
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ measurement of sulfur isotope ratios in sulfide samples with LA-ICP-MS/MS using N2O and He reaction gas†
Estida
Eensoo
 *a,
Päärn
Paiste
a,
Kärt
Paiste
ab,
David A.
Fike
b and
Jennifer L.
Houghton
b
*a,
Päärn
Paiste
a,
Kärt
Paiste
ab,
David A.
Fike
b and
Jennifer L.
Houghton
b
aDepartment of Geology, University of Tartu, Tartu, 50411, Estonia. E-mail: estida@ut.ee; estida.eensoo@gmail.com
bDepartment of Earth, Environmental & Planetary Sciences, Washington University in St Louis, St Louis, MO 63130, USA
First published on 4th July 2025
Abstract
Sulfur isotope signatures (δ34S) in sulfide minerals such as pyrite and pyrrhotite may reflect the specific geological conditions at their genesis. Understanding the δ34S variability can help track (bio)–geochemical processes, from ore formation to finding evidence of early life. However, as sulfide mineral growth can occur at various stages of rock history, traditional bulk S isotope analysis can incorporate mixed geochemical signals generated by unrelated processes. In situ analytical techniques can be used to investigate compositional changes in δ34S caused by early environmental or secondary processes. In this study, we aim to characterize δ34S variability in pyrite and pyrrhotite using laser ablation inductively coupled plasma tandem mass spectrometry (LA-ICP-MS/MS) while introducing a mixture of N2O and He in the reaction chamber to remove polyatomic interferences at m/z = 32 and m/z = 34. Alongside tuning the respective laser and ICP parameters, we employ a self-developed signal-smoothing device consisting of coiled thermoplastic elastomer (TPE) tubing and a cyclonic spray chamber to achieve better signal stability. In this way, we propose a new, fast, in situ screening approach for measuring the δ34S of sulfides.
1. Introduction
Pyrite (FeS2) and pyrrhotite (FeS), as part of the sulfide minerals category, are widely found in various geological environments, and they serve as valuable indicators in exploring geochemical processes. The genesis of pyrite occurs under a diverse set of conditions, from sediment deposition to multiple stages of metamorphism, resulting in petrographically distinct pyrites with diagnostic sulfur isotope and trace element compositions. Pyrrhotite may also appear in early or late stages of rock formation; however, it is metastable in low-temperature sedimentary environments where it converts to pyrite.1 These iron sulfides exhibit δ34S compositions that reflect the changes in the chemistry of mineralizing fluids and allow differentiation between biogenic and abiogenic sulfur cycling processes.2 Therefore, the sulfur isotopes in pyrite and pyrrhotite are powerful tools for investigating processes such as sulfide-gold-ore formation,3–5 tracking oil generation and migration in petroleum reservoirs,6,7 as well as for searching for evidence of early life in Earth and beyond,2,8 and biogeochemical processes throughout Earth's history.9Regardless of the specific formation pathway, the genesis of FeS2 and FeS typically occurs in diffusively limited environments where the mineralizing fluid's properties affect mineral crystallization and chemical composition. Since the expression of S isotope fractionations is sensitive to open-vs-closed system conditions, heterogeneous δ34S signatures can form on the micro-scale and be preserved in iron sulfides that precipitate over time in evolving environmental conditions.9–11 In addition, late-stage processes can introduce secondary sulfur- and metal-bearing fluids into sedimentary systems, thereby inducing the precipitation of multiple generations of genetically and isotopically distinct sulfide species.9,12–14 In addition, as the temperature rises during metamorphism, pyrite can transform into pyrrhotite, and this process can reverse during the cooling stages of metamorphism.15 On a bulk scale, isotopic fractionations accompanying the conversion of pyrite to pyrrhotite or vice versa are small;16,17 however, isotopic zoning within individual crystals can develop.15,18 Consequently, specific environmental conditions and late-stage alteration can lead to significant pyrite and pyrrhotite compositional variability, undetectable by traditional bulk extraction methods. During the commonly applied chromium-reducible sulfur (CRS) and acid-volatile sulfur (AVS) extractions,19 the sulfur isotope variability is homogenized as the different components of a sample are combined. In contrast, petrographic investigations combined with high-resolution sulfur isotope analysis allow for a more comprehensive approach to assessing the processes and factors influencing mineral growth, recrystallization, and alteration.
The traditional CRS and AVS extraction techniques are complemented by high-resolution techniques capable of δ34S measurements, such as secondary ion mass spectrometry (SIMS) and laser ablation inductively coupled mass spectrometer (LA-ICP-MS), allowing for finer detailed analysis with minimal sample preparation.20 Even though SIMS offers superior spatial resolution and high precision and accuracy in measuring sulfur isotope ratios,21–23 it is less accessible, more complex to operate, and unsuitable for large sample quantities.24 Alternatively, rapid developments in the field of LA-ICP-MS25–27 have made it a viable, time- and cost-efficient alternative for in situ sulfide δ34S measurements. Especially the introduction of multiple collector ICP-MS (MC-ICP-MS) has enabled high precision and rapid isotope ratio measurements, and thus, significant effort has been placed into improving such instruments' application in geological research.26,28,29 On the other hand, tandem ICP-MS/MS, although favoured for its affordability, versatility, speed, and efficiency in routine elemental analyses, has received relatively little attention in stable isotope measurements due to its comparatively lower precision. In sulfur isotope analysis, the presence of isobaric effects at mass-to-charge ratios m/z = 32 and m/z = 34, mainly from O2 and at m/z = 32 from NO have limited the application of tandem systems. Despite this drawback, the possibility of using so interferences at the masses of interest shows promise in expanding the LA-ICP-MS/MS application in determining δ34S of iron sulfide minerals.27 Here, we build upon the idea to improve the application of LA-ICP-MS/MS as a fast-screening method for pyrite and pyrrhotite δ34S measurements in geosciences.
2. Experimental
2.1 Samples and in-house reference materials
We selected five sulfide samples with unknown δ34S compositions. A vein pyrite collected from the Maksovo open-pit quarry near the village of Shunga, where the rocks from the Paleoproterozoic Zaonega formation are exposed (Onega Basin, Karelia, Russia; coordinates 62°35′35′′N, 34°56′52′′E). The Zaonega formation's coarse (mm-sized individual crystals) anhedral pyrite from a laminated quartz vein shows vein selvage overgrowth texture, suggesting it grew relatively late in vein development. Additionally, the study involved four Paleoproterozoic graphitic schist samples containing pyrrhotite from the Uljaste küla drill core (Ida-Viru County, Estonia, coordinates 59°21′25.19′′N 26°47′. 7.79′′E), at depths of 227.8 m, 249.9 m, 305.1 m, 317 m, and 334.5 m. The unknown pyrite sample in this article is referred to as Zf-pyrite, and the pyrrhotite samples are called Uljaste pyrrhotites.During the experiments, we applied an in-house pyrite reference material provided by D. Fike (Washington University in St Louis) and obtained from Ward's Science (Rochester, NY, USA). Previously conducted isotopic ratio mass spectrometry (IRMS) analysis assigned Ward's pyrite a δ34S value of −1.0 ± 0.2‰ (V-CDT).22 SIMS analysis has shown a fragment-to-fragment reproducibility of ±1.9‰ and intra-fragment reproducibility of ±2.8‰ for Ward's pyrite.22 We utilized a mm-sized porphyroblast Balmat pyrite obtained from J. Valley (University of Wisconsin–Madison) as a quality control pyrite sample between pyrite's analytical runs. The Balmat sample is well characterized and previously proposed as a potential S isotope reference material.20 The Balmat sample has an IRMS δ34S = 15.1 ± 0.2‰ (V-CDT), intra-crystal reproducibility within 0.1‰20, and inter-crystal variability δ34S in the range of 14.4–14.9‰.26 However, δ34S values from 13.8 to 16.4‰ have also been reported for this pyrite.20,26,30–32 During the tuning procedures for the ICP-MS/MS instrument, an approximately four cm-sized cubic hydrothermal pyrite crystal from the Huanzala mine (Huánuco, Peru coordinates 9°55′04′′S 76°59′50′′W) was used. A pyrrhotite obtained from Ward's Science and provided by D. Fike, was also used as in-house reference material with a preliminary working value of δ34S = −0.11 ± 0.5 (V-CDT) ‰ determined by IRMS analysis.
The Zf-pyrite and the Huanzala pyrite rock specimens, which subsequently cut into smaller approximately 1 cm3 sections to allow mounting into the ablation cell. The samples underwent a systematic grinding and polishing process to prepare an even and well-polished surface for analysis. Initially, the samples were ground using silicon carbide grinding papers from Buehler Metallography with index P = 2500 and P = 4000, followed by a series of diamond polishes using 9 μm, 6 μm, 3 μm, and 1 μm solutions. Afterward, we cleaned the samples with ethanol to remove any possible residues. The individual grains of Ward's pyrrhotite and Balmat's pyrite were embedded in a round (d = 25 mm) epoxy mount. The Ward's pyrite sample was an irregularly shaped rock (∼1 cm in diameter); therefore, to prepare it for LA-ICP-MS/MS analysis, it was also embedded in epoxy. The sample mounts were ground and polished, following the same steps as the Zf and Huanzala pyrite. In addition, the Estonian Geological Survey provided the Uljaste pyrrhotite samples, which were polished thick sections.
2.2 IRMS analysis
We determined the 34S/32S ratios in pyrite and pyrrhotite samples from powdered specimens obtained through micro-drilling (Minimo One Series Ver.2 micro grinder, with 1 mm diamond-coated drill bit) at adjacent locations. Specifically, we drilled a grid of 16 spots (4 × 4) equally spaced from each other on the crosscut 4 × 4 cm Huanzala pyrite sample, covering the sample surface (Fig. S1, in ESI†). Additionally, five replicates were drilled on the surface of Ward's pyrite, and two replicates were sampled on Zf-pyrite (including one from the vein and one from non-vein Zf-pyrite). Similarly, for each of the Uljaste pyrrhotite samples, we extracted two adjacent replicates from the pyrrhotite veins. The limited number of pyrrhotite replicates was due to the small areas of pyrrhotite in the Uljaste samples. Prior characterization of the Uljaste samples was conducted using reflective light microscopy and a ZEISS EVO MA15 scanning electron microscope (SEM) at the Department of Geology, University of Tartu. The SEM images were used to identify optimal drilling locations.Around 300 μg of the drilled powdered material was loaded into tin capsules with excess V2O5. We measured the 34S/32S ratios using a Thermo Flash HT Element Analyzer connected with Thermo Delta B plus IRMS via ConFlo IV open split device at the University of Tartu. The sulfur isotope compositions are expressed in standard delta notation as per mil (δ34S in ‰) deviations from the Vienna Canyon Diablo Troilite (V-CDT) standard33 as follows:
| δ34SVCDT (‰) = [(34S/32S)sample/(34S/32S)VCDT − 1] × 1000 | (1) |
To calibrate the S isotopic ratios (IR) values, international standards composed of BaSO4 such NBS-127 and IAEA-SO-6 were included in each analytical run. Four aliquots of each international standard were analysed-two at the beginning and two at the end of each analytical run to assess the instrumental stability over time. The δ34S value of NBS-127 is 20.3 ± 0.2‰, while the operating value of the IAEA-SO-6 reference material is −34.1 ± 0.2‰.
2.3 LA-ICP-MS/MS analysis
We performed the sulfur isotope measurements at the University of Tartu utilizing a Cetac LSX-213 G2+ laser system with a HelEx II fast-washout two-volume large format cell using 800 mL per min helium as carrier gas. The LSX-213 G2+ system features an Nd: YAG laser (Neodymium-Doped Yttrium Aluminium Garnet). Typically, we ablated the samples at locations adjacent to the micro-drilled craters created by IRMS (see Fig. 1 below). | ||
| Fig. 1 Microscopic images of the Uljaste 304.5 pyrrhotite sample showing the micro-drilled crater's location and the LA-ICP-MS/MS spots. IRMS spot d = 1 mm, LA-ICP-MS spot d = 100 μm. | ||
The pyrite and pyrrhotite samples and standards are ablated in the same analytical run and placed evenly in the sample holder (12 cm × 12 cm × 1 cm). Subsequently, the composition of the ablated material was analysed using an Agilent 8800 quadrupole ICP-MS/MS.
Configuration A: this setup included a straight 20 cm polypropylene-based thermoplastic elastomer (TPE tube from Pharmed® BPT) with an internal diameter of 1.52 mm connected to the “squid” device and the ICP.
Configuration B: this setup consisted of the same TPE tubing coiled 5 times around a cylinder with a 7 mm outer diameter (O.D) attached to the “squid” device.
Configuration C: the same TPE tubing without coiling was connected to the “squid” device paired with a cyclonic spray chamber (Agilent Isomist Spray Chamber).
Configuration D: the configuration combined the TPE tubing coiled 5 times around a cylinder with 7 mm O.D., with the “squid” and the cyclonic spray chamber (see Fig. 2).
 | ||
| Fig. 2 Signal smoothing configuration D connecting to “squid” compartment, composed of 5 times coiled TPE tubing around a cylinder (O.D. 7 mm) and a cyclonic spray chamber, which connects to the ICP-MS system. | ||
During these experiments, the sulfur signal was measured as two primary reaction products: SO+ and SO2+. Sulfur ions were detected at specific mass-to-charge (m/z) ratios: m/z = 48 for 32S16O+ and m/z = 50 for 34S16O+ in case of SO+, and m/z = 64 for 32S16O2+ and m/z = 66 for 34S16O2+ in case of SO2+.
The laser parameters for measuring sulfur signals as SO+ reaction products included the spot size set at 40 μm, fluence at 10 J cm−2, and repetition rate of 10 Hz. For measuring sulfur signals as SO2+ reaction product, configurations included a 50 μm spot size and fluence of 19.5 J cm−2, while maintaining the repetition rate at the same value. Each ablation ran for 90 seconds: 20 s for warming up the laser, followed by the next 40 s for ablation, and 30 seconds for cooling down the laser. On the ICP-MS/MS, the sampling depth was changed between SO and SO2-based experiments, corresponding to 6.0 mm for measuring SO+ and 7.0 mm for SO2+ reaction products. We tuned the ICP-MS/MS to maintain the 32S16O+ and 32S16O2+ signal counts of reaction product below 106 counts per second (cps), while dwell times were at 0.0465 s for each product ion mass. During this experiment, the 4th cell gas's operating flow was at 40%. The ablation targeted nine evenly spaced spots on the surface of the Huanzala pyrite (100 μm apart) for each configuration. To characterize the outcomes of this experiment, we use the reproducibility of the non-drift corrected S isotope ratios, measurement uncertainty, and washout time.
2.3.1.1 Reproducibility of the non-drift corrected sulfur isotope ratios. Background-corrected S signals from nine ablations for each signal-smoothing configuration provided the data needed to calculate reproducibility. Data extraction was done through Iolite 3.62 software, where we defined sections for individual baselines and sample signals. Additionally, we used the “Mean without Outlier Rejection” option to allow the software to generate sulfur isotope ratios. The reproducibility is presented as the relative standard deviation in ‰ without applying additional drift correction (eqn (2)).
 | (2) |
![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) : mean value of sulfur isotope ratios. n: number of measurements (n = 9).
: mean value of sulfur isotope ratios. n: number of measurements (n = 9).
2.3.1.2 Measurement uncertainty. We calculated the measurement uncertainty to assess the stability of the non-drift corrected sulfur isotope ratios during the ablation of the nine spots for each signal-smoothing configuration. For each spot, we then calculated the standard deviation of these uncorrected isotope ratios, expressed in ‰. The overall measurement uncertainty for each configuration was calculated by taking the mean standard deviation of the nine spots (eqn (3)).
 | (3) |
2.3.1.3 Washout. Our primary goal was to obtain a smooth ablation profile by keeping the time between analyses (e.g., washout) as short as possible. By washout, we refer to the time it takes for the signal to decrease to the initial background level at the end of ablation. To calculate the washout, we take the ratio of the average counts per second (cps) between two distinct time intervals (A) 3 seconds before turning off the laser, and (B) 8–10 seconds after turning off the laser or, alternatively, between A and (C) 18–20 seconds after turning off the laser. Washout is especially troublesome for ablating sulfides due to the slow desorption of sulfur species from the inlet line.25,34 The so-called memory effects associated with S species cause longer delays between analyses and increase the fraction of time spent on data acquisition.
In the first set of experiments, we investigated the reaction product distribution for pure N2O, increasing the gas flow rate from 10% to 100% in 5% increments. In the second set, we fixed the N2O flow rates at 5%, 10%, 15%, and 20% while progressively adding He from 1 to 7 mL min−1 during a continuous ten-minute line scan. All ICP-MS/MS tuning parameters remained consistent between the two experiments, except for the energy discrimination. For the N2O experiment alone, the energy discrimination value was set at −3 V, and for the N2O–He experiment at −7 V, to achieve the highest signal output for the main reaction products. Following each change in N2O or He gas flow rate, we allowed the signal to stabilize for 30 seconds. The reported signals in this experiment result from recording a 20 seconds online signal measurement and calculating its average.
Combination 1: both S reaction product signals are maintained in pulse mode for spot analysis.
Combination 2: 34S16O2 in pulse mode, while 32S16O in analog mode for line scans.
Combination 3: 34S16O2 in pulse mode, while 32S16O in analog mode for spot analysis.
During these experiments, both monitored masses had dwell times of 0.05 s with a total sampling period of 0.114 s. Parameters that were tuned the same during the analysis in different detection modes were reflected power (1100 V), sampling depth (5 mm), and wait time offset (5 s). Additional ICP-MS/MS tuning parameters that differed during our analytical modes were He flow rate (2.5 mL min−1 in pulse–pulse, 4.5 mL min−1 in pulse-analog), 4th gas flow (15% in pulse–pulse and 5% in pulse-analog), and energy discrimination (−3 V in pulse–pulse and −7 V in pulse-analog). For pulse–pulse mode experiments, the 32S16O signal intensity was in the range of 8–9 × 105 cps for all samples, while for pulse-analog mode experiments, the 34S16O2 signal was tuned to the same range. We adjusted the laser parameters for each pyrite and pyrrhotite sample to achieve a smooth ablation signal profile. Pyrite and pyrrhotite samples usually display similar ablation characteristics within the respective group (see Table 1). During the ablation of Ward's pyrrhotite, hidden cracks became visible on the sample surface. To mitigate their impact, pre-ablation was conducted to expose and exclude these areas from subsequent ablations.
| Sample type | Analysis mode | Spot size (μm) | Fluence (J cm−2) | Repetition rate (Hz) | Scan speed (μm s−1) |
|---|---|---|---|---|---|
| a Laser firing time consisted of 20 s of gas background, 40 s of ablation, and 30 s wash out. | |||||
| Pyrite samples | Spot | 50 | 10 | 5 | — |
| Lines | 40 | 8 | 10 | 10 | |
| Pyrrhotite samples | Spot | 100 | 10 | 5 | — |
| Lines | 100 | 8 | 8 | 10 | |
The normalization of the δ34S is done by following the sample-standard-bracketing approach (SSB) applying both Ward's pyrite and Ward's pyrrhotite.35 Before the 34S/32S measurements, we conditioned the plasma by ablating 50 spots randomly allocated on the Huanzala pyrite. After stabilizing the plasma, the sequence began with the ablation of five spots on Ward's pyrite standard, followed by five spots on the Zf-pyrite, five spots on the pyrrhotite standard, and lastly, five spots on the Uljaste pyrrhotite. This cycle of ablating 5 spots of the standards before samples was repeated four times.
| Parameter (unit) | Assigned value |
|---|---|
| Forward power (V) | 1170 |
| Nebulizer gas flow (mL min−1) | 1.10 |
| Extract lenses 1 & 2 (V) | −24.0 & −210.0 |
| He flow rate (mL min−1) | 4.5 |
| 4th reaction gas flow rate (%) | 5 |
| Sampling depth (mm) | 5.0 |
| Energy discrimination (V) | −7 |
| Wait time offset (ms) | 5 |
Initial data reduction of LA-ICP-MS/MS data for all experiments was performed using Iolite 3.62 software. Each ablation signal was manually integrated whereby the most stable ablation region was selected, excluding regions with signal spikes. Additionally, the sample backgrounds were manually defined to exclude any spikes. Furthermore, every crater was visually investigated after ablation, and signals from craters that revealed cracked surfaces or mixed ablation of sulfides and surrounding matrix were excluded further data processing (Fig. S4, in ESI†). Afterwards, the software algorithm calculates the raw isotopic ratio for each spot. We have selected the “mean with no outlier rejection” option so the software calculates the average using all data points. The generated signals were exported for post processing in Microsoft Excel. For δ34S calculations, a second or third order polynomial fit was used for mass bias correction by using the signals of Ward's pyrite and pyrrhotite as in-house reference materials. The polynomial curve was applied to account for the instrumental mass bias, where we applied the correction function to the samples measured isotope ratios. The final δ34S is presented as the average δ34S of n-spots relative to Vienna Canyon Diablo Troilite (eqn (1)), while the uncertainties are expressed as one sigma (1σ) standard deviation.
3. Results and discussion
3.1 IRMS assigned sulfur isotopic composition of the samples
Additionally, the micro-drilled δ34S values for the Zf-pyrite are −11.1‰ and −9.87‰ (n = 2). The studied Zf-pyrite's δ34S composition is within range of [−20 to 0] ‰ of the δ34S values obtained from metamorphosed strata at the contacts of mafic magmatic intrusions with sedimentary host rocks of the FAR-DEEP 12AB core, located ∼3 km to the north of the Maksovo quarry.36
3.2 The effect of signal-smoothing devices
For all configurations, the measurement uncertainty and reproducibility based on uncorrected isotope ratios of nine spot analyses are presented in Table 3.| Interface configuration | Reproducibility RSD in ‰ (n = 9) | Measurement uncertainty (±1σ, n = 9) in ‰ | Washout A/B | Washout A/C |
|---|---|---|---|---|
| Configuration A for SO | 4.3 | 1.8 | 68.1 | 70.8 |
| Configuration A for SO2 | 9.1 | 2.1 | 22.7 | 22.9 |
| Configuration B for SO | 2.9 | 1.7 | 64.0 | 66.1 |
| Configuration B for SO2 | 2.4 | 2.0 | 22.5 | 22.7 |
| Configuration C for SO | 2.0 | 1.7 | 50.7 | 58.0 |
| Configuration C for SO2 | 3.2 | 2.2 | 20.5 | 21.9 |
| Configuration D for SO | 1.9 | 1.7 | 48.9 | 58.8 |
| Configuration D for SO2 | 1.8 | 1.8 | 20.5 | 22.0 |
Adding coils to the TPE tubing and the cyclonic spray chamber improves the stability of the signal, resulting in better reproducibility and measurement uncertainty between the spot analyses. With maximal smoothing (configuration D), reproducibility for SO+ is reduced from 4.3 to 1.9‰ and from 9.1 to 1.8‰ for SO2+, compared to the minimal smoothing in configuration A. In this study, the primary function of the cyclonic spray chamber is to act as a mixing chamber, homogenizing the aerosol before it enters the plasma. The coiled tubing serves a specific function: it is used to remove larger particles from the laser aerosol flow through centrifugal force. These larger particles can contribute to significant elemental and isotopic fractionation in the plasma due to incomplete ionization.26 However, compared to SO+, the SO2+ signal to background ratio as indicated by Washout A/C is lower affecting the observed measurement uncertainty.
In Fig. 3, the representative washout profiles are shown. For experimental configurations A and B, the washout between intervals A/B and A/C is comparable, and the baseline signal is achieved 10 seconds after the ablation ends. By looking at Fig. 3, in the 62–63 s range, it can be seen that the TPE tubing coiling in C configuration introduces a slight delay in signal drop. Adding the cyclonic spray chamber in configurations C and D further increases the washout times and background signal, as seen by the difference between A/B and A/C interval values compared to configurations A and C (Fig. 3 and Table 3). An increase in the background signal for configurations C and D could arise from the connections between the added cyclonic spray chamber and air permeation through the connections. Although the washout time was increased, background equivalent signals were achieved 20 s after the end of ablation. To reduce possible sample carryover effects, regular cleaning of the sample introduction system using high flows of inert gas or air is conducted.
 | ||
| Fig. 3 Signal tailings after applying signal-smoothing configurations. Only one representative signal is shown in the picture. | ||
For subsequent measurements of sulfur isotope ratios of the unknown samples and standard materials, we increased the number of coils to seven on the TPE tubing, which was the maximum number attainable with the tubing length used in the experiments. We noticed that seven coils lowered the measurement uncertainty of configuration D to 0.7‰ (Table 4).
| Analysis mode on configuration D with 7 coils | Reproducibility RSD in ‰ (n = 9) | Measurement uncertainty (±1σ, n = 9) in ‰ | Washout A/B | Washout A/C |
|---|---|---|---|---|
| Pulse–pulse spots | 2.2 | 1.9 | 17.4 | 18.2 |
| Pulse-analog lines | 5.5 | 4.8 | 27.8 | 29.0 |
| Pulse-analog spots | 3.6 | 0.9 | 16.7 | 17.2 |
| Natural pyrrhotite sample experiment | 2.0 | 0.7 | 19.3 | 19.7 |
3.3 The effects of N2O and N2O + He on sulfur signal
The effect of N2O as a reaction gas on the SO+ and SO2+ reaction product signal intensities were tested over a range of N2O gas flows (from 10 to 100% 4th cell gas flow) while laser ablation parameters remained constant:The main interferences arising from the use of N2O as reaction gas on the SO+ reaction product are N2O+ and possibly O3+ ions and for SO2+ reaction products the O4+ ion cluster (O2+· O2).37
The N2O was selected as a reaction gas owing to its enhanced reaction efficiency towards sulfur, when compared to more commonly used O2 gas.38
The 32S16O+ signal intensity was the highest at 20% N2O (∼300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cps), and from 15% N2O, the 32S16O2 signal intensity surpassed that of 32S16O+ and reached peak intensity (∼550000 cps) at 35% N2O (Fig. 4). Although highest SO2+ product ions signal was achieved at 35% N2O flow rate, the background signal at m/z = 64 increases with N2O flow rate. This trend is likely due to the enhanced formation of the O4+ ion cluster (O2+· O2)37 caused by higher N2O gas density in the collision-reaction cell. As the disassociation energy required for breaking the NN–O bond is 1.7 eV, with increased N2O flow the O2 availability increases and follows the reaction principle as eqn (4):38
000 cps), and from 15% N2O, the 32S16O2 signal intensity surpassed that of 32S16O+ and reached peak intensity (∼550000 cps) at 35% N2O (Fig. 4). Although highest SO2+ product ions signal was achieved at 35% N2O flow rate, the background signal at m/z = 64 increases with N2O flow rate. This trend is likely due to the enhanced formation of the O4+ ion cluster (O2+· O2)37 caused by higher N2O gas density in the collision-reaction cell. As the disassociation energy required for breaking the NN–O bond is 1.7 eV, with increased N2O flow the O2 availability increases and follows the reaction principle as eqn (4):38
O2+ + 2O2 ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) O4+ + O2 O4+ + O2 | (4) |
 | ||
| Fig. 4 The reaction efficiency and background signals under different flow rates of N2O gas as signal intensities of 32S16O and 32S16O2 product ions. | ||
Increase in the O4+ ion formation during this ion–gas reaction pathway have been shown to increase with increasing gas density in the reaction chamber in SIFT instruments.39
Based on signal smoothing experiments, it was, however, evident that the signal-to-background ratio for the 32S16O2+ reaction product was approximately 3 times lower than that of the 32S16O+ reaction product under comparable conditions, expressed as the A/C ratio in Table 3. The reason behind this difference could be due to a higher formation rate of 16O4+ ions compared to 14N218O+ ions under the tested conditions. Therefore, we tested how the addition of He to the collison reaction cell at different N2O gas flows would influence the reaction efficiency of 32S16O+ and 32S16O2+ product ion production.
The investigation's purpose was to see if using an inert gas in addition to a reactive gas changes could change the reaction dynamics to affect the product ion distribution and formation rate. In our case, He was used as a collision gas.
The combination of N2O and He as cell gases significantly attenuates the 32S16O2 signal, reducing detector counts to below 200000 cps across all N2O and He flow rate configurations (Fig. 5). In contrast, the 32S16O signal amplifies considerably, and the optimal He flow rate depends on the N2O flow rate. The optimal conditions for 32S16O+ product ion generation are achieved when using a mixture of a 5% N2O flow rate and a He flow rate of 4.5 mL min−1. Notably, for SO+ product ions at a 20% N2O flow rate with a He flow rate of less than 3 mL min−1, the combination of reactive and inert gases yields superior results relative to the use of pure N2O. The 32S16O signal intensity is approximately 350000 cps (Fig. 5), compared to about 300000 cps when using N2O alone at the optimum 20% concentration (Fig. 4). Under the optimized analytical conditions for SO+ and SO2+ ion formation, the background signal at m/z = 64 (for SO2+) was 2 times higher than at m/z = 48 (for SO+). Owing to the higher signal intensity, 32S16O-based signals exhibited approximately 3 times higher signal-to-background ratio than their 32S16O2-based counterparts when other analytical conditions were identical.
 | ||
| Fig. 5 The reaction efficiency under different He and N2O gas flow rates as signal intensities of 32S16O and 32S16O2 product ions. | ||
3.4 Effect of detection mode and sample matrix on δ34S measurements
We selected two in-house reference materials- Ward's pyrite and pyrrhotite-as reference materials to correct for the instrumental mass bias during measurement of sulfur isotopic ratios in our natural sulfide samples. In addition, we investigated measuring the sulfur signals in different detection modes. These modes included maintaining the 32S16O+ and 34S16O2+ reaction product signal in pulse detection mode during spot analysis and then changing analysis conditions so that the 32S16O signal would be collected in analog mode for spot and line ablations. The measured sulfur isotope ratios were normalized using Wards pyrite (Fig. 6) and pyrrhotite (Fig. 7) in-house reference materials and compared to the values obtained by IRMS analysis. For samples with single IRMS analysis result, error of 0.2‰ was used in Fig. 6–8. When multiple analysis results were available, experimentally determined 1σ values were applied. | ||
| Fig. 6 Sulfur isotope ratios of pyrite and pyrrhotite samples in different detection modes normalized by Ward's pyrite. | ||
 | ||
| Fig. 7 Sulfur isotope ratios of pyrite and pyrrhotite samples in different detection modes normalized by Ward's pyrrhotite. | ||
For Zf-pyrite, a slight negative bias is observed in pulse–pulse mode when normalized with pyrite (−0.12‰). In contrast, pulse-analog mode for line scans shows positive bias (0.82‰), which becomes even more pronounced when using pyrrhotite as the reference material (2.73‰). Additionally, in pulse-analog mode spot analysis, the bias is inconsistent—positive when normalized with pyrite and negative when normalized with pyrrhotite.
For Uljaste pyrrhotite sample a positive bias was observed in case of both normalization procedures (Fig. 6 and 7). It was noticed that the variabilities obtained for the Zf-pyrite and Uljaste samples are within the uncertainty range of the IRMS control value when normalizing with either pyrrhotite or pyrite reference materials.
The results show that Ward's pyrite sample exhibits a smaller bias (−0.07‰) in pulse–pulse mode when normalized with pyrrhotite. However, considering the standard deviation values, normalization with Ward's pyrite provides better precision (standard deviation of 0.35‰, compared to 1.07‰ with Ward's pyrrhotite).
Conversely, the Ward's pyrrhotite sample aligns well with IRMS values across all detection modes when normalized with pyrrhotite. In contrast, the best precision is achieved in pulse–pulse mode under this normalization (standard deviation 1σ = 1.32‰ compared to 1.65‰ in pulse-analog lines and 1.62‰ in pulse-analog spots). When normalized with Wards pyrite the 1σ deviations were between 1.45 to 1.88‰ and a larger bias was observed. However, in pulse–pulse mode the bias was minimal (−0.05‰)
In the subsequent experiments, we used the pulse–pulse mode for analysis. The pyrite samples were normalized using Ward's pyrite, while pyrrhotite samples were normalized with Ward's pyrrhotite.
3.5 Measurements of the natural pyrite and pyrrhotite samples
The sulfur isotope ratios δ34S of pyrite and pyrrhotite samples were derived by applying the methodologies outlined for signal smoothing, reaction gas combinations, and detector modes. We present the δ34S values from multiple spot analyses within the same textural regions of individual pyrite and pyrrhotite samples, along with their associated uncertainties calculated as 1σ standard deviation. The isotope ratios for the Uljaste pyrrhotite samples are illustrated in Fig. 8, and for the Balmat and Zf-pyrite are presented in Table 5.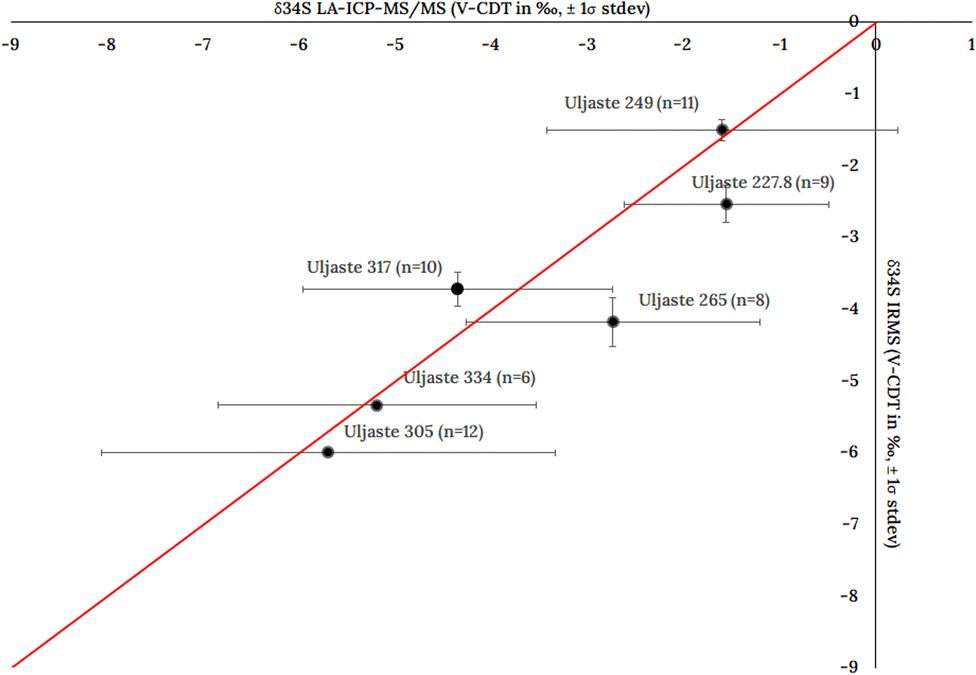 | ||
| Fig. 8 Comparison of the LA-ICP-MS with the IRMS control values for Uljaste pyrrhotite samples. Number of analysis (n) is for LA-ICP-MS/MS dataset. | ||
| Sulfide samples | LA-ICP-MS/MS value | 1σ standard deviation | IRMS | 1σ standard deviation | Bias |
|---|---|---|---|---|---|
| ZF pyrite (n = 7) | −10.29 | 1.70 | −10.49 | 0.87 | 0.21 |
| Balmat pyrite (n = 9) | 15.40 | 0.98 | 15.10 | 0.20 | 0.29 |
| Ward's pyrite (n = 10) | −0.96 | 0.35 | −1.19 | 0.15 | 0.23 |
| Ward's pyrite * (n = 10) | −1.26 | 1.07 | −1.19 | 0.15 | 0.05 |
| Ward's pyrrhotite (n = 10) | −0.16 | 1.88 | −0.11 | 0.5 | 0.05 |
| Ward's pyrrhotite* (n = 10) | −0.10 | 1.32 | −0.11 | 0.5 | 0.01 |
The Uljaste 249, Uljaste 334 and Uljaste 305 samples generally follow the 1:1 correlation line, suggesting that the δ34S values obtained from LA-ICP-MS/MS and IRMS are consistent. On the other hand, Uljaste 227.8 and Uljaste 265 samples exhibit positive bias (0.98‰ and 1.45‰, respectively), while Uljaste 317 has a negative bias (−0.62‰). Even though, generally, the measured average δ34S values for the pyrrhotite samples fall within 1 to 1.5‰ of the IRMS obtained control values, the variability is higher compared to the IRMS results. This variability could be due to the variation of the δ34S in natural pyrrhotite samples. As the drill core diameter used to drill and collect the sample material subjected to IRMS is 10 times larger than the laser beam diameter used to ablate the pyrrhotite samples, larger variations in results would be expected for LA-ICP-MS/MS analysis. Initially 12 ablation spots were marked on each sample. The final number of spots used in calculations in some of the samples was reduced because the laser drifted from the original location, resulting in ablations falling on cracked surfaces or surrounding matrix thus being excluded (Fig. S4 in ESI†).
The Balmat pyrite shows a good agreement with the reported values with a slight positive bias and a relatively low standard deviation compared to the Zf-pyrite. Lowest observed standard deviation (0.35‰) was observed for isotopically homogenous Ward's pyrite when using Ward's pyrrhotite for normalisation. The standard deviation achieved on Wards pyrrhotite when normalised using Ward's pyrite was 1.32‰. Accuracy, expressed as bias, was below 0.3‰ for all 4 samples (Table 5).
The difference in precision of the results between the Balmat and Zf-pyrite could be due to the sample homogeneity. Balmat is a widely characterized sample with consistent results in its applications. On the other hand, the Zf-pyrite is a more heterogeneous natural pyrite sample, and the significant variability could be attributed to the inhomogeneous distribution of the S isotope ratios. On the other hand, the two reference materials Ward's pyrite and pyrrhotite differ slightly from their IRMS values with a low standard deviation.
4. Conclusions
This study presents a novel method for characterizing sulfur isotope ratios in sulfide minerals, focusing on pyrite and pyrrhotite, using LA-ICP-MS/MS. Our approach advances in situ δ34S determinations using quadrupole-based ICP-MS systems, which face limitations due to polyatomic interferences at m/z = 32 and m/z = 34 and low sulfur sensitivity. When using pure N2O as reaction gas at a flow rate of 35% of the 4th cell gas flow rate, the highest sensitivity was achieved for the SO2+ reaction product. Adding a He flow to the reaction cell alongside N2O, the primary reaction product shifted from SO2+ to SO+. The optimal gas flow rate to achieve the highest product ion signal intensity was 5% flow rate on the 4th cell gas using N2O and 4.5 mL per min He. By using a mixture of N2O and He in the collision cell while keeping other analytical parameters constant, an increase of 1.4 times in the primary production signal relative to using only N2O can be achieved. Owing to the higher signal intensity, 32S16O-based signals exhibited approximately 3 times higher signal-to-background ratio than their 32S16O2-based counterparts when other analytical conditions were identical.Through the combined use of a “squid”, a coiled TPE tubing and a cyclonic spray chamber, the highest signal smoothing effects were achieved as observed by the lowest measurement uncertainty. The signal smoothing compartments such as coiled TPE tubing and the cyclonic spray chamber mitigate the issue of large particles being produced using a Nd: YAG laser. Although increasing the number of signal smoothing components increased the washout time, background equivalent signals were achieved 20 s after the end of ablation.
By comparing the results of natural pyrite and pyrrhotite samples normalized using Wards pyrite and pyrrhotite in-house reference materials, matrix dependent bias was observed. The bias was smaller than the measurement uncertainty of the analysis, but for the analysis of the unknown samples, the use of matrix matched reference material is preferred. To minimize the effects of the isotopic fractionation arising from different sources, the sample-standard bracketing approach with ablation of the matrix matched standards under the same analytical conditions was applied.
Under optimized analytical conditions with signal collection in pulse mode detection only, good alignment with IRMS control values for natural samples was achieved. For Uljaste pyrrhotite samples, our measurement values were within ±1.5‰ of IRMS values, with a 1σ standard deviation of 1.1–2.4‰. For isotopically homogenous sulfide samples, the 1σ standard deviation was in the range of 0.35–1.88‰ and the method uncertainty can conservatively be assumed to be in the range of 1–1.5‰. Conversely, accuracy for the isotopically homogenous samples was within ±0.3‰ of reference values.
This study provides a promising approach for characterizing sulfur isotopic ratio variability in natural samples using LA-ICP-MS/MS. Although there are limitations in precision as compared to the LA-MC-ICP-MS technique, this method can be applied to identify samples with sufficiently variable sulfur isotope ratios. Most promisingly, for samples where microbial induced isotopic fractionation, with δ34S ranging from [−50 to 0] ‰, can be expected. Owing to the lower instrument capital costs, the developed method can also be considered as a more available or affordable pre-screening tool, prior to more detailed δ34S investigations.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
E. Eensoo: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft, writing – review & editing. P. Paiste: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, supervision, writing – review & editing. K. Paiste: conceptualization, funding acquisition, project administration, supervision, writing – review & editing. D. Fike: resources, writing – review & editing. J. Houghton: resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No. 894831. We thank J. Valley for sharing pieces of the Balmat pyrite.Notes and references
- D. Rickard and G. W. Luther, Chemistry of iron sulfides, Chem. Rev., 2007, 107(2), 514–562, DOI:10.1021/cr0503658.
- R. O. Rye and H. Ohmoto, Sulfur and carbon isotopes and ore genesis: a review, Econ. Geol., 1974, 69(6), 826–842, DOI:10.2113/gsecongeo.69.6.826.
- R. R. Large, V. V. Maslennikov, F. Robert, L. V. Danyushevsky and Z. Chang, Multistage sedimentary and metamorphic origin of pyrite and gold in the giant sukhoi log deposit, Lena Gold province, Russia, Econ. Geol., 2007, 102(7), 1233–1267, DOI:10.2113/gsecongeo.102.7.1233.
- H. V. Thomas, R. R. Large, S. W. Bull, V. Maslennikov, R. F. Berry, R. Fraser, S. Froud and R. Moye, Pyrite and pyrrhotite textures and composition in sediments, laminated quartz veins, and reefs at bendigo gold mine, Australia: insights for ore genesis, Econ. Geol., 2011, 106(1), 1–31, DOI:10.2113/econgeo.106.1.1.
- A. Amrani, Organosulfur compounds: molecular and isotopic evolution from biota to oil and gas, Annu. Rev. Earth Planet. Sci., 2014, 43(42), 733–768, DOI:10.1146/annurev-earth-050212-124126.
- C. Cai, H. Li, K. Li and D. Wang, Thermochemical sulfate reduction in sedimentary basins and beyond: a review, Chem. Geol., 2022, 607, 121018, DOI:10.1016/j.chemgeo.2022.121018.
- H. Ohmoto, T. Kakegawa and D. R. Lowe, 3.4-billion-year-old biogenic pyrites from barberton, South Africa: sulfur isotope evidence, Science, 1993, 262(5133), 555–557, DOI:10.1126/science.11539502.
- D. T. Johnston, J. Farquhar, K. S. Habicht and D. E. Canfield, Sulphur Isotopes and the search for life: strategies for identifying sulphur metabolisms in the rock record and beyond, Geobiology, 2008, 6(5), 425–435, DOI:10.1111/j.1472-4669.2008.00171.x.
- D. A. Fike, A. S. Bradley and C. V. Rose, Rethinking the ancient sulfur cycle, Annu. Rev. Earth Planet. Sci., 2015, 43(1), 593–622, DOI:10.1146/annurev-earth-060313-054802.
- H. Cui, K. Kitajima, M. J. Spicuzza, J. H. Fournelle, A. Denny, A. Ishida, F. Zhang and J. W. Valley, Questioning the biogenicity of neoproterozoic superheavy pyrite by SIMS, Am. Mineral., 2018, 103(9), 1362–1400, DOI:10.2138/am-2018-6489.
- V. Pasquier, R. N. Bryant, D. A. Fike and I. Halevy, Strong local, not global, controls on marine pyrite sulfur isotopes, Sci. Adv., 2021, 7(9), eabb7403, DOI:10.1126/sciadv.abb7403.
- H. G. Machel, Bacterial and thermochemical sulfate reduction in diagenetic settings — old and new insights, Sediment. Geol., 2001, 140(1–2), 143–175, DOI:10.1016/S0037-0738(00)00176-7.
- A. Lecomte, M. Cathelineau, R. Michels, C. Peiffert and M. Brouand, Uranium mineralization in the alum shale formation (Sweden): evolution of a U-rich marine black shale from sedimentation to metamorphism, Ore Geol. Rev., 2017, 88, 71–98, DOI:10.1016/j.oregeorev.2017.04.021.
- K. Paiste, D. A. Fike, K. Kirsimäe, C. Jones and A. Lepland, Testing the global significance of the sulfur isotope record of the ca. 2.0 Ga zaonega formation: a micro-scale S isotope investigation, Geochim. Cosmochim. Acta, 2022, 331, 86–104, DOI:10.1016/j.gca.2022.05.021.
- J. R. Craig, The metamorphism of pyrite and pyritic ores: an overview, Mineral. Mag., 1993, 57(386), 3–18, DOI:10.1180/minmag.1993.057.386.02.
- A. J. Hall, A. J. Boyce and A. E. Fallick, Iron sulphide sulfides in metasediments: isotopic support for a retrogressive pyrrhotite to pyrite reaction, Chem. Geol. Isot. Geosci. Sect., 1987, 65(3–4), 305–310, DOI:10.1016/0168-9622(87)90010-8.
- N. H. S. Oliver, T. C. Hoering, T. W. Johnson, D. Rumble and W. C. Shanks, Sulfur isotopic disequilibrium and fluid-rock interaction during metamorphism of sulfidic black shales from the waterville-augusta Area, Maine, USA, Geochim. Cosmochim. Acta, 1992, 56(12), 4257–4265, DOI:10.1016/0016-7037(92)90266-L.
- T. Wagner and A. J. Boyce, Pyrite metamorphism in the devonian hunsruck slate of Germany: insights from laser microprobe sulfur isotope analysis and thermodynamic modeling, Am. J. Sci., 2006, 306(7), 525–552, DOI:10.2475/07.2006.02.
- D. E. Canfield, R. Raiswell, J. T. Westrich, C. M. Reaves and R. A. Berner, The use of chromium reduction in the analysis of reduced inorganic sulfur in sediments and shales, Chem. Geol., 1986, 54(1–2), 149–155, DOI:10.1016/0009-2541(86)90078-1.
- D. E. Crowe and R. G. Vaughan, Characterization and use of isotopically homogeneous standards for in situ laser microprobe analysis of 34S/32S ratios, Am. Mineral., 1996, 81(1–2), 187–193, DOI:10.2138/am-1996-1-223.
- M. Chaussidon, F. Albarède and S. M. F. Sheppard, Sulphur isotope variations in the mantle from ion microprobe analyses of micro-sulphide sulfide inclusions, Earth Planet. Sci. Lett., 1989, 92(2), 144–156, DOI:10.1016/0012-821X(89)90042-3.
- R. N. Bryant, C. Jones, M. R. Raven, M. L. Gomes, W. M. Berelson, A. S. Bradley and D. A. Fike, Sulfur isotope analysis of microcrystalline iron sulfides using secondary ion mass spectrometry imaging: extracting local paleo-environmental information from modern and ancient sediments, Rapid Commun. Mass Spectrom., 2019, 33(5), 491–502, DOI:10.1002/rcm.8375.
- C. Jones, D. A. Fike and K. M. Meyer, Secondary ion mass spectrometry methodology for isotopic ratio measurement of micro-grains in thin sections: true grain size estimation and deconvolution of inter-grain size gradients and intra-grain radial gradients, Geostand. Geoanalytical Res., 2019, 43(1), 61–76, DOI:10.1111/ggr.12247.
- L. R. Riciputi, B. A. Paterson and R. L. Ripperdan, Measurement of light stable isotope ratios by SIMS, Int. J. Mass Spectrom., 1998, 178(1–2), 81–112, DOI:10.1016/S1387-3806(98)14088-5.
- P. R. Craddock, O. J. Rouxel, L. A. Ball and W. Bach, Sulfur isotope measurement of sulfate and sulfide by high-resolution MC-ICP-MS, Chem. Geol., 2008, 253(3–4), 102–113, DOI:10.1016/j.chemgeo.2008.04.017.
- S. E. Gilbert, L. V. Danyushevsky, T. Rodemann, N. Shimizu, A. Gurenko, S. Meffre, H. Thomas, R. R. Large and D. Death, Optimisation of laser parameters for the analysis of sulphur isotopes in sulphidesulfide minerals by laser ablation ICP-MS, J. Anal. Spectrom., 2014, 29(6), 1042–1051, 10.1039/C4JA00011K.
- O. Peter, F. Melcher and C. Walkner, In situ measurement of sulfur isotopic composition (δ34S) in sphalerite using LA-(QQQ)-ICP-MS, 2017 Search PubMed.
- P. R. D. Mason, J. Košler, J. C. M. De Hoog, P. J. Sylvester and S. Meffan-Main, In situ determination of sulfur isotopes in sulfur-rich materials by laser ablation multiple-collector inductively coupled plasma mass spectrometry (LA-MC-ICP-MS), J. Anal. Spectrom., 2006, 21(2), 177–186, 10.1039/B510883G.
- F. Börner, M. Keith, J. L. Bücker, P. Voudouris, R. Klemd, K. Haase, M. Kutzschbach and F. Schiperski, In situ trace element and S isotope systematics in pyrite from three porphyry-epithermal prospects, limnos island, Greece, Front. Earth Sci., 2022, 10, 916107, DOI:10.3389/feart.2022.916107.
- D. E. Crowe, J. W. Valley and K. L. Baker, Micro-analysis of sulfur-isotope ratios and zonation by laser microprobe, Geochim. Cosmochim. Acta, 1990, 54(7), 2075–2092, DOI:10.1016/0016-7037(90)90272-M.
- M. J. Whitehouse, B. S. Kamber, C. M. Fedo and A. Lepland, Integrated Pb- and S-isotope investigation of sulphidesulfide minerals from the early archaean of Southwest Greenland, Chem. Geol., 2005, 222(1–2), 112–131, DOI:10.1016/j.chemgeo.2005.06.004.
- R. Kozdon, N. T. Kita, J. M. Huberty, J. H. Fournelle, C. A. Johnson and J. W. Valley, In situ sulfur isotope analysis of sulfide minerals by SIMS: precision and accuracy, with application to thermometry of ∼3.5 Ga pilbara cherts, Chem. Geol., 2010, 275(3–4), 243–253, DOI:10.1016/j.chemgeo.2010.05.015.
- G. Skrzypek, C. E. Allison, J. K. Böhlke, L. Bontempo, P. Brewer, F. Camin, J. F. Carter, M. M. G. Chartrand, T. B. Coplen, M. Gröning, J.-F. Hélie, G. Esquivel-Hernández, R. A. Kraft, D. A. Magdas, J. L. Mann, J. Meija, H. A. J. Meijer, H. Moossen, N. Ogrinc, M. Perini, A. Possolo, K. M. Rogers, A. Schimmelmann, A. Shemesh, D. X. Soto, F. Thomas, R. Wielgosz, M. R. Winchester, Z. Yan and P. J. H. Dunn, Minimum requirements for publishing hydrogen, carbon, nitrogen, oxygen and sulfur stable-isotope delta results (IUPAC technical report), Pure Appl. Chem., 2022, 94(11–12), 1249–1255, DOI:10.1515/pac-2021-1108.
- C. E. Rees, Sulphur isotope measurements using SO2 and SF6, Geochim. Cosmochim. Acta, 1978, 42(4), 383–389, DOI:10.1016/0016-7037(78)90269-7.
- Z.-Y. Zhu, S.-Y. Jiang, C. L. Ciobanu, T. Yang and N. J. Cook, Sulfur isotope fractionation in pyrite during laser ablation: implications for laser ablation multiple collector inductively coupled plasma mass spectrometry mapping, Chem. Geol., 2017, 450, 223–234 CrossRef CAS.
- K. Paiste, A. Pellerin, A. L. Zerkle, K. Kirsimäe, A. R. Prave, A. E. Romashkin and A. Lepland, The pyrite multiple sulfur isotope record of the 1.98 Ga zaonega formation: evidence for biogeochemical sulfur cycling in a semi-restricted basin, Earth Planet. Sci. Lett., 2020, 534, 116092 CrossRef CAS.
- J.-H. Yang and D. C. Conway, Bonding in ion clusters. I. O4+, J. Chem. Phys., 1964, 40(6), 1729–1735 CrossRef CAS.
- S. T. Lancaster, T. Prohaska and J. Irrgeher, Characterisation of gas cell reactions for 70+ elements using N2O for ICP tandem mass spectrometry measurements, J. Anal. At. Spectrom., 2023, 38(5), 1135–1145, 10.1039/D3JA00025G.
- A. B. Raksit, Reactions of O2+, O4+ and O2+·H2O ions with neutral molecules, Int. J. Mass Spectrom. Ion Process., 1986, 69(1), 45–65, DOI:10.1016/0168-1176(86)87041-0.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ja00166h |
| This journal is © The Royal Society of Chemistry 2025 |