
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn integrated microfluidic platform for on-site qPCR analysis: food allergen detection from sample to result†
Anne-Gaëlle
Bourdat
 *,
Remco
den Dulk
,
Bastien
Serrano
,
François
Boizot
,
Gervais
Clarebout
,
Xavier
Mermet
,
Raymond
Charles
,
Jean
Porcherot
,
Armelle
Keiser
,
Manuel
Alessio
,
Patricia
Laurent
,
Nicolas
Sarrut
and
Myriam
Cubizolles
*,
Remco
den Dulk
,
Bastien
Serrano
,
François
Boizot
,
Gervais
Clarebout
,
Xavier
Mermet
,
Raymond
Charles
,
Jean
Porcherot
,
Armelle
Keiser
,
Manuel
Alessio
,
Patricia
Laurent
,
Nicolas
Sarrut
and
Myriam
Cubizolles
Univ. Grenoble Alpes, CEA Leti, F-38000 Grenoble, France. E-mail: anne-gaelle.bourdat@cea.fr
First published on 4th October 2024
Abstract
Improving food safety is crucial in the context of a “One Health” approach. To guarantee product quality and safety, the food industry, which has a very high turnover rate, needs short time-to-result analyses. Therefore, user-friendly systems at the point-of-need are necessary, presenting relevant analytical information and fulfilling the current regulations. To answer these challenges, a microfluidic platform integrating sample preparation and subsequent multiplex qPCR detection has been developed for on-site testing. The system consists of a fully automated instrument driving a microfluidic cartridge dedicated to the detection of multiple allergens in complex food matrices. The first part of the microfluidic cartridge contains pumps, reservoirs, valves and a filter to achieve DNA extraction, concentration and purification. Multiplex qPCR detection is carried out in the second part of the cartridge including a negative control chamber and five chambers for target analyte detection. The in-house developed instrument contains all functions to autonomously drive the microfluidic cartridge: pneumatic control for fluid actuation, thermal control for qPCR amplification and an optical system using three fluorescent wavelengths for multiplex detection of the target analytes and controls. We demonstrate the simultaneous detection of four different allergens – gluten, sesame, soy and hazelnut – from various complex food matrices. The turn-around-time from sample to result is close to two hours and controls in place validate the obtained results. For gluten, a direct comparison with ELISA shows that the regulatory threshold of 20 ppm is comfortably fulfilled. Moreover, all results are in agreement with external laboratory analyses performed in parallel on the same samples. Our findings confirm that the system can be used safely on-site without the risk of cross contamination between the various samples being analysed. In conclusion, our microfluidic platform offers a robust method for on-site allergen management.
Introduction
The WHO highlights that a “One Health” approach is critical to address health threats, in particular improving food safety and environmental monitoring. More specifically, new challenges to food safety will continue to emerge, largely due to (i) evolution in food diversity, production and supply, including the importation of foods on an unprecedented scale, (ii) food contamination due to evolution in the environment (e.g. allergens, bacteria, toxins, plants…), and (iii) evolution in consumer preferences and habits.1–3 In most cases, central lab testing remains the gold standard despite a strong urge to shorten the time-to-result. Raw material suppliers are requested to provide analysis certificates that establish clearly allergen safety in their products.4 Waiting one week for a central lab analysis for these suppliers results in storage expenses and requires a large place for these bulky materials. In addition, this storage increases the risk of allergen contamination between various batches of raw materials. Lastly, one-week analysis is not compatible with perishable goods. Therefore, the food industry, having a very high turnover rate to guarantee product quality and safety, would benefit tremendously from a short time-to-result. Indeed, successful examples of commercial point-of-need analytical systems exist for certain applications such as diagnosis of COVID-19,5–7 diagnostic systems (cobas® Liat® system (Roche Diagnostics) and BioFire® FilmArray® Panels (bioMérieux)). Nevertheless, some domains, such as the food industry, are still behind in this respect.Challenges for allergen detection involve the great variety of complex sample matrices and targets, the trace levels needed to be detected (concentration levels on the order of ppm, for example, the gluten regulatory threshold of 20 ppm), and the analysis of large sample volumes required for representativity.8 In addition, allergen contamination is sometimes limited to specific ‘hot spots’ rather than being distributed homogeneously throughout the whole sample, particularly when analysing whole grains for gluten.9 Therefore, to avoid false positive/negative results in allergen detection, a large sample amount (i.e. several grams or millilitres) is mandatory. To ensure that a representative test will be obtained, conventional methods use homogenization (i.e. thorough grinding to a powder) of a sufficient amount of the food matrix, typically around 5 g.10–13 Usually only 5% of this homogenized sample is actually used for the extraction of the target analytes.
Methods for detecting allergenic substances can be categorized into two types: protein-based and DNA-based. Immunoassays quantify allergenic proteins responsible for the immune reaction in sensitive people, whereas DNA-based techniques like PCR identify the genetic material of the allergenic food, serving as a surrogate for the allergen. DNA amplification techniques such as the polymerase chain reaction (PCR) are commonly used because of their speed, efficiency and simplicity.14 These amplification techniques are reliable, highly specific and sensitive and show an excellent limit of detection down to 1 mg kg−1 or 1 ppm.2,15–17 However, quantitative PCR (qPCR) requires a high degree of purification in order to detect and quantify fragmented and low-concentration DNA in highly processed foods.18 Furthermore, food matrices that have complex compositions19 may contain PCR inhibitors such as fat and polyphenols, which are present in high amounts for example in chocolate, ham and green vegetables.20–24 In addition, DNA presents an elevated stability upon thermal treatment, pH alteration or partial hydrolysis, processes that are frequently used in the food industry. It should be noted that food processing can increase or decrease or leave unaffected the allergenic properties of food proteins.10,25–27 Complex food matrices are more challenging for immunoassay methods than for biomolecular methods.28–30 Indeed, numerous studies have shown that different pre-processing methods affect ELISA results.
All those challenges need to be taken into account in microfluidic approaches to ensure efficient concentration and purification. Therefore combining on-site both rigorous sample preparation (SP) and subsequent analyte detection in fully automated systems is mandatory. In addition, considering that future analytical methods will require sensitivity, specificity, robustness, repeatability, quantification and a high dynamic range for any food matrix (e.g. fat, sterilized foods), orthogonal approaches may be required to ensure the safety and health of allergic individuals.
To meet the demand for point-of-need methods in the food industry, various microfluidic approaches are employed to detect food allergens. In our laboratory, a microfluidic technology based on pneumatically collapsible chambers was used for the development of an immunoassay for gluten detection exhibiting a dynamic range of 10–30 ppm with good sensitivity (2 ppm) and specificity.31 Passive microfluidics also demonstrated its usefulness: simple, fast and accurate electrochemical detection of one peanut allergen has been demonstrated using a microfluidic paper-origami based nano-aptasensor, integrating aptamers as bioreceptors.32 Also for peanut allergens, Ma et al. developed a microfluidic droplet device for amplifying peanut DNA.33 Multiplex allergen detection was also addressed, for example using circular fluorescence probe-mediated isothermal amplification (CFPA) in a microfluidic device to detect food allergen genes,34 or using colorimetric LAMP in a microfluidic device for detecting the allergen genes of peanut, sesame, and soybeans.35 Recently, Natsuhara et al. designed a microfluidic device with passive stop valves to achieve sequential liquid dispensing into an array of 10 microchambers. Using a colorimetric LAMP assay, they demonstrated simultaneous detection of multiple food allergens including controls without cross contamination between the microchambers.36
To provide integrated point of need devices, microfluidic approaches have to ensure efficient concentration and purification of either the protein allergens or the corresponding DNA. Indeed, sample preparation is a prerequisite of microfluidic devices exhibiting sample-to-result capabilities.9 Nevertheless, very few examples of integrated microfluidic allergen sample preparation are described in the literature. Magnetic beads can be used prior to use of the microfluidic device for efficient extraction of protein allergens.37 DNA adsorption, purification and extraction from a complex food matrix (plants seeds) was demonstrated prior to in-tube LAMP detection for peanut and soybeans, using a paper-based microfluidic chip.38 Although great achievements in detection have been made, the complete integration of multiple functions that combine both sample preparation and subsequent analyte detection in fully automated systems for allergen management by food companies is still required.9,39
In this paper, we demonstrate a generic fully automated microfluidic system suitable for on-site testing from sample to result. The system includes a sample preparation module for DNA sample extraction, concentration and purification from complex food matrices that is combined with a multiplex detection module based on qPCR. An over-representation step of low-concentred target allergens in heterogeneous food matrices is performed40 to be able to analyse a much larger sample volume than conventional methods and thus ensure good sample representativity. DNA extraction, concentration and purification is achieved in one part of the microfluidic cartridge, using integrated microfluidic pumps, valves, reservoirs, and a filter membrane. The second part of the microfluidic cartridge is dedicated to multiplex qPCR detection, including a negative control chamber and five chambers for target analyte detection. As a proof of concept, we focused our work on the detection of gluten, sesame, soy and hazelnut. We show that our on-field deployable system can easily deal with different complex food matrices (namely flour, spread, seed mix, flavouring mix or canned food) for effective detection of all allergens. All of those matrices may naturally contain allergens and/or were spiked with a known amount of allergenic food. In a fully automated way, DNA preparation and multiplex qPCR detection for gluten, soy, sesame and hazelnut are successfully demonstrated within a single microfluidic cartridge. The developed method proves to be robust since the controls in place validate the results and the outcomes are in agreement with external laboratory analyses (using gold reference methods) performed in parallel on the same samples.
Experimental
Microfluidic system
The instrument and cartridges are developed and realised in-house as described in detail below. The microfluidic system, as shown in Fig. 1, consists of an autonomous instrument (Fig. 1A–C) that drives the microfluidic cartridge operations (Fig. 1D–G) in order to automatically realise all of the protocol steps. A generic fluid pneumatic actuation pilots the microfluidic cartridge.41 In the developed instrument, the use of three fluorescence signals allows the inclusion of a microfluidic filling control and an internal positive control in each of the six qPCR chambers. Data analysis of the qPCR fluorescence signals is done using three different fluorescence wavelengths to obtain valid quantification cycle (Cq) values and avoid false positives or false negatives (Fig. 2) as described in the corresponding section below. | ||
| Fig. 1 Instrument and microfluidic cartridge. (A) In-house developed instrument (25 × 25 × 50 cm3) containing all functions to drive the microfluidic cartridge. (B) Detailed view of the microfluidic cartridge, fully equipped with all reservoirs, placed on the instrument. (C) Schematic representation of the instrument showing the main technical modules: pneumatic control for operation of valves and pumps, thermal control driving a Peltier element for qPCR cycling, optics for fluorescence imaging and a clamping area for pneumatic and thermal interfacing between cartridge and instrument. (D) Photographs of microfluidic cartridges containing either only the sample preparation circuit (top left) or only the qPCR circuit (bottom right). (E) Photograph of the complete microfluidic cartridge. (F) Schematic representation of the complete microfluidic cartridge showing the fluidic path (blue lines), the valves (blue and black circles), Luer ports (L1–L5), pumps (P1–P2), stretchable reservoirs (R1–R2), filters (F1–F2), qPCR chambers (C1–C6) and pneumatic control lines (uncolored). (G) Exploded view of the cartridge assembly showing the 5 layers: 1) top layer with Luer ports for reservoirs, 2) fluidic layer, 3) pneumatic layer with filters and elastic valve membranes, 4) bottom layer with the qPCR chambers, and 5) adhesive film sealing the cartridge. | ||
 | ||
| Fig. 2 Interpretation of the fluorescence signals for qPCR analysis. The main fluorescence signal representing an allergen qPCR result (C) is interpreted with the aid of two supplementary fluorescence signals: a fluidic control (A) and an internal qPCR control (B). For each qPCR cycle those three fluorescence signals are obtained, of which the supplementary signals (ROX, green fluorescence and Cy5, red fluorescence) are used to validate or invalidate or correct the main signal (FAM, blue fluorescence). The curves in the graphs are schematic examples of cases that can be encountered with best case examples in green, acceptable cases in orange and examples of cases that invalidate the qPCR assay in red. (A) The fluidic control represents a constant number of fluorescent molecules present in the qPCR master mix, which would normally stay constant at an expected level (A1). A sudden (A2) or gradual (A3) decrease in the signal typically indicates bubble formation or leakage of liquid from the chamber. The data of such a change in the fluidic control signal can be used to correct the other fluorescence signals. A very low level (or complete absence) of fluorescence signal (A4) typically indicates the qPCR chamber is not filled correctly, which invalidates the qPCR assay. (B) The internal qPCR control represents an independent qPCR reaction that takes place regardless of the allergen content of the sample. It would normally show an exponential curve with an expected Cq value between 32 and 38 (B1). A slightly shifted curve (B2) typically indicates a low and therefore acceptable inhibition of the qPCR reaction. The amount of shift could be used to correct the main signal, but when the amount of inhibition is too large (B3) the qPCR assay is invalidated. The complete absence of amplification results in a flat curve, which invalidates the qPCR assay as well. (C) The allergen qPCR signal represents the actual qPCR assay to determine the quantity of targeted allergen in the sample. All curves represent a reliable result when both the fluidic control and the internal qPCR control are valid. The first curve (C1) represents a higher allergen content compared to the second curve (C2), which represents a lower allergen content. The third curve (C3) represents a non-detectable allergen content, because the Cq value is larger than 42. The fourth curve (C4) shows no sign of amplification and therefore represents a non-detectable allergen content as well. | ||
Microfluidic cartridge
The microfluidic cartridge (84 × 54 × 6 mm3) is International Organization for Standardization (ISO) 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 916: 2022 compliant.42 It is composed of four layers of cyclic olefin copolymer (COC) (Fig. 1G) that are structured by high-precision micro milling (Datron M7HP). The top layer (layer 1) contains openings for stretchable reservoirs and Luer ports into which cut off syringes can be plugged to be used as fluid reservoirs. The fluidic layer (layer 2) contains the fluidic channels that interconnect the inlet/outlet ports, the various valves, pumps and filters and the qPCR chambers that are located in the bottom layer. The pneumatic layer (layer 3) contains the pneumatic channels to actuate valves and pumps. The elastic membranes for the valves and pumps, as well as the filter membranes are sandwiched between the fluidic and pneumatic layer at designated locations. The valves are actuated by deforming their elastic membranes by compressed air or vacuum to respectively close or open the valve. The pumps are actuated in the same way, where a negative pressure fills the pump volume with liquid and a positive pressure empties the pump volume. The stretchable reservoirs R1 and R2 (Fig. 1F) are also covered with an elastic membrane, which has several advantages. First, it allows for large variations in the volume. Second, the reservoir can be emptied completely without leaving much residue. Last, and most importantly, the reservoir is closed, which prevents contamination of the environment of the system. The qPCR chambers are located in the bottom layer (layer 4) to provide good thermal contact with the heating element of the instrument. The four COC layers are bonded by thermal bonding and the bottom layer is closed off with an adhesive film (layer 5, MicroAmpTM, Applied Biosystems) to hermetically seal the qPCR chambers. Primers and probes for each allergen target (FAM fluorescence) and for the internal control (Cy5 fluorescence) as well as DNA for the internal control are dried overnight under vacuum in the qPCR chambers before sealing the cartridge with the adhesive film. Cartridges are stored at room temperature in the dark prior to use. In order to test the sample preparation circuit and the DNA amplification circuit separately from each other, two preliminary cartridges were developed (Fig. 1D) before fabricating the complete cartridge (Fig. 1E).
916: 2022 compliant.42 It is composed of four layers of cyclic olefin copolymer (COC) (Fig. 1G) that are structured by high-precision micro milling (Datron M7HP). The top layer (layer 1) contains openings for stretchable reservoirs and Luer ports into which cut off syringes can be plugged to be used as fluid reservoirs. The fluidic layer (layer 2) contains the fluidic channels that interconnect the inlet/outlet ports, the various valves, pumps and filters and the qPCR chambers that are located in the bottom layer. The pneumatic layer (layer 3) contains the pneumatic channels to actuate valves and pumps. The elastic membranes for the valves and pumps, as well as the filter membranes are sandwiched between the fluidic and pneumatic layer at designated locations. The valves are actuated by deforming their elastic membranes by compressed air or vacuum to respectively close or open the valve. The pumps are actuated in the same way, where a negative pressure fills the pump volume with liquid and a positive pressure empties the pump volume. The stretchable reservoirs R1 and R2 (Fig. 1F) are also covered with an elastic membrane, which has several advantages. First, it allows for large variations in the volume. Second, the reservoir can be emptied completely without leaving much residue. Last, and most importantly, the reservoir is closed, which prevents contamination of the environment of the system. The qPCR chambers are located in the bottom layer (layer 4) to provide good thermal contact with the heating element of the instrument. The four COC layers are bonded by thermal bonding and the bottom layer is closed off with an adhesive film (layer 5, MicroAmpTM, Applied Biosystems) to hermetically seal the qPCR chambers. Primers and probes for each allergen target (FAM fluorescence) and for the internal control (Cy5 fluorescence) as well as DNA for the internal control are dried overnight under vacuum in the qPCR chambers before sealing the cartridge with the adhesive film. Cartridges are stored at room temperature in the dark prior to use. In order to test the sample preparation circuit and the DNA amplification circuit separately from each other, two preliminary cartridges were developed (Fig. 1D) before fabricating the complete cartridge (Fig. 1E).
Instrument hardware and software
An autonomous instrument of about 25 × 25 × 50 cm3 that contains hardware components for fluid driving, thermal control and optical detection drives the microfluidic cartridge. Fig. 1 shows a photo of the instrument and a schematic drawing indicating the main technical modules. Fluid driving is based on pneumatic actuation of elastic membranes forming either valves or pumps. The instrument is therefore equipped with 3 pressure controllers (SMC, ITV series), 32 solenoid valves (SMC, VV061 series) and a dedicated homemade electronics board. The cartridge holder forms a pneumatic interface between the 32 pneumatic control lines of the instrument and the pneumatic channels on the cartridge. By placing the cartridge onto the instrument, the 32 pneumatic connections are made instantly in a single step.41 Thermal control is realised through a Peltier heating element in direct contact with the bottom of the cartridge, covering a zone of 20 × 20 mm2 right under the qPCR chambers. A dedicated electronics board (TEC Microsystems, DX5100) ensures proportional–integrative–derivative (PID) control of the Peltier element using a Pt1000 temperature sensor that is integrated in the heating area. Optical detection is based on fluorescence imaging. The instrument has a cover to make sure that the imaging takes place in the dark, to avoid the interference of ambient light on the fluorescence signals. A monochrome CMOS-type camera (IDS, UI-3060CP) takes images of the qPCR area. To multiplex the actual allergen qPCR detection with two supplementary controls, this system has been designed to detect the three fluorophores of interest, while remaining compact for on-site experiments. The fluorescence measurements were intended for the detection of the following probes (center wavelength/bandwidth): FAM excitation 472 nm/30 nm, emission 512 nm/25 nm (blue); ROX excitation 559 nm/34 nm, emission 600 nm/14 nm (green); and Cy5 excitation 632 nm/22 nm, emission 676 nm/29 nm (red). It is equipped with three LED sources (Thorlabs), each with a dominant wavelength centered on the fluorophore absorption peak. Each of these three sources is coupled to a high-core multimode optical fiber (Thorlabs), a collimator and a spectral filter to illuminate the total surface qPCR area with sufficient power density to excite the fluorophores. Fluorescence emissions are collected by an optical system comprising an achromatic doublet (Thorlabs), a filter wheel (Thorlabs) equipped with three spectral filters (Semrock) and a tube lens (Thorlabs) for imaging the area of the 6 qPCR chambers on the camera. The spectral filters used to collect fluorescence emissions have been specifically chosen so that their spectral bands recover only the signal emitted by the associated fluorophore. This is particularly the case for ROX and Cy5, for which absorption curves overlap, and therefore the bandwidth of the ROX emission filter is chosen short enough to avoid collecting the Cy5 emission. This choice of spectral filter pairs (excitation/emission) eliminates the need for digital processing to correct fluorescence signal levels. An LED driver (Mightex) controls the intensity of each of the LEDs individually and thus enables together with the filter wheel to switch wavelength as desired. A USB hub integrated in the instrument connects all elements by a single cable to a PC from which everything can be controlled via an in-house developed software application (uFlu factory). Using Python scripts, a fully autonomous functioning of the complete protocol is achieved.Protocol steps
The procedure for extraction and purification of the allergen DNA was adapted from state of the art protocols.40,43,44 The first step of the protocol consists of the extraction of DNA from 10 g of food matrix using 80 mL of lysis buffer. Because of this large volume, which is needed to achieve a high representativity of the sample, this step takes place outside the microfluidic cartridge. A fraction of the extract is transferred into reservoir L1 of the microfluidic cartridge (Fig. 1F) through a Whatman GD/X syringe filter to avoid clogging due to large particles. Allergen DNA was extracted from various food matrices such as flour, spread, seed mix, flavouring mix and canned food (French cassoulet). In addition, surface sampling (wet wipe) was used to analyse the presence of allergens on a surface, following the same extraction protocol with lysis buffer. Allergen free matrices were spiked with a known concentration of wheat flour, hazelnut, sesame or soy to evaluate the analytical performance (e.g. limit of detection, accordance with regulations). Some samples were spiked with two or three of those allergens to perform multiplex analyses. Some matrices naturally contain various amounts of these allergens.The remainder of the sample preparation procedure consists of the purification of the extracted DNA and all steps are executed automatically by the microfluidic system following the instructions of a predefined Python script. Fig. 1F shows a map of the fluidic circuit (blue lines) with all elements labelled for reference. After placing the cartridge on the instrument the script closes all valves to allow the user to manually fill the fluid reservoirs attached to the Luer ports as follows. L1 is filled with the filtered sample of crude extracted DNA. A minimum of about 150 μL is required, but the system automatically aliquots the exact volume of 105 μL needed for the protocol. L2 is filled with 250 μL of ethanol (96%) and L3 with 250 μL of ethanol (70%). L4 is filled with 100 μL of Luna qPCR master mix (New England Biolabs Luna® Universal Probe qPCR Master Mix M3004E), which contains a passive reference dye (ROX) and Hot Start Taq DNA Polymerase. L5 is left empty and serves as a waste reservoir. First, chamber C1 is filled with qPCR mix from L4 by applying a negative pressure above the hydrophobic filter F2. After its upstream and downstream valves have been closed, an image is taken to verify if the chamber has been filled correctly using the fluorescent signal of the passive reference dye (ROX). Then, 105 μL of sample is pumped from L1 to reservoir R1 by one single stroke of pump P1. Subsequently, 105 μL of ethanol (96%) is pumped from L2 to reservoir R1 by one single stroke of pump P1 so that the sample is mixed with ethanol in an exact 1-to-1 ratio. In order to improve mixing, and thus promote the precipitation of DNA, the liquid is pumped in back-and-forth movements between R1 and P1 a couple of times before an incubation of 3 minutes takes place in reservoir R1. After incubation, the liquid containing precipitated DNA is pumped from R1 over the filter F1 towards the waste reservoir L5 by several strokes of pump P1. Then, the complete content of 250 μL of ethanol (70%) is pumped from L3 over the filter F1 towards the waste reservoir L5 by several strokes of pump P1. Initially, part of the volume is passed via R1 to recover any leftover traces of the DNA sample, then the remainder of the volume is passed directly over the filter and finally air from the empty L3 reservoir is pumped over the filter to eliminate residual ethanol acting as a PCR inhibitor. The precipitated DNA is now retained on the filter while all the excess liquid has been removed towards the waste reservoir L5. The qPCR master mix is then pumped from L4 over the filter F1 to reservoir R2 by several strokes of pump P2. The pellet of precipitated DNA on the filter is dissolved by the qPCR master mix and, in order to improve this dissolution, the liquid is pumped in back-and-forth movements between R2 and P2 a couple of times before leaving the qPCR master mix with the purified DNA in reservoir R2. Then the qPCR chambers C2–C6 are filled with the liquid in R2 by applying a negative pressure on the hydrophobic filters F2. After closing all of the upstream and downstream valves, an image is taken to verify if all the chambers have been filled correctly using the fluorescent signal of the passive reference dye (ROX). Thermal cycling is then started with an activation phase of 3 min at 95 °C for the Hot Start polymerase followed by 50 cycles of the following profile: 30 sec at 95 °C and 40 sec at 60 °C at which temperature the three fluorescence images (FAM, Cy5, ROX) are recorded sequentially.
qPCR data analysis
In order to make the qPCR detection robust against various artefacts, the fluorescent detection uses three different fluorescent dyes that are all present simultaneously in each chamber. The first supplementary fluorescence signal represents the fluidic control. It is an ROX dye (green) that is present in the qPCR master mix as a passive reference at a known quantity, which would normally stay constant at an expected level. Any fluidic artefact such as incomplete filling, bubble formation or leakage would affect this baseline signal. The second supplementary fluorescence signal represents the internal qPCR control, a TaqMan probe labelled with a Cy5 dye (red) that is specific to a chosen DNA sequence other than the targeted allergens. Because a known quantity of this chosen DNA sequence was dried in each qPCR chamber, at least one amplification will always take place in each chamber. It is thus a positive control from which the amount of inhibition, related to the purity of the DNA sample, can be estimated. The main fluorescence signal representing the allergen qPCR result is a TaqMan probe labelled with FAM dye (blue). As illustrated in Fig. 2, showing different cases of potential artefacts, the allergen qPCR result is only interpreted when both the fluidic control and internal qPCR control are valid. The fluorescence images, taken at each of the 50 cycles of the qPCR assay, are treated as follows. Six regions of interest (ROI) defined as the exact locations of the six chambers are used to measure the average fluorescence intensity for each chamber. These values are then plotted against cycle number to obtain an amplification curve from which the Cq values are determined by finding the maximum value of the second derivative. Signal conditioning, such as the moving average, and some additional criteria, such as a minimum fluorescence level or a minimum cycle number, were used to be robust against imperfections of the fluorescence signal. Finally, the results were displayed for each chamber as “valid” or “invalid” based on the controls and for each allergen as “detected” or “not detectable” based on the Cq values.Results and discussion
Performance of the sample preparation assay in the microfluidic cartridge
The first part of our work was focused on the integration of a sample preparation assay into the microfluidic cartridge, leading to a cartridge containing only the sample preparation circuit (Fig. 1D). Three different food matrices (seed mix, millet flour and sunflower spread) were spiked with 200 ppm of wheat flour in order to obtain contaminated samples with 20 ppm of gluten. In addition, a non-spiked sunflower spread was used as a gluten-free matrix. The 10 factor correspondence between the ppm of wheat flour and ppm of gluten was determined by ELISA (RIDASCREEN® Gliadin kit, R-Biopharm), i.e. 10 ppm of wheat flour correspond to 1 ppm of gluten. Following a short millifluidic sample pretreatment, the sample preparation circuit in the microfluidic cartridge effectuates the following major steps: precipitation, purification, concentration and elution of the DNA leading to an automated sample preparation procedure of 20 minutes. Each food matrix was processed using a representative number of replicates to assess the validity of these sample preparation steps (n > 10) (Fig. 3A). Then, qPCR analysis of the microfluidic prepared DNA was performed in-tube using a commercial device (QS5, Thermo Fischer) using three fluorescent wavelengths (ROX, Cy5 and FAM). Internal qPCR controls (IC) show a low and therefore acceptable level of inhibition with Cq values comprised between 34.6 and 36.5, thus validating the efficacy of the purification level of our sample preparation method. For gluten detection, Cq values were found between 29.4 and 30.3 with a variation of below 5%. The gluten-free sample presents a very high Cq value of 46.8, thus confirming the absence of gluten in this food matrix. To complete these results, we investigated a large range of gluten contamination varying from 10 to 5000 ppm by spiking gluten-free sunflower spread with various amounts of wheat flour (Fig. 3B). Using the same approach as described above, the results of the 20 samples that were analysed show that the Cq values varied from 32.4 at 10 ppm down to 24.3 at 5000 ppm, following a reasonable linear regression (R2 = 0.93). Moreover, for all of the spiked samples the presence of gluten was confirmed even at low concentrations. The International Food Standard (IFS), namely the Codex Alimentarius,4 defines the 20 ppm gluten threshold as a limit: below 20 ppm of gluten a food is considered to be “gluten-free”, and between 20 and 100 ppm of gluten a food is classified to have “low-gluten content”. Thus, with a limit of detection of 10 ppm of wheat flour - corresponding to 1 ppm gluten – our sample preparation method indeed meets the sensitivity requirement of the IFS.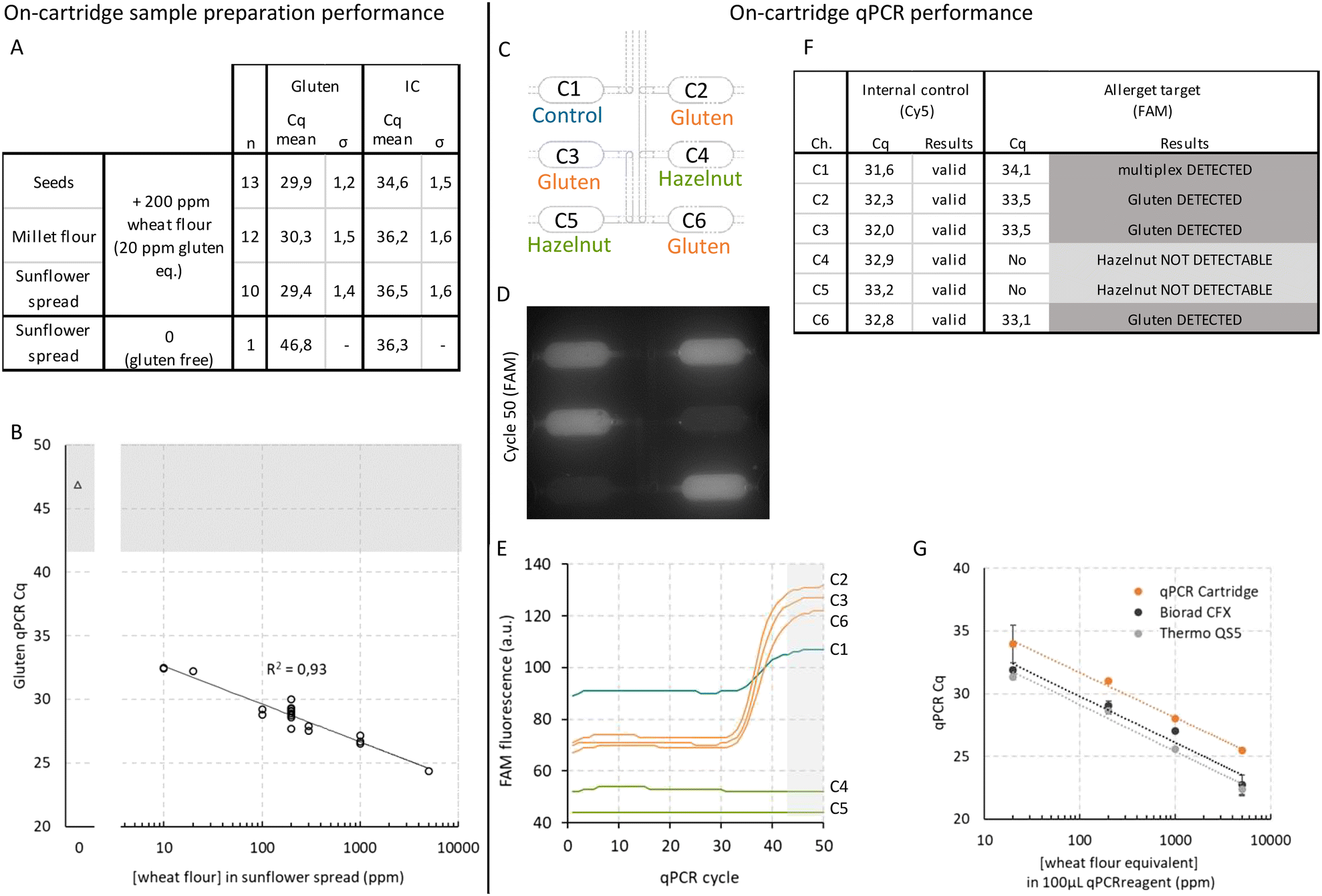 | ||
| Fig. 3 Results demonstrating the functionality of the sample preparation cartridge (A and B) and the qPCR cartridge (C–G) separately (see Fig. 1D). (A) On-cartridge sample preparation followed by in-tube qPCR for gluten detection in seeds, flour or spread food matrices (only sunflower spread is gluten free, all other matrices are low gluten <20 ppm). (B) On-cartridge sample preparation and in-tube qPCR for gluten detection in sunflower spread samples spiked with 10 to 5000 ppm wheat flour (10 ppm n = 2, 20 ppm n = 1, 100 ppm n = 2, 200 ppm n = 9, 300 ppm n = 2, 1000 ppm n = 3, 5000 ppm n = 1). (C) Schematic representation of qPCR chambers each with specific allergen target. Chamber 1 contains multiple allergen targets for qPCR control. (D) Fluorescence image (FAM) of the corresponding qPCR chambers after 50 cycles. (E) Corresponding qPCR amplification curves. (F) Corresponding measured Cq values and results for gluten and hazelnut detection using DNA purified from wheat (equivalent to 2 ppm gluten) as well as Cq values of internal control validating the results. (G) Comparison of qPCR Cq values for on-cartridge and commercial instruments using DNA purified from wheat (CFX and QS5: 20 ppm n = 3, 200 ppm n = 2, 1000 ppm n = 1, 5000 ppm n = 4. qPCR cartridges: 20 ppm n = 9, 200 ppm n = 3, 1000 ppm n = 3, 5000 ppm n = 3). | ||
Performance of the qPCR assay in the microfluidic cartridge
The second part of our work was focused on the integration of the qPCR assay into the microfluidic cartridge, leading to a cartridge containing only the qPCR circuit (Fig. 1D). Prior to testing the amplification in the cartridge, the thermal profile of the system was assessed and the results showed that the Peltier module controls the temperature in a satisfactory manner, as measured by the Pt1000 (Fig. S1†). Primers and probes were dried inside the qPCR chambers and different targets were distributed over the chambers as follows (Fig. 3C). Chambers C2, C3 and C6 contain dried reagents for gluten detection, chambers C4 and C5 contain dried reagents for hazelnut detection, while chamber C1 contains dried reagents for multiple allergen targets. Furthermore, each chamber includes control DNA for the internal control test as detailed in Fig. 2B. To evaluate the performance of the qPCR assay in the microfluidic cartridge, we used DNA purified from wheat using standard laboratory preparation protocols. We chose a rather low concentration of DNA, corresponding to an equivalent of 2 ppm of gluten, to ensure sufficient sensitivity. All chambers were filled with PCR reagents containing wheat DNA, even chamber C1, mimicking the case of a potential DNA contamination of the qPCR reagents to evaluate the function of this chamber.Fig. 3D shows the raw fluorescence image (FAM) after 50 cycles for all qPCR chambers before any signal conditioning. One can easily observe that chambers C1, C2, C3 and C6, containing the gluten primers and probes, show fluorescence, whereas chambers C4 and C5, containing only hazelnut primers and probes, stay dark at the end of the amplification reaction. Fig. 3E represents the corresponding qPCR amplification curves resulting from the image analysis as described earlier. As expected, the amplification curve of chamber C1 presents an increase in fluorescence signal after 30 cycles in the presence of the target DNA. This validates undoubtedly that this chamber containing multiple allergen targets can be used as a negative control. In further experiments, chamber C1, which is completely isolated from the other chambers, was filled with qPCR mix only, so that it could be used as a control for the absence of DNA contamination in the qPCR mix. The curves for chambers C2, C3 and C6 also clearly show that amplification occurs as expected in the presence of the targeted DNA. At the same time, the fluorescence of chambers C4 and C5 (hazelnut) does not vary over time, as expected, indicating the absence of non-specific amplification despite the presence of non-target primers and probes. It is also important to note that no migration of fluorescence is detected between chambers whether amplification has occurred or not. Indeed, each chamber behaves as an independent reactor despite the fact that these five chambers belong to the same fluidic circuit, a point which is of utmost importance for simultaneous detection of multiple targets in the same sample.36 Moreover, the internal qPCR control, as represented by the red (Cy5) fluorescent signal, shows correct amplification regardless of the presence or absence of a target amplification in each chamber, which thus validates the obtained results (Fig. 3F). Data analysis of the FAM fluorescence signals leads to Cq values that correspond to the following results as expected: the positive control is validated since a multiplex is detected in chamber C1, gluten is detected in chambers C2, C3 and C6, and hazelnut is not detectable in chambers C4, C5. Cq values for gluten detection ranged from 33.1 to 34.1 validating the detection of an equivalent of around 2 ppm of gluten (20 ppm of wheat flour). To further assess the performance of our microfluidic qPCR assay, we compared our microfluidic results to the ones obtained in-tube using two different commercial instruments (CFX, Biorad and QS5, Thermo Fisher). Fig. 3G shows the Cq values obtained with various concentrations of wheat purified DNA with the three qPCR systems. In addition, data shown on Fig. 3G demonstrated similar slopes between on-chip amplification and commercial systems, validating that PCR efficacy is the same: this indicates that the temperature is identical on-chip and in tube. Compared to the qPCR assays in commercial real-time qPCR instruments, one can conclude that our results obtained from our microfluidic qPCR assay are very similar in terms of dynamic range and sensitivity, and also in their duration of about 1 h 45 min. However, our microfluidic qPCR assay has the advantage of being automatically coupled with sample preparation. Compared to previously published more rapid (60 min) isothermal amplification methods described for allergen detection,34–36 our device offers the advantage of an internal control in each reaction chamber amplified simultaneously with the target allergen. In addition, our integrated and automated system demonstrates that qPCR can be achieved without the need of highly trained personnel and laboratory equipment.
Gluten detection using the complete microfluidic cartridge
After successfully implementing the sample preparation assay and qPCR assay independently in two microfluidic cartridges as described above, we combined both protocols in one single microfluidic cartridge (Fig. 1E), resulting in a full protocol of 2 hours 10 minutes from sample-to-result without any user intervention. This 2 h 10 min time-to-results protocol is similar to other sample preparation and biomolecular amplification microfluidic studies on food allergens,35 with the great advantage of presenting a fully integrated protocol directly from sample to results. In addition, commercial sample-to-answer systems used in central laboratories45 present comparable turnaround times, with manual sample preparation steps of one hour followed by PCR lasting 1 h 30 min. Four different food matrices (sunflower spread, millet flour and canned food) were prepared with various gluten contents and each sample was tested in a complete microfluidic cartridge. Following a short millifluidic sample pretreatment, the sample preparation circuit of the cartridge allowed for precipitation, purification, concentration and elution of the DNA, which was then amplified in the qPCR microchambers by thermal cycling. Fig. 4A shows the resulting amplification curves representing the fluorescence signal with baseline subtraction of chamber 2 (containing the gluten target) for all four cartridges. The curve corresponding to sample 1 (gluten-free sunflower spread) shows a slight increase in fluorescence at around 45 cycles. Since this weak increase is situated above the Cq value of 42 (see Fig. 2C), the gluten content of sample 1 should be considered as not detectable. For the three other samples, much lower Cq values were found, which indicates that all of those three samples should be considered as gluten positive. To further assess these results, we made a direct comparison with a recently developed microfluidic architecture to perform ELISA assays31 by analysing the same four samples on both microfluidic systems. As presented in Fig. 4B, the results of both systems are found to be in agreement, except for sample 2. In this particular case of very low gluten content (0.4 ppm of gluten), the qPCR resulted in “detected”, whereas the ELISA reported “not detectable”. This is, however, in agreement with the fact that the limit of detection of the microfluidic ELISA is at 2 ppm and confirms that qPCR is a more sensitive method than ELISA.46,47 For the low gluten content sample (millet flour) and the high gluten content sample (cassoulet) the results are consistent with both methods reporting “detected” as expected. To provide an additional independent comparison to these results obtained with novel microfluidic systems, we submitted the same four samples to an external laboratory for routine analysis by both qPCR and ELISA. The reported results from the external laboratory were received after one week and are identical to the results obtained with the microfluidic systems in only 2 h 10 min. This illustrates the added value of our microfluidic approach. | ||
| Fig. 4 Gluten detection using complete microfluidic cartridges with four different gluten content samples. (A) qPCR amplification curves obtained with complete microfluidic cartridges. FAM fluorescence measured in chamber 2 (baseline subtracted) inside four different cartridges. (B) Comparison of in-house and external laboratory analysis for both qPCR and ELISA. In-house ELISA results are obtained using the microfluidic platform and cartridges previously described.45 | ||
The comparison of qPCR and ELISA in the context of food allergen detection is not an obvious one, since both methods target different macromolecules. Allergic reactions in sensitive patients are due to the presence of proteins that trigger an immune response, and a non-exhaustive list of proteins is known for this. For example, currently twelve different proteins are known for hazelnut allergenicity of which six were identified in 2003, three in 2008, one in 2013, one in 2019, and one in 2022.48 As a consequence, immunoassay tests need to evolve continuously to take into account new discoveries. It should be noticed that the FDA uses two different types of ELISA kits to test food samples before confirming any test result.49 On the contrary, DNA does not trigger any immune response but reflects the presence of a particular food (e.g. hazelnut, soy, sesame). It is a more stable molecule than proteins and therefore rarely degrades to induce failing of a qPCR test. Therefore, it appears valuable to use DNA analysis to complement protein analysis, as mentioned in other studies.28,47,50,51
When comparing the analytical performance of qPCR and ELISA, we show that our microfluidic qPCR is more sensitive than our microfluidic ELISA, with gluten detected in samples containing as little as 0.4 ppm of gluten (4 ppm wheat flour), whereas the ELISA limit of detection is 2 ppm.31 Furthermore, ELISA is a quantitative method presenting a fine resolution allowing one to distinguish between 10, 20 and 30 ppm of gluten, but over a limited dynamic range (2–60 ppm).31 If a sample is out of this dynamic range (high concentration), it is considered as positive, but it has to be diluted and analysed again to obtain quantification. In comparison, qPCR presents an extended dynamic range from 0.4 ppm up to 500 ppm of gluten without any dilution of the sample. However, the results are qualitative (“detected” or “not detectable”) even if a clear correlation can be observed between the concentration of the allergen and the measured Cq value.
Multiplex allergen detection using the complete microfluidic cartridge
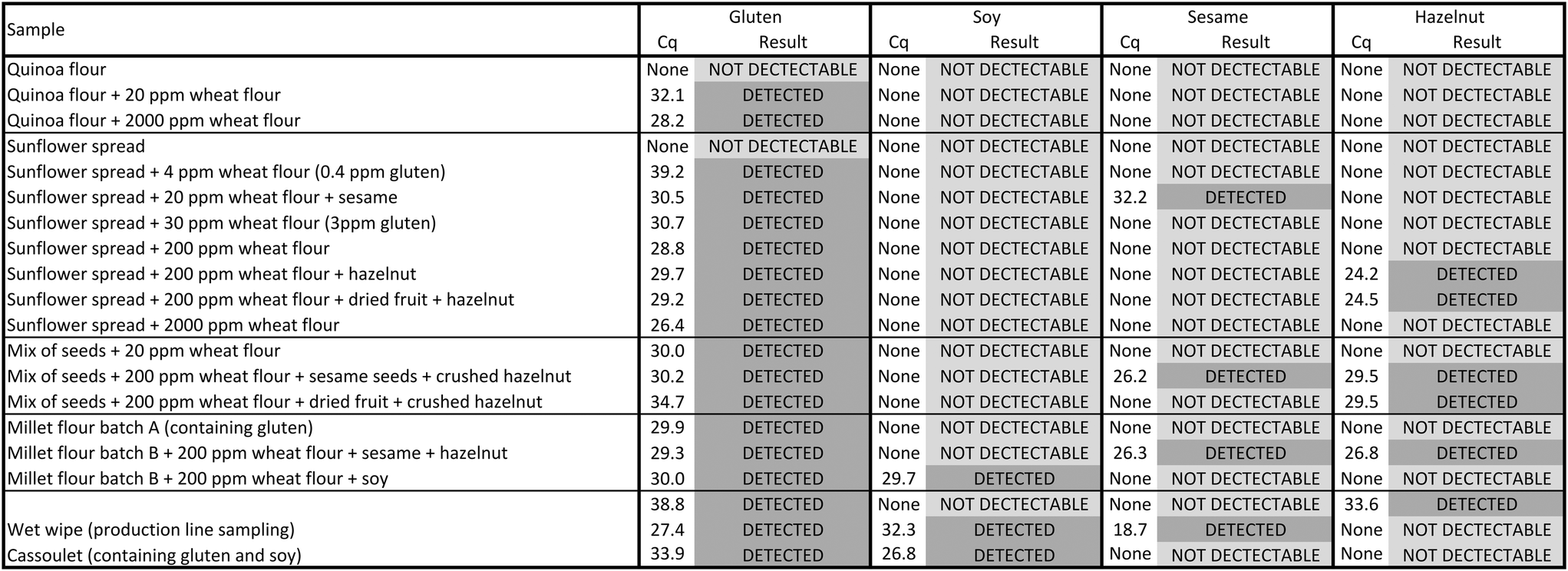
We focused the last part of our work on the detection of multiple allergens simultaneously from the same sample. Thanks to the different qPCR microfluidic chambers, we were able to target four different allergens simultaneously: gluten, soy, sesame and hazelnut. We decided to use a wide range of food matrices (flour, spread, seed mix, flavouring mix, canned food - French cassoulet) and included surface sampling (wet wipe) to address the cleaning process of a food production line. We used raw samples and also the same samples spiked with the various targeted allergens for a total of 20 processed food samples from industrial production lines. Table 1 summarises all the obtained outcomes with their Cq values and corresponding result.
|
Gluten, soy, sesame and hazelnut are not detectable in raw quinoa flour nor in raw sunflower spread. Subsequently, these samples were spiked with various amounts of allergen. As expected, quinoa flour spiked with wheat flour leads to gluten detection with Cq values of 32.1 and 28.2 for 20 ppm and 2000 ppm, respectively. No other allergens were detected. Sunflower spread was spiked either with only wheat flour from 2000 ppm down to 4 ppm, or with both wheat flour and a high amount of other allergen (sesame seeds or crushed hazelnut) to evaluate the impact on gluten detection. Gluten is detected in all the samples spiked with only wheat flour, even at very low concentrations, such as 4 ppm (corresponding to 0.4 ppm gluten), with a Cq value of 39.2. Moreover, the presence of allergens other than gluten in huge amounts in those samples does not alter the gluten detection. Gluten is unambiguously detected in spiked wheat flour samples containing sesame seeds or crushed hazelnut. Furthermore, sesame and hazelnut are also correctly detected in those samples. Moreover, the addition of dried fruit,24 containing glucose susceptible to affect PCR amplification does not alter the gluten detection either in a sample containing 200 ppm wheat flour and crushed hazelnut. In addition, we can observe a clear trend between the Cq values and the amount of spiked wheat flour in sunflower spread. Finally, we investigated several potentially challenging samples, namely a flavouring mix (containing Provence herbs, fried onions, dried tomatoes, parsley, red paprika and sunflower seeds), a canned food matrix (highly processed French cassoulet) and a surface sampling wet wipe (used on a contaminated surface). With the controls in place validating the outcome, this demonstrates that our platform is robust when working with those challenging samples. The results show that the flavouring mix presents gluten and hazelnut. Logically, French cassoulet contains gluten and soy, as is mentioned on its label. Finally, the wet wipe from a production line sampling reveals the presence of gluten, soy and sesame.
Numerous studies report matrix effects on allergen detection by PCR.24,52–55 Indeed, enzymatic reactions like PCR are sensitive to inhibitors that originate from the sample or from the extraction/purification process. For plant studies, PCR inhibition has been demonstrated to occur from contamination by polysaccharides and polyphenols, such as cellulose, starch, glucose, dextran sulfate, pectin, flavanol, gallic acid, resveratrol, and secoisolariciresinol. Many steps in the PCR process can be affected by those inhibitors, leading to reduced sensitivity or even false negatives. Numerous modes of inhibition include: (i) competitive binding of inhibitor and template to the Taq polymerase, preventing the enzymatic reaction; (ii) nucleic acid cross-linking/interaction that hampers nucleic acid isolation by changing the chemical properties; (iii) magnesium cofactor depletion leading to decreased enzyme activity.24 With the food samples we tested, we observe matrix effects. Nevertheless, gluten is always correctly detected, even in seed mix and millet flour spiked with wheat flour in the presence of sesame seeds, crushed hazelnut, dried fruit, spices or soy. For all of the tested food matrices the expected results were obtained on the four targeted allergens. Moreover, the controls in place (negative control, fluidic control and internal positive control) validate consistently the outcome of the full process from sample preparation to qPCR detection.
Based on all these results (Table 1), we can conclude that the developed method is robust: the controls in place validate the outcomes of all 20 samples and the results are in agreement with the actual known presence of allergens. All samples (n = 12) containing 200 ppm (or less than 200 ppm) of wheat flour, corresponding to the 20 ppm gluten regulation threshold criteria, are clearly detected in all food matrices. We also addressed the method specificity and we observed no inhibition nor cross reactivity for gluten detection in the presence of other allergens or in challenging samples containing fat, polyphenols, spices and/or aromatic herbs.20–23,56–58 In addition, during all experiments we observed systematically that the control chamber C1 never presents any amplification for gluten, sesame, soy or hazelnut. This confirms the absence of environmental cross contamination despite the numerous food samples that were manipulated in the same room, which is most probably thanks to the confined format of our microfluidic cartridge. This demonstrates that our integrated microfluidic system can be used safely on-site.
Point-of-need systems for both diagnosis and food safety require methods that need to be certified, because the associated results are crucial for patient health.59 Therefore, the full analytical chain, from the raw sample up to the result display on-site, is mandatory for certification. Currently, no point of need instruments address the food industry market. In that context, our system is able to respond to the following constraints: detect trace of targets at the ppm level, address a variety of samples and analyse multiple targets. In clinics, commercial biomolecular diagnosis point-of-need devices (cobas® Liat® system (Roche Diagnostics) and BioFire® FilmArray® Panels (bioMérieux)) deal with various human specimens i.e. swabs, blood cultures or soft/watery stools. Similarly, our system is able to prepare and analyse multiple agro-food samples such as raw materials, wet wipes or canned foods. Diagnosis requires simultaneous testing of multiple targets for comprehensive results and appropriate therapy management (BioFire® FilmArray® Panels (bioMérieux)). In the same spirit, our system currently performs the detection of four different allergens at the same time to initiate and ensure multiplexing implementation.
Conclusions
Our work focuses on the development of a microfluidic platform for the integration of complex biological protocols. In the perspective of future industrialization of the system, microfluidic cartridges are made of COC, a material that can be easily manufactured by injection molding for large volume production. Our microfluidic cartridge includes both sample preparation and multiplex biomolecular detection with internal controls and is driven by an autonomous instrument for easy-to-use on-site operation. The microfluidic architecture has been designed to perform DNA extraction, purification, concentration, elution in PCR reagents, and finally qPCR amplification that takes place in six microchambers containing specific dried primers and probes for each allergen and for controls. The instrument drives the cartridge in a fully automated way from sample to result without any intervention of the user, taking care of all fluidic handling, thermal cycling and fluorescent imaging to obtain results in close to two hours.We evaluated the system in the context of food safety for on-site allergen management. We illustrate that four allergens (i.e. gluten, sesame, hazelnut and soy) are unambiguously detected in various food matrices. The same microfluidic platform could be used to detect other allergens of interest, just by adapting the embedded primers and probes. Here, we pay particular attention to gluten and we fulfil comfortably the regulation threshold criteria of 20 ppm of gluten, even in challenging samples. In addition, we demonstrate on various food matrices that our PCR results are in agreement with (or even more sensitive than) another orthogonal analysis method, namely ELISA, as well as being in accordance with independent external laboratory analyses (PCR and ELISA). Considering that future analytical methods will require sensitivity, specificity, robustness, repeatability, quantification and high dynamic range, orthogonal methods are mandatory to ensure the safety and health of allergic individuals. Based on our previous study31 and taken together with these new results, our global approach could pave the way for on-site allergen management, with the development of a validated and accredited method,60 including both ELISA and PCR analysis as complementary approaches.
Data availability
Data for the article cannot be made available due to legal confidentiality requirements asked for by our industrial partner.Author contributions
(AGB) supervision, conceptualization, methodology, investigation, validation, data curation, formal analysis, visualization, project administration, writing – original draft, writing – review & editing, funding acquisition; (RDD) conceptualization, methodology, investigation, software, validation, data curation, formal analysis, visualization, writing – original draft, writing – review & editing; (BS) conceptualization, investigation, methodology, validation, data curation, formal analysis; (FB) investigation, methodology, resources; (GC) investigation, validation, data curation; (XM) investigation, resources; (RC) investigation, resources; (JP) resources, software, data curation; (AK) investigation, resources; (MA) investigation, resources; (PL) investigation, validation; (NS) conceptualization, investigation, methodology, validation, software; (MC) project administration, supervision, conceptualization, methodology, investigation, validation, data curation, formal analysis, visualization, writing – original draft, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Charles-Elie Goujon for the development of the electronic cards used in the instrument and also Florine Varin, Olivier Ducloux, Olivier Fuchs and Fabrice Navarro for grant writing and funding acquisition. This work was supported by the Groupe Dusaussoy, the “région Hauts-de-France” and Bpifrance. The authors also acknowledge CEA for internal funding.Notes and references
- N. T. Salihah, M. M. Hossain, H. Lubis and M. U. Ahmed, J. Food Sci. Tech. Mys., 2016, 53, 2196–2209 CrossRef CAS PubMed.
- V. Blaschke, A. Berten, H. Sprenger, J. Zagon and M. Winkel, J. Agric. Food Chem., 2023, 71, 12029–12042 CrossRef CAS PubMed.
- Centers for Disease Control and Prevention, Food Safety, https://www.cdc.gov/food-safety/index.html, (accessed June 20, 2024).
- Codex Alimentarius International Food Standard - CODEX STAN 118-1979, 2015.
- H. Zhu, H. Zhang, S. Ni, M. Korabečná, L. Yobas and P. Neuzil, TrAC, Trends Anal. Chem., 2020, 130, 115984 CrossRef CAS PubMed.
- X. Dong, L. Liu, Y. Tu, J. Zhang, G. Miao, L. Zhang, S. Ge, N. Xia, D. Yu and X. Qiu, TrAC, Trends Anal. Chem., 2021, 143, 116377 CrossRef CAS PubMed.
- S. Sachdeva, R. W. Davis and A. K. Saha, Front. Bioeng. Biotechnol., 2021, 8, 602659 CrossRef PubMed.
- R. Mu, N. Bu, J. Pang, L. Wang and Y. Zhang, Foods, 2022, 11, 3727 CrossRef CAS PubMed.
- W. Su, D. Liang and M. Tan, Trends Food Sci. Technol., 2021, 110, 213–225 CrossRef CAS.
- G. M. Sharma, A. Chatim, M. Ferguson and K. M. Williams, J. Food Sci., 2019, 84, 2357–2363 CrossRef CAS PubMed.
- I. D. B. Slot, M. G. E. G. Bremer, I. van der Fels-Klerx and R. J. Hamer, Cereal Chem., 2015, 92, 513–521 CrossRef CAS.
- B. T. Aksoy and Ö. A. Sönmezoğlu, Int. J. Life Sci. Biotechnol., 2022, 5, 546–561 CrossRef CAS.
- P. Stefanova, M. Taseva, T. Georgieva, V. Gotcheva and A. Angelov, Biotechnol. Biotechnol. Equip., 2013, 27, 3803–3810 CrossRef CAS.
- M. Słowianek and I. Majak, Biotechnol. Food Sci., 2011, 76, 39–44 Search PubMed.
- U. Jabeen, A. Ali, S. Ullah, R. Mushtaque, S. W. H. Naqvi, J. Uddin, A. Khan and A. Al-Harrasi, Rev. Fr. Allergol., 2023, 63, 103620 CrossRef.
- R. E. Poms, C. L. Klein and E. Anklam, Food Addit. Contam., 2004, 21, 1–31 CrossRef CAS PubMed.
- T. Bauer, K. Kirschbaum, S. Panter, M. Kenk and J. Bergemann, J. AOAC Int., 2011, 94, 1863–1873 CrossRef CAS PubMed.
- A. Al-Shaibany, H. Gargouri and H. H. Kacem, Int. Food Res. J., 2022, 29, 828–842 CAS.
- C. Anandharamakrishnan, J. A. Moses and S. Priyanka, in Food Digestion and Absorption: Its Role in Food Product Development, 2023 Search PubMed.
- M. H. Rezadoost, M. Kordrostami and H. H. Kumleh, 3 Biotech, 2016, 6, 61 CrossRef PubMed.
- N. Gryson, K. Dewettinck and K. Messens, Eur. Food Res. Technol., 2007, 226, 247–254 CrossRef CAS.
- T. Pan, Y. Wu, S. He, Z. Wu and R. Jin, Curr. Opin. Food Sci., 2022, 43, 36–42 CrossRef CAS.
- L. Rossen, P. Norskov, K. Holmstrom and O. Rasmussen, Int. Food Res. J., 1992, 17, 37–45 CAS.
- B. Easparro, Z. Morehouse, C. Proctor and J. Atwood, FASEB J., 2018, 32(supplement), 522.12 Search PubMed.
- A. Gomaa and J. I. Boye, Food Res. Int., 2013, 52, 483–489 CrossRef CAS.
- P. Moreira, J. Costa, C. Villa, I. Mafra, A. T. S. C. Brandão, C. Dias, A. F. Silva, C. M. Pereira and R. Costa, Anal. Chim. Acta, 2023, 1259, 341168 CrossRef CAS PubMed.
- K. C. M. Verhoeckx, Y. M. Vissers, J. L. Baumert, R. Faludi, M. Feys, S. Flanagan, C. Herouet-Guicheney, T. Holzhauser, R. Shimojo, N. van der Bolt, H. Wichers and I. Kimber, Food Chem. Toxicol., 2015, 80, 223–240 CrossRef CAS PubMed.
- T. Holzhauser, P. Johnson, J. P. Hindley, G. O'Connor, C.-H. Chan, J. Costa, C. K. Faeste, B. J. Hirst, F. Lambertini, M. Miani, M.-C. Robert, M. Roeder, S. Ronsmans, Z. Bugyi, S. Tomoskozi and S. D. Flanagan, Food Chem. Toxicol., 2020, 145, 111709 CrossRef CAS PubMed.
- C. Villa, M. B. M. Moura, C. S. S. Teixeira, J. Costa and I. Mafra, Nutrients, 2023, 15, 482 CrossRef CAS PubMed.
- A. C. Eischeid, Food Addit. Contam.: Part A, 2022, 39, 1797–1805 CrossRef CAS PubMed.
- C. Parent, P. Laurent, C.-E. Goujon, X. Mermet, A. Keiser, F. Boizot, R. Charles, L. Audebert, Y. Fouillet and M. Cubizolles, Lab Chip, 2022, 22, 3147–3156 RSC.
- H. Jiang, Q. Guo, C. Zhang, Z. Sun and X. Weng, Food Chem., 2021, 365, 130511 CrossRef CAS PubMed.
- S.-Y. Ma, Y.-C. Chiang, C.-H. Hsu, J.-J. Chen, C.-C. Hsu, A.-C. Chao and Y.-S. Lin, J. Sens., 2019, 2019, 4712084 Search PubMed.
- Y. Liu, X. Fang, X. Sun, B. Niu and Q. Chen, Food Anal. Methods, 2021, 14, 453–464 CrossRef.
- D. Yuan, J. Kong, X. Li, X. Fang and Q. Chen, Sci. Rep., 2018, 8, 8682 CrossRef PubMed.
- D. Natsuhara, S. Misawa, R. Saito, K. Shirai, S. Okamoto, M. Nagai, M. Kitamura and T. Shibata, Sci. Rep., 2022, 12, 12852 CrossRef CAS PubMed.
- H.-Y. Lin, C.-H. Huang, J. Park, D. Pathania, C. M. Castro, A. Fasano, R. Weissleder and H. Lee, ACS Nano, 2017, 11, 10062–10069 CrossRef CAS PubMed.
- X. Sun, Y. Liu, B. Niu, Q. Chen and X. Fang, PLoS One, 2022, 17, e0266775 CrossRef CAS PubMed.
- G. M. S. Ross, D. Filippini, M. W. F. Nielen and G. I. Salentijn, Anal. Chim. Acta, 2020, 1140, 190–198 CrossRef CAS PubMed.
- CEA - COMMISSARIAT A L ENERGIE ATOMIQUE & AUX ENERGIES ALTERNATIVES, EP3831959, 2021.
- B. Gilquin, M. Cubizolles, R. Den Dulk, F. Revol-Cavalier, M. Alessio, C.-E. Goujon, C. Echampard, G. Arrizabalaga, A. Adrait, M. Louwagie, P. Laurent, F. P. Navarro, Y. Coute, M.-L. Cosnier and V. Brun, Anal. Chem., 2021, 93, 683–690 CrossRef CAS PubMed.
- ISO 22916, https://www.iso.org/standard/74157.html, (accessed June 20, 2024).
- R. P. Hearn and K. E. Arblaster, Biochem. Mol. Biol. Educ., 2010, 38, 161–166 CrossRef CAS PubMed.
- F. Guan, Y. Jin, J. Zhao, J. Ai and Y. Luo, Food Anal. Methods, 2019, 12, 100–107 CrossRef.
- Lyophilized qPCR food allergen test kit manufacturers - Bioside, Lyophilized qPCR food allergen test kit manufacturers, https://www.bioside.it/lyophilized-multiplex-real-time-qpcr-test-kit-manufacturers/lyophilized-multiplex-qpcr-food-allergen-test-kit-manufacturers/, (accessed September 13, 2024).
- D. Houhoula, S. Papatheodorou, D. Moschou, S. Pappa, N. Tsaatazoglou, S. Koussissis, J. Tsaknis, V. Lougovois, J. Impe and E. Tsakali, J. Food Res., 2019, 8, 71–76 CrossRef CAS.
- A. C. Eischeid, S. R. Stadig and P. Rallabhandi, Food Addit. Contam.: Part A DOI:10.1080/19440049.2021.1874061.
- World Health Organization and International Union of Immunological Societies (WHO/IUIS) Allergen Nomenclature Sub-Committee, https://www.allergen.org/search.php?allergensource=Corylus+avellana, (accessed June 20, 2024).
- US Food and Drug Administration, Food Allergies, https://www.fda.gov/food/food-labeling-nutrition/food-allergies, (accessed June 20, 2024).
- R. Madrid, A. Garcia-Garcia, P. Cabrera, I. Gonzalez, R. Martin and T. Garcia, Foods, 2021, 10, 440 CrossRef CAS PubMed.
- L. Tuppo, I. Giangrieco, M. Tamburrini, C. Alessandri, A. Mari and M. A. Ciardiello, Foods, 2022, 11, 878 CrossRef CAS PubMed.
- C. Villa, J. Costa, C. Gondar, M. B. P. P. Oliveira and I. Mafra, Food Chem., 2018, 262, 251–259 CrossRef CAS PubMed.
- J. Costa, C. Villa, L. Grazina and I. Mafra, Food Chem., 2022, 397, 133778 CrossRef CAS PubMed.
- J. Costa, I. Silva, C. Villa and I. Mafra, J. Food Compos. Anal., 2023, 115, 105042 CrossRef CAS.
- E.-M. Ladenburger, M. Dehmer, R. Grünberg, H.-U. Waiblinger, D. Stoll and J. Bergemann, J. AOAC Int., 2018, 101, 170–184 CrossRef CAS PubMed.
- E. T. Aksun Tumerkan, Molecules, 2022, 27, 5674 CrossRef CAS PubMed.
- M. Koprinarova, J. Food Prot., 2021, 84, 1309–1314 CrossRef CAS PubMed.
- S. K. Verma and N. Biswas, Sci. Rep., 2020, 10, 11513 CrossRef CAS PubMed.
- US Food and Drug Administration, FDA Certification | Medical Devices - IAS USA, https://ias-certification.com/fda-certification-in-usa/, (accessed September 24, 2024).
- C. Tramuta, L. Decastelli, E. Barcucci, F. Ingravalle, S. Fragassi, S. Lupi and D. M. Bianchi, Foods, 2022, 11, 643 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00570h |
| This journal is © The Royal Society of Chemistry 2025 |