | 1.4 (298 K and 50 bar) | — | 108 (2012) | |
| Reduced graphene oxide–Mg nanocomposite | — | — | 6.5 (473 K and 15 bar) | Recyclability tested up to five times | 109 (2016) | |
| P-Doped graphene | 288.4 | (1.019) | 3.66 (298 K and 4 MPa) | 1.03 wt% capacity loss after four adsorption–desorption cycles at 298 K and 4 MPa | 110 (2022) | |
| 5% Pd-doped graphene | — | — | 7.16 (298 K, 60 bar) | — | 106 (2016) | |
| 5% Ni-doped graphene | — | — | 1.18 (298 K, 60 bar) | — | 106 (2016) | |
| Ni/Pd alloy doped reduced graphene oxide (rGO) | — | — | 2.65 (298 K, 4 MPa) | — | 111 (2023) | |
| N-Doping of Pd decorated graphene | — | — | 1.92 (298 K, 4.4 MPa) | Enhancement of hydrogen uptake capacity by almost 272% | 112 (2012) | |
| Al-Doped graphene | — | — | 5.13 (300 K and 0.1 GPa) | Computational | 113 (2010) | |
| Palladium-phosphide-modified P-doped graphene | 217.22 | 0.188 | 3.79 (298 K, 4 MPa) | Calcinated at 500 °C | 114 (2023) | |
| Ni-B-rGO (nickel decorated boron doped reduced graphene oxide) | 85 | 0.36 cc g−1 | 6.9 (77 K and 20 bar) | — | 115 (2023) | |
| B-rGO (boron-doped reduced graphene oxide) | 119 | 0.98 cc g−1 | 8.2 (77 K and 20 bar) | — | 115 (2023) | |
| Activated carbon | Nitrogen doped activated carbon | 1646 | 0.70 | 2.14 (77 K and 0.93 bar) | — | 116 (2023) |
| Biomass-derived carbon (cigarette butt) | 4300 | 2.09 P/P0 is 0.99 | 8.1 (77 K and 20 bar) | KOH activated | 102 (2017) | |
| Activated carbon | 2451 | 1.14 | 5.9 (77 K, 4 MPa) | — | 117 (2011) | |
| Cellulose acetate derived AC | 3771 | 1.75 | 7.0(77 K and 20 bar) |
KOH activation at 700 °C | 53 (2017) | |
| Activated carbon (furfural) | 2179 | 1.03 | 5.4 (77 K and 20 bar) | F-1/4-700 | 118 (2011) | |
| Activated carbon (glucose) | 2121 | 1.00 | 5.3 (77 K and 20 bar) | G-1/4-700 | 118 (2011) | |
| Activated carbon (starch) | 2194 | 1.01 | 5.6 (77 K and 20 bar) | S-1/4-700 | 118 (2011) | |
| Activated carbon (cellulose) | 2370 | 1.08 | 5.6 (77 K and 20 bar) | C2-1/4-700 | 118 (2011) | |
| Activated carbon (eucalyptus sawdust) | 2252 | 1.03 | 5.6 (77 K and 20 bar) | E1/4-700 | 118 (2011) | |
| Activated carbon monolith (ACM) | — | (1.04) | 2.97 (77 K and 4 MPa) | Compressed monolith samples have slightly higher hydrogen storage than powder samples | 119 (2008) | |
| AX-21_33 | 3363 | — | 57.1 mg g−1 (77 K and 2.5 MPa) | — | 96 (2016) | |
| Polythiophene-derived activated carbon | 3000 | 1.75 | 5.71 (77 K, 20 bar) | Chemically activated with KOH | 120 (2011) | |
| Commercial AC (MSC30) | 3305 | 1.596 | 5.78 (77 K and 4 MPa) | — | 121 (2021) | |
| Commercial AC MSP20X | 2363 | 0.935 | 4.74 (77 K and 4 MPa) | — | 121 (2021) | |
| Commercial AC SA20 | 1737 | 1.190 | 3.24 (77 K and 4 MPa) | — | 121 (2021) | |
| Commercial AC SA1500 | 2204 | 1.331 | 4.04 (77 K and 4 MPa) | — | 121 (2021) | |
| Commercial AC TH90I | 1204 | 0.495 | 2.90 (77 K and 4 MPa) | — | 121 (2021) | |
| Commercial AC Filtrasorb400 | 983 | 0.469 | 2.34 (77 K and 3 MPa) | — | 121 (2021) | |
| Biomass derived carbon (corn cob) | 3012 | 1.7 | 2.0 (77 K, 1 atm and 0.6) | Chemically activated using KOH, K2CO3, or NaOH | 122 (2010) | |
| Biomass derived carbon (corn cob) | 3708 | 1.99 | 5.80 (77 K and 4 MPa) | — | 123 (2014) | |
| Biomass-derived carbon (tamarind seeds) | 1785 | 0.94 | 4.73 (298 K, 4 MPa) | KOH chemical activation | 124 (2015) | |
| Biomass derived carbon (rice husk) | 2537 | 1.31 | 1 (303 K, 25 MPa) | — | 125 (2013) | |
| Biomass derived carbon (coffee bean waste) | 2070 | — | 4.0 (77 K, 4 MPa) | Chemically activated using KOH | 126 (2011) | |
| Pt-Doped activated carbon (AX-21) | 2518 | 1.22 | 1.2 (298 K and 10 MPa) | 5.6 wt% Pt doping | 127 (2007) | |
| Biomass derived carbon (Posidonia oceanica) | 2810 | 0.48 | 6.3 (77 K and 8 MPa) | — | 128 (2020) | |
| Biomass derived carbon (wooden chips) | 2835 | 0.73 | 6.4 (77 K and 8 MPa) | — | 128 (2020) | |
| Biomass derived carbon (Meluca) | 1806 | 0.82 | 2.16 (77 K and 1.0 MPa) | — | 128 (2020) | |
| Biomass derived carbon (hemp stem) | 3018 | 1.73 | 2.94 (77 K and 0.1 MPa) | KOH activation at 4.5:1 ratio at 800 °C for 1.5 hours |
129 (2017) | |
| Pt-Doped biomass derived carbon (olive pomace) | 1128 | 0.47 | 0.53 (298 K and 180 bar) | +1.10 wt% Pt | 130 (2017) | |
| Horse-chestnut derived activated carbon | 2270 | — | 4.01 (77 K and 20 bar) | ZnCl2 activating agent at 600 °C | 131 (2024) | |
| Pd-Doped biomass derived carbon (olive pomace) | 1074 | 0.45 | 2.46 (77 K and 40 bar) | +1.73 wt% Pd | 130 (2017) | |
| C–Ni nanocomposite (agarose) | 300 | Total pore volume: 0.18 cc g−1 | 0.73 (298 K and 20 bar) | Pyrolysis at 400 °C | 132 (2024) | |
| Biomass derived carbon (bagasse) | 2580 | 1.27 | 2.62 (77 K, 1 bar) | KOH activation | 133 (2020) | |
| Biomass derived carbon (cornstalk) | 2464 | 1.21 | 2.61 (77 K, 1 bar) | KOH activation | 133 (2020) | |
| Biomass derived carbon (pine powder) | 2243 | 1.34 | 2.13 (77 K, 1 bar) | KOH activation | 133 (2020) | |
| Pt-doped activated carbon (commercial AC) | — | — | 0.9 (298 K and 10 MPa) | Hydrogen capacity increased three-fold when compared to undoped carbon | 134 (2007) | |
| Pt/Pd doped activated carbon (corncob-derived) | 2955 | 1.65 | 1.65 (298 K and 180 bar) | 2.5% Pt and 2.5% Pd hybrid doped | 135 (2014) | |
| Ni deposited on activated carbon | 1957 | 1.090 | 1.6 (303 K and 5 MPa) | — | 136 (2012) | |
| Biomass derived carbon (sucrose) | 631.4 | 0.43 | 2.3 (123 K and 45 bar) | KOH:C mass ratio of 4:1 |
137 (2024) | |
| Biomass derived (saw dust) | 3951 | 2.47 | 5.5 (77 K and 20 bar) | KOH activated at 800 °C | 138 (2015) | |
| Biomass derived carbon (spider silk) | 2730 | 1.56 | 3.7 (77 K and 25 bar) | — | 139 (2023) | |
| Activated carbon (peanut shell) | 3728.06 | 2.21 | 6.36 (77 K and 40 bar) | — | 140 (2024) | |
| Activated carbon (beer lees) | 2408 | 1.505 | 2.92 (77 K and 1 bar) | — | 141 (2018) | |
| Activated carbon (coconut shell) | 2800 | 2.17 | 0.85 (298 K, 100 bar) | — | 142 (2007) | |
| Activated carbon (onion peel) | 3150 | 1.64 | 3.67 (77 K, 1 bar) | — | 143 (2020) | |
| Activated carbon (sword bean shell) | 2838 | 1.54 | 2.63 (77 K, 1 bar) | KOH activation | 144 (2019) | |
| Biomass derived carbon (Polypodium vulgare feedstock) | 1234 | 0.57 | 2.73 (298 K, 45 bar) | — | 145 (2024) | |
| Biomass derived carbon (walnut shells) | 1163 | 0.491 | 2.6 (77 K, 1 bar) | — | 146 (2024) | |
| Biomass derived carbon (pine tree bark) | 351.45 | — | 0.73 (77 K and 90 bar) | LiCl was used as an activating agent at 900 °C | 147 (2023) | |
| Template derived carbons | Cr-MOF derived carbon | 628 | 0.54 | 0.9 (77 K, 1 bar) | — | 148 (2016) |
| MOF-5 derived carbon | 2393 | 1.13 | 2.7 (77 K, 1 bar) | — | 148 (2016) | |
| Zeolite β derived carbon | 3150 | 1.13 | 2.5 (77 K and 1 atm) | Chemical vapour deposition | 101 (2007) | |
| Zeolite-templated carbon | 3600 | — | 5.5 (77 K, 2.4 MPa) | — | 149 (2012) | |
| ZIF-derived (Basolite Z1200) carbon | 3188 | 1.94 | 6.2 (77 K and 20 bar) | After activation with KOH | 150 (2012) | |
| Zeolite Y template derived carbon | 2160 | 1.26 | 4.9 (77 K and under 20 bar) | — | 151 (2010) | |
| Pt-Functionalised silica-templated carbon | 712 | 0.84 | 1.1 (77 K and 1.8 MPa) | — | 152 (2008) | |
| ZIF-templated carbon material | 3323 | 2.32 | 6.5 (77 K and 20 bar) | Compaction under force of 5 tonnes | 153 (2014) | |
| Ni-Doped silica templated carbon | 560 | — | 2.2 (77 K and 10 bar) | — | 154 (2015) | |
| Zn-Doped silica templated carbon | 568 | — | 4.4 (77 K and 10 bar) | — | 154 (2015) | |
| USY zeolite template using sucrose precursor | 1200 | — | 3.65 (77 K and 20 bar) | — | 155 (2014) | |
| Zeolite templated CNT | 334 | — | 1.2 (308.15 K and 1 bar) | — | 156 (2022) | |
| NH4Y zeolite template and furfuryl precursor | 1886 | 0.866 (0.222) | 0.92 (173.15 K and 1 bar) | Carbonization temperature of 750 °C & time of 3 h | 157 (2013) | |
| Zeolite 13× template and furfuryl precursor | 3332 | 1.66 | 6.2 (77 K and 2 bar) | Synthesis involves impregnation of furfuryl alcohol, then chemical vapour deposition (CVD) of ethylene at 700 °C | 158 (2013) | |
| Zeolite EMC-2 template and acetonitrile precursor | 3360 | 1.71 | 5.0 (77 K and 20 bar) | Prepared at temperature of 750 °C | 159 (2011) | |
| Silica nanoparticles from rice husk as a template material and glycerol as carbon precursor | 749 | 1.44 | 2.41 (77 K and 7.3 MPa) | Isosteric heat of adsorption 6.8 kJ mol−1 | 160 (2013) | |
| Silica nanoparticles from rice husk as a template material and sucrose as carbon precursor | 644 | 0.85 | 1.42 (77 K and 1.9 MPa) | Isosteric heat of adsorption 7.3 kJ mol−1 | 160 (2013) | |
| Pt-Exchanged zeolite-templated carbons | 2179 | 1.20 | 4.51 (77 K and 20 bar) | Acetonitrile as carbon precursor at 800 °C | 161 (2011) | |
| Pt impregnated zeolite-templated carbons | 2798 | 1.48 | 4.27 (20 bar at 77 K) | Acetonitrile as carbon precursor at 850 °C | 161 (2011) | |
| Al-doped zeolite Y template carbon | 1833 | 1.06 | 3.9 (77 K and 20 bar) | Acetonitrile precursor at 800 °C | 162 (2011) | |
| Silica-derived carbon | 1975 | 3.07 | 0.16 (173.15 K and 1 bar) | 650 °C for 3 h | 163 (2014) | |
| Co doped ZIF-67 templated carbon | 1500 | 0.63 | 0.77 (30 bar and 300 K) | — | 164 (2023) | |
| Pt-Doped zeolite EMC-2 templated carbon | 2047 | 1.01 | 4.58 (77 K and 20 bar) | Prepared using acetonitrile precursor | 165 (2020) | |
| Zeolite EMC-2 templated carbon | 3360 | 1.71 | 6.33 (77 K and 20 bar) | — | 165 (2020) | |
| Carbon nanotubes | MWCNT | 729.4 | — | 3.46 (298 K and 12.79 kPa) | — | 166 (2021) |
| SWCNT | — | — | 2.91 (298 K, 0.174 MPa) | Molecular dynamics | 167 (2024) | |
| Sulfur decorated SWCNT | 764 | 0.78 | 1.70 (77 K and 9 MPa) | Endohedrally encapsulated sulfur | 168 (2023) | |
| Fe@f-MWCNTs (carboxylate functionalised nanotubes) | 18 | — | 0.55 (253 K, 70 bar) | Fe@f-MWCNTs prepared in DMF | 169 (2017) | |
| Cu@f-MWCNTs (carboxylate functionalise nanotubes) | 6 | — | 0.68 (253 K, 70 bar) | Cu@f-MWCNTs Prepared in DMF | 169 (2017) | |
| MWNT with DyNi2 catalyst | — | — | 3.3 (373 K and 94.7 bar) | — | 170 (2008) | |
| Aryl-functionalised MWCNT | — | — | 0.45 (77 K, 2.5 MPa) | — | 171 (2021) | |
| SWCNT | 700 | — | 0.8 (298 K, 3 MPa) | — | 172 (2010) | |
| MWCNT | 236.39 | 0.9563 | 0.2215 (77 K and 100 kPa) | Ball-milling at 700 rpm for 6 h | 173 (2010) | |
| SnO2 MWCNT composites | — | — | 2.03 (373 K, and 5 bar) | Ratio of MWCNT to SnO2 is 5 wt% | 174 (2017) | |
| TiO2 decorated SWNT | — | — | 0.45 (298 K and 30 atm) | — | 175 (2014) | |
| Pd-SWNT | — | — | 0.40 (298 K and 30 atm) | — | 175 (2014) | |
| SWCNT | 700 | — | 0.06 (298 K and 30 atm) | — | 175 (2014) | |
| Ni/MWCNT | 160 | 1.6331 | 2.27 (298 K and 8 MPa) | Synthesized by high energy ball milling for 8 h | 176 (2014) | |
| Pt-MWCNT | 160 | — | 2.9 (1.67 MPa and 298 K) | — | 84 (2007) | |
| Pristine MWCNT | 200 | — | 0.075 (1.67 MPa and 298 K) | — | 84 (2007) | |
| Activated MWCNTs | 479 | 1.073 | 0.54 (77 K and 1 bar) | Activated at 900 °C | 177 (2012) | |
| HiPco™ single-walled carbon nanotubes (SWNTs) | — | — | 0.43 (298 K and 8 MPa) | — | 178 (2003) | |
| MWCNTs | 729.4 | — | 3.46 (298 K and 12.79 kPa) | — | 166 (2021) | |
| Boron and nitrogen co-doped CNT | 87.7 | 1.96 (77 K and 16 bar) | Chemical vapor deposition synthesis | 179 (2020) | ||
| Li loaded activated MWCNT | 174.19 | 0.19 | 1.33 (298 K and 21 bar) | — | 180 (2020) | |
| Co loaded activated MWCNT | 150.28 | 0.17 | 1.06 (298 K and 21 bar) | — | 180 (2020) | |
| Activated MWCNT | 359.18 | 0.27 | 0.82 (298 K and 21 bar) | MWCNT:KOH ratio of 1:5, 800 °C, and 1 h of activation |
180 (2020) | |
| Pt doped MWNT | 497 | — | 0.55 (77 K and 25 bar) | 181 (2020) | ||
| P-CNTs | — | — | 0.65 (298 K and 80 bar) | — | 182 (2018) | |
| F-CNTs | — | — | 0.89 (298 K and 80 bar) | — | 182 (2018) | |
| Pd-Functionalised CNT | — | — | 1.7 (77 K and 1 atm) | — | 81 (2012) | |
| PVP capped Pd-CNT | — | — | 4 (77 K and 1 atm) | — | 81 (2012) | |
| MWCNT | 189 | — | 0.281 (77 K and 1 bar) | — | 183 (2021) | |
| MWCNT-OH | 153 | — | 0.321 (77 K and 1 bar) | — | 183 (2021) | |
| MWCNT-O-Schiff base | 119 | — | 0.370 (77 K and 1 bar) | — | 183 (2021) | |
| MWCNT-O-Schiff base-Cu | 116 | — | 0.396 (77 K and 1 bar) | — | 183 (2021) | |
| Ni-MWCNTs | — | — | 0.298 (298 K and 20 bar) | — | 184 (2018) | |
| MWCNT | 441.3 | 0.2 (298 K and 100 bar) | 185 (2014) | |||
| γ-Ray irradiated MWCNTs | — | — | 1.2 (373.15 K and 1 atm) | 150 kGy | 186 (2017) | |
 | ||
| Fig. 6 Scatter plot of temperature (K) and pressure (bar) versus excess storage capacity (wt%) for (a) graphene materials, (b) activated carbon, (c) template-derived carbons, and (d) carbon nanotubes taken from reported data. (e) Overlay of scatter plots (a)–(d). Plotted from the data reported in references listed in Table 3. | ||
In the following sections, we will see that there exists great variability in reported hydrogen storage capacities of different types of carbon materials. Additionally, methods of improving the practical hydrogen uptake properties have been explored using various methods, such as by incorporating metals and other composite materials for enhancing thermodynamic properties. Table 3 summarises these approaches for various types of carbon materials like graphene, activated carbon, template derived carbons, carbon nanotubes and other types.
3.1. Graphene-based carbon materials
Graphene consists of a two-dimensional lattice of hexagonally arranged carbon, organised in a one or few-layered structures.187 Chemically, each carbon atom in graphene forms three covalent bonds with neighboring atoms, forming a 2-D plane of sp2 hybridized lattice. The perpendicular delocalized π-electrons along the plane govern the interactions between graphene layers, and give rise to its unique electrical properties, excellent thermal stability and mechanical strength.188 Theoretically, it has a surface area-to-mass ratio of 2630 m2 g−1 which makes it suitable for application of hydrogen storage.189,190 However, the actual surface area of graphene falls within a range of 150–600 m2 g−1 due to the tendency towards agglomeration of graphene layers.191 DFT calculations have demonstrated that van der Waals interactions between hydrogen and graphene play a major role in the adsorption mechanism.192 In addition, it has been shown that morphological factors like the curvature of graphene planes, defects, and inter-layer spacing greatly affect the hydrogen storage capacity of graphene.191 It was predicted by DFT calculations that the gravimetric capacity of graphene is optimal when the inter-layer spacing is between 0.6 nm and 0.8 nm.193 Hydrogenated graphene is one of the methods for hydrogen storage and was explored by various research groups, with a theoretical maximum capacity of 7.7 wt%.194 The main idea behind the hydrogenation of graphene, otherwise known as “graphane”, bypasses the limitation posed by the weak interaction of hydrogen with graphene via physisorption, by increasing the amount of hydrogen on the graphene surface by Birch reduction.194,195 Subrahmanyam et al. have explored this by post-synthetically modifying graphene via Birch reduction to form sp3 C–H bonds. These bonds can be broken upon heating or irradiation, leading to the release of chemisorbed hydrogen with a storage capacity of 5 wt%.196 In another study, Morse et al. have also investigated hydrogenated graphene (HG) showing a hydrogen storage capacity of 2.7 wt% and 2.8 wt%, for the pelletized and powder samples, respectively.194 As shown in Fig. 5, thermogravimetric analyses coupled with mass spectrometry (TGA-MS) analysis in this study (Fig. 7) indicate the evolution of hydrogen as a result of its thermal decomposition. | ||
| Fig. 7 (a) Photograph of compressed hydrogenated graphite (HG) in a TGA pan. (b) TGA for HG pellets and HG powder. (c) Differential thermogravimetric analysis (DGA) for HG pellet and powder. (d) TGA-MS profile for m/z of 2 of HG pellets and powder samples. This figure has been reproduced from ref. 194 with permission from Elsevier, copyright 2021. | ||
Unmodified graphene usually has an uptake capacity of less than 1 wt% at ambient temperature and pressures ranging from 1 to 10 bar.197 Srinivas et al. have synthesised graphene nanosheets with a gravimetric storage of 0.1 wt% at ambient temperature and pressure of 10 bar.104 Moreover, the highest experimental H2 uptake capacity recorded for graphene was 3.1 wt% at 100 bar and 25 °C.198 Hence, although being a promising candidate material for hydrogen storage, on its own, graphene practically possesses very low hydrogen interactions to be used in practical applications. Various modifications have been explored to further enhance the hydrogen storage capacity of graphene which involves the decoration of graphene layer surfaces with various metals. Numerous reports have proved that dispersing alkali and transition metal nanoparticles greatly enhances the hydrogen uptake capacity by strengthening the binding of hydrogen to the material.199 This method utilizes a phenomenon known as the spillover effect, in which molecular hydrogen dissociates due to the presence of a catalyst, followed by the migration of hydrogen atoms onto the adjacent fragment of the substrate materials and their diffusion into the substrate surface.200 In a study, Zhou et al. have developed Pd-graphene composite sheets with a measured hydrogen uptake of 8.67 wt% at a pressure of 6 MPa and ambient temperature.106 As shown in Fig. 8a, the amount of Pd dispersed in the composite greatly affects the hydrogen storage capacity. This was attributed to the structural change caused by the amount of decorator metal on the graphene structure. As shown in Fig. 8b, the 1% Pd:graphene ratio resulted in a higher D/G band ratio in the Raman spectra, accounting for more disorder and resulting in higher hydrogen uptake. In addition to experimental data, theoretical calculations have also predicted incredible hydrogen storage capacity for metal-decorated graphene. Ab initio calculations by Ao et al. on Al-decorated graphene predicted an impressive hydrogen storage capacity of 13.79% under ambient conditions.113 Other computer analyses investigated storage capacities for Si, Li, and Ca decorated graphene.201,202
 | ||
| Fig. 8 (a) Hydrogen uptake capacities of 1% Pd/graphene and 5% Pd/graphene functionalized graphene at various charging pressures. (b) Raman spectroscopy of graphene, 1% Pd/graphene, and 5% Pd/graphene, showing D and G bands at ∼1355.38 cm−1 and ∼1588.46 cm−1, respectively. This figure has been reproduced from ref. 106 with permission from American Chemical Society, copyright 2016. | ||
Heteroatom doping in metal-decorated graphene has opened up new avenues for achieving even higher hydrogen storage capacities. Heteroatom doping involves the introduction of atoms into the graphene chemical structure including either chemically substituting carbon with atoms like P, S, and N into the graphene lattice or the addition of halogen atoms through the formation of a C–X bond.203 Alongside metal nanoparticle decoration, both approaches can introduce defects into the graphene structure and allow for fine-tuning of the electronic and chemical properties of graphene, leading to synergistic effects, by (i) increasing the surface area for adsorption, (ii) introducing defects that would minimize nanoparticle agglomeration, and (iii) altering the charge density of the graphene surface, resulting in increased enthalpy for better hydrogen adsorption, all of which can significantly enhance hydrogen uptake capacity.203–205 Vinayan et al. have explored the fabrication of Pd functionalized N-doped solar exfoliated graphene (SG) composites.206 In their work, Pd/N-SG was found to have a higher hydrogen uptake capacity of 4.3 wt% at 25 °C and pressure of 4 MPa, compared to 0.68 wt%, 1.06 wt%, and 3.57 wt% for SG, N-SG, and Pd-SG, respectively. Moreover, the metal-decorated graphene composites displayed 90% adsorption of hydrogen at room temperature, demonstrating high reversibility. In another study the Pd3P decorated P-doped graphene composite demonstrated an uptake capacity of 3.66 wt% at 4 MPa and 25 °C, with effective 4 cycles of adsorption and desorption.110 DFT calculations have also investigated the surface functionalization of N-doped holey graphene with light metals like Na, K, and Mg,207 and estimated the highest loading capacity of 6.5 wt% and 5.5 wt% for Mg and Na functionalization, respectively. Other methods of utilizing graphene involve its use as a support in composites for other hydrogen storage materials. Cho et al. have explored incorporating N- and B-doped graphene for the nanoencapsulation of Mg nanoparticles.208 In their research, the graphene functionalized composites showed enhanced adsorption kinetics of hydrogen into Mg nanoparticles by the reduction of activation energy. Furthermore, introduction of heteroatom doping together with the existing structural defects of the graphene lattice aided in charge transfer, contributing to faster kinetics and higher hydrogen loading.209 Zhang et al. have synthesized fluorinated graphene composites of LiBH4,210 which showed superior desorption kinetics when compared to bare LiBH4, including lowering the desorption temperature by 120 °C and decreasing the activation energy of dehydrogenation from 180.10 kJ mol−1 to 130.87 kJ mol−1. In another study, researchers have used graphene nanoplatelet (GNP) structures that are composed of up to 100 graphene layers, along with Mg nanoparticles, as composites to solve the agglomeration issue of metal nanoparticles.211 The study has reported that by controlling the size of the GNP, the tendency towards agglomeration of Mg nanoparticles can be controlled, with smaller GNP particles resulting in higher agglomeration. As indicated from previous examples from the literature, the use of precious metals like Pd can greatly enhance the ambient storage capacity due to the possible spillover effect. However, on a practical basis, this can hinder the potential use of such composites due to material cost and poor abundance. Chen et al. have fabricated Ni/Pd alloy-doped graphene composites to reduce the dependence on Pd alone for doping.111 Through DFT calculations, the composites have shown a H2 storage capacity of 2.65 wt% at room temperature and pressure of 4 MPa.
Apart from monolayer graphene configurations, three-dimensional (3D) graphene materials are generating increasing interest as hydrogen storage materials. With an SSA that can reach as high as 3400 m2 g−1,212 3-D graphene structures have an intricate interconnected network of graphene sheets which provides a high surface area for interactions with hydrogen. Liu et al. have synthesized three dimensional porous hierarchical graphene using polymethyl methacrylate (PMMA) microspheres as a template, by the deposition of negatively charged GO-COOH nanosheets on positively charged PMMA (Fig. 9).213 The nickel-functionalized 3D graphene exhibited a surface area of 925 m2 g−1 and a pore volume of 0.58 cm3 g−1 showing a maximum hydrogen capacity of 4.22 wt% and 1.95 wt% at 77 K and 25 °C at 0.5 MPa, respectively. In 2015, Klechikov et al. reported activated graphene scaffolds, with an ultra-high surface area of more than 3000 m2 g−1 and a pore volume of 2.2 cm3 g−1.212 The activation treatment using high temperature annealing of reduced graphene oxide (r-GO) resulted in the development of the 3D scaffold structure with high defects and interconnected layers, giving rise to an uptake capacity of 4.23 wt% of H2 at 12 MPa and 193 K, with a maximum saturation capacity of 7.48 wt% at 77 K. Jung et al. have explored using interconnected 3D graphene foams with modified porosity and Pt functionalization, achieving an uptake capacity of 3.19 wt% at 25 °C and a pressure of 10 MPa.83 Other studies have also explored hydrogen storage using graphene aerogels and MgH2 graphene aerogel (GA) composites.214
 | ||
| Fig. 9 (a) Schematic illustrating the synthesis for three-dimensional porous hierarchical graphene. (b and c) SEM images of three-dimensional porous hierarchical graphene. This figure has been reproduced from ref. 213 with permission from Elsevier, copyright 2018. | ||
As can be seen from the above examples, both theoretical and experimental studies on the adsorptive capacities of graphene materials show its potential as a hydrogen storage material, when compared to other materials. Graphene can show relatively high surface areas and good uptake capacities when modified with not only precious metals like Pd, but also inexpensive dopants, such as Ca, Li, and Na.
3.2. Carbon nanotubes
Carbon nanotubes (CNT) are cylindrical structures of rolled graphene. There are various allotropes of CNTs, such as single-walled carbon nanotubes (SWCNTs) which are made from a single sheet of rolled graphene, and multi-walled carbon nanotubes (MWCNTs) which are formed as a result of multiple concentric layers of carbon nanotubes with interlayer distances being around 3.3 Å.215 CNTs have been reported with a wide range of surface areas from 50 to 1300 m2 g−1.190 In 1997, Dillon et al. demonstrated the potential of SWCNT materials for the reversible storage of hydrogen, showing an uptake capacity ranging from 5 to 10 wt% at ambient temperature and 0.04 MPa.216 Since then, carbon nanotubes have been extensively studied due to their unique morphology, high SSA, high tensile strength and thermal stability. However, discrepancies were reported in later studies for hydrogen storage capacities of CNTs, showing less than 1.7 wt% hydrogen storage capacity under 12 MPa pressure at room temperature.217 This variability among results might be dependent on external and internal factors involving the measurement techniques, devices, and synthesis methods.218 Furthermore, these inconsistencies highlighted the need for a better understanding of the mechanisms of hydrogen adsorption in CNTs. Both theoretical and experimental data suggest the involvement of physisorption and chemisorption as plausible adsorption mechanisms.185 However, despite the uncertainty surrounding the interaction of hydrogen with CNTs, it is typically regarded that the hydrogen molecules are adsorbed as a monolayer on the interior surface (endohedral), exterior surface (exohedral), or between the spaces left between bundles of CNTs.219 Additionally, like other carbon materials, chemical surface composition, surface area, and temperature greatly influence the hydrogen storage capacity. As shown in Fig. 10, grand canonical Monte Carlo (GCMC) simulations showed the effect of temperature on hydrogen adsorption capacity of carbon nanotubes, predicting higher hydrogen uptake capacity at lower operating temperatures.219 | ||
| Fig. 10 Grand canonical Monte Carlo simulations showing the sites of hydrogen molecules around carbon nanotube taken at conditions of 100 bar pressure at 77 K (left), 175 K (middle), and 293 K (right). This figure has been reproduced from ref. 219 with permission from Elsevier, copyright 2011. | ||
Despite the challenges, various ways were investigated to improve the textural and chemical properties of CNTs to achieve higher H2 storage capacities.218 For example, modification of CNTs by chemical and physical activation showed enhanced porosity and introduced beneficial defects. As illustrated in Fig. 11, Chen et al. have shown that following KOH-treatment, MWCNTs showed a significant increase in H2 storage capacity from 0.71 to 4.47 wt% at ambient temperature and pressure of 0.1 MPa.220
 | ||
| Fig. 11 Schematic showing the effect of the activation process on the structural morphology of CNTs. This figure has been reproduced from ref. 220 with permission from Elsevier, copyright 2007. | ||
Another form of modification reported involves the use of irradiation. For example, Silambarasan et al. have investigated the effect of γ-radiation on the structure of MWCNTs, and it was found that higher doses of around 100–150 kGy of γ-radiation are sufficient to show increased ID/IG ratios in their Raman spectra indicating introduction of structural defects via the Compton effect, leading to an increased hydrogen storage capacity of up to 1.2 wt% at temperature of 100 °C and ambient pressure.186 Ball-milling is another way of introducing defects in the CNTs. The sheer and frictional forces introduce defects by shortening the length, increasing the surface area and increasing the amorphous nature (higher ID/IG), leading to improved hydrogen uptake.173 Liu et al. have obtained a six-fold increase in hydrogen storage capacity at ambient room temperature and pressure of about 8–9 MPa for MWCNTs when treated with ball milling for 10 hours.221
In addition to physical methods that enhance the morphology of CNTs, metal-doping has also been used to improve hydrogen storage capacity and increase the hydrogen–adsorbate interaction. Like other carbon materials, doping with metals facilitates enhanced hydrogen adsorption.222 DFT calculations by Liu et al. on the effect of Li-doping of CNTs have found a high uptake capacity of 13.45 wt%, which can be attributed to Kubas binding.223 Other groups have also concluded higher storage capacity when compared to undoped MWCNTs at 0.075 wt% and 2.9 wt% for MWCNTs and Pt-MWCNTs, respectively.84 Doping of CNTs with metals like Cu, Mg, Ni, Ti and Al has been also reported.169,218 Nanoconfinement and incorporation into composites have also been explored for CNTs. For example, Khalilov et al. have investigated the nanoconfinement of transition metals in SWCNTs endohedrally through molecular dynamic (MD) simulations.167 The addition of Ni atoms to the inner structure of SWCNTs contributed to an increase of gravimetric uptake from 1.86 to 2.91 wt% when compared to unfunctionalized SWCNTs at ambient temperatures and pressures of 0.17 MPa. Other than transition metals, the hydrogen capacity of CNTs can be enhanced by endohedral doping with heteroatoms, like sulfur.168 Other types of CNT composites involve the incorporation of metal oxides. Vellingiri et al. have reported an uptake of 2.03 wt% at 100 °C and 5 bar through the use of SnO2 impregnated MWCNTs (Fig. 12).174 The addition of SnO2 contributed to the formation of defect sites. Additionally, the SnO2 particles can provide sites for catalytic activity that can facilitate hydrogen adsorption. A number of studies have concluded that hydrogen interacts with SnO2 and other metal oxide CNT composites via electrostatic and weak chemisorptive forces.175,224
 | ||
| Fig. 12 SEM images of (a) pristine MWCNTs and (b) MWCNT:SnO2 composites. This figure has been reproduced from ref. 174 with permission from Elsevier, copyright 2018. | ||
Besides nanoparticles, CNTs can be used as support materials for polymer composites in applications of hydrogen storage, acting as support materials for polymers such as polyaniline, polyvinylpyrrolidone (PVP) and polypyrrole.225,226
Despite the discrepancies surrounding CNTs for hydrogen storage, studying this class of materials can provide significant understanding of hydrogen–carbon interactions. Additionally, they exhibit the potential to be used alongside other materials as composites for modification of textural, physical, and chemical properties for hydrogen storage materials. In general, MWCNTs and SWCNT have shown an uptake capacity of up to 3.5 wt% at pressures up to 140 bar.201
In terms of adsorptive capacity, it is thought that carbon nanotubes offer no practical advantages when compared with other porous materials like MOFs and activated carbon, due to weak thermodynamics.74 The average enthalpy of adsorption for SWCNTs of diameter 0.8 nm is 8–9 kJ mol−1, decreasing to 5.9 kJ mol−1 with a larger diameter of 1.2 nm.227 As highlighted by Cheng et al.,228 the successful use of carbon nanotubes for practical hydrogen storage requires (i) finding methods of mass production of CNTs, (ii) development of effective pretreatment and modification methods for improving hydrogen storage, (iii) studying the cyclability and kinetic behaviour of adsorption of hydrogen, and (iv) use of theoretical interpretation to guide the pathway for developing CNTs.
3.3. Activated carbon
Unlike the ordered structure of graphene and CNTs, activated carbons (ACs) are made up of intricate pore networks of graphite crystallites and amorphous carbon. These disordered structures give rise to ultra-high surface areas often exceeding 3000 m2 g−1, slit-shaped pore morphology, and a variety of pore volumes ranging from the macro, meso, to microporous range.229 ACs have been reported to show the highest adsorption storage capacities reported to date among the different types of carbon materials between 77 K and 300 K.230,231 What makes this class of carbon materials unique as hydrogen storage materials is their availability and tunable properties; ACs can be synthesized from the pyrolysis and activation of precursors from a wide range of carbon-rich materials either from natural (biomass derived) or unnatural sources, including coal, petrochemical materials, peat, polymeric materials, and various agricultural derivatives like wood, bamboo, and food waste.232 Lignocellulosic biomass is one of the most popular precursors for synthesizing activated carbon. Most of the carbon in biomass materials exists in different forms, including lignin, hemicellulose, and cellulose, and their composition in the initial biomass precursor affects the final morphology of the activated carbon, including its surface area and micropore volume.233 Pyrolysis is the thermal decomposition of precursor materials into carbon in the absence of oxygen.234 Besides pyrolysis, ACs can be activated via both physical and chemical methods to help enhance the porous and textural properties for better hydrogen storage. Physical activation involves high temperature gasification of the carbon material with an oxidizing gas (usually CO2 or H2O). Meanwhile for chemical activation, the raw material is treated first with an oxidizing agent at high temperature. Several chemical activation agents (occasionally called porogens), such as KOH, ZnCl2, H2SO4, H3PO4, and NaOH, are usually used. Studies have proved chemical activation to be more efficient, yielding a higher surface area and optimal microporosity.48,235 Together, pyrolysis and activation temperature and conditions were shown to play crucial roles in establishing ultra-high surface areas and desirable micro porosity.236 For example, in a study, the effect of carbonization temperature and activation conditions on lignin-derived activated carbon derived from a feedstock precursor using a physical activation method was explored.237 The use of higher carbonization temperatures was shown to result in reduced BET surface areas, as the porous network of micropores is further widened by the activation process, leading to the formation of larger micropores, mesopores, resulting in an overall decrease in surface area. Hence, lower carbonization temperature can sometimes achieve maximal microspore volumes and limit the pore widening effect of activation. The results from optimization of conditions yielded a surface area of 1409 m2 g−1, with a total H2 uptake of 1.9 wt% at 77 K and 0.1 MPa, using a low carbonization temperature to retain micropores, followed by a high activation temperature of 350 °C and 1000 °C, respectively. Similarly, this trend was also observed in other studies as activation was shown to develop and enhance the pore structures in coal-derived activated carbon.238It was shown that in addition to porosity, other factors like surface morphology, composition and functionalization are important in determining the adsorptive capacity of AC. Exceptional 8.1 wt% excess uptake and 9.4 wt% total hydrogen uptake at 77 K and 20 bar pressure were achieved using AC derived from cigarette butts.102 The AC samples were synthesized by hydrothermal carbonization, followed by chemical activation with KOH, resulting in an ultra-high surface area of 4300 m2 g−1, 90% of which is a result of micropores. It was concluded via elemental analysis that the activation process yielded high oxygen content in the final AC product, mainly in the form of –OOH, –C–OH and HO–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional groups. The surface composition of the AC contributed greatly in enhancing the hydrogen uptake capacity, as a result of the high binding energy between hydrogen and the oxygen-containing functional groups. In another study, KOH activated carbon derived from a bamboo precursor achieved a surface area of 3208 m2 g−1 and a micropore composition of 1.01 cm3 g−1.239 The maximum hydrogen uptake capacities at 77 K were 6.6 wt% at 40 bar and 2.74 wt% at 1 bar, respectively.
O functional groups. The surface composition of the AC contributed greatly in enhancing the hydrogen uptake capacity, as a result of the high binding energy between hydrogen and the oxygen-containing functional groups. In another study, KOH activated carbon derived from a bamboo precursor achieved a surface area of 3208 m2 g−1 and a micropore composition of 1.01 cm3 g−1.239 The maximum hydrogen uptake capacities at 77 K were 6.6 wt% at 40 bar and 2.74 wt% at 1 bar, respectively.
Sun et al. have used corncob to synthesise porous carbon (Fig. 13) which was studied using different activation methods including two-step KOH activation with ultrasonication treatment. The obtained activated carbon showed a surface area of 3012 m2 g−1 and a pore volume of 1.7 cm3 g−1. Hydrogen adsorption studies showed an uptake of 2.0 wt% at a temperature and pressure of 77 K and 1 bar, respectively.
 | ||
| Fig. 13 (a and b) SEM images of activated carbon derived from corncob using various chemical activation methods. This figure has been reproduced from ref. 122 with permission from Elsevier, copyright 2010. | ||
Other studies also have explored different biomass sources like tamarind seeds, olive pomace, and coffee bean wastes. Activated carbons from fungi-based chars were also explored, with an achieved surface areas of 1600–2500 m2 g−1 and hydrogen storage capacity of 2.4 wt% at 1 bar and −196 °C and a saturated uptake ranging from 4.2–4.7 wt% at −196 °C and 35 bar.240 Peng et al. have explored different methods of chemical and physical activation of porous carbon derived from common biomass waste precursors like bagasse, cornstalk and pine powder.133 The resulting carbons were activated using CO2, ZnO2, and KOH. As shown with other literature reports, KOH was shown to have the best effect in developing a high micropore volume and surface area. Hydrogen adsorption studies have shown an uptake capacity of 2.62 wt%, 2.61 wt% and 2.13 wt%, for bagasse, cornstalk, and pine powder carbons, respectively. Additionally, as shown in Fig. 14, the adsorption isotherm showed type I adsorption indicating that the formed hydrogen monolayer reaches saturation at higher pressure, a typical behaviour of microporous materials.
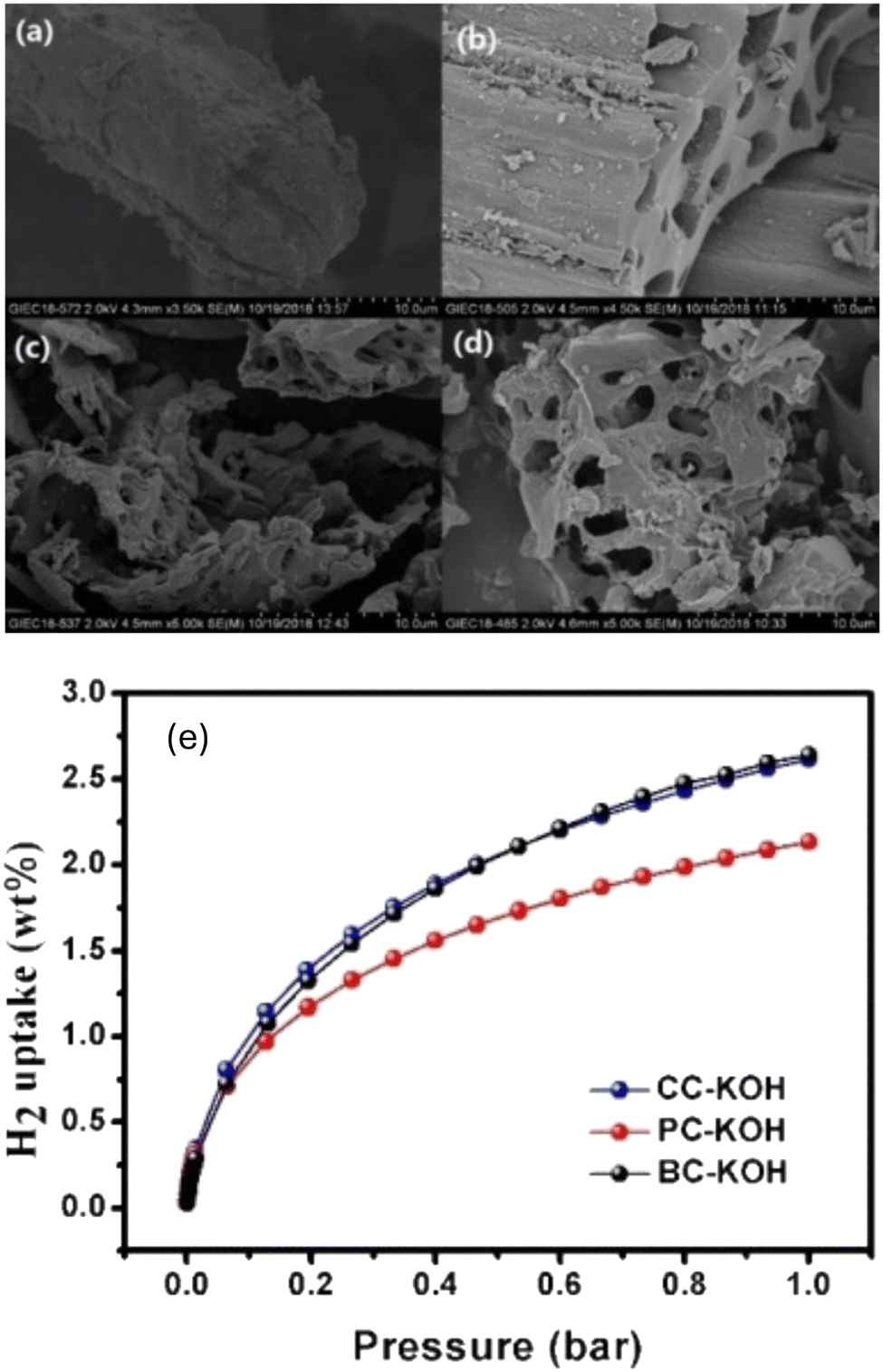 | ||
| Fig. 14 SEM image of carbon derived from cornstalk (CC), after (a) thermal activation at 800 °C, (b) ZnCl2 activation, (c) H3PO4 activation and (d) KOH activation. (e) H2 adsorption for the KOH activated carbons derived from bagasse, cornstalk and pine powder shown. This figure has been reproduced from ref. 133 with permission from John Wiley and Sons, copyright 2020. | ||
Functionalization of AC results in enhanced hydrogen storage capacity and improved kinetics.241 A number of studies have investigated doping AC with metals like Pt, Cu, Ni and Pd to enhance hydrogen uptake capacity via the spillover effect.134–136,242
Flamina et al. have synthesised Ni-functionalised carbon nanocomposites with enhanced hydrogen storage.132 The composites were synthesised by preparation of aerogel by freeze drying a mixture of agarose and nickel acetate salt. The freeze-dried composites were then pyrolyzed at temperatures of 400, 600 and 800 °C to prepare Ni–C nanocomposites of Ni particles and nanotextured carbon. The carbon composite synthesised at 400 °C had a BET surface area of 300 m2 g−1, which is lower than the non-functionalised carbon. Despite the lower surface area, the composite showed 6.5 times more hydrogen storage capacity of 0.73 wt% at 298 K and 20 bar, which was attributed to the spill-over effect.
3.4. Template-derived carbon
Template-derived carbon is produced as a result of the pyrolysis of a template precursor. Unlike activated carbon, template derived carbons often have controlled hierarchical pore morphology and narrow pore size distribution depending on the synthesis method and precursors used. Template derived carbons can be synthesized via hard templating and soft templating methods. An example of hard-templating is shown in Fig. 15 involving the impregnation of a sacrificial template with carbon rich molecules by chemical vapor deposition (CVD) followed by carbonization. This results in a porous 3D negative structure of the template. Hossain et al. and others have used sucrose as a carbon source for synthesizing porous carbon,137,155,243 and several studies have explored the use of furfuryl alcohols.158,244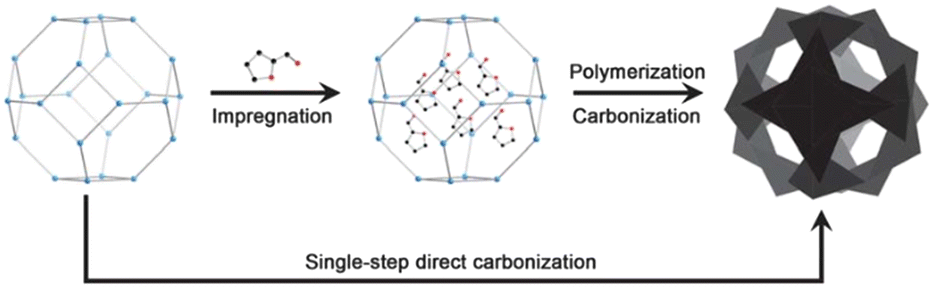 | ||
| Fig. 15 Scheme showing the steps of hard-template synthesis of templated carbon. This figure has been reproduced from ref. 245, with permission from Royal Society of Chemistry, copyright 2013. | ||
For example, choosing a silica-based template resulted in the formation of mesoporous carbon, while carbonization of zeolites formed porous carbon with a higher micropore volume showing better hydrogen storage capacity.246 Hydrogen adsorption capacities in template-derived carbon are among the highest reported to date for various types of carbon materials. For example, zeolite 13× derived carbon with a surface area of 3332 m2 g−1 and a pore volume of 1.66 cm3 g−1, respectively, showed a hydrogen uptake of 7.3 wt% at 20 bar and 77 K, with an estimated maximum uptake capacity of 9.22 wt%.158 Zeolite-β derived carbon materials with a surface area of 3200 m2 g−1 and a pore volume of up to 2.41 cm3 g−1 showed a hydrogen adsorption capacity of 6.9 wt% and a projected maximum of 8.33 wt% at 77 K and 2 MPa.101 A high hydrogen adsorption capacity of 5.5 wt% at 77 K and 30 MPa was achieved via zeolite templated-carbon (ZTC), with the surface area reaching 3600 m2 g−1.149 These high hydrogen uptake capacities are attributed to the higher micropore volume of zeolite-derived carbons.
Cai et al. have used sucrose as a carbon precursor on an ultra-stable Y (USY) zeolite template shown in Fig. 16.155 The powdery zeolite was first mixed with sucrose at a ratio of 1:1 in an aqueous solution along with H2SO4. After a series of mixing and heating up to 160 °C, the resulting composite mixture was then heated under N2 at a heating rate of 5 °C min−1 up to until 900 °C for four hours resulting in the porous carbon (UCs-90-2) shown in Fig. 14. The template derived carbon showed a BET surface area of 1219 m2 g−1 and a total pore volume value of 1.068 cm3 g−1 with a hydrogen uptake capacity of 3.65 wt% at 77 K and 20 bar.
 | ||
| Fig. 16 SEM images of (a) USY zeolite and (b) template derived carbon UCs-90-2. This figure has been reproduced from ref. 155 with permission from Elsevier, copyright 2014. | ||
As for carbons derived from silica-based templates, several materials have been investigated for potential for storage of hydrogen, yielding mesoporous carbon that showed relatively low hydrogen storage capacity.160,163
Another template source for carbon involves the use of MOFs as precursors. MOFs are a class of crystalline three-dimensional structures of metal nodes interconnected by organic ligands, forming potential voids.247 Due to their high microporosity, with surface areas reaching up to ∼15000 m2 g−1, MOF-derived carbons have been widely studied for hydrogen storage. Zn-based MOFs, IRMOF-1, IRMOF-3, and IRMOF-8 were used for the synthesis of MOF-derived carbon (MDC).248 Carbon derived from IRMOF-1 with a terephthalic acid linker resulted in the highest BET surface area of 3174 m2 g−1 and high ultra-microporosity when compared to 1678 and 1978 m2 g−1 for IRMOF-3 and IRMOF-8, respectively. Hydrogen uptake for IRMOF-1 derived carbon reached an exceptional 3.25 wt% at 77 K and 0.1 MPa. For MOF-derived carbons, the aromatic nature of the linker provides a good source of carbon, in addition, it has been shown that the metal-to-linker ratio affects the extent of porosity and surface area.249 A positive correlation between porosity and metal-to-linker ratio was observed for Zn-based MOFs; after carbonization, a Zn/C ratio equal to 0.07 and 0.25 yielded a surface area of 900 m2 g−1 and 1800 m2 g−1, respectively. Meanwhile an opposite trend was seen in a Cr-based MOF due to the possible formation of Cr-oxides and carbides after pyrolysis.148 Zeolitic imidazolate frameworks (ZIF) are another subgroup of MOFs that have imidazolate linkers. Mokaya et al. have synthesised ZIF-derived carbon from Basolite Z1200. The resulting carbon after activation had a surface area of up to 3200 m2 g−1 and a pore volume of 1.94 cm3 g−1, and an excellent hydrogen uptake capacity of 6.2 wt% at 2 MPa and 77 K was observed.150 Jiang et al. have also worked on using ZIF-8 as a template together with furfuryl alcohol as a precursor for synthesizing porous carbon, showing a BET surface area of 3405 m2 g−1 and a pore volume of 2.58 cm3 g−1 with a hydrogen uptake of 2.77 wt% at 77 K and 1 bar.244
Other forms of template derived carbon include carbide-derived carbons (CDC) which are synthesised by removing the metallic component from the carbide structure by chlorination to yield the carbon material with a uniquely narrow PSD and high microporosity. KOH activated porous carbon derived from Zr–C has shown a surface area of up to 2800 m2 g−1 and a hydrogen uptake of 6.2 wt% at 77 K and 2 MPa. Additionally, at 1 bar, activated CDC have shown an uptake of 2.7 wt% of hydrogen, which is one of the highest values to be recorded for porous carbon materials at this pressure.250 Other forms of precursors have also been investigated for microporous carbon structures, including polynuclear complexes.251,252
3.5. Other carbon structures
Besides the different types of carbon mentioned above, more miscellaneous forms of carbon have been explored due to their unique morphology and properties. Carbon nanospheres are hollow nanostructures of carbon that possess a high surface area-to-volume ratio, additionally, their hollow morphologies make them good candidates for nanoencapsulation. Carbon nanospheres have been investigated for hydrogen storage by Baca et al. at 313 K and a pressure of 4.5 MPa, and Pt-doped carbon nanospheres showed a hydrogen uptake of 0.48 wt%.253 In another study, CO2 activated nanospheres derived from starch showed a high surface area and pore volume of 3350 m2 g−1 and 1.75 cm3 g−1, respectively. The nanospheres showed a high hydrogen uptake of 6.4 wt% at 77 K and pressure of 2 MPa.254Carbon nanorods are elongated carbon structures that are the by-product of CNT synthesis. Studies have explored their potential for hydrogen storage through temperature programmed reduction (TPR) measurements showing an uptake of 0.14 wt%.255 Carbon nanofibers (CNFs) are an allotrope of carbon synthesised via the deposition of gaseous hydrocarbons like methane and acetylene onto a metal catalyst. This creates carbon structures with varied morphology depending on the shape of the catalyst used, including herringbone, tubular morphology, and card deck (stacked). In 1998, Chambers et al. reported the synthesis and characterisation of graphite carbon nanofibers with herringbone arrangement and a questionably high hydrogen uptake of about 67 wt%.256 However, no one has been able to replicate these results successfully. Fullerenes are a group of peculiarly shaped nanostructures composed of hexagonal and pentagonal carbon meshes.257 Fullerenes can have ellipsoidal, spherical and tube-like shapes. However, the most stable structure of fullerene is C60, otherwise known as Buckminsterfullerene or buckyballs.258 Fullerenes have been studied via computer simulations for their use as hydrogen adsorbents. Scandium bound fullerenes have been investigated using DFT calculations with an estimated maximum hydrogen storage capacity of 9 wt%.259 Other studies have calculated a maximum hydrogen storage capacity of 8 wt% for Ti-decorated C60 under ambient conditions.260 Despite the high theoretical uptake capacity, most of these materials are yet to be tested experimentally. In the previous sections, we have highlighted widely studied types of porous carbon for hydrogen storage application. Studies on graphene have led to providing crucial insight in understanding H2–adsorbate interactions, and have shown an adsorption capacity of up to 3 wt%. Graphene can also be modified into 3D structures including aerogels and foams that can be incorporated into composites. Templated carbon has the advantage of high tunability and high surface area. While activated carbon can be produced from waste biomass pyrolysis and can achieve high surface areas and storage capacities, they offer less tunability than templated carbon. Each type of carbon and their preparation methods offer a range of advantages and disadvantages for hydrogen storage applications.
4. Life cycle assessment of carbon materials
While porous carbon materials can offer promising applications across various industries including the hydrogen energy sector due to their exceptional properties, their production and use come with significant environmental challenges. Life cycle assessment (LCA) is a tool that can be used for assessing the environmental impact of products to allow their development and optimisation, while encompassing factors like cost and environmental load such as waste and emissions along the life cycle of a product.261 The aim of LCA is to (i) reduce the consumption of non-renewable resources, (ii) decrease the emission and the release of harmful waste products, (iii) improve the product's efficiency, and (iv) improve the fate of the product at end-of-life (EOL) and its potential for reusability and recyclability.As for CNTs, optimistic projections show an annual production of 7000 tons by 2025,262 with a composition of 20% single-walled carbon nanotubes (SWCNTs) and 80% multi-walled carbon nanotubes (MWCNTs), respectively. The materials needed for CNT synthesis required a carbon precursor which may be solid or gaseous alongside a metal catalyst. Additionally, various synthesis methods exist, including electric arc discharge, laser ablation, chemical vapor deposition (CVD), and high-pressure carbon monoxide (HiPco). The most common and oldest method to synthesise CNTs is electric arc discharge, where electric arc vaporises a hollow graphite anode packed with a mixture of transition metals and graphite powder, along with inert gas flow. Flowrate, gas pressure and metal concentration can be optimised to obtain a high yield of CNTs of up to 70%.263 CVD is another common method for CNT synthesis that produces a higher yield of CNTs (95–99%), with greenhouse gas emissions (GHG) ranging from 28.55 to 0.02 kg of CO2 emitted per one gram of material synthesised.264 Economic assessment done by Isaacs et al. has calculated the manufacturing costs per 1 g of SWCNT for arc discharge, CVD, and HiPco processes to be $1906 g−1, $1706 g−1, and $485 g−1, respectively.265 In addition to great variability in synthesis methods, cost and emissions produced, most methods of CNT synthesis are energy intensive. For example, the manufacturing and production of CNTs requires from 2 to 100 times more energy than materials like aluminium.266 Moreover, CNT production emits 10000 times more GHG than other carbonaceous materials like graphite.267
Additionally, the environmental impact of CNT manufacture and production is substantial. LCA primarily associates CNT production to climate change, acidification, and depletion of inorganic resources.268 Gavankat et al. have analysed the environmental impact of a commercial SWCNT plant showing a high global warming potential of 153 and 818 kg of CO2 emitted per gram of CNT, synthesised by the fluidized bed CVD and CoMoCAT method, respectively.269 As for graphene carbons, the source of carbon material and the method of synthesis greatly impact their cost and environmental impact. It was calculated that using conventional methods, a kilogram of graphene oxide costs around $50–990,270 whereas synthesising graphene using flash-joule technology from biomass waste as a source of carbon can reduce its cost to around $5.41–8.84 per kg, in addition to mitigating the environmental impact resulting from the production of graphene by 10 fold when compared to conventional production methods.271
Similarly, activated carbon showcases varying potential for environmental improvement, with its processing location and choice of feedstock influencing global warming impacts and significantly affecting environmental footprints.272,273 Global warming impact can be reduced by 60% when the material is processed in a country with a low-carbon electricity system.272
As for synthesis steps, LCA done on activated carbon derived from biomass suggests that recycling of KOH involved in the activation process could reduce environmental emissions, lowering greenhouse gas (GHG) emissions to 83.76% as well as reducing production cost.274 Additionally, a pre-carbonisation step has a higher environmental impact than single-step activation.275
Although LCA provides a valuable framework for evaluating and mitigating environmental impacts, guiding the development of sustainable practices in the synthesis, manufacturing and production, the field of LCA for nanomaterials is still at its infancy and has many challenges. Most studies lack end-of-life analysis, while others focus only on the cradle to grave approach. Hischier and Walser have identified key issues, including defining the functional unit and impact assessment values and have suggested strategies to improve LCA studies by involving the collection of inventory data, addressing emissions, and establishing toxicity factors while properly defining the goal and scope of LCA studies.276
LCA of adsorbents for hydrogen storage application displays the environmental and economic implications of various materials used in storage systems, emphasizing the need for sustainable choices. In addition to environmental impact, hydrogen economy involves the integration of multiple sectors including heating, electrical and transportation.277 Besides material discovery and optimisation, energy and mass transfer processes should be also considered throughout the system.278,279 In addition, for viable and sustainable implementation, production of hydrogen from low-carbon and renewable resources like water electrolysis and photocatalysis,280–282 and biogas sources,283 as well as exergy analysis to evaluate the usable amount of energy must be considered.284,285
5. Challenges, current trend, and future outlook
Based on the large number of studies, the use of porous materials for hydrogen storage is promising. However, many challenges need to be addressed to integrate porous carbon and other porous materials into real world applications. As mentioned earlier, many challenges already exist for synthesis and material development. As highlighted by the US DOE, these materials should achieve the set goals of high gravimetric capacity, recyclability, and reasonable operating temperatures and pressure. However, not only are we facing challenges in the material development phase, but also in other steps along the material designing and manufacturing process including large-scale manufacturing and sustainability as highlighted under life cycle assessment. As we have seen from numerous reports, the discovery of materials for hydrogen storage is guided by experimental as well as theoretical predictions from computer simulations like DFT and GCMC. Despite their limitations,286–289 they have been proven to be helpful in understanding the interactions between carbon and hydrogen. Computational modelling coupled with other emerging prediction tools, such as machine learning, is being employed increasingly to improve the material selection and discovery process.237,290 Machine learning (ML) is a subset of artificial intelligence (AI) that uses statistical methods to process huge amounts of data and identify patterns that might not be visible using conventional methods. It can be categorized into supervised learning, unsupervised learning, semi-supervised learning and reinforcement learning.291 Recently, ML has been increasingly employed to aid in the process of porous material discovery and optimisation.292 In the context of porous materials for hydrogen storage, Giappa et al. have used a bottom up approach for predicting hydrogen storage capacity. Ab initio calculations of binding energy have been coupled with machine learning techniques to explore the effect of potential ligand functionalisation on hydrogen storage capacity of MOFs.293 Calculations have shown that the functional groups like –NH2 and –OPO3H2 enhance the H2 binding enthalpy by 16% and 26%, respectively, and –OSO3H2 functionalisation has predicted an enhancement of 81% when compared to the non-functionalised benzene ring. GCMC simulations were used under cryogenic and ambient conditions to simulate the hydrogen adsorption capacity of three IRMOFs using the strongest binding functional groups from ab initio calculations. In all cases, the results show an increased volumetric hydrogen uptake, which is in line with the increased binding energies from ab initio studies. Finally, ML was successfully used to predict binding energies using supervised machine learning. The study demonstrates the power of combining advanced computational methods with machine learning techniques to predict and optimize material properties in the field of hydrogen storage. In another study, GCMC was used as a validation technique for deep-learning models predicting deliverable hydrogen storage capacities in MOFs.294 Light Gradient Boosting Machine (LightGBM) and Random Forest (RF) ML models were employed using a database of 219 experimental samples of MOFs. In addition to a pre-processing of data down to 183 samples, the study has revealed that changes in H2 uptake were mostly affected by enthalpy of adsorption, followed by the surface area of MOFs.295 In another study, the random forest model and Shapley additive explanations (SHAP) were used on 68 samples and 1745 data points of activated carbon,296 and interesting structure–property relationships of activated carbon were revealed. It was found that the pressure and BET SSA are the most significant predictors of hydrogen uptake, while the pore volume had the smallest influence. Interestingly, oxygen content had a positive impact with enhanced uptake by about 0.6 wt%. Rahimi et al. investigated the optimization of properties of activated carbon for better hydrogen storage using machine learning techniques.297 The study has demonstrated the influence of micropore volume and pore widening on hydrogen storage. Davoodi et al. evaluated a database of 2072 records from 68 porous carbon samples, each with eleven identified dependent and independent variables using four ML learning models (as shown in Fig. 17).298 The authors have highlighted that machine learning can handle large database sets, learn and improve their predictions, and provide insight on hydrogen storage mechanisms in addition to identifying influential factors for enhancing hydrogen storage capacity. However, it can still suffer from disadvantages like requirement of expertise in data analysis and resources. Despite the challenges that are limited by its complexity and data quality,299 machine learning can enable rapid screening, prediction, and optimization of the material discovery process and can significantly reduce the time and cost associated with it.300 As more experimental and computational data become available through experimental and theoretical studies, contributing towards a larger database and more reliable data, the application of ML in this field is expected to grow as a validation and optimisation tool for material discovery, alongside experimental and computational techniques. | ||
| Fig. 17 The use of machine learning can be effective in predicting hydrogen uptake values of porous carbon materials, using models like least-squares-support-vector machine (LSSVM), generalized-regression neural network (GRNN), extreme-learning machine (ELM), and adaptive-neuro-fuzzy-inference system (ANFIS). A database of 2072 records of hydrogen storage data is collected from the literature, including the BET surface area, ultrapore volume, hydrogen storage capacity, temperature, and pressure. This figure has been reproduced from ref. 298 with permission from Elsevier, copyright 2023. | ||
Another challenge in this field is identified as the wide discrepancy of practice among researchers, in measurement and reporting hydrogen uptake capacities. This particularly arises due to factors like variability in synthesis conditions, measurement errors, and lack of standard protocols in reporting data. For example, most available literature studies reporting hydrogen storage capacities correlate good uptake with a change in BET surface area. However, it was shown that the BET method has its own limitations when it comes to measuring the surface area of highly microporous materials.301 To overcome these problems, a series of recommendations by Broom and Hirscher, and others were suggested for the accurate measurement, calibration and avoidance of error. Suggestions have been made for reporting and presenting isotherm data, to ensure reproducibility and eliminate interlaboratory differences.302,303 Existing efforts for the standardisation and ease of access for adsorption data include the suggestion made by Evans et al. for establishing a decentralised adsorption database library for porous materials using proposed Adsorption Information File (AIF).304 Moreover, there is a need for reference materials with well-characterised and established synthesis and measurement protocols that can act as benchmark materials for calibration.305 These recommendations can also contribute towards the standardisation of reporting hydrogen storage capacities of various materials. As a routine practice, besides addressing the challenges faced in the material development phase, it would be also beneficial to consider lifecycle analysis, carbon balance and cost production analysis at the early phase of the material selection process, for a sustainable approach.306
6. Conclusion
Due to the challenges associated with physical properties of hydrogen, its efficient storage is one of the key barriers for successful implementation of hydrogen economy on a wider scale. Many physical storage methods have been introduced including compression and liquefaction. However, there is a growing interest in adsorption-based hydrogen storage in porous materials due to higher efficiency and low-pressure requirement. In this review, some of the key parameters for hydrogen storage in porous materials are discussed, including definitions of excess, net, and total as well as engineering and usable capacities. The effectiveness of hydrogen storage on different types of carbon has been discussed and it was observed that in addition to operating conditions (e.g., pressure and temperature) the storage capacity can be affected by factors intrinsic to the materials, such as the surface area, pore size and surface chemistry, due to the presence of intermediary interactions like Kubas binding and the spillover effect. Porous carbon comes in the form of many different allotropes with unique morphologies and arrangements including graphene and nanotubes. They are versatile and attractive for hydrogen storage due to their high surface area and thermal and mechanical stability. In this review a wide range of porous carbon materials have been examined, ranging from graphene to activated carbon, concluding that in general carbon materials with higher surface areas and micropore volumes show high hydrogen storage capacities, with template-derived carbons and activated carbons showing the highest storage capacities reported to date. Besides the high surface area, various modifications and optimizations can further enhance hydrogen storage capacity, particularly using strategies that either (1) enhance the structural and morphological features of the material by introducing defects and/or increasing the surface area, or (2) incorporate functional groups, heteroatoms, or doped nanoparticles to make hydrogen adsorption kinetics under ambient conditions more favorable. Besides the synthetic strategies, the comparative overview of the performance of different classes of porous carbon along with the importance of LCA, and the reflection on the emerging trend of use of machine learning in this review can be useful for material selection and further improvement for developing superior porous carbons for hydrogen storage applications.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Lila A. M. Mahmoud: writing – original draft. Jemma L. Rowlandson, David J. Fermin, Valeska P. Ting, and Sanjit Nayak: supervision; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
VPT thanks the Engineering and Physical Sciences Research Council (EPSRC) for funding via an EPSRC Research Fellowship (EP/R01650X/1). SN thanks the EPSRC Centre for Doctoral Training in Composites Science, Engineering and Manufacturing (EP/S021728/1).References
- M. Meinshausen, N. Meinshausen, W. Hare, S. C. B. Raper, K. Frieler, R. Knutti, D. J. Frame and M. R. Allen, Nature, 2009, 458, 1158–1162 CrossRef CAS.
- M. Shahbaz, C. Raghutla, K. R. Chittedi, Z. Jiao and X. V. Vo, Energy, 2020, 207, 118162 CrossRef.
- C.-J. Winter, Int. J. Hydrogen Energy, 2009, 34, S1–S52 CrossRef CAS.
- K. Mazloomi and C. Gomes, Renewable Sustainable Energy Rev., 2012, 16, 3024–3033 CrossRef CAS.
- M. D. Allendorf, V. Stavila, J. L. Snider, M. Witman, M. E. Bowden, K. Brooks, B. L. Tran and T. Autrey, Nat. Chem., 2022, 14, 1214–1223 CrossRef CAS PubMed.
- K. T. Møller, T. R. Jensen, E. Akiba and H.-w. Li, Prog. Nat. Sci.: Mater. Int., 2017, 27, 34–40 CrossRef.
- A. Züttel, Mater. Today, 2003, 6, 24–33 Search PubMed.
- US. DOE, Target Explanation Document: Onboard Hydrogen Storage for Light-Duty Fuel Cell Vehicles, https://www.energy.gov/eere/fuelcells/downloads/target-explanation-document-onboard-hydrogen-storage-light-duty-fuel-cell, (accessed November 2024).
- G. Nazir, A. Rehman, S. Hussain, S. Aftab, K. Heo, M. Ikram, S. A. Patil and M. Aizaz Ud Din, Adv. Sustainable Syst., 2022, 6, 2200276 CAS.
- A. Züttel, Sci. Nat., 2004, 91, 157–172 CrossRef PubMed.
- F. Zhang, P. Zhao, M. Niu and J. Maddy, Int. J. Hydrogen Energy, 2016, 41, 14535–14552 CAS.
- R. Folkson, in Alternative Fuels and Advanced Vehicle Technologies for Improved Environmental Performance, ed. R. Folkson and S. Sapsford, Woodhead Publishing, 2nd edn, 2022, pp. 151–171, DOI:10.1016/B978-0-323-90979-2.00013-5.
- M. R. Usman, Renewable Sustainable Energy Rev., 2022, 167, 112743 CrossRef CAS.
- S. Z. S. Al Ghafri, S. Munro, U. Cardella, T. Funke, W. Notardonato, J. P. M. Trusler, J. Leachman, R. Span, S. Kamiya, G. Pearce, A. Swanger, E. D. Rodriguez, P. Bajada, F. Jiao, K. Peng, A. Siahvashi, M. L. Johns and E. F. May, Energy Environ. Sci., 2022, 15, 2690–2731 Search PubMed.
- Q. Cheng, R. Zhang, Z. Shi and J. Lin, Int. J. Lightweight Mater. Manuf., 2024, 7, 269–284 CAS.
- A. Gil, Catal. Today, 2023, 423, 114016 CrossRef CAS.
- M. Hirscher and B. Panella, J. Alloys Compd., 2005, 404–406, 399–401 CrossRef CAS.
- Y. Xia, Z. Yang and Y. Zhu, J. Mater. Chem. A, 2013, 1, 9365–9381 RSC.
- T. K. A. Hoang and D. M. Antonelli, Adv. Mater., 2009, 21, 1787–1800 CrossRef CAS.
- ‘chemisorption’ in IUPAC Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry, 3rd edn, 2006, Online version 3.0.1, 2019, DOI:10.1351/goldbook.C01048.
- Y. Zhang, S. Wu, L. Wang and X. Zhang, Frontiers in Energy, 2023, 17, 72–101 CrossRef.
- in Metal-Catalysed Reactions of Hydrocarbons, ed. G. C. Bond, Springer US, Boston, MA, 2005, pp. 93–152, DOI:10.1007/0-387-26111-7_3.
- A. Zaluska, L. Zaluski and J. O. Ström-Olsen, J. Alloys Compd., 2000, 307, 157–166 CrossRef CAS.
- S. Meduri and J. Nandanavanam, Mater. Today: Proc., 2023, 72, 1–8 CAS.
- S.-i. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 Search PubMed.
- X. L. Zhang, Y. F. Liu, X. Zhang, J. J. Hu, M. X. Gao and H. G. Pan, Mater. Today Nano, 2020, 9, 100064 Search PubMed.
- P. Khadilkar, N. S. Samudre and S. Krishnamurty, J. Energy Storage, 2024, 84, 110833 CrossRef.
- C. Chung, J. Ihm and H. Lee, J. Korean Phys. Soc., 2015, 66, 1649–1655 CrossRef CAS.
- C. V. J. Skipper, A. Hamaed, D. M. Antonelli and N. Kaltsoyannis, Dalton Trans., 2012, 41, 8515–8523 RSC.
- B. Sakintuna, F. Lamari-Darkrim and M. Hirscher, Int. J. Hydrogen Energy, 2007, 32, 1121–1140 CrossRef CAS.
- D. P. Broom, in Hydrogen Storage Materials: The Characterisation of Their Storage Properties, ed. D. P. Broom, Springer London, London, 2011, pp. 183–234, DOI:10.1007/978-0-85729-221-6_6.
- S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard, III, J. Am. Chem. Soc., 2008, 130, 11580–11581 CrossRef CAS PubMed.
- E. Klontzas, E. Tylianakis and G. E. Froudakis, Nano Lett., 2010, 10, 452–454 CrossRef CAS PubMed.
- N. B. McKeown, P. M. Budd and D. Book, Macromol. Rapid Commun., 2007, 28, 995–1002 CrossRef CAS.
- D. Ramimoghadam, E. M. Gray and C. J. Webb, Int. J. Hydrogen Energy, 2016, 41, 16944–16965 CrossRef CAS.
- X. Yin, A. Alsuwaidi and X. Zhang, Microporous Mesoporous Mater., 2022, 330, 111633 CrossRef CAS.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CAS.
- J. Lin, Y. Zhong, L. Tang, L. Wang, M. Yang and H. Xia, Nano Sel., 2022, 3, 320–347 CAS.
- K. Polak-Kraśna, R. Dawson, L. T. Holyfield, C. R. Bowen, A. D. Burrows and T. J. Mays, J. Mater. Sci., 2017, 52, 3862–3875 Search PubMed.
- X. Ma, R. Chen, K. Zhou, Q. Wu, H. Li, Z. Zeng and L. Li, ACS Sustainable Chem. Eng., 2020, 8, 11721–11728 CAS.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1(3), 15018 CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 Search PubMed.
- P. M. Budd, K. J. Msayib, C. E. Tattershall, B. S. Ghanem, K. J. Reynolds, N. B. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263–269 CrossRef CAS.
- S. Rochat, K. Polak-Kraśna, M. Tian, T. J. Mays, C. R. Bowen and A. D. Burrows, Int. J. Hydrogen Energy, 2019, 44, 332–337 CrossRef CAS.
- Z. Liu, C. Zang, Z. Ju, D. Hu, Y. Zhang, J. Jiang and C. Liu, J. Cleaner Prod., 2020, 247, 119565 CrossRef CAS.
- Q. Zhang, S. Yan, X. Yan and Y. Lv, Sci. Total Environ., 2023, 902, 165944 CrossRef CAS PubMed.
- N. B. McKeown, Sci. China: Chem., 2017, 60, 1023–1032 CrossRef CAS.
- Z. Heidarinejad, M. H. Dehghani, M. Heidari, G. Javedan, I. Ali and M. Sillanpää, Environ. Chem. Lett., 2020, 18, 393–415 CrossRef CAS.
- D. Kobina Sam, H. Li, Y.-T. Xu and Y. Cao, J. Ind. Eng. Chem., 2024, 135, 17–42 CrossRef CAS.
- R. Grünker, V. Bon, P. Müller, U. Stoeck, S. Krause, U. Mueller, I. Senkovska and S. Kaskel, Chem. Commun., 2014, 50, 3450–3452 RSC.
- O. K. Farha, A. Özgür Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS.
- L. S. Blankenship, N. Balahmar and R. Mokaya, Nat. Commun., 2017, 8, 1545 CrossRef PubMed.
- Y. Li, Q. Guo, Z. Ding, H. Jiang, H. Yang, W. Du, Y. Zheng, K. Huo and L. L. Shaw, Chem. Eng. J., 2024, 485, 149665 CrossRef CAS.
- X. L. Zou, G. Zhou, W. H. Duan, K. Choi and J. Ihm, J. Phys. Chem. C, 2010, 114, 13402–13407 CrossRef CAS.
- Z. P. Ke, Y. Y. Cheng, S. Y. Yang, F. Li and L. F. Ding, Int. J. Hydrogen Energy, 2017, 42, 11461–11468 CrossRef CAS.
- N. Du, G. P. Robertson, J. Song, I. Pinnau and M. D. Guiver, Macromolecules, 2009, 42, 6038–6043 CrossRef CAS.
- B. Satilmis, M. Lanč, A. Fuoco, C. Rizzuto, E. Tocci, P. Bernardo, G. Clarizia, E. Esposito, M. Monteleone, M. Dendisová, K. Friess, P. M. Budd and J. C. Jansen, J. Membr. Sci., 2018, 555, 483–496 CrossRef CAS.
- W. Han, C. Zhang, M. Zhao, F. Yang, Y. Yang and Y. Weng, J. Membr. Sci., 2021, 636, 119544 CrossRef CAS.
- C. Y. Loh, R. Huang, R. Bell and M. Xie, RSC Sustainability, 2023, 1, 2287–2295 RSC.
- D. DeSantis, J. A. Mason, B. D. James, C. Houchins, J. R. Long and M. Veenstra, Energy Fuels, 2017, 31, 2024–2032 CrossRef CAS.
- Y. Wang, H. Wang, Y. Liu, M. Peng, H. Fan and H. Meng, Chin. Chem. Lett., 2024, 110189, DOI:10.1016/j.cclet.2024.110189.
- K. S. W. Sing, in Studies in Surface Science and Catalysis, ed. F. Rodriguez-Reinoso, J. Rouquerol, K. S. W. Sing and K. K. Unger, Elsevier, 1991, vol. 62, pp. 1–9 Search PubMed.
- C. Nguyen and D. D. Do, Carbon, 2001, 39, 1327–1336 CrossRef CAS.
- F. Shabir, M. Sultan, T. Miyazaki, B. B. Saha, A. Askalany, I. Ali, Y. Zhou, R. Ahmad and R. R. Shamshiri, Renewable Sustainable Energy Rev., 2020, 119, 109630 CrossRef CAS.
- V. P. Ting, A. J. Ramirez-Cuesta, N. Bimbo, J. E. Sharpe, A. Noguera-Diaz, V. Presser, S. Rudic and T. J. Mays, ACS Nano, 2015, 9, 8249–8254 CrossRef CAS.
- Y. Gogotsi, C. Portet, S. Osswald, J. M. Simmons, T. Yildirim, G. Laudisio and J. E. Fischer, Int. J. Hydrogen Energy, 2009, 34, 6314–6319 CrossRef CAS.
- G. Sethia and A. Sayari, Carbon, 2016, 99, 289–294 CrossRef CAS.
- L. Zhang, M. D. Allendorf, R. Balderas-Xicohténcatl, D. P. Broom, G. S. Fanourgakis, G. E. Froudakis, T. Gennett, K. E. Hurst, S. Ling, C. Milanese, P. A. Parilla, D. Pontiroli, M. Riccò, S. Shulda, V. Stavila, T. A. Steriotis, C. J. Webb, M. Witman and M. Hirscher, Prog. Energy, 2022, 4, 042013 CrossRef CAS.
- P. International Union of and C. Applied, DOI:10.1351/goldbook.I03313.
- A. F. Kloutse, R. Zacharia, D. Cossement, R. Chahine, R. Balderas-Xicohténcatl, H. Oh, B. Streppel, M. Schlichtenmayer and M. Hirscher, Appl. Phys. A: Mater. Sci. Process., 2015, 121, 1417–1424 CrossRef CAS.
- A. Nuhnen and C. Janiak, Dalton Trans., 2020, 49, 10295–10307 RSC.
- J. J. Purewal, H. Kabbour, J. J. Vajo, C. C. Ahn and B. Fultz, Nanotechnology, 2009, 20, 204012 CrossRef CAS.
- S. K. Bhatia and A. L. Myers, Langmuir, 2006, 22, 1688–1700 CrossRef CAS.
- X. Gao, Z. Zhong, L. Huang, Y. Mao, H. Wang, J. Liu, L. Ouyang, L. Zhang, M. Han, X. Ma and M. Zhu, Nano Energy, 2023, 118, 109038 CrossRef CAS.
- S. Schaefer, A. Jeder, G. Sdanghi, P. Gadonneix, A. Abdedayem, M. T. Izquierdo, G. Maranzana, A. Ouederni, A. Celzard and V. Fierro, Int. J. Hydrogen Energy, 2020, 45, 30767–30782 CrossRef CAS.
- H. Shen, H. Li, Z. Yang and C. Li, Green Energy Environ., 2022, 7, 1161–1198 CrossRef CAS.
- S. Khoobiar, J. Phys. Chem., 1964, 68, 411–412 CrossRef CAS.
- N. M. Briggs, L. Barrett, E. C. Wegener, L. V. Herrera, L. A. Gomez, J. T. Miller and S. P. Crossley, Nat. Commun., 2018, 9, 3827 CrossRef PubMed.
- G. M. Psofogiannakis and G. E. Froudakis, J. Am. Chem. Soc., 2009, 131, 15133–15135 CrossRef CAS PubMed.
- P. Singh, M. V. Kulkarni, S. P. Gokhale, S. H. Chikkali and C. V. Kulkarni, Appl. Surf. Sci., 2012, 258, 3405–3409 CrossRef CAS.
- C.-H. Chen, T.-Y. Chung, C.-C. Shen, M.-S. Yu, C.-S. Tsao, G.-N. Shi, C.-C. Huang, M.-D. Ger and W.-L. Lee, Int. J. Hydrogen Energy, 2013, 38, 3681–3688 CrossRef CAS.
- H. Jung, K. T. Park, M. N. Gueye, S. H. So and C. R. Park, Int. J. Hydrogen Energy, 2016, 41, 5019–5027 CrossRef CAS.
- R. Zacharia, S.-U. Rather, S. W. Hwang and K. S. Nahm, Chem. Phys. Lett., 2007, 434, 286–291 CrossRef CAS.
- J. E. Sharpe, N. Bimbo, V. P. Ting, A. D. Burrows, D. Jiang and T. J. Mays, Adsorption, 2013, 19, 643–652 CrossRef CAS.
- F. D. Minuto, R. Balderas-Xicohténcatl, A. Policicchio, M. Hirscher and R. G. Agostino, Int. J. Hydrogen Energy, 2018, 43, 14550–14556 CrossRef CAS.
- A. L. Myers and P. A. Monson, Langmuir, 2002, 18, 10261–10273 CrossRef CAS.
- P. A. Parilla, K. Gross, K. Hurst and T. Gennett, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 201 CrossRef.
- S. Gumma and O. Talu, Langmuir, 2010, 26, 17013–17023 CrossRef CAS PubMed.
- D. P. Broom, Adsorption, 2023, 30, 1565–1591 CrossRef.
- K. Murata, M. El-Merraoui and K. Kaneko, J. Chem. Phys., 2001, 114, 4196–4205 CrossRef CAS.
- J. Goldsmith, A. G. Wong-Foy, M. J. Cafarella and D. J. Siegel, Chem. Mater., 2013, 25, 3373–3382 CrossRef CAS.
- S. Rochat, K. Polak-Kraśna, M. Tian, L. T. Holyfield, T. J. Mays, C. R. Bowen and A. D. Burrows, J. Mater. Chem. A, 2017, 5, 18752–18761 RSC.
- P. Schmieder, M. Grzywa, D. Denysenko, M. Hambach and D. Volkmer, Dalton Trans., 2015, 44, 13060–13070 RSC.
- D. P. Broom, C. J. Webb, G. S. Fanourgakis, G. E. Froudakis, P. N. Trikalitis and M. Hirscher, Int. J. Hydrogen Energy, 2019, 44, 7768–7779 CrossRef CAS.
- M. Schlichtenmayer and M. Hirscher, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 379 CrossRef.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- K. M. Thomas, Catal. Today, 2007, 120, 389–398 CrossRef CAS.
- K. A. G. Amankwah and J. A. Schwarz, Int. J. Hydrogen Energy, 1991, 16, 339–344 CrossRef CAS.
- M. S. Reza, C. S. Yun, S. Afroze, N. Radenahmad, M. S. A. Bakar, R. Saidur, J. Taweekun and A. K. Azad, Arab J. Basic Appl. Sci., 2020, 27, 208–238 Search PubMed.
- Z. Yang, Y. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673–1679 CrossRef CAS PubMed.
- L. S. Blankenship and R. Mokaya, Energy Environ. Sci., 2017, 10, 2552–2562 RSC.
- I. A. Baburin, A. Klechikov, G. Mercier, A. Talyzin and G. Seifert, Int. J. Hydrogen Energy, 2015, 40, 6594–6599 CrossRef CAS.
- G. Srinivas, Y. Zhu, R. Piner, N. Skipper, M. Ellerby and R. Ruoff, Carbon, 2010, 48, 630–635 CrossRef CAS.
- C. X. Guo, Y. Wang and C. M. Li, ACS Sustainable Chem. Eng., 2013, 1, 14–18 CrossRef CAS.
- C. Y. Zhou and J. A. Szpunar, ACS Appl. Mater. Interfaces, 2016, 8, 25933–25940 CrossRef CAS.
- C.-C. Huang, N.-W. Pu, C.-A. Wang, J.-C. Huang, Y. Sung and M.-D. Ger, Sep. Purif. Technol., 2011, 82, 210–215 CrossRef CAS.
- S. H. Aboutalebi, S. Aminorroaya-Yamini, I. Nevirkovets, K. Konstantinov and H. K. Liu, Adv. Energy Mater., 2012, 2, 1439–1446 CrossRef CAS.
- E. S. Cho, A. M. Ruminski, S. Aloni, Y.-S. Liu, J. Guo and J. J. Urban, Nat. Commun., 2016, 7, 10804 CrossRef CAS.
- C. Jin, J. Li, K. Zhang, Habibullah, G. Xia, C. Wu, Y. Wang, W. Cen, Y. Chen, Y. Yan and Y. Chen, Nano Energy, 2022, 99, 107360 CrossRef CAS.
- Y. Chen, Habibullah, G. Xia, C. Jin, Y. Wang, Y. Yan, Y. Chen, X. Gong, Y. Lai and C. Wu, Inorganics, 2023, 11, 251 CrossRef CAS.
- V. B. Parambhath, R. Nagar and S. Ramaprabhu, Langmuir, 2012, 28, 7826–7833 CrossRef CAS.
- Z. M. Ao and F. M. Peeters, Phys. Rev. B, 2010, 81, 205406 CrossRef.
- Y. W. Chen, Habibullah, G. H. Xia, C. A. Jin, Y. Wang, Y. G. Yan, Y. G. Chen, X. F. Gong, Y. Q. Lai and C. L. Wu, Materials, 2023, 16, 4219 CrossRef CAS PubMed.
- A. Flamina, R. M. Raghavendra, A. Gupta and A. Subramaniam, Appl. Surf. Sci., 2023, 13, 100371 Search PubMed.
- A. Morandé, P. Lillo, E. Blanco, C. Pazo, A. B. Dongil, X. Zarate, M. Saavedra-Torres, E. Schott, R. Canales, A. Videla and N. Escalona, J. Energy Storage, 2023, 64, 107193 Search PubMed.
- W. Zhao, V. Fierro, C. Zlotea, E. Aylon, M. T. Izquierdo, M. Latroche and A. Celzard, Int. J. Hydrogen Energy, 2011, 36, 5431–5434 CAS.
- M. Sevilla, A. B. Fuertes and R. Mokaya, Energy Environ. Sci., 2011, 4, 1400–1410 CAS.
- M. Jordá-Beneyto, D. Lozano-Castelló, F. Suárez-García, D. Cazorla-Amorós and A. Linares-Solano, Microporous Mesoporous Mater., 2008, 112, 235–242 Search PubMed.
- M. Sevilla, A. B. Fuertes and R. Mokaya, Int. J. Hydrogen Energy, 2011, 36, 15658–15663 CAS.
- P. Ramirez-Vidal, R. L. S. Canevesi, G. Sdanghi, S. Schaefer, G. Maranzana, A. Celzard and V. Fierro, ACS Appl. Mater. Interfaces, 2021, 13, 12562–12574 CAS.
- Y. Sun and P. A. Webley, Chem. Eng. J., 2010, 162, 883–892 CAS.
- D. Wang, Z. Geng, C. Zhang, X. Zhou and X. Liu, J. Energy Chem., 2014, 23, 601–608 CrossRef.
- T. Ramesh, N. Rajalakshmi and K. S. Dhathathreyan, J. Energy Storage, 2015, 4, 89–95 CrossRef.
- A. Minoda, S. Oshima, H. Iki and E. Akiba, J. Alloys Compd., 2013, 580, S301–S304 CrossRef CAS.
- H. Akasaka, T. Takahata, I. Toda, H. Ono, S. Ohshio, S. Himeno, T. Kokubu and H. Saitoh, Int. J. Hydrogen Energy, 2011, 36, 580–585 CrossRef CAS.
- Y. Li and R. T. Yang, J. Phys. Chem. C, 2007, 111, 11086–11094 CrossRef CAS.
- R. Pedicini, S. Maisano, V. Chiodo, G. Conte, A. Policicchio and R. G. Agostino, Int. J. Hydrogen Energy, 2020, 45, 14038–14047 CrossRef CAS.
- J. Zhang, J. Gao, Y. Chen, X. Hao and X. Jin, Results Phys., 2017, 7, 1628–1633 CrossRef.
- N. Bader and A. Ouederni, J. Energy Storage, 2017, 13, 268–276 CrossRef.
- A. Turkyilmaz, K. Isinkaralar, M. Dogan, B. K. Kizilduman and Z. Bicil, Sustainable Chem. Pharm., 2024, 40, 12 Search PubMed.
- A. Flamina, R. M. Raghavendra and A. Subramaniam, Carbon, 2024, 119657, DOI:10.1016/j.carbon.2024.119657.
- Z. Peng, Y. Xu, W. Luo, C. Wang and L. Ma, ChemistrySelect, 2020, 5, 11221–11228 CrossRef CAS.
- Y. Li, R. T. Yang, C.-J. Liu and Z. Wang, Ind. Eng. Chem. Res., 2007, 46, 8277–8281 CrossRef CAS.
- Z. Geng, D. Wang, C. Zhang, X. Zhou, H. Xin, X. Liu and M. Cai, Int. J. Hydrogen Energy, 2014, 39, 13643–13649 CrossRef CAS.
- M. Z. Figueroa-Torres, C. Domínguez-Ríos, J. G. Cabañas-Moreno, O. Vega-Becerra and A. Aguilar-Elguézabal, Int. J. Hydrogen Energy, 2012, 37, 10743–10749 CrossRef CAS.
- P. Ruz, S. Banerjee, T. Das, A. Kumar, V. Sudarsan, A. K. Patra and P. U. Sastry, J. Porous Mater., 2024, 31, 81–96 CrossRef CAS.
- B. Adeniran and R. Mokaya, Nano Energy, 2015, 16, 173–185 CrossRef CAS.
- R. Muhammad, Y. C. Nah and H. Oh, J. CO2 Util, 2023, 69, 102401 CrossRef CAS.
- X. Li, H. Tian, S. Yan, H. J. Shi, J. B. Wu, Y. L. Sun, Y. Q. Xing, H. C. Bai and H. Zhang, Int. J. Hydrogen Energy, 2024, 50, 324–336 CrossRef CAS.
- M. S. Balathanigaimani, M. B. Haider, D. Jha, R. Kumar, S. J. Lee, W. G. Shim, H. K. Shon, S. C. Kim and H. Moon, J. Nanosci. Nanotechnol., 2018, 18, 2196–2199 CrossRef CAS PubMed.
- H. Jin, Y. S. Lee and I. Hong, Catal. Today, 2007, 120, 399–406 CrossRef CAS.
- N. M. Musyoka, B. K. Mutuma and N. Manyala, RSC Adv., 2020, 10, 26928–26936 RSC.
- T. Chen, Y. Zhou, L. Luo, X. Wu, Z. Li, M. Fan and W. Zhao, Electrochim. Acta, 2019, 325, 134941 CrossRef CAS.
- J. Serafin, B. Dziejarski, C. Solis, P. Ramírez de la Piscina and N. Homs, Fuel, 2024, 363, 130975 CrossRef CAS.
- V. Lionetti, C. Poselle Bonaventura, G. Conte, O. De Luca, A. Policicchio, T. Caruso, G. Desiderio, M. Papagno and R. G. Agostino, Int. J. Hydrogen Energy, 2024, 61, 639–649 CrossRef CAS.
- E. Çetingürbüz and A. Turkyilmaz, Ind. Crops Prod., 2023, 203, 117171 CrossRef.
- T. Segakweng, N. M. Musyoka, J. W. Ren, P. Crouse and H. W. Langmi, Res. Chem. Intermed., 2016, 42, 4951–4961 CrossRef CAS.
- N. P. Stadie, J. J. Vajo, R. W. Cumberland, A. A. Wilson, C. C. Ahn and B. Fultz, Langmuir, 2012, 28, 10057–10063 CrossRef CAS.
- A. Almasoudi and R. Mokaya, J. Mater. Chem., 2012, 22, 146–152 RSC.
- Y. Xia, Z. Yang and R. Mokaya, Chem. Vap. Deposition, 2010, 16, 322–328 CrossRef CAS.
- R. Campesi, F. Cuevas, R. Gadiou, E. Leroy, M. Hirscher, C. Vix-Guterl and M. Latroche, Carbon, 2008, 46, 206–214 CrossRef CAS.
- A. Almasoudi and R. Mokaya, J. Mater. Chem. A, 2014, 2, 10960–10968 RSC.
- J. M. Juárez, M. B. Gómez and O. A. Anunziata, Int. J. Energy Res., 2015, 39, 941–953 Search PubMed.
- J. Cai, M. Yang, Y. Xing and X. Zhao, Colloids Surf., A, 2014, 444, 240–245 CrossRef CAS.
- M. Kapsi, C. M. Veziri, G. Pilatos, G. N. Karanikolos and G. E. Romanos, Int. J. Hydrogen Energy, 2022, 47, 36850–36872 CrossRef CAS.
- R. J. Konwar and M. De, Microporous Mesoporous Mater., 2013, 175, 16–24 CrossRef CAS.
- E. Masika and R. Mokaya, Prog. Nat. Sci.: Mater. Int., 2013, 23, 308–316 CrossRef.
- Y. Xia, R. Mokaya, D. M. Grant and G. S. Walker, Carbon, 2011, 49, 844–853 CrossRef CAS.
- N. F. Attia, S. M. Lee, H. J. Kim and K. E. Geckeler, Microporous Mesoporous Mater., 2013, 173, 139–146 CrossRef CAS.
- N. Alam and R. Mokaya, Microporous Mesoporous Mater., 2011, 142, 716–724 CrossRef CAS.
- N. Alam and R. Mokaya, Microporous Mesoporous Mater., 2011, 144, 140–147 CrossRef CAS.
- R. J. Konwar and M. De, J. Anal. Appl. Pyrolysis, 2014, 107, 224–232 CrossRef CAS.
- P.-S. Tseng, L.-X. Chang, Y.-S. Ou, C.-M. Chou, C.-S. Tsao, Y. Wu, J.-P. Chou, P.-J. Chen and C.-Y. Wang, Appl. Surf. Sci., 2023, 638, 158097 CrossRef CAS.
- Z. Yang, Q. Jia, B. Chen, X. Gou, Y. Zhu and Y. Xia, Int. J. Hydrogen Energy, 2020, 45, 25086–25095 CrossRef CAS.
- E. Mosquera-Vargas, R. Tamayo, M. Morel, M. Roble and D. E. Díaz-Droguett, Heliyon, 2021, 7, e08494 CrossRef CAS.
- U. Khalilov, U. Uljayev, K. Mehmonov, P. Nematollahi, M. Yusupov and E. Neyts, Int. J. Hydrogen Energy, 2024, 55, 604–610 CrossRef CAS.
- C. D. Brewster, L. R. Terry, H. V. Doan, S. Rochat and V. P. Ting, Energy Adv., 2023, 2, 398–409 RSC.
- M. Konni, A. S. Dadhich and S. Babu Mukkamala, Int. J. Hydrogen Energy, 2017, 42, 953–959 CrossRef CAS.
- R. B. Rakhi, K. Sethupathi and S. Ramaprabhu, Int. J. Hydrogen Energy, 2008, 33, 381–386 CrossRef CAS.
- Ü. Çalışır, B. Çiçek and M. Doğan, ullerenes, Nanotubes Carbon Nanostruct., 2021, 29, 899–906 CrossRef.
- A. M. Rashidi, A. Nouralishahi, A. A. Khodadadi, Y. Mortazavi, A. Karimi and K. Kashefi, Int. J. Hydrogen Energy, 2010, 35, 9489–9495 CrossRef CAS.
- J. H. Lee, K. Y. Rhee and S. J. Park, Int. J. Hydrogen Energy, 2010, 35, 7850–7857 CrossRef CAS.
- L. Vellingiri, K. Annamalai, R. Kandasamy and I. Kombiah, Int. J. Hydrogen Energy, 2018, 43, 848–860 CrossRef CAS.
- M. M. Larijani and S. Safa, Acta Phys. Pol., A, 2014, 126, 732–735 CrossRef CAS.
- S.-u. Rather and K. S. Nahm, Mater. Res. Bull., 2014, 49, 525–530 CrossRef CAS.
- S.-Y. Lee and S.-J. Park, J. Solid State Chem., 2012, 194, 307–312 CrossRef CAS.
- H. Kajiura, S. Tsutsui, K. Kadono, M. Kakuta, M. Ata and Y. Murakami, Appl. Phys. Lett., 2003, 82, 1105–1107 CrossRef CAS.
- S. V. Sawant, S. Banerjee, A. W. Patwardhan, J. B. Joshi and K. Dasgupta, Int. J. Hydrogen Energy, 2020, 45, 13406–13413 CrossRef CAS.
- M. Aghababaei, A. A. Ghoreyshi and K. Esfandiari, Int. J. Hydrogen Energy, 2020, 45, 23112–23121 CrossRef CAS.
- A. Sharma, Int. J. Hydrogen Energy, 2020, 45, 2967–2974 CrossRef CAS.
- R. S. Rajaura, S. Srivastava, P. K. Sharma, S. Mathur, R. Shrivastava, S. S. Sharma and Y. K. Vijay, Nano-Struct. Nano-Objects, 2018, 14, 57–65 CrossRef CAS.
- Ü. Çakır, F. Kestel, B. K. Kızılduman, Z. Bicil and M. Doğan, Diamond Relat. Mater., 2021, 120, 108604 Search PubMed.
- S. Kaskun and M. Kayfeci, Int. J. Hydrogen Energy, 2018, 43, 10773–10778 CAS.
- S. H. Barghi, T. T. Tsotsis and M. Sahimi, Int. J. Hydrogen Energy, 2014, 39, 1390–1397 CAS.
- D. Silambarasan, V. J. Surya, K. Iyakutti, K. Asokan, V. Vasu and Y. Kawazoe, Appl. Surf. Sci., 2017, 418, 49–55 CAS.
- S. Dwivedi, Int. J. Hydrogen Energy, 2022, 47, 41848–41877 CrossRef CAS.
- Y. S. Choi, J. M. Yoo and B. H. Hong, in Graphene for Flexible Lighting and Displays, ed. T.-W. Lee, Woodhead Publishing, 2020, pp. 5–26, DOI:10.1016/B978-0-08-102482-9.00002-2.
- S. K. Tiwari, S. Sahoo, N. Wang and A. Huczko, J. Sci.: Adv. Mater. Devices, 2020, 5, 10–29 Search PubMed.
- A. Peigney, C. Laurent, E. Flahaut, R. R. Bacsa and A. Rousset, Carbon, 2001, 39, 507–514 CrossRef CAS.
- Y. Okamoto and Y. Miyamoto, J. Phys. Chem. B, 2001, 105, 3470–3474 CrossRef CAS.
- F. Costanzo, P. L. Silvestrelli and F. Ancilotto, J. Chem. Theory Comput., 2012, 8, 1288–1294 CrossRef CAS PubMed.
- S. Patchkovskii, J. S. Tse, S. N. Yurchenko, L. Zhechkov, T. Heine and G. Seifert, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10439–10444 CrossRef CAS.
- J. R. Morse, D. A. Zugell, E. Patterson, J. W. Baldwin and H. D. Willauer, J. Energy Storage, 2021, 494, 229734 CAS.
- L. Kaczmarek, T. Warga, P. Zawadzki, M. Makowicz, B. Bucholc and P. Kula, Int. J. Hydrogen Energy, 2019, 44, 23149–23159 CrossRef CAS.
- K. S. Subrahmanyam, P. Kumar, U. Maitra, A. Govindaraj, K. P. S. S. Hembram, U. V. Waghmare and C. N. R. Rao, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2674–2677 CrossRef CAS.
- O. K. Alekseeva, I. V. Pushkareva, A. S. Pushkarev and V. N. Fateev, Nanotechnol. Russ., 2020, 15, 273–300 CrossRef CAS PubMed.
- A. Ghosh, K. S. Subrahmanyam, K. S. Krishna, S. Datta, A. Govindaraj, S. K. Pati and C. N. R. Rao, J. Phys. Chem. C, 2008, 112, 15704–15707 CrossRef CAS.
- I. Lopez-Corral, E. German, G. P. Brizuela, A. Juan and M. A. Volpe, Int. J. Hydrogen Energy, 2010, 35, 2377–2384 CrossRef CAS.
- H. Cheng, L. Chen, A. C. Cooper, X. Sha and G. P. Pez, Energy Environ. Sci., 2008, 1, 338–354 RSC.
- K. Jastrzębski and P. Kula, Materials, 2021, 14, 2499 CrossRef PubMed.
- J. Long, J. Y. Li, F. Nan, S. Yin, J. J. Li and W. L. Cen, Appl. Surf. Sci., 2018, 435, 1065–1071 CrossRef CAS.
- S. Ghotia, T. Rimza, S. Singh, N. Dwivedi, A. K. Srivastava and P. Kumar, J. Mater. Chem. A, 2024, 12, 12325–12357 RSC.
- P. A. Denis, ACS Omega, 2022, 7, 45935–45961 CrossRef CAS PubMed.
- S. J. Lee, J. Theerthagiri, P. Nithyadharseni, P. Arunachalam, D. Balaji, A. Madan Kumar, J. Madhavan, V. Mittal and M. Y. Choi, Renewable Sustainable Energy Rev., 2021, 143, 110849 CrossRef CAS.
- B. P. Vinayan, R. Nagar and S. Ramaprabhu, J. Mater. Chem. A, 2013, 1, 11192 RSC.
- E. Anikina, S. R. Naqvi, H. Bae, H. Lee, W. Luo, R. Ahuja and T. Hussain, Int. J. Hydrogen Energy, 2022, 47, 10654–10664 CrossRef CAS.
- Y. Cho, S. Kang, B. C. Wood and E. S. Cho, ACS Appl. Mater. Interfaces, 2022, 14, 20823–20834 CrossRef CAS.
- B. P. Tarasov, A. A. Arbuzov, A. A. Volodin, P. V. Fursikov, S. A. Mozhzhuhin, M. V. Lototskyy and V. A. Yartys, J. Alloys Compd., 2022, 896, 162881 CrossRef CAS.
- W. Zhang, G. Xu, L. Chen, S. Pan, X. Jing, J. Wang and S. Han, Int. J. Hydrogen Energy, 2017, 42, 15262–15270 CrossRef CAS.
- E. Ruse, M. Buzaglo, I. Pri-Bar, L. Shunak, R. Nadiv, S. Pevzner, O. Siton-Mendelson, V. M. Skripnyuk, E. Rabkin and O. Regev, Carbon, 2018, 130, 369–376 CrossRef CAS.
- A. Klechikov, G. Mercier, T. Sharifi, I. A. Baburin, G. Seifert and A. V. Talyzin, Chem. Commun., 2015, 51, 15280–15283 RSC.
- Y. Liu, Z. Zhang and T. Wang, Int. J. Hydrogen Energy, 2018, 43, 11120–11131 CrossRef CAS.
- D. D. Peng, Z. M. Ding, L. Zhang, Y. K. Fu, J. S. Wang, Y. Li and S. M. Han, Int. J. Hydrogen Energy, 2018, 43, 3731–3740 CrossRef CAS.
- A. Züttel and S.-I. Orimo, MRS Bull., 2002, 27, 705–711 CrossRef.
- A. C. Dillon, K. M. Jones, T. A. Bekkedahl, C. H. Kiang, D. S. Bethune and M. J. Heben, Nature, 1997, 386, 377–379 CrossRef CAS.
- C. Liu, Y. Chen, C.-Z. Wu, S.-T. Xu and H.-M. Cheng, Carbon, 2010, 48, 452–455 CrossRef CAS.
- J. Lyu, V. Kudiiarov and A. Lider, Nanomaterials, 2020, 10, 255 CrossRef CAS.
- G. E. Froudakis, Mater. Today, 2011, 14, 324–328 CrossRef CAS.
- C.-H. Chen and C.-C. Huang, Int. J. Hydrogen Energy, 2007, 32, 237–246 CrossRef CAS.
- F. Liu, X. Zhang, J. Cheng, J. Tu, F. Kong, W. Huang and C. Chen, Carbon, 2003, 41, 2527–2532 CrossRef CAS.
- A. Reyhani, S. Z. Mortazavi, S. Mirershadi, A. Z. Moshfegh, P. Parvin and A. N. Golikand, J. Phys. Chem. C, 2011, 115, 6994–7001 CrossRef CAS.
- W. Liu, Y. H. Zhao, Y. Li, Q. Jiang and E. J. Lavernia, J. Phys. Chem. C, 2009, 113, 2028–2033 CrossRef CAS.
- D. Silambarasan, V. J. Surya, K. Iyakutti and V. Vasu, Int. J. Hydrogen Energy, 2014, 39, 391–397 CrossRef CAS.
- N. F. Attia, M. M. Menemparabath, S. Arepalli and K. E. Geckeler, Int. J. Hydrogen Energy, 2013, 38, 9251–9262 CrossRef CAS.
- D. Silambarasan, V. Vasu and K. Iyakutti, IEEE Trans. Nanotechnol., 2014, 13, 261–267 CAS.
- H. Cheng, A. C. Cooper, G. P. Pez, M. K. Kostov, P. Piotrowski and S. J. Stuart, J. Phys. Chem. B, 2005, 109, 3780–3786 CrossRef CAS PubMed.
- H.-M. Cheng, Q.-H. Yang and C. Liu, Carbon, 2001, 39, 1447–1454 CrossRef CAS.
- M. Jordá-Beneyto, F. Suárez-García, D. Lozano-Castelló, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2007, 45, 293–303 CrossRef.
- M. Mohan, V. K. Sharma, E. A. Kumar and V. Gayathri, Energy Storage, 2019, 1, e35 CrossRef CAS.
- L. Zhou, Int. J. Hydrogen Energy, 2006, 31, 259–264 CrossRef CAS.
- N. Kostoglou, C. Koczwara, S. Stock, C. Tampaxis, G. Charalambopoulou, T. Steriotis, O. Paris, C. Rebholz and C. Mitterer, Chem. Eng. J., 2022, 427, 131730 CrossRef CAS.
- J. Deng, T. Xiong, H. Wang, A. Zheng and Y. Wang, ACS Sustainable Chem. Eng., 2016, 4, 3750–3756 CrossRef CAS.
- S. S. A. Syed-Hassan and M. S. M. Zaini, Korean J. Chem. Eng., 2016, 33, 2502–2512 CrossRef CAS.
- M. Sankir and N. D. Sankir, Hydrogen Storage Technologies, John Wiley & Sons, Incorporated, Newark, United States, 2018 Search PubMed.
- A. C. Lua, Biomass Convers. Biorefin., 2020, 10, 523–533 CrossRef.
- J. L. Rowlandson, J. Coombs OBrien, K. J. Edler, M. Tian and V. P. Ting, C, 2019, 5, 82 CAS.
- C. Zhao, L. Ge, X. Li, M. Zuo, C. Xu, S. Chen, Q. Li, Y. Wang and C. Xu, Energy, 2023, 273, 127177 CrossRef CAS.
- W. G. Zhao, L. Luo, H. Y. Wang and M. Z. Fan, BioResources, 2017, 12, 1246–1262 CAS.
- J. Wang, I. Senkovska, S. Kaskel and Q. Liu, Carbon, 2014, 75, 372–380 CrossRef CAS.
- A. Morande, P. Lillo, E. Blanco, C. Pazo, A. B. Dongil, X. Zarate, M. Saavedra-Torres, E. Schott, R. Canales, A. Videla and N. Escalona, J. Energy Storage, 2023, 64, 107193 CrossRef.
- J. Hu, Q. Gao, Y. Wu and S. Song, Int. J. Hydrogen Energy, 2007, 32, 1943–1948 CrossRef CAS.
- M. S. Hossain, T. Furusawa and M. Sato, Inorg. Chem. Commun., 2023, 148, 110376 CrossRef CAS.
- H.-L. Jiang, B. Liu, Y.-Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS.
- W. Chaikittisilp, K. Ariga and Y. Yamauchi, J. Mater. Chem. A, 2013, 1, 14–19 RSC.
- Y. D. Xia, Z. X. Yang, X. L. Gou and Y. Q. Zhu, Int. J. Hydrogen Energy, 2013, 38, 5039–5052 CrossRef CAS.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- S. J. Yang, T. Kim, J. H. Im, Y. S. Kim, K. Lee, H. Jung and C. R. Park, Chem. Mater., 2012, 24, 464–470 CrossRef CAS.
- G. Sdanghi, G. Sdanghi, G. Maranzana, A. Celzard and V. Fierro, in Hydrogen Storage Technologies, 2018, ch. 9, pp. 263–320, DOI:10.1002/9781119460572.
- M. Sevilla, R. Foulston and R. Mokaya, Energy Environ. Sci., 2010, 3, 223–227 RSC.
- P. A. Kobielska, R. Telford, J. Rowlandson, M. Tian, Z. Shahin, A. Demessence, V. P. Ting and S. Nayak, ACS Appl. Mater. Interfaces, 2018, 10, 25967–25971 CrossRef CAS PubMed.
- G. Yushin, R. Dash, J. Jagiello, J. E. Fischer and Y. Gogotsi, Adv. Funct. Mater., 2006, 16, 2288–2293 CrossRef CAS.
- M. Baca, K. Cendrowski, W. Kukulka, G. Bazarko, D. Moszyński, B. Michalkiewicz, R. J. Kalenczuk and B. Zielinska, Nanomaterials, 2018, 8, 639 CrossRef PubMed.
- Y. Li, D. Li, Y. Rao, X. Zhao and M. Wu, Carbon, 2016, 105, 454–462 CrossRef CAS.
- A.-A. G. A. Islam, J. Janaun, M. S. Mastuli and Y. H. Taufiq-Yap, Mater. Lett., 2016, 179, 57–60 CrossRef.
- A. Chambers, C. Park, R. T. K. Baker and N. M. Rodriguez, J. Phys. Chem. B, 1998, 102, 4253–4256 CrossRef CAS.
- P. Jin and X. Gu, in Handbook of Fullerene Science and Technology, ed. X. Lu, T. Akasaka and Z. Slanina, Springer Singapore, Singapore, 2021, pp. 1–30, DOI:10.1007/978-981-13-3242-5_22-1.
- S. Margadonna and K. Prassides, in Encyclopedia of Materials: Science and Technology, ed. K. H. J. Buschow, R. W. Cahn, M. C. Flemings, B. Ilschner, E. J. Kramer, S. Mahajan and P. Veyssière, Elsevier, Oxford, 2001, pp. 3379–3383, DOI:10.1016/B0-08-043152-6/00603-3.
- Y. Zhao, Y.-H. Kim, A. C. Dillon, M. J. Heben and S. B. Zhang, Phys. Rev. Lett., 2005, 94, 155504 CrossRef.
- T. Yildirim, J. Íñiguez and S. Ciraci, Phys. Rev. B, 2005, 72, 153403 CrossRef.
- I. V. Muralikrishna and V. Manickam, in Environmental Management, ed. I. V. Muralikrishna and V. Manickam, Butterworth-Heinemann, 2017, pp. 57–75, DOI:10.1016/B978-0-12-811989-1.00005-1.
- S. Temizel-Sekeryan, F. Wu and A. L. Hicks, Int. J. Life Cycle Assess., 2021, 26, 656–672 CrossRef CAS.
- N. M. Mubarak, E. C. Abdullah, N. S. Jayakumar and J. N. Sahu, J. Ind. Eng. Chem., 2014, 20, 1186–1197 CrossRef CAS.
- H. Y. Teah, T. Sato, K. Namiki, M. Asaka, K. Feng and S. Noda, ACS Sustainable Chem. Eng., 2020, 8, 1730–1740 CrossRef CAS.
- J. A. Isaacs, A. Tanwani, M. L. Healy and L. J. Dahlben, J. Nanopart. Res., 2010, 12, 551–562 CrossRef.
- V. K. K. Upadhyayula, D. E. Meyer, M. A. Curran and M. A. Gonzalez, J. Cleaner Prod., 2012, 26, 37–47 CrossRef CAS.
- G. Wernet, C. Bauer, B. Steubing, J. Reinhard, E. Moreno-Ruiz and B. Weidema, Int. J. Life Cycle Assess., 2016, 21, 1218–1230 CrossRef.
- M. L. Healy, L. J. Dahlben and J. A. Isaacs, J. Ind. Ecol., 2008, 12, 376–393 CrossRef CAS.
- S. Gavankar, S. Suh and A. A. Keller, J. Ind. Ecol., 2015, 19, 51–60 CrossRef CAS.
- K. He, Z.-Y. Zhang and F.-S. Zhang, Waste Manage., 2021, 124, 283–292 CrossRef CAS PubMed.
- C. Jia, M. Pang, Y. Lu, Y. Liu, M. Zhuang, B. Liu, J. Lu, T. Wei, L. Wang, T. Bian, M. Wang, F. Yu, L. Sun, L. Lin, T. Teng, X. Wu, Z. He, J. Gao, J. Luo, S. Zhang, L. Feng, X. Yin, F. You, G. Li, L. Zhang, Y.-G. Zhu, X. Zhu and Y. Yang, One Earth, 2022, 5, 1394–1403 CrossRef.
- N. Arena, J. Lee and R. Clift, J. Cleaner Prod., 2016, 125, 68–77 CrossRef CAS.
- A. Vilén, P. Laurell and R. Vahala, J. Environ. Manage., 2022, 324, 116356 CrossRef PubMed.
- Y. Wang, J. Wang, X. Zhang, D. Bhattacharyya and E. M. Sabolsky, Energies, 2022, 15, 351 CrossRef CAS.
- C. Li, K. Sun, Y. Sun, Y. Shao, G. Gao, L. Zhang, S. Zhang and X. Hu, Chem. Eng. J., 2024, 480, 148176 CrossRef CAS.
- R. Hischier and T. Walser, Sci. Total Environ., 2012, 425, 271–282 CrossRef CAS PubMed.
- Q. Hassan, S. Algburi, A. Z. Sameen, H. M. Salman and M. Jaszczur, Int. J. Hydrogen Energy, 2024, 50, 310–333 CrossRef CAS.
- A. R. Razmi, A. R. Hanifi and M. Shahbakhti, Renewable Energy, 2023, 215, 118996 CrossRef CAS.
- L. Esposito, M. van der Wiel and C. Acar, Int. J. Hydrogen Energy, 2024, 79, 579–593 CrossRef CAS.
- P. Muthukumar, A. Kumar, M. Afzal, S. Bhogilla, P. Sharma, A. Parida, S. Jana, E. A. Kumar, R. K. Pai and I. P. Jain, Int. J. Hydrogen Energy, 2023, 48, 33223–33259 CrossRef CAS.
- Y. Ghorbani, S. E. Zhang, G. T. Nwaila, J. E. Bourdeau and D. H. Rose, J. Cleaner Prod., 2024, 434, 140414 CrossRef CAS.
- M. Younas, S. Shafique, A. Hafeez, F. Javed and F. Rehman, Fuel, 2022, 316, 123317 CrossRef CAS.
- S. Di Micco, F. Romano, E. Jannelli, A. Perna and M. Minutillo, Int. J. Hydrogen Energy, 2023, 48, 31457–31467 CrossRef CAS.
- H. Caliskan, I. Dincer and A. Hepbasli, Appl. Therm. Eng., 2013, 61, 784–798 Search PubMed.
- A. Khosravi, R. N. N. Koury, L. Machado and J. J. G. Pabon, Energy, 2018, 148, 1087–1102 CrossRef.
- D. P. Broom, C. J. Webb, K. E. Hurst, P. A. Parilla, T. Gennett, C. M. Brown, R. Zacharia, E. Tylianakis, E. Klontzas, G. E. Froudakis, T. A. Steriotis, P. N. Trikalitis, D. L. Anton, B. Hardy, D. Tamburello, C. Corgnale, B. A. van Hassel, D. Cossement, R. Chahine and M. Hirscher, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 151 CrossRef.
- D. Caviedes and I. Cabria, Int. J. Hydrogen Energy, 2022, 47, 11916–11928 CrossRef CAS.
- K. M. Thomas, Dalton Trans., 2009, 1487–1505, 10.1039/b815583f.
- X. J. Wu, Y. G. Wang, Z. J. Cai, D. M. Zhao and W. Q. Cai, Chem. Eng. Sci., 2020, 226, 10 Search PubMed.
- H. V. Thanh, S. Ebrahimnia Taremsari, B. Ranjbar, H. Mashhadimoslem, E. Rahimi, M. Rahimi and A. Elkamel, Energies, 2023, 16, 2348 CrossRef CAS.
- A. I. Osman, W. Abd-Elaziem, M. Nasr, M. Farghali, A. K. Rashwan, A. Hamada, Y. M. Wang, M. A. Darwish, T. A. Sebaey, A. Khatab and A. H. Elsheikh, Environ. Chem. Lett., 2024, 22, 1703–1740 CrossRef CAS.
- K. Salehi, M. Rahmani and S. Atashrouz, Int. J. Hydrogen Energy, 2023, 48, 33260–33275 CrossRef CAS.
- R. M. Giappa, E. Tylianakis, M. Di Gennaro, K. Gkagkas and G. E. Froudakis, Int. J. Hydrogen Energy, 2021, 46, 27612–27621 CrossRef CAS.
- S. Shekhar and C. Chowdhury, Surf. Interfaces, 2024, 14, 100166 CrossRef.
- M. Seyyedattar, S. Zendehboudi, A. Ghamartale and M. Afshar, Int. J. Hydrogen Energy, 2024, 69, 158–172 CrossRef CAS.
- M. I. Maulana Kusdhany and S. M. Lyth, Carbon, 2021, 179, 190–201 CrossRef CAS.
- M. Rahimi, M. H. Abbaspour-Fard and A. Rohani, J. Cleaner Prod., 2021, 329, 129714 CrossRef CAS.
- S. Davoodi, H. Vo Thanh, D. A. Wood, M. Mehrad, M. Al-Shargabi and V. S. Rukavishnikov, Sep. Purif. Technol., 2023, 316, 123807 CrossRef CAS.
- A. I. Osman, M. Nasr, M. Farghali, A. K. Rashwan, A. Abdelkader, A. A. H. Al-Muhtaseb, I. Ihara and D. W. Rooney, Environ. Chem. Lett., 2024, 22, 1005–1071 CrossRef CAS.
- M. H. Mobarak, M. A. Mimona, M. A. Islam, N. Hossain, F. T. Zohura, I. Imtiaz and M. I. H. Rimon, Appl. Surf. Sci., 2023, 18, 100523 CrossRef.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- C. Zlotea, P. Moretto and T. Steriotis, Int. J. Hydrogen Energy, 2009, 34, 3044–3057 CrossRef CAS.
- D. P. Broom and M. Hirscher, ChemPhysChem, 2021, 22, 2141–2157 CrossRef CAS PubMed.
- J. D. Evans, V. Bon, I. Senkovska and S. Kaskel, Langmuir, 2021, 37, 4222–4226 CrossRef CAS PubMed.
- A. Hruzewicz-Kołodziejczyk, V. P. Ting, N. Bimbo and T. J. Mays, Int. J. Hydrogen Energy, 2012, 37, 2728–2736 CrossRef.
- G. M. Kahilu, S. Bada and J. Mulopo, Waste Dispos. Sustain. Energy., 2023, 5, 125–149 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |