
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A microfluidic system for the cultivation of cyanobacteria with precise light intensity and CO2 control: enabling growth data acquisition at single-cell resolution†
Lennart
Witting
 ab,
Johannes
Seiffarth
ab,
Birgit
Stute
a,
Tim
Schulze
c,
Jan Matthis
Hofer
d,
Katharina
Nöh
a,
Marion
Eisenhut
c,
Andreas P. M.
Weber
d,
Eric
von Lieres
ab and
Dietrich
Kohlheyer
*a
ab,
Johannes
Seiffarth
ab,
Birgit
Stute
a,
Tim
Schulze
c,
Jan Matthis
Hofer
d,
Katharina
Nöh
a,
Marion
Eisenhut
c,
Andreas P. M.
Weber
d,
Eric
von Lieres
ab and
Dietrich
Kohlheyer
*a
aIBG-1: Institute of Bio- and Geosciences, Forschungszentrum Jülich GmbH, Jülich, Germany. E-mail: d.kohlheyer@fz-juelich.de
bComputational Systems Biotechnology (AVT.CSB), RWTH Aachen University, Aachen, Germany
cFaculty of Biology/Computational Biology, University of Bielefeld, Bielefeld, Germany
dInstitute of Plant Biochemistry, Heinrich-Heine University Düsseldorf, Düsseldorf, Germany
First published on 30th September 2024
Abstract
Quantification of cell growth is central to any study of photoautotrophic microorganisms. However, cellular self-shading and limited CO2 control in conventional photobioreactors lead to heterogeneous conditions that obscure distinct correlations between the environment and cellular physiology. Here we present a microfluidic cultivation platform that enables precise analysis of cyanobacterial growth with spatio-temporal resolution. Since cyanobacteria are cultivated in monolayers, cellular self-shading does not occur, allowing homogeneous illumination and precise knowledge of the photon-flux density at single-cell resolution. A single chip contains multiple channels, each connected to several hundred growth chambers. In combination with an externally applied light gradient, this setup enables high-throughput multi-parameter analysis in short time. In addition, the multilayered microfluidic design allows continuous perfusion of defined gas mixtures. Transversal CO2 diffusion across the intermediate polydimethylsiloxane membrane results in homogeneous CO2 supply, with a unique exchange-surface to cultivation-volume ratio. Three cyanobacterial model strains were examined under various, static and dynamic environmental conditions. Phase-contrast and chlorophyll fluorescence images were recorded by automated time-lapse microscopy. Deep-learning trained cell segmentation was used to efficiently analyse large image stacks, thereby generating statistically reliable data. Cell division was highly synchronized, and growth was robust under continuous illumination but stopped rapidly upon initiating dark phases. CO2-Limitation, often a limiting factor in photobioreactors, was only observed when the device was operated under reduced CO2 between 50 and 0 ppm. Here we provide comprehensive and precise data on cyanobacterial growth at single-cell resolution, accessible for further growth studies and modeling.
1 Introduction
Cyanobacteria are a morphologically diverse ancient group of bacteria that have emerged more than 2.5 billion years ago. They were the first organisms performing oxygenic photosynthesis, thus having a major impact on the evolution of life on earth. During oxygenic photosynthesis, light energy delivered by impinging photons is captured and stored as chemical energy in organic substances, thereby CO2 is used as a carbon source. As a side product, O2 is generated.1Cyanobacteria are subject to many fundamental research studies and are of biotechnological relevance. They were identified as potential producers of biofuels,1 high value chemicals like pigments, proteins, and vitamins. Furthermore, they can be used for waste water treatment and the degradation of oil components. Cyanobacterial biomass can also be used as food supplement for humans or as a feed additive in aqua culture.2,3
Lab-scale cultivation devices for cyanobacteria commonly include microtiterplates, shake flasks, bubble columns, flat-panel reactors, stirred tank reactors and tubular reactors. Of these, only microtiterplates are applicable for automated high-throughput screening.4 This is especially important when cultivating phototropic organisms due to their slow growth in comparison to many heterotrophic bacteria. Microtiterplates can be illuminated homogeneously5 or with differing light intensity between individual wells, to further increase experimental throughput.6,7 Nevertheless, all the above-mentioned systems suffer from limited process control and fundamental physical restrictions such as inefficient CO2 supply and self-shading of cells. Cultivation parameters such as light intensity and dissolved CO2 are device specific and hardly comparable among different systems.4,8 However, present research on cellular metabolism and modelling as well as model-based reactor scale-up requires growth data to be as precise as possible. Microfluidic systems have the potential for acquiring non-biased growth data at high throughput.4,9
Over the last decade, microfluidic cultivation devices, mainly fabricated from polydimethylsiloxane (PDMS), have emerged as powerful tools for systematically evaluating growth and physiology of microorganisms. E.g. we have previously used our technology to investigate growth characteristics of heterotrophic bacteria such as Corynebacterium glutamicum10 and Escherichia coli11,12 at single-cell resolution. Furthermore, microfluidics can be used to mimic and investigate the impact of bioreactor inhomogeneities and intrinsic cell-to-cell heterogeneity on microbial growth.13,14 Microfluidic cultivation structures designed to restrict cell growth to monolayers, enable homogeneous illumination, thereby eliminating the problem of cellular self-shading and light scattering. In addition, the commonly used chip material PDMS is highly gas-permeable, enabling efficient CO2 supply during cultivation.15
PDMS based microfluidic systems for photoautotrophic organisms are predominately used to study medium composition under homogeneous illumination.16–20 A few publications focus on testing multiple CO2 concentration in one experiment while keeping the light intensity constant.21,22 Some publications demonstrate the investigation of multiple light intensities in parallel, thus enabling high-throughput experimentation:
Kim et al. proposed a laminated (four layers) microfluidic PDMS chip enabling cultivation of microalgae under eight different light intensities and day–night cycles. Their approach is versatile, enables single-cell resolution, and precise data acquisition at high-throughput. Nevertheless, their innovative device is technically complex and not well suited for regular application.23
Graham et al. placed a PDMS chip on an illumination unit consisting of a liquid crystal display with a LED backlight. Imaging of cyanobacteria at single-cell resolution was not performed and growth rates were derived from the increasing chlorophyll fluorescence in the cultivation sites. The system allows controlling the light intensity and spectrum for each of the 238 microreactors individually. It was used to investigate the effects of light intensity, wavelength and duty cycle on the growth of Synechococcus elongatus PCC7942.24
Liu et al. modified a light microscope by placing a half-moon mask into the diascopic light path. The resulting light-intensity gradient illuminated an array of 64 microhabitats on a chip. The green algae Chlamydomonas reinhardtii was cultivated and growth was quantified by episcopic imaging of the chlorophyll fluorescence.25,26
While all of the above mentioned systems allow high-throughput cultivation with multiple light intensities, they do not restrict growth to monolayers nor do they enable CO2 control. Only the device presented by Kim et al.27 allows the observation of single cells.
Here we present a platform for the cultivation of cyanobacteria at single-cell resolution, in which a ringlight is used for controlled illumination. The applied spectrum can be varied within the photosynthetic active range of illumination and by half shading the ringlight we were able to create a linear light-intensity gradient within the physiological relevant range of light intensities. Therefore, enabling high-throughput experimentation. The system was designed with a focus on applicability, to enable use by non-specialist. The performance of the system for dynamic environmental control is demonstrated by the cultivation of S. elongatus UTEX2973 under decreasing CO2 concentrations and under a 12 h day 12 h night cycle. To the best of our knowledge, this is the first platform allowing precise analysis of the influence of light intensity and CO2 on cyanobacteria at single-cell resolution.
2 Materials & methods
2.1 Technical
A Nikon Ti-Eclipse (Nikon; Japan) inverted microscope was used for time-lapse microscopy and was modified for phototrophic cultivations. During the cultivation experiments four different illumination settings were available, as shown in Fig. 1: (1C) in diascopic imaging mode, phase-contrast microscopy was performed. (1D) In episcopic imaging mode, chlorophyll fluorescence was imaged using the following fluorescence filter cube: Ex 514/30 nm; DM 561; Em 629/56 nm (Nikon; Japan). Phase contrast and fluorescence images were acquired using a DS-Fi3 camera (Nikon; Japan) and a Plan Apo λ 100× Oil Ph3 DM objective. Additionally, the microscope was equipped with a Plan Apo λ 2× objective (Nikon; Japan) and an Andor Zyla scientific camera (Oxford Instruments; Great-Britain). The 2× objective and the Andor Zyla camera were exclusively used for the calibration of the light-intensity gradient (see section 2.6).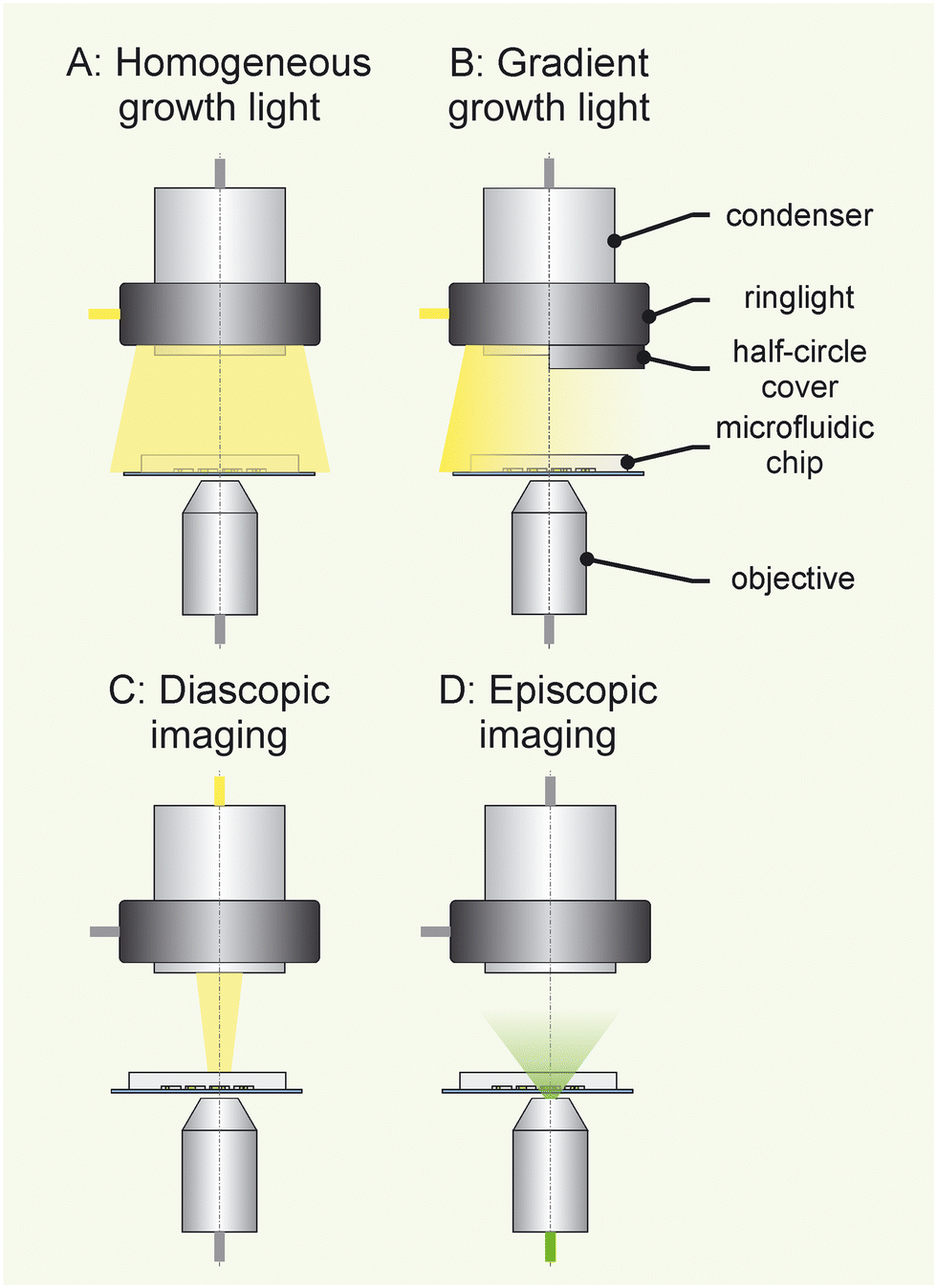 | ||
| Fig. 1 Overview over the illumination system for cyanobacteria cultivation. A ringlight is mounted around the microscope condenser providing controlled illumination for photosynthesis. A: The ringlight enables homogeneous illumination of the microfluidic chip. B: A linear light-intensity gradient is generated when shading half of the ringlight emitter-surface with a half-circle cover. C: During time-lapse imaging the ringlight illumination is temporary turned off to avoid optical interference. Then, the diascopic light path is used for phase-contrast imaging. D: For fluorescence imaging the episcopic light path is used. | ||
A Spectra Tune Lab light engine (LEDMOTIVE; Spain) served as the growth light-source, enabling the generation of a custom spectrum within the photosynthetic active range of light (400–700 nm). The impact of the lamp's waste heat was minimised by installing the lamp outside the microscope incubator (see ESI† material). A Coldvision, A08360 ringlight (SCHOTT; Germany) was mounted to the microscopes condenser lens (long working distance, LWD = 0.52) and connected via a flexible wave guide to the Spectra Tune Lab light engine. The software μwave (LEDMOTIVE; Spain) was used to control the lamp during timelapse imaging. The Spectra Tune Lab can illuminate the microfluidic chip with a maximum of 384 μE m−2 s−1 during homogeneous growth-light illumination (Fig. 1A), with data shown in the ESI† material. A light-intensity gradient along the microfluidic chip was generated, when half of the ringlight emitter-surface was covered with a custom-made aluminum cover plate (Fig. 1B). The ringlight illumination for growth was temporally interrupted by closing a Lambda SC optical shutter (Sutter Instrument; USA), to avoid optical interference during time-lapse imaging. A microscope incubator NL 2000 and controller TempController 2000-2 (PECON; Germany) ensured stable temperature throughout the entire cultivation. BG11 growth medium28,29 (see ESI† material) was supplied by a Nemesys syringe pump (CETONI; Germany). Reproducible light-intensity measurements were achieved by installing a LI-190R Terrestrial Quantum Sensor (LI-COR Biosiences; USA) into the objective revolver using a 3D printed connector. This connector was mounted to an PMMA thread compatible with the objective revolver of the Ti-E microscope. The connector was designed in SolidWorks 2016 (Dassault systemes; France) and printed with a Form 3B (Formlabs; USA) using Tough 2000 V1 resin. The 3D CAD files are available in the ESI.†
2.2 Laboratory-scale cultivation
The cyanobacteria strains used in this work are listed in Table 1. Cyanobacteria were cultivated at 30 °C and constantly shaken at 110 rpm in 100 mL shake flasks filled with 30 mL BG11 medium. Shake flasks were illuminated with approximately 20 μE m−2 s−1 using a simple LED growth light array mounted ontop of the shaking incubator. Every 14 days 2 mL of culture were propagated into fresh BG11 medium.Cyanobacteria were then transferred into the MC-1000 OD Multi-Cultivator (Photon Systems Instruments; Czeck Republic) (MC) for growth experiments. The MC allows online monitoring of the culture's optical density (OD) at 680 and 720 nm. Culture tubes were filled with 50 mL of BG11 medium and cells were inoculated to an OD720 of 0.1. The OD720 correlates linearly to biomass in the range from 0.05–0.4. The calculation of the growth rate included the following steps: i.) all OD720 values under 0.05 and over 0.4 were cut off. ii.) The natural logarithm of the cutoff OD720 over time was formed. ii.) A linear model was fitted onto the natural logarithm using numpy.34 iv.) The slope of the linear model is the growth rate.
A CO2-Controller 2000 (PECON; Germany) connected to the MC enabled aeration of the culture with defined CO2 concentrations. Data was plotted and analyzed with a custom python notebook available at: https://github.com/JuBiotech/Supplement-to-Witting-et-al.-2024.
2.3 Device fabrication
The cultivation chip was designed using CleWin5 (WieWeb software; Netherlands). As shown in Fig. 2A, the layout includes four parallel channel arrays. Each channel array is 24 mm long and incorporates 8 × 40 cultivation chambers. Cultivation chambers are 60 × 60 μm in size with a height of 1.029 μm. Cultivations under homogeneous (Fig. 1A) illumination were performed using a shorter chip design, including channel arrays of 3.72 mm in length and a chamber height of 0.985 μm. Aqueous growth medium was supplied continuously by adjacent channels with a height of 10 μm. | ||
| Fig. 2 Microfluidic cultivation chip layout. A: Cultivation chips of approximately 1 mm thickness (the cultivation layer) were used for experiments with CO2 control to minimize the thickness of the gas exchange membrane. The second chip (the gas-control layer) was bonded onto the cultivation layer enabling the perfusion of air with defined CO2 concentrations. B: Each chamber is connected to medium supply channels (10 μm height) resulting in a diffusive mass transfer within the chamber. The chambers have a surface area of 60 μm × 60 μm and a height of approximately 1 μm. C: The cultivation layer contains four 24 mm long arrays, each containing 8 rows of 40 evenly distributed growth chambers. | ||
The replication master for PDMS moulding was manufactured at the Helmholtz Nano Facility.35 It consists of a 4′′ silicon wafer carrying two layers of SU-8 photoresist. In short, SU-8 lithography included wafer dehydration, photoresist spincoating, pre and post-exposure baking, maskless resist exposure using a DWL 66+ maskless lithography system (Heidelberg Instruments; Germany), resist developing in a bath, and final hard baking. Detailed process parameters are listed in the ESI.†
The SYLGARD™184 Silicone Elastomer Kit was used for chip moulding. The process was performed as follows: PDMS monomer (Dow Corning; USA) and curing agent were mixed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, degassed to remove air bubbles, poured onto the silicon wafer, baked at 80 °C for 2 h and released from the mold. The molded PDMS chips were separated by manual cutting and subsequently washed in n-pentane and acetone to remove uncured monomers.
:1 ratio, degassed to remove air bubbles, poured onto the silicon wafer, baked at 80 °C for 2 h and released from the mold. The molded PDMS chips were separated by manual cutting and subsequently washed in n-pentane and acetone to remove uncured monomers.
The replication master for the gas layer was designed in SolidWorks 2016 (Dassault systemes; France). The replication master was printed using a Form 3B (Formlabs; USA) printer and the Model V3 resin. The CAD File of the replication master is provided in the ESI† material. The design includes a 30 × 9.76 × 0.55 mm channel for homogeneous gas supply. Moulding of the gas layer included mixing of PDMS with curing agent in a 10:1 ratio, degassing, PDMS casting and baking at 80 °C for 2 h. Cultivation chips of approximately 5 mm thickness were used for experiments without CO2 control. When CO2 control was required, the cultivation chips were approximately 1 mm thick. This resulted in a thin gas exchange membrane between the cultivation chip and the gas layer.
The cultivation chips and the gas layer were prepared in advance and stored until further usage. The final assembly of the cultivation device was carried out on the day the experiment was inoculated: i) cultivation without CO2 control: the cultivation chip was treated with oxygen plasma and bonded onto a 175 μm thick glass slide. ii) Cultivation including CO2 control: the gas layer was first bonded onto the cultivation chip, thereby enabling gas supply to the cultivation chambers as well as additional mechanical stability when handling the thin PDMS chips. Finally, the cultivation chip was bonded onto the glass slide. For a detailed video description and more information about the plasma bonding as well as the general procedure the reader is referred to Grünberger et al.36
2.4 Microfluidic cyanobacteria cultivation
The platform presented in this work allows carrying out different experimental modes: microfluidic cultivations can be performed either with or without CO2 control, depending on the chip configuration. Without the additional gas control layer, CO2 availability depends on the surrounding atmosphere. The microfluidic cultivation chip can be illuminated homogeneously or a light-intensity gradient can be applied. The light-intensity can be constant, but also dynamic profiles, for example day–night cycles can be applied.Before starting an experiment, cyanobacteria were precultivated in the MC. Therefore, cyanobacteria were inoculated to an OD720 of 0.1 and cultivated for approximately 24 h. Prior to inoculation the tubing for BG11 medium supply and outflow were connected. BG11 medium was perfused at a flow rate of 200 nL min−1. After the cell inoculation, growth chambers containing cyanobacteria were selected manually for time-lapse imaging. Pictures were taken every 1 hour for experiments with homogeneous growth-light illumination and every 2 hours in experiments with gradient growth light-illumination. For microfluidic experiments with CO2 control, a premixed synthetic air bottle containing 200 ppm CO2 was used. Final CO2 concentrations were achieved by mixing defined volume flow rates of the synthetic air, N2 and O2 using red-y-smart thermal mass flow controllers (Vögtlin; Germany). Gas was perfused through the gas layer in countercurrent to medium flow. All experiments were performed at 37 °C. The Spectra Tune Lab light engine was set to emit Planck's radiation distribution at 5800 K, mimicking the spectrum emitted by the sun.37 Day–night rhythms were programmed in μwave and started simultaneously with the time-lapse sequence.
2.5 Growth rate modelling
A variety of models describing the relationship between growth rate and light-intensity are described in literature.38 The hyperbolic tangent model39 and the shape-modified monod kinetic model40 worked well to describe our data. Of these, the hyperbolic tangent model was chosen for data fitting, because for all the therein described parameters a physical interpretation can be derived (see ESI† material). Finally, for each graph an excel file including the raw data will be provided to enable further modelling. Data fitting was performed in OriginPro 2020. | (1) |
 | (2) |
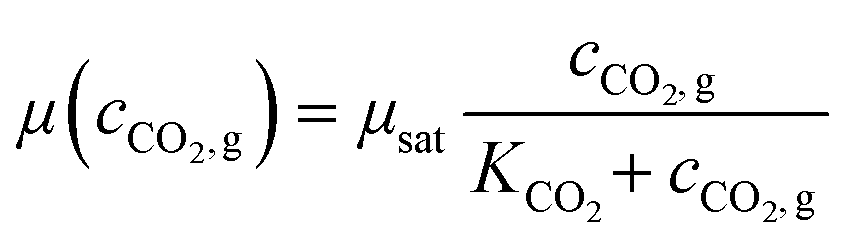 | (3) |
2.6 Growth-light calibration
For direct light-intensity measurements of the homogeneous and gradient illumination (data shown in Fig. 4A), the sensor spot of a Li-180 Spectrometer (Li-Cor Biosciences; USA) was mounted on the X–Y-Stage and during measurements it was moved relative to the ringlight. Therefore, the ringlight and the Li-180 sensor were mounted at a comparable distance as between the ringlight and the microfluidic chip.The light-intensity gradient was calibrated prior to each experiment to assign a specific light-intensity for all cultivation chambers. An exemplary calibration is illustrated in detail in the ESI† material. Each calibration procedure included the following three main steps:
i.) Using the LI-190R Terrestrial Quantum Sensor under homogeneous illumination of the growth light, the photon flux density (PFD) in the photosynthetic active range of illumination [μE m−2 s−1] was measured at various power settings [%] in the light engine's control software. A linear correlation was found between power setting and the resulting PFD.
ii.) Instead of the cultivation chip, a microscopy calibration slide of homogeneous color and density (Chroma Technology, USA) was mounted in the same optical plane. Using the 2× objective, the microscope was focused on the top surface of this calibration slide. Then bright-field images of the calibration slide were taken with the Zyla camera under homogeneous growth-light illumination at various power settings. A linear correlation between the light engine's power settings and the averaged camera grey-values was found. By replacing the power levels with the corresponding PFD values from i), a linear correlation between camera pixel grey values and PFD can be derived.
iii.) Finally, the half-circle cover was installed to generate the light-intensity gradient. Bright-field images of the calibration slide under gradient illumination at specific power settings were taken. During capture, no additional microscopy illumination was applied. The light-intensity gradient illumination resulted in linearly increasing camera grey values, resolving the gradient at camera resolution. By replacing the grey values with the corresponding PFD from ii.), the light-intensity gradient can now be described as linear function of PFD over position. The microscope's objective can not be moved relative to the ringlight. Hence, when a cultivation chip is later placed in the microscope the knowledge of the linear relation of the PFD in dependence on X-position allows to assign specific light-intensity values to each growth chamber.
2.7 Semi-automated cell segmentation
Prior to image analysis, some chambers were manually discarded for three main reasons: i.) the inoculated cell did not grow. ii.) The inoculated cell was washed out of the chamber after the start of the experiment. iii.) Poor image quality due to loss of focus. Tables displaying the number of observed and analysed chambers can be found in the ESI.†Image preprocessing was done in Fiji.41 Image sequences were registered with Correct 3D drift,42 rotated, cropped, exported as *.tif files and subsequently uploaded onto our OMERO server43 for storage and further processing. In order to train the deep learning cell segmentation model, 20 exemplary image crops were selected using microbeSEG44 and annotated semi-automatically using the ObiWan-Microbi tool.45 Based on the annotated data, the Omnipose46 cell segmentation model was finally fine-tuned. Using the trained model, cell instances were segmented, from which cell counts and cell areas were derived. For applying the cell segmentation procedures and deriving growth data, Jupyter notebook processing and analysis workflows were designed. For more details on cell segmentation and data analysis the reader is refereed to other publications.45,47 Calculation of growth rates according to an exponential model included the following steps: i.) manually selecting the timeframe between the start of exponential growth and the overflow of the chamber. ii.) Forming of the natural logarithm of the cell area or cell count. iii.) Fitting a linear model onto the natural logarithm within the selected timeframe using numpy.34 iv.) The growth rate is the slope of the linear model. An exemplary analysis script for the growth rate calculation in form of a Jupyter notebook is available at: https://github.com/JuBiotech/Supplement-to-Witting-et-al.-2024. All generated .tif image stacks and the therefrom derived growth rates are publicly available at: https://zenodo.org/records/13220517.
3 Results & discussion
3.1 Microfluidic cultivation under homogeneous illumination
The cyanobacterium S. elongatus UTEX2973 was successfully grown under homogeneous growth-light illumination at light intensities between 10 and 140 μE m−2 s−1. A total of 11670 images containing 1817542 cells were segmented and analysed. Several independent experiments were performed and the entire cultivation time under homogeneous growth-light illumination lasted roughly 32 days. Detailed growth data of two exemplary microbial colonies is shown in Fig. 3C–E. The dependency of the growth rate on light intensity is shown in Fig. 3F. The number of biological replicates for each light intensity is indicated in Fig. 4B. Information on how many chambers were discarded prior to image analysis is shown in the ESI.† In image based single-cell analysis, the colony averaged growth rate can be derived from the cell number or the projected cell area over time, with both results shown in Table 2. The growth rate derived from cell number over time was slightly higher than the rate based on the projected cell area. This difference is a result of the decreasing cell size over the cultivation time, as shown by the plot of mean cell area over time in Fig. 3D. In the following, growth rate calculations are based solely on cell area over time.
 | ||
| Fig. 3 Cyanobacterial growth at different light-intensities under homogeneous and constant growth-light illumination. A: Data was acquired by time-lapse microscopy, recording phase contrast and chlorophyll fluorescence images. B: Images were preprocessed in Fiji before cell instance segmentation was performed using a deep learning model that was trained on annotated sample images. DL based cell segmentation was performed on phase-contrast images to derive cell number (and area) over time from which growth rates were determined using an exponential growth model. Video examples of the time-lapse microscopy and cell segmentation can be found in the ESI† material. C: Total cell area of segmented cells over the cultivation time. D: Mean cell area per frame over cultivation time. E: Number of segmented cells over the cultivation time. F: Colony based growth analysis derived from image data at single-cell resolution of UTEX2973 under homogeneous illumination in comparison to laboratory-scale MC cultivation (n = 2). The microfluidic device was operated without CO2 control. The ambient air had a CO2 concentration of approximately 400 ppm. The MC cultivations were performed with ambient and with CO2 enriched air. | ||
 | ||
| Fig. 4 A: Light-intensity profiles across the light cone emitted by the ringlight (homogeneous illumination mode and longitudinal to the light-intensity gradient). B: Growth data of UTEX2973 from microfluidic cultivations under light-intensity gradient illumination and under homogeneous illumination for comparison. Under light-intensity gradient illumination, each data point resembles growth inside distinct chambers from a single, continuously performed experiment (cultivation time approximately 4 days). Data points obtained during homogeneous illumination, include standard deviation and the number of replicates (n = analyzed chambers). These replicates were obtained from multiple chambers on the same chip, but the corresponding light-intensity was varied over several independent experiments (cultivation time approximately 32 days). C: Growth data of three different cyanobacteria strains under gradient growth-light illumination during microfluidic cultivation. Each datapoint represents a single growth chamber. | ||
| Parameter | 140 μE m−2 s−1 | 20 μE m−2 s−1 |
|---|---|---|
| Cell count based growth rate [h−1] | 0.105 | 0.064 |
| Cell area based growth rate [h−1] | 0.093 | 0.050 |
Synchronous cell division of Synechococcus as previously reported by other authors,48,49 was also observed in our microfluidic growth chambers and is evident from the sawtooth pattern when plotting the mean single-cell area over time (Fig. 3D). This typical sawtooth pattern results from the repetitive cell division cycle. It is characterized by the almost linear increase in mean cell size, followed by a sudden decrease as image-based cell segmentation eventually results in two independent, but smaller cells at the end of each cell division cycle. As cell numbers increase, cell division seems less synchronous, as indicated by the less pronounced sawtooth pattern with increasing cultivation time. Clearly, the continuous decrease in cell size also contributes to a less pronounced sawtooth pattern.
The growth data in Fig. 3F is typical for phototropic organisms. In the light-limited region, a linear growth rate dependence on the light intensity is observed.3 When further increasing the light-intensity, in the light-saturated region, the growth rate reaches a plateau and is limited either by the maximum CO2 uptake rate or the CO2 availability in the culture medium.50 By increasing the CO2 concentration, higher growth rates can be achieved to some extent, until no further growth-rate increase is achieved. The parameters derived from fitting eqn (1) onto the data in Fig. 3F are shown in Table 3.
| Experiment | μ max (h−1) | α ((h μE m−2 s−1)−1) | I 1/2 (μE m−2 s−1) |
|---|---|---|---|
| Cell area | 0.087 | 2.16 × 10−3 | 22.11 |
| Cell count | 0.096 | 2.49 × 10−3 | 21.14 |
| MC ambient CO2 | 0.064 | 9.46 × 10−4 | 37.01 |
| MC 3% CO2 | 0.103 | 8.71 × 10−4 | 65.30 |
Supplying ambient air, the maximum growth rate of UTEX2973 cultivated inside the microfluidic chip, was higher than the growth rate of this strain cultivated in the laboratory scale MC. The effective CO2 concentration was obviously higher under microfluidic conditions. Inside the MC cultivation tubes, CO2 is provided by bubbling air through the culture broth with inefficient mass transfer. Therefore, supplying ambient air during MC cultivation, results in a lower effective CO2 concentration and CO2 limited growth in the light saturated region. In consequence, additional MC cultivation were performed at 3% (30000 ppm) and 5% CO2 aeration, enabling higher maximum growth rates. Though, no further growth rate increase was observed at 5% in comparison to 3%, indicating saturated photosynthetic capacity in the MC (data in ESI†).
Comparing growth performances, the slope of ‘growth rate over light intensity’ (Table 3) was steeper in the microfluidic device. Thus in the microfluidic device the maximum growth rate of UTEX2973 was reached at lower light-intensities than in the MC. Noteworthy, light-intensities between different cultivation systems for photoautotrophic organisms are often not comparable. In our microfluidic system, the light intensity was measured directly below the cultivation chip, with only the 175 μm thick glass slide in-between the cultivation plane and the intensity sensor. This means, that during microfluidic cultivation, cells growing in monolayers are homogeneously and continuously exposed to this specific predetermined light intensity. However, in conventional photobioreactors, microorganisms are exposed to heterogeneous illumination due to cellular self-shading and light scattering,51 which results in a lower effective light intensity available for photosynthesis.
3.2 Characterization of the linear light-intensity gradient
The data presented in Fig. 3F was derived when performing distinct cultivation experiments over roughly 32 days. In order to accelerate experimental throughput, a light-intensity gradient longitudinal to the microfluidic chip was applied, enabling simultaneous cultivation at various light intensities. The light-intensity gradient results from covering half of the ringlight and has a linear range of 3 cm (Fig. 4A) with intensities in the physiological relevant range for the cultivation of cyanobacteria (Table 4). In contrast, cultivations with homogeneous growth-light illumination were performed using a shorter chip layout (3.72 mm length). Consequently the diversion of the light intensity for homogeneous growth-light illumination shown in (Fig. 4A) is not relevant.The gradient was calibrated before each experiment, to assign a specific light intensity to each column of cultivation chambers. An example calibration is shown in the ESI† material. Therein all correlations showed R2 values over 0.99.
3.3 Microfluidic cultivation with the linear light-intensity gradient
The application of the light-intensity gradient to enable experimentation at high-throughput, was demonstrated by cultivating the cyanobacterial model strains UTEX2973, PCC7942 and PCC6803. Thereby, the required experimental time was reduced to roughly 4 days for the cultivation of all three organisms. The derived model parameters are shown in Table 5.| Strain | μ max (h−1) | α ((h μE m−2 s−1)−1) | I 1/2 (μE m−2 s−1) |
|---|---|---|---|
| PCC7942 | 0.157 | 2.10 × 10−3 | 40.97 |
| UTEX2973 | 0.086 | 2.77 × 10−3 | 17.12 |
| PCC6803 | 0.061 | 2.77 × 10−3 | 12.10 |
The growth performance, exemplary shown for UTEX2973 in Fig. 4B, was comparable between homogeneous (Table 3) and gradient growth-light illumination (Table 5). The layout of the microfluidic chip incorporates four parallel chamber arrays enabling several organisms to be cultivated simultaneously. The uppermost two arrays were inoculated with UTEX2973. The third array was inoculated with PCC7942, the last array was inoculated with PCC68703. Per array 20 growth chambers were selected for imaging. The number of therefrom discarded chambers is shown in the ESI.† In comparison, PCC7942 showed the highest maximum growth rates, while PCC6803 showed the highest initial slope and the lowest half maximum intensity (Fig. 4C). Significant growth-rate variances in the orthogonal direction of the gradient were not observed (ESI† material). The data is comparable to previously reported growth rates.19,24
3.4 Dynamic environmental control
After characterizing and validating the linear light-intensity gradient, dynamic environmental control was implemented. Therefore, UTEX2973 was cultivated under a 12 h day 12 h night cycle, while simultaneously applying the light-intensity gradient at day time. As shown in Fig. 5A, the cells responded rapidly to the changing light conditions. Day growth-rates were μ = 0.096 ± 0.005 h−1 for 90 μE m−2 s−1 and μ = 0.47 ± 0.01 h−1 for 20 μE m−2 s−1. The maximum growth rate was slightly lower on the first day (0.084 h−1) than on the following days (0.088 h−1, 0.093 h−1 and 0.091 h−1 for days 2, 3 and 4). It is reasonable to attribute the lower growth rates on the first day to a primary adaptation phase after inoculation. No changes in chlorophyll fluorescence were observed. | ||
| Fig. 5 Microfluidic cultivation with dynamic environmental control. A: UTEX2973 was cultivated under a 12 h day 12 h night cycle. Total cell-area per frame plotted over time for a chamber illuminated with 90 μE m−2 s−1 and a chamber with 20 μE m−2 s−1. B: Daytime growth-rates plotted over the light intensity. Each datapoint represents growth within a growth chamber illuminated at a distinct light intensity C: growth rate over light intensity for the cultivation of UTEX2973 under decreasing CO2 concentrations. CO2 concentration was altered after 24 h. | ||
In order to evaluate the effects of the CO2 concentration on cell growth, UTEX2973 was cultivated under stepwise decreasing CO2 concentrations starting from 100 ppm to 0 ppm in a single experiment. Each CO2 concentration (100, 50, 15 and 0 ppm) was applied for 24 h, resulting in 4 days cultivation time in total. The light-intensity gradient was simultaneously applied.
The CO2 mass transfer in the double-layer cultivation chip was simulated with COMSOL Multiphysics 6.2. using mass transfer constants from Maki et al.53 The simulations confirm that a homogeneous distribution of CO2 in the gas layer can be assumed. The simulation also shows, that perfusing the CO2 depleted air in countercurrent to the medium flow quickly reduces the CO2 concentration in the aqueous phase. Data is shown in the ESI† material.
Here, growth rates were calculated for each 24 h interval separately. However, for some growth chambers growth rates could not be calculated for each interval. Detailed information is shown in the ESI.† Evaluated growth rates plotted against the light intensity are shown in Fig. 5C. Eqn (1) was fitted onto the data for each CO2 concentration and the resulting model parameters are shown in Table 6. As expected, negligible growth was measurable at 0 ppm CO2, therefore, the mean value is calculated instead. Eqn (3) was fitted to the data in Table 6, to model the dependency of μmax on the CO2 partial pressure. The modeling yielded a μsat of 0.094 h−1 and a KCO2 of 6.5 ppm.
| CO2 [ppm] | μ max (h−1) | α ((h μE m−2 s−1)−1) | I 1/2 (μE m2 s−1) |
|---|---|---|---|
| 100 | 0.087 | 3.56 × 10−3 | 13.50 |
| 50 | 0.085 | 3.26 × 10−3 | 14.34 |
| 15 | 0.065 | 4.31 × 10−3 | 8.33 |
| 0 | −0.0027 ± 0.01 | — | — |
During microfluidic cultivation, CO2 containing gas is continuously supplied via the top PDMS layer resulting in diffusive gas exchange occurring across the interfacial PDMS layer. This thin interface acts as a gas permeable membrane between the gas-supply channel and the underlying monolayer growth chambers, resulting in a high ‘gas exchange-surface’ to ‘monolayer cultivation-volume’ ratio of 0.97 μm−1. During cultivation, each individual cyanobacteria is constantly aerated (and illuminated) over roughly half of its cell's surface area. In addition, the growth medium is supplied continuously, resulting in very homogeneous growth conditions.
Therefore, for our microfluidic cultivation, we can assume the following: as long as gas is supplied continuously at relatively high flow-rates, concentration changes in the gas supply can be neglected. Thus, during photoautotrophic growth, the resulting CO2 concentration in the growth chamber is at equilibrium with the supplied gas (according to Henry's law54), and is not limited by the actual mass-transfer rates. Inefficient mass transfer in process aeration, often a growth limiting aspect at larger cultivation scales, did not limit continuous single-cell growth under the applied growth conditions. As evident from Fig. 5C, the maximum photosynthetic capacity of UTEX2973 lies below 50 ppm CO2 under the current conditions, since no further maximum growth-rate increase was observed at 100 ppm CO2.
Our analytical performance puts us in a unique position, allowing us to consider our single-cell resolution based growth data as a standard to characterise other technical cultivation systems, such as the laboratory-scale MC. Consequently, eqn (3) was used to calculate the effective CO2 availability to evaluate the gas transfer efficiency in the MC. Count based growth rates seem to be more comparable to the MC, but eqn (3) was calculated from area based growth rates. To resolve this issue the growth rate at ambient air aeration (≈400 ppm CO2 in the supply gas) in the MC (μ = 0.064 h−1) was transformed into an equivalent area based growth rate (μarea = 0.056 h−1) using the model parameters in Table 3. This transformed growth rate was used in eqn (3) to derive the effective CO2 concentration cCO2 = 9.60 ppm. According to Henry's law,54 this is equivalent to a concentration of 0.0108 mg L−1 CO2 in the culture medium of the MC (calculation in ESI† material).
The initial slope of the growth curve characterises the dependency between light intensity and growth rate in the light-limited region of phototropic growth. It is reasonable to assume that the light intensity in the microfluidic device is the actual light intensity impinging on the cells. This allows us to quantify the deviation between the incident light intensity (given by the MC device manufacturer) and the effective light intensity within the culture medium of the MC. Thus, the initial slope at 15 ppm in the microfluidic device was compared to the initial slope at ambient air in the MC, resulting in the conclusion that the effective light intensity in the MC is 4.6 times lower than the incident light intensity as given by the manufacturer.
4 Conclusions
Microfluidic based single-cell analysis is a highly multidisciplinary field with many technological and biological hurdles. This is one of the reasons why many innovative systems, are still too laborious, resulting in hardly any real applications. Simplicity is a key consideration to ensure the new methodology is accessible to shortly trained non-technical specialists. In conclusion, we would like to emphasize the following three aspects of our novel cultivation platform for phototrophic organisms: i) the modular design, ii) the comprehensive cyanobacterial growth dataset, and iii) our device as an enabling technology. In more detail:i) The present device was developed in intensive collaboration between bio-, micro- and software engineers together with experts from biological sciences, considering applicability and device simplicity of high priority. Our modular system allows microbial cultivation in defined growth chambers during time-lapse microscopy. Additional environmental controls can be added when required. Functional elements, e.g. gas supply, can be easily placed on the standardized gas-permeable PDMS cultivation chip, resulting in efficient diffusive gas transfer through the intermediate membrane.
Cyanobacteria were cultivated in monolayers in the microfluidic device. In contrast to conventional photo-bioreactors, all cells are exposed to the same light intensity. The growth-light source can be a simple LED or, as in this publication, a light engine with spectral control. A linear light-intensity gradient was created by covering half of the ringlight to increase cultivation throughput. To the best of our knowledge, the device presented in this paper is the first to combine multi-parameter growth screening under light intensity and CO2 control. Other advantages of our device are it's simple, straightforward design and it's capability to image cyanobacteria at single-cell resolution.
In image analysis at single-cell resolution, time-lapse microscopy generates large image-data stacks, but unfortunately image analysis is often developed case specifically, resulting in inefficiency and high manual workload. Our generic DL-based cell segmentation has been retrained for the studied cyanobacteria and all image analysis tasks have been automated in python, allowing large datasets to be analyzed and visualized efficiently in a short time with reproducible accuracy. The analysis specifications can be easily modified and adapted to specific biological requirements. In summary, we consider our approach as an enabling platform technology and the device is currently being used in biological studies.
ii) Although the initial goal of our device validation was to analyze model cyanobacterial strains for proof-of-principle purposes only, the project has generated an extensive image dataset at single-cell resolution. Over 2.8 million individual cyanobacteria were segmented and further analyzed. We also demonstrated the long-term stability of our system by continuously culturing cyanobacteria for up to 172 hours. The implementation of the light-intensity gradient reduced the experimental effort from 32 days of multiple single experiments to only 4 days of continuous cultivation, resulting in comparable results. The data contains detailed information on cell size and morphology, position resolved in space and time under a wide range of growth conditions; far more details than previously published datasets.
iii) In addition to the visualized plots, the full datasets will be available and can be used for further modeling and research. We see great potential in using our approach to perform single-cell mass balances of photoautotrophically generated cellular biomass, accurately identify light compensation points, study cell maintenance under controlled limiting conditions and investigation of the cyanobacteria carbon concentration mechanism. Furthermore, we see potential of our precise growth data to be used in bioreactor optimization. E.g. modeling of light regimes, CO2 distribution and particle trajectories can benefit from precise correlations of the light-intensity and the CO2 concentration to growth rates, hence reducing experimental effort during scale-up.
Data availability
All .tif image stacks are publicly available at: https://doi.org/10.5281/zenodo.13220517. The code for analyzing image stacks and the Multi-Cultivator data is available at: https://github.com/JuBiotech/Supplement-to-Witting-et-al.-2024.Author contributions
LW: investigation, formal analysis, visualization, writing – original draft. JS: software, visualisation, writing – review & editing. BS: investigation. TS: writing – review & editing. JH: writing – review & editing. KN: funding acquisition, writing – review & editing. ME: funding acquisition, writing – review & editing. AW: funding acquisition, writing – review & editing. EL: supervision. DK: project administration, funding acquisition, conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
LW, TS, and JH, were funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – SFB1535 – Project ID 458090666. DK was partly funded by the Helmholtz organisation “Earth and Environment” Innovation Pool project “High CO2 – metabolic responses and bioeconomic opportunities”. JS and KN were funded by the President's Initiative and Networking Funds of the Helmholtz Association of German Research Centres [EMSIG ZT-I-PF-04-044]. We thank Dr. Katharina Pflüger-Grau and Franziska Kratzl for providing us with Synechococcus elongatus PCC7942 cscB.Notes and references
- T. Matsunaga, T. Yoshino, Y. Liang, M. Muto and T. Tanaka, Marine Microalgae, in Springer Handbook of Marine Biotechnology, ed. S.-K. Kim, Springer, 2015, ch. 5 Search PubMed.
- R. Abed, S. Dobretsov and K. Sudesh, J. Appl. Microbiol., 2009, 106, 1–12 CrossRef CAS PubMed.
- C. Posten, Integrated bioprocess engineering, De Gruyter, Berlin, 2018 Search PubMed.
- P. Benner, L. Meier, A. Pfeffer, K. Kruger, J. E. O. Vargas and D. Weuster-Botz, Bioprocess Biosyst. Eng., 2022, 45, 791–813 CrossRef CAS PubMed.
- H. Morschett, D. Schiprowski, C. Müller, K. Mertens, P. Felden, J. Meyer, W. Wiechert and M. Oldiges, Biotechnol. Bioeng., 2017, 114, 122–131 CrossRef CAS PubMed.
- M. Chen, T. Mertiri, T. Holland and A. S. Basu, Lab Chip, 2012, 12, 3870 RSC.
- J. Heo, D.-H. Cho, R. Ramanan, H.-M. Oh and H.-S. Kim, Biochem. Eng. J., 2015, 103, 193–197 CrossRef CAS.
- J. Degen, A. Uebele, A. Retze, U. Schmid-Staiger and W. Trösch, J. Biotechnol., 2001, 92, 89–94 CrossRef CAS PubMed.
- H. Morschett, V. Loomba, G. Huber, W. Wiechert, E. von Lieres and M. Oldiges, FEMS Microbiol. Lett., 2018, 365(1), 1–9 CrossRef PubMed.
- A. Grünberger, N. Paczia, C. Probst, G. Schendzielorz, L. Eggeling, S. Noack, W. Wiechert and D. Kohlheyer, Lab Chip, 2012, 12, 2060 RSC.
- C. Probst, A. Grünberger, W. Wiechert and D. Kohlheyer, J. Microbiol. Methods, 2013, 95, 470–476 CrossRef PubMed.
- D. Lindemann, C. Westerwalbesloh, D. Kohlheyer, A. Grünberger and E. von Lieres, RSC Adv., 2019, 9, 14040–14050 RSC.
- S. Täuber, C. Golze, P. Ho, E. von Lieres and A. Grünberger, Lab Chip, 2020, 20, 4442–4455 RSC.
- A. Grünberger, W. Wiechert and D. Kohlheyer, Curr. Opin. Biotechnol., 2014, 29, 15–23 CrossRef PubMed.
- S. Halldorsson, E. Lucumi, R. Gomez-Sjoberg and R. M. T. Fleming, Biosens. Bioelectron., 2015, 63, 218–231 CrossRef CAS PubMed.
- C. Westerwalbesloh, C. Brehl, S. Weber, C. Probst, J. Widzgowski, A. Grünberger, C. Pfaff, L. Nedbal and D. Kohlheyer, PLoS One, 2019, 14, 1–13 CrossRef PubMed.
- S. Bae, C. W. Kim, J. S. Choi, J.-W. Yang and T. S. Seo, Anal. Bioanal. Chem., 2013, 405, 9365–9374 CrossRef CAS PubMed.
- G. Zheng, Y. Wang, Z. Wang, W. Zhong, H. Wang and Y. Li, Mar. Pollut. Bull., 2013, 72, 231–243 CrossRef CAS PubMed.
- C. S. Luke, J. Selimkhanov, L. Baumgart, S. E. Cohen, S. S. Golden, N. A. Cookson and J. Hasty, ACS Synth. Biol., 2016, 5, 8–14 CrossRef CAS PubMed.
- Y.-J. Eu, H.-S. Park, D.-P. Kim and J. Wook Hong, Biomicrofluidics, 2014, 8, 024113 CrossRef PubMed.
- Z. Xu, Y. Wang, Y. Chen, M. H. Spalding and L. Dong, Biomicrofluidics, 2017, 11, 064104 CrossRef PubMed.
- H. Abdulla Yusuf, S. M. Z. Hossain, A. A. Khamis, H. T. Radhi, A. S. Jaafar and P. R. Fielden, Arabian J. Sci. Eng., 2021, 46, 6765–6774 CrossRef.
- H. S. Kim, T. L. Weiss, H. R. Thapa, T. P. Devarenne and A. Han, Lab Chip, 2014, 14, 1415–1425 RSC.
- P. J. Graham, J. Riordon and D. Sinton, Lab Chip, 2015, 15, 3116–3124 RSC.
- F. Liu, L. Gaul, F. Shu, D. Vitenson and M. Wu, Lab Chip, 2022, 22(17), 3138–3146 RSC.
- F. Liu, L. Gaul, A. Giometto and W. Mingming, Sci. Rep., 2024, 14(1), 1–13 CrossRef PubMed.
- B. J. Kim, L. V. Richter, N. Hatter, C.-K. Tung, B. A. Ahner and M. Wu, Lab Chip, 2015, 15, 3687–3694 RSC.
- M. M. Allen, J. Phycol., 1968, 4, 1–4 CrossRef CAS PubMed.
- R. Rippka, J. Deruelles, J. B. Waterbury, M. Herdman and R. Y. Stanier, Microbiology, 1979, 111, 1–61 Search PubMed.
- J. J. Yu, M. Liberton, P. F. Cliften, R. D. Head, J. M. Jacobs, R. D. Smith, D. W. Koppenaal, J. J. Brand and H. B. Pakrasi, Sci. Rep., 2015, 5(1), 1–10 CAS.
- R. Y. Stanier, R. Kunisawa, M. Mandel and G. Cohen-Bazire, Bacteriol. Rev., 1971, 35, 35 CrossRef PubMed.
- F. Brandenburg, H. Schoffman, S. Kurz, U. Krämer, N. Keren, A. P. Weber and M. Eisenhut, Plant Physiol., 2017, 173, 1798–1810 CrossRef CAS PubMed.
- D. C. Ducat, J. A. Avelar-Rivas, J. C. Way and P. A. Silver, Appl. Environ. Microbiol., 2012, 78, 2660–2668 CrossRef CAS PubMed.
- C. R. Harris, K. J. Millman, S. J. van der Walt, R. Gommers, P. Virtanen, D. Cournapeau, E. Wieser, J. Taylor, S. Berg, N. J. Smith, R. Kern, M. Picus, S. Hoyer, M. H. van Kerkwijk, M. Brett, A. Haldane, J. F. del Río, M. Wiebe, P. Peterson, P. Gérard-Marchant, K. Sheppard, T. Reddy, W. Weckesser, H. Abbasi, C. Gohlke and T. E. Oliphant, Nature, 2020, 585, 357–362 CrossRef CAS PubMed.
- W. Albrecht, J. Moers and B. Hermanns, J. Large Scale Res. Facil., 2017, 3(A112), 1–9 Search PubMed.
- A. Grünberger, C. Probst, A. Heyer, W. Wiechert, J. Frunzke and D. Kohlheyer, J. Visualized Exp., 2013, e50560 Search PubMed.
- X. G. Zhu, S. P. Long and D. R. Ort, Curr. Opin. Biotechnol., 2008, 19, 153–159 CrossRef CAS PubMed.
- E. Lee, M. Jalalizadeh and Q. Zhang, Algal Res., 2015, 12, 497–512 CrossRef.
- A. D. Jassby and T. Platt, Limnol. Oceanogr., 1976, 21, 540–547 CrossRef CAS.
- T. T. Bannister, Limnol. Oceanogr., 1979, 24, 76–96 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- A. Parslow, A. Cardona and R. J. Bryson-Richardson, J. Visualized Exp., 2014, 51086 Search PubMed.
- C. Allan, J.-M. Burel, J. Moore, C. Blackburn, M. Linkert, S. Loynton, D. MacDonald, W. J. Moore, C. Neves, A. Patterson, M. Porter, A. Tarkowska, B. Loranger, J. Avondo, I. Lagerstedt, L. Lianas, S. Leo, K. Hands, R. T. Hay, A. Patwardhan, C. Best, G. J. Kleywegt, G. Zanetti and J. R. Swedlow, Nat. Methods, 2012, 9, 245–253 CrossRef CAS PubMed.
- T. Scherr, J. Seiffarth, B. Wollenhaupt, O. Neumann, M. P. Schilling, D. Kohlheyer, H. Scharr, K. Nöh and R. Mikut, PLoS One, 2022, 17, e0277601 CrossRef CAS PubMed.
- J. Seiffarth, T. Scherr, B. Wollenhaupt, O. Neumann, H. Scharr, D. Kohlheyer, R. Mikut and K. Nöh, SoftwareX, 2024, 26, 1–6 CrossRef.
- K. J. Cutler, C. Stringer, T. W. Lo, L. Rappez, N. Stroustrup, S. Brook Peterson, P. A. Wiggins and J. D. Mougous, Nat. Methods, 2022, 19, 1438–1448 CrossRef CAS PubMed.
- K. Kasahara, M. Leygeber, J. Seiffarth, K. Ruzaeva, T. Drepper, K. Nöh and D. Kohlheyer, Front. Microbiol., 2023, 14, 1–13 Search PubMed.
- B. M. C. Martins, A. K. Tooke, P. Thomas and J. C. W. Locke, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E11415–E11424 CAS.
- K. Moore, S. Altus, J. Tay, J. Meehl, E. Johnson, D. Bortz and J. Cameron, Nat. Microbiol., 2020, 5(5), 757–767 CrossRef CAS PubMed.
- A. Richmond, Handbook of microalgal culture : applied phycology and biotechnology, Wiley Blackwell, Chichester, West Sussex, U.K., 2nd edn, 2013 Search PubMed.
- V. Loomba, G. Huber and E. von Lieres, Biotechnol Biofuels, 2018, 11(149), 1–11 Search PubMed.
- H. S. Kim, T. P. Devarenne and A. Han, Lab Chip, 2015, 15, 2467–2475 RSC.
- A. J. Maki, M. Peltokangas, J. Kreutzer, S. Auvinen and P. Kallio, Chem. Eng. Sci., 2015, 137, 515–524 CrossRef CAS.
- J. J. Carroll, J. D. Slupsky and A. E. Mather, J. Phys. Chem. Ref. Data, 1991, 20, 1201–1209 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4lc00567h |
| This journal is © The Royal Society of Chemistry 2025 |