
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated on-line monitoring of a lithium hydroxide production process using micro-discharge OES†
Bastian
Wiggershaus
 *a,
Miisamari
Jeskanen
b,
Aappo
Roos
b,
Toni
Laurila
b and
Carla
Vogt
*a
*a,
Miisamari
Jeskanen
b,
Aappo
Roos
b,
Toni
Laurila
b and
Carla
Vogt
*a
aTU Bergakademie Freiberg, Institute of Analytical Chemistry, Lessingstraße 45, Freiberg 09599, Germany. E-mail: Carla.Vogt@chemie.tu-freiberg.de
bSensmet Oy, Customer Application Center, Otakaari 7, Espoo 02150, Finland
First published on 17th December 2024
Abstract
The fast, precise and continuous on-line analysis of highly saline process solutions is challenging for conventional laboratory techniques like ICP-OES or ICP-MS due to the necessity of high plasma gas flow rates (Ar and He), a high power consumption and the limited resistance of the sensitive spectrometer technique to harsh on-site conditions like dust, vibrations or temperature fluctuations. Therefore, an on-site and on-line method with comparable performance despite such conditions would be preferable. In this study we used the Micro-Discharge Optical Emission Spectroscopy (μDOES) for the given challenge of monitoring fully automated on-site and on-line production of lithium hydroxide, which is an essential precursor for the battery industry. The technology is based on creating a micro-plasma directly inside the aqueous sample without any carrier gas by using electrodes and high voltage pulses and thus enabling optical emission spectroscopy on-site. After optimisation of several parameters like sample conductivity, signal integration settings or selection of emission lines, measurements were carried out at an industrial pilot plant. The entire process chain was monitored, starting with the leaching of the calcined lithium-containing ore, through several intermediate products to the end product lithium hydroxide monohydrate of battery grade. The individual process steps were measured continuously (10–20 h), simplifying the monitoring of the process and allowing trends in the concentrations of the elements Li, Na, K, Ca, Mg and Rb to be identified. Reference measurements were performed using laboratory ICP-OES and/or ion chromatography to verify the results. Micro-discharge OES proved to be useful for a fast and precise on-site and on-line analysis of saline solutions with good long-term stability and a high agreement with the used reference methods, resulting in deviations below 10% for the most important components Li, Na and K.
1 Introduction
1.1 Lithium hydroxide as a battery precursor
Lithium-ion batteries are rechargeable power sources used in various devices, from smartphones to electric vehicles.1,2 The main components of these batteries include the anode, cathode, electrolyte, and separator.3–7 The anode is typically made of graphite, while the cathode is composed of lithium cobalt oxide (LCO), lithium iron phosphate (LFP), lithium nickel manganese cobalt oxide (NMC) or other lithium-based materials.1,3,4,7 The electrolyte, often a lithium salt dissolved in an organic solvent, facilitates the movement of lithium ions between the anode and cathode during charging and discharging.4–6 The separator is a thin porous membrane that prevents contact between the anode and cathode while allowing ions to pass through.5,8 Lithium hydroxide (LiOH) and carbonate (Li2CO3) play a crucial role in the production of cathode materials for lithium-ion batteries. Although both lithium compounds can be used, the hydroxide form offers some advantages. Lithium hydroxide is preferred for high-nickel cathode materials used in batteries for vehicles with long driving ranges, because it enables a higher packing density, better crystallinity, structural purity and can be used at a lower synthesis temperature.9 Lithium hydroxide can be extracted both from brines and ores.10 The extraction from ores like spodumene requires various steps, whereby the raw mineral is first crushed and ground. Since α-spodumene is chemically very resistant, it must be converted into the thermodynamically less stable β-spodumene by heating it in a rotary kiln at 1100 °C. This step is classically followed by roasting the β-spodumene with concentrated sulfuric acid (H2SO4) at 250 °C generating lithium sulphate (Li2SO4).10 Depending on the industrial process used, further steps are necessary, which may differ in detail but generally include leaching of the previously obtained intermediate product with subsequent filtration, purification and carbonation.11 Finally, lithium carbonate is converted into lithium hydroxide monohydrate.1.2 Advantages of process analysis
The monitoring of industrial processes is becoming increasingly important and has a positive influence on various phases of process development. The process analytical technology (PAT) offers various tools for the investigation and optimisation of chemical productions and is applied in various fields such as the petrochemical industry or pharmaceutical industry.12 In principle, measurements can be carried out off-, at-, on- or in-line,13 whereby the measurement time for the former can be over a day and for the latter a few seconds.14 Various spectroscopic methods such as UV-visible spectroscopy,15 infrared spectroscopy16 or Raman spectroscopy17 have been used successfully for process monitoring for several decades. During process development, on-site and on-line analysis techniques can be used to increase process understanding.18 For example, the data obtained support modelling or adjustments of process parameters. PAT is also used to verify the process in later stages of development and for process control.18 This makes it easy to check whether the experiments can be repeated over a longer period of time or whether they can be carried out at different locations. Other advantages include time savings compared to off-line analysis, automated and secure sampling as well as a significantly higher measurement frequency. Challenges include the requirement for high sensitivity, but also the need for the analytical methods to be robust against typical fluctuations that can occur in the process.18 In principle, the use of PAT can be divided into two categories: in the first case, the analysis methods are used as a support to control the process, carry out continuous optimisations or quickly identify problems. In the second case, the traditional off-line laboratory analyses are replaced by an on-line method.18The aim of this study was to investigate the on-site and on-line capabilities of the Micro-Discharge Optical Emission Spectroscopy (μDOES) to replace classic laboratory methods such as titration, inductively coupled plasma optical emission spectroscopy (ICP-OES) or ion chromatography (IC), whereby the complete process chain of the lithium hydroxide monohydrate production at a pilot plant was selected as a proof of concept. Multiple process steps were monitored, starting with the leaching of the calcined lithium-containing ore, through several intermediate products to the end product lithium hydroxide monohydrate of battery grade. In addition to Li, other elements were present in significantly higher (Na and K) or lower concentrations (Ca, Mg, and Rb), typically in the mg L−1 to g L−1 range. The main challenges were to generate precise and correct analytical results despite fluctuating measurement conditions on site (temperature of the environment and samples, dust and vibrations) as well as changing matrices (pH value, anions, and ionic strength/conductivity) and concentration ratios in the course of the process chain. The analysis method used here is based on optical emission spectroscopy, whereby excited atoms and ions emit electromagnetic radiation at their characteristic wavelengths, and was described in more detail in previous work.19
The main differences from the established ICP-OES are that a micro-plasma is generated directly inside the aqueous process sample by applying high-voltage pulses to electrodes (Fig. 1). The plasma generation is based on a so called in-liquid corona discharge. Using a high-voltage electronic setup consisting of a capacitor bank, switch card and ballast, up to 2 kW of electrical power is concentrated into a small volume of less than 1 mm3 around the cathode, which is located directly within the process solution. Consequently, the solution heats up rapidly, leading to in situ air bubble formation and finally to a partial breakdown. As a result, a non-equilibrium water vapor plasma primarily consisting of H+, OH+ and electrons is generated. During a typical analysis, approximately 1000–3000 micro-plasma pulses, each lasting for about 1 ms, are generated and averaged. Unlike most existing micro plasma techniques, micro-ischarge OES is characterized by the direct generation of a micro-plasma in the aqueous solution using two rod electrodes and high-voltage pulses, without the need for any carrier gas, such as argon. In contrast to the well-known solution cathode glow discharge20 (SCGD), both electrodes are immersed directly in the sample solution (with no air gap), eliminating the need for a waste reservoir to establish the electrical connection. Furthermore, the system is fully automated for on-site and on-line measurements including sampling, data evaluation as well as cleaning and micro-discharge OES is significantly more environmentally friendly due to the complete absence of plasma gases (Ar, He, and N2) and a much lower power consumption.19
 | ||
| Fig. 1 By applying high-voltage pulses to the electrodes, a micro-plasma is generated by an in-liquid corona discharge due to a partial breakdown as a result of the sample heating up instantaneously and in situ formation of air bubbles.19 | ||
2 Experimental
2.1 Instrumentation
The industrial pilot plant and the various process steps were operated by the project partner K-UTEC AG SALT TECHNOLOGIES. On-site experiments were carried out using a μDOES® Online Multi-metal Process Liquid Analyser including an automatic dilution unit (Sensmet Oy, Finland). The analyser was equipped with tungsten electrodes (99.95%) and the cathode–anode distance was approx. 2 cm. Four Czerny–Turner spectrometers with CMOS sensors were utilised covering a wavelength range from approx. 200 to 835 nm at a resolution of 0.1 nm. The emitted light was transferred from the measurement cell to the detector unit using an optical fibre in radial view, whereby the light beam was divided into four partial beams, one per spectrometer. The four individual spectra were combined into one by the instrument software. For each measurement, 50 pulse series with 50 pulses each were generated, resulting in 2500 pulses total. The parameter settings (Table 1) were chosen based on previous investigations.19| Parameter | Value |
|---|---|
| Plasma discharge energy (J) | 2.0 |
| Electrical conductivity (μS cm−1) | 2500 |
| Number of pulses | 2500 (50 × 50) |
| Frequency (Hz) | 750 |
| Dilution ratio | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000/25000/50000 000/25000/50000 |
2.2 Materials and reagents
For calibration, certified ICP element standard solutions containing the elements Li, Na, K, Mg, Ca and Rb were used (Merck, 10.000 mg L−1 respectively). Ultrapure water (Merck Milli-Q®, 18.2 MΩ cm−1) was used to dilute the solutions and sulphuric acid (Merck, analytical grade, 1%) to adjust the conductivity. Nitric acid (Merck, analytical grade, 65%) was used to completely dissolve the solid end product LiOH·H2O, which was obtained in the last process step. All sample tubes (except for the waste) and the mixing chamber for dilution and conductivity adjustment were made of PTFE.2.3 Setup and measurement procedure
In process steps I-III, the sample was taken from the overflow of the respective clarifier (Fig. 2b). The highly concentrated salt solution/suspension was then automatically diluted using the dilution unit (Fig. 2c). | ||
| Fig. 2 On-site setup and sampling point exemplary for the first process step (A: addition of the calcined lithium ore, B: clarifier with the product, C: analyser, dilution unit and ultrapure water supply, and D: analyser front view with operating software). | ||
The dilution was carried out in several stages with ultrapure water, whereby the dilution ratio was either 10000, 25000 or 50000, depending on the process step. The main criteria for the dilution ratio were the resulting electrical conductivity of the sample and the concentration range, which should be preferably within the optimum measuring range of μDOES. Next, the diluted sample was transported to the analyser and the conductivity was adjusted stepwise and precisely with 1% sulphuric acid until the target value of 2500 μS cm−1 was reached. After dilution and conductivity adjustment, the measurement was carried out by generating a sequence of micro-plasmas and acquiring the emission spectra. Each sample was measured for 1 min, 2500 plasmas were generated and the corresponding emission spectra were averaged. This corresponds to one measurement point in on-line monitoring. As the system in this case was operated in stop-flow mode, one sample at a time was taken from the process, diluted and analysed. Depending on the dilution ratio, the total time required was approx. 10 min per sample, whereby the measurement itself took only 1 min. The continuous measurement sequence was designed in a way that an ultrapure water measurement was carried out after every 3 samples for a blank control. Process step IV (purification of lithium hydroxide monohydrate) was carried out batch-wise to obtain the solid end product. Prior to analysis, the samples were dissolved using nitric acid and diluted using ultrapure water (cf. ESI† for more details). The subsequent procedure was analogous to process steps I-III. Additionally, after each measurement series (up to 20 h) the rinsing and cleaning of the system were carried out with ultrapure water prior to the next process step.
2.4 Data evaluation
For spectral evaluation the analyser software SenSpec™ was used. Suitable emission lines were selected for each element, whereby the most intensive and interference-free lines were chosen. The respective lines were analysed either in single pulse mode (2500 pulses individually integrated and averaged) or pulse series mode (50 pulses of a series integrated together and averaged over 50 pulse series). Since hydrogen is ubiquitously present in aqueous samples it can be used as an internal standard. All emission lines investigated here (Table 2) were normalised with a selected hydrogen emission line (H 486.1 nm), which allows residual plasma fluctuations due to various factors such as slight fluctuations at the applied voltage, the surface condition of the electrode over time or the electrical conductivity to be compensated.| Element | Wavelengtha in nm | |
|---|---|---|
| Single pulse mode | Pulse series mode | |
| a Wavelength ranges including the background to the left/right of the emission line for correction are indicated. | ||
| Li | 811.6–813.3 | 609.7–610.6 |
| Na | 817.5–820.2 | 589.3–590.0 |
| K | 765.7–770.4 | 765.7–770.4 |
| Ca | 442.2–445.8 | 396.6–397.2 |
| Mg | 516.4–518.5 | 277.5–279.2 |
| Rb | 779.0–781.0 | 779.0–781.0 |
| H (internal standard) | 485.0–487.0 | 485.0–487.0 |
The intensity of the emission lines and thus reaching the saturation limit depend on the integration mode of the spectrometer.19 In single pulse mode, all intensities are reduced, and the more sensitive main lines of the respective elements can be used. In pulse series mode, on the other hand, these are quickly saturated so that less sensitive emission lines must be used.
2.5 Reference measurements
Based on selected process samples, various reference measurements were carried out to verify the micro-discharge OES results. The samples were collected manually throughout the process at different stages and aliquoted, ensuring that each method was using exactly the same sample for analysis. The reference samples were taken at regular intervals based on the sampling plan of the industrial partner. Depending on the process step, the sampling schedule varied. In addition to the real-time on-line monitoring during the process (approx. 10 -minute intervals), the samples collected were analysed batch-wise using μDOES after each process step was completed. ICP-OES reference measurements were performed using two different spectrometers: a Thermo Scientific iCAP 6500 Duo (RF-Power 1250 W, plasma gas 14 L min−1, auxiliary gas 1.0 L min−1, nebulizer gas 0.6 L min−1, 25 rpm) and a Spectro Blue EOP (RF-Power 1400 W, plasma gas 15 L min−1, auxiliary gas 1.2 L min−1, nebulizer gas 0.8 L min−1, 30 rpm). Ion chromatography measurements were also carried out using a Thermo Scientific Dionex Easion (IonPac CS12A column 4 × 250 mm, an eluent of 0.02 M methanesulfonic acid with 0.02 M tetrabutylammonium hydroxide at a flow rate of 1 mL min−1, 10 μL injection volume, suppressor, conductivity detector). Depending on the total sample volume available, all or only a selection of the reference methods was used.3 Results and discussion
The individual process steps were monitored on-line in real-time and additionally selected samples were analysed batch-wise for comparison with laboratory results of the ICP-OES and ion chromatography measurements. As the samples originate from an industry project, all samples were anonymised and numbered, starting always from 1 for each process step. In the following section, only the results of the most important components Li, Na and K are discussed in detail. Further results (Ca and Mg) can be found in the ESI.†3.1 Exemplary spectra of the process samples
All results presented in this study are based on optical emission spectra. In contrast to classical ICP-OES, the spectra are dominated by atomic lines and less crowded.Comparing the single pulse mode with the pulse series mode (Fig. 3), it is noticeable that the intensities of all emission lines differ significantly, both from the solvent components like hydrogen (H 486.1 nm) or the OH rotational lines at approx. 300–310 nm and from the analytes such as sodium (Na 589.5 nm) or potassium (K 766.4 nm). Depending on the process step and the concentrations present in the salt solution, both integration modes have their application. The longer integration time is suitable for low concentrations (trace elements) or less sensitive lines and the shorter integration time for high concentrations (main components) or more sensitive emission lines.
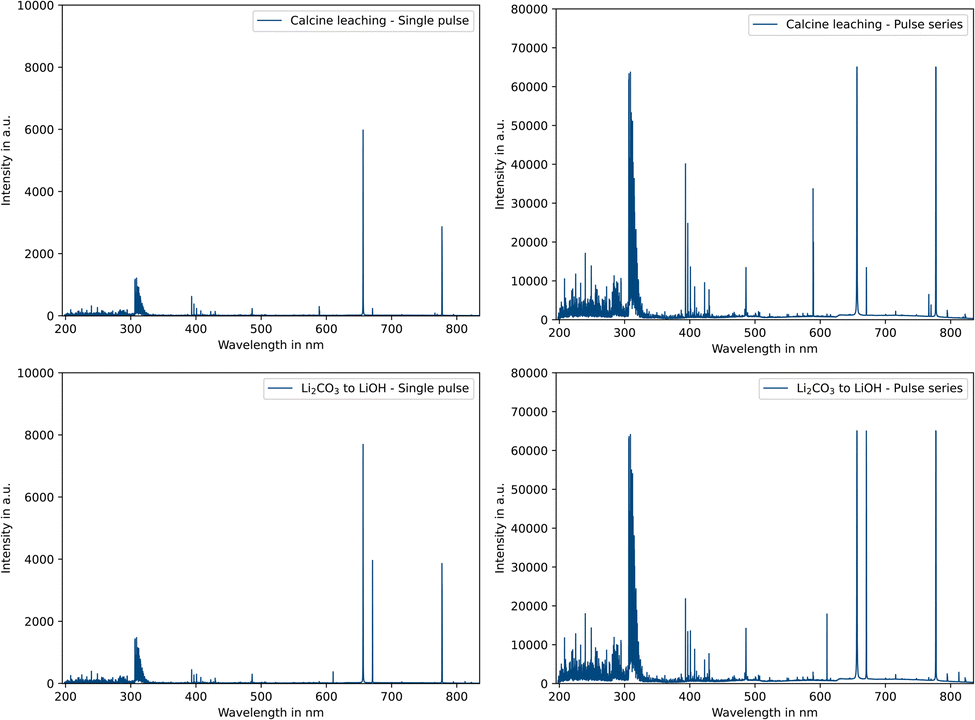 | ||
| Fig. 3 Optical emission spectra of selected process samples comparing single pulse (left) and pulse series integration modes (right). | ||
3.2 Calcine leaching
The first process step consisted of leaching a calcined ore mixture, which was supplied as a fine yellowish powder (Fig. 2a). In a total of approx. 13 h, over 800 kg of the starting material was fed into the process using a conveyor belt with a defined dosing rate. In the first reactor, the ore was leached using a leaching solution consisting mainly of H2O (and small amounts of LiOH, Na2SO4 and K2SO4 from previous pilot campaign runs) at slightly elevated temperatures, whereby insoluble components (such as CaSO4) remained at the bottom as solids (eqn (1)).| Calcined ore + leaching solution → Li2NaK(SO4)2aq + Solid residue | (1) |
The overflow suspension was passed on to another reactor for additional leaching, was finally transported into a clarifier in which the solid particles could sediment further and the overflow was taken for the analysis. First, it can be seen that the lithium (dark blue), magnesium (brown) and rubidium (yellow) concentrations remained constant over the entire measurement period of about 10 h (Fig. 4). The ultrapure water measurements after every three samples show that no memory effects occurred and the blank levels are reached immediately. Na (light blue) and K (orange) are at a constant level in the first five hours of the process step, with K showing slightly higher concentrations than Na. In the second half of the experiment, it is clearly visible that the levels of both elements increase significantly, and for example Na increases from approx. 4500 mg L−1 at the beginning to 10000 mg L−1 towards the end of the measurement series. This effect can be traced back to a change in the leaching solution used and the resulting reduction in pH value (from ≈11 to 9).
 | ||
| Fig. 4 Real-time monitoring of the ore leaching process based on the main components Li, Na and K as well as the minor components Ca, Mg and Rb. | ||
Furthermore, two clear Ca “spikes” (green) can be recognised (approx. hours 3.5 and 4.5), which are most likely due to solid CaSO4 particles that have not sedimented and have thus entered the plasma. The use of a filter system in future work would be conceivable in order to minimise the number of particles even further. However, the disadvantage would be increased cleaning requirements once the filter capacity is reached. It should be noted that particles in no way interfere with the measurement of the other elements and are not harmful to the analyser.
The comparison of the results with the laboratory ICP-OES data shows overall good agreement between the two methods for the most important component lithium in the first process step (Fig. 5).
 | ||
| Fig. 5 Comparison of the micro-discharge OES lithium results with laboratory ICP-OES data based on selected samples of the ore leaching process step (on-line μDOES: n = 1 continuous process and ICP-OES: n = 5). | ||
For Na and K, it is clear that the two measurement modes applied for the μDOES system differ from each other. In the pulse series mode, the results for sodium (Fig. 6) deviate in some cases by up to 40% and for potassium (Fig. 7) up to 50% from the ICP-OES reference results.
 | ||
| Fig. 6 Comparison of the micro-discharge OES sodium results with laboratory ICP-OES data based on selected samples of the ore leaching process (on-line μDOES: n = 1 continuous process and ICP-OES: n = 5). | ||
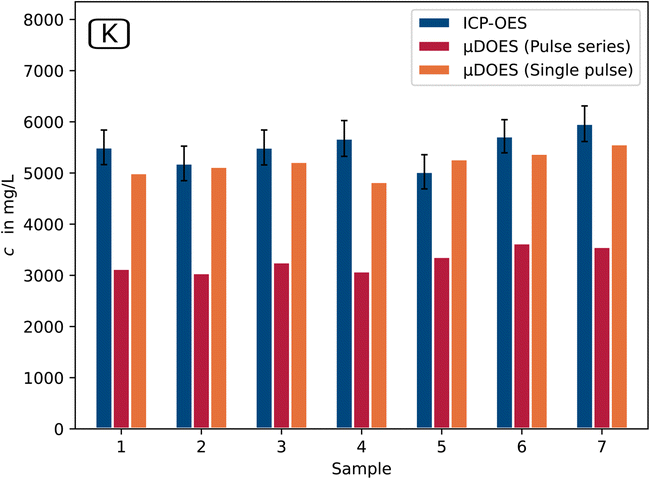 | ||
| Fig. 7 Comparison of the micro-discharge OES potassium results with laboratory ICP-OES data based on selected samples of the ore leaching process step (on-line μDOES: n = 1 continuous process and ICP-OES: n = 5). | ||
This is due to non-linearities and self-absorption effects that occur particularly strongly in this measurement mode.19 In the single pulse mode, on the other hand, values are achieved that agree very well with the ICP-OES values and in most cases deviate by less than 10% for both elements.
3.3 Li2CO3 precipitation
The previously obtained solution containing Li2SO4 was then used for lithium carbonate precipitation. A total of three reactors were set up as a cascade in order to utilise the counter current principle. The lithium-containing solution was introduced at slightly elevated temperatures and a sodium carbonate solution was added in stages, whereupon lithium carbonate was precipitated (eqn (2)).| 2Li+ + CO32− → 2Li2CO3↓ | (2) |
The comparison with ion chromatography (Fig. 8) shows that there is good agreement for the six selected samples analysed. Only sample 5 appears to have a significantly higher concentration when measured with the μDOES system. This could be due to particles which entered the plasma as the samples were not filtered prior to the measurement.
 | ||
| Fig. 8 Comparison of Li concentrations measured by micro-discharge OES and ion chromatography using selected samples of the lithium carbonate precipitation process step (batch-wise μDOES: n = 3 and IC: n = 1 due to long measurement time). | ||
3.4 Conversion of Li2CO3 to LiOH
The next main step consisted of converting the previously precipitated and washed lithium carbonate into lithium hydroxide. For this purpose, a Ca(OH)2 suspension was prepared in excess at a high temperature. The solid lithium carbonate was then continuously fed to the Ca(OH)2 suspension, whereupon it reacted to form the hydroxide (eqn (3)).| 2Li+ + CO32− + Ca2+ + 2OH− → 2LiOHaq + CaCO3↓ | (3) |
The resulting lithium hydroxide solution was fed as overflow into a thickener in which the solid particles could sediment. In this process step, particular care was taken to keep reactors and vessels covered and closed in order to counteract water evaporation at the greatly increased temperatures and to minimise the intake of CO2, which would trigger the reverse reaction of lithium hydroxide to lithium carbonate.
In the initial hours of the experiment, the lithium concentration was approx. 2 g L−1. In the course of the process, however, the concentration increases to 10 g L−1 as more lithium carbonate was added to the calcium hydroxide suspension (Fig. 9). This real-time on-line monitoring allows the process to be terminated once a certain target value has been reached, thus saving valuable process time.
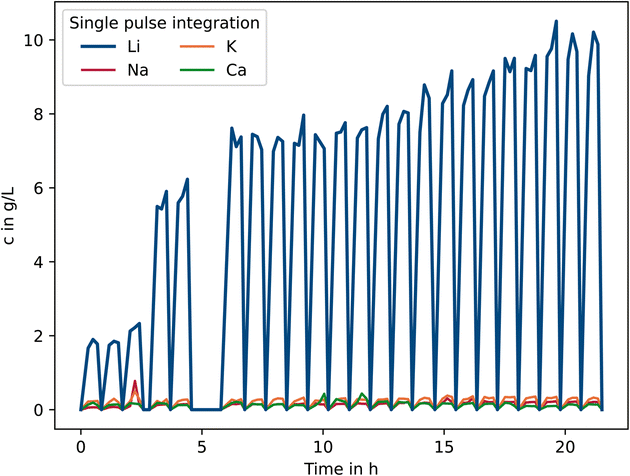 | ||
| Fig. 9 Increase in lithium concentration with increasing process time and addition of lithium carbonate to the Ca(OH)2 suspension. | ||
A very good agreement between micro-discharge OES and ion chromatography was again found for the selected samples analysed (Fig. 10).
 | ||
| Fig. 10 Lithium concentration in mg L−1 of the obtained LiOH solution comparing μDOES and ion chromatography (batch-wise μDOES: n = 3 and IC: n = 1). | ||
3.5 Purification of LiOH·H2O
Finally, the lithium hydroxide solution obtained previously was purified, and this step was carried out batch-wise.The process route consisted of cyclic recrystallisation with hot solution, vacuum filtration and cooling crystallisation to obtain solid lithium hydroxide monohydrate of battery grade. As can be seen (Fig. 11), the theoretical target value for lithium (16.54% Li in LiOH·H2O) is well achieved by both analysis methods. Trace elements like Ca, Mg, Al, Fe or Zn could not be quantified using micro-discharge OES as such high matrix-analyte-ratios (up to 300000:1) were not possible to measure at the time of the measurement campaign, but Al (36.1–45.6 mg kg−1) and Fe (0.52–0.67 mg kg−1) were determined using laboratory ICP-OES. Further development work is on-going at this point in order to be able to also carry out the last step of the process chain, purity control of the end products, in a fully comprehensive manner.
 | ||
| Fig. 11 Lithium concentration in % of the final product LiOH·H2O comparing μDOES and ICP-OES results with the theoretical value (batch-wise μDOES: n = 3). | ||
4 Conclusions
Within the scope of this study, micro-discharge optical emission spectroscopy was utilized to monitor the fully automated on-site and on-line industrial production of lithium hydroxide monohydrate. The entire process chain was successfully analysed at a pilot plant, starting with the leaching of the calcined lithium-containing ore, through several intermediate products to the end product LiOH·H2O of battery grade. Continuous measurements of the individual process steps (10–20 h) were carried out, simplifying the monitoring of the process and allowing trends in the element concentrations to be identified. This is particularly useful for adjusting the dosing rates of individual components in order to further optimize the process. The reference measurements carried out using laboratory ICP-OES and ion chromatography confirmed the results of micro-discharge OES. In most cases, the deviations were below 10% for the most important elements (Li/Na/K) using the single pulse mode, which represents very good values, especially considering that the analysis is carried out continuously (24/7) directly on-site/on-line and with an enormous time saving compared to the classic laboratory analysis. In addition, micro-discharge OES is environmentally friendly due to the absence of carrier/plasma gases like Ar and low energy consumption.19 Disadvantages include the more complex planning and integration of the analysis method into an industrial process. The use of a filtering system should be considered for the next on-site measurements to remove any remaining solid particles. Differences in results between μDOES and the reference methods for which the samples were filtered in advance could thus be further reduced. The aim of future work will also be to further reduce the dilution time, which accounts for a large part of the total measurement time, or to make the dilution completely obsolete. Preliminary results using a reversed polarity to generate micro plasmas at the anode and modified electrodes have already shown promise for extending the linearity range up to 1000 mg L−1, which would result in significantly lower dilution ratios, reduced ultrapure water consumption and faster measurements. In addition, the trace element analysis of the end product lithium hydroxide monohydrate in which very high matrix analyte ratios are present, would be feasible. Another drawback is that only cations can be analysed at the moment, but anions are often required for balancing equations and process control. Thus, the utilisation of a spectrometer in the wavelength range up to 1000 nm is currently being tested in order to quantify the non-metals chlorine (837.6 nm) and sulphur (921.3 nm) which are present as chloride and sulphate anions in the process solutions.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Bastian Wiggershaus: conceptualization, data curation, formal analysis, investigation, methodology, software, validation, visualization, writing – original draft, Miisamari Jeskanen: data curation, methodology, software, validation, writing – review & editing, Aappo Roos: data curation, formal analysis, methodology, supervision, validation, writing – review & editing, Toni Laurila: conceptualization, funding acquisition, resources, supervision, writing – review & editing, and Carla Vogt: conceptualization, funding acquisition, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Parts of this work were funded by K-UTEC AG SALT TECHNOLOGIES. We acknowledge Mr Daniel Schneider for his assistance in sampling and performing the IC measurements and Ms. Johanna Helminen for her contribution to the μDOES measurements.References
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Li-ion battery materials: present and future, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- B. Scrosati and J. Garche, Lithium batteries: Status, prospects and future, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- G. E. Blomgren, The Development and Future of Lithium Ion Batteries, J. Electrochem. Soc., 2017, 164, A5019–A5025 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Challenges for Rechargeable Li Batteries, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- M. Winter, B. Barnett and K. Xu, Before Li Ion Batteries, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- K. Xu, Nonaqueous liquid electrolytes for lithium-based rechargeable batteries, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS.
- B. Xu, D. Qian, Z. Wang and Y. S. Meng, Recent progress in cathode materials research for advanced lithium ion batteries, Mater. Sci. Eng., R, 2012, 73, 51–65 CrossRef CAS.
- S. S. Zhang, A review on the separators of liquid electrolyte Li-ion batteries, J. Power Sources, 2007, 164, 351–364 CrossRef CAS.
- B. Fitch, M. Yakovleva and S. Meiere, Lithium Hydroxide Based Performance Improvements for Nickel Rich Ncm Layered Cathode Material, Meet. Abstr., 2016,(MA2016-02), 469 CrossRef.
- L. Talens Peiró, G. Villalba Méndez and R. U. Ayres, Lithium: Sources, Production, Uses, and Recovery Outlook, JOM, 2013, 65, 986–996 CrossRef.
- C. Dessemond, F. Lajoie-Leroux, G. Soucy, N. Laroche and J.-F. Magnan, Spodumene: The Lithium Market, Resources and Processes, Minerals, 2019, 9, 334 CrossRef CAS.
- W. Chew and P. Sharratt, Trends in process analytical technology, Anal. Methods, 2010, 2, 1412 RSC.
- D. C. Hassell and E. M. Bowman, Process Analytical Chemistry for Spectroscopists, Appl. Spectrosc., 1998, 52, 18A–29A CrossRef CAS.
- S. Kueppers and M. Haider, Process analytical chemistry-future trends in industry, Anal. Bioanal. Chem., 2003, 376, 313–315 CrossRef CAS PubMed.
- K. A. Bakeev, Process Analytical Technology. Spectroscopic Tools and Implementation Strategies for the Chemical and Pharmaceutical Industries, Wiley, Chichester, West Sussex, 2nd edn, 2010 Search PubMed.
- J. Ryczkowski, IR spectroscopy in catalysis, Catal. Today, 2001, 68, 263–381 CrossRef CAS.
- I. R. Lewis, Handbook of Raman Spectroscopy. From the Research Laboratory to the Process Line, CRC Press, New York, 4th edn, 2001, vol. 28 Search PubMed.
- L. L. Simon, H. Pataki, G. Marosi, F. Meemken, K. Hungerbühler, A. Baiker, S. Tummala, B. Glennon, M. Kuentz, G. Steele, H. J. M. Kramer, J. W. Rydzak, Z. Chen, J. Morris, F. Kjell, R. Singh, R. Gani, K. V. Gernaey, M. Louhi-Kultanen, J. O'Reilly, N. Sandler, O. Antikainen, J. Yliruusi, P. Frohberg, J. Ulrich, R. D. Braatz, T. Leyssens, M. von Stosch, R. Oliveira, R. B. H. Tan, H. Wu, M. Khan, Des O'Grady, A. Pandey, R. Westra, E. Delle-Case, D. Pape, D. Angelosante, Y. Maret, O. Steiger, M. Lenner, K. Abbou-Oucherif, Z. K. Nagy, J. D. Litster, V. K. Kamaraju and M.-S. Chiu, Assessment of Recent Process Analytical Technology (PAT) Trends: A Multiauthor Review, Org. Process Res. Dev., 2015, 19, 3–62 CrossRef CAS.
- B. Wiggershaus, M. Jeskanen, A. Roos, C. Vogt and T. Laurila, Trace element analysis in lithium matrices using micro-discharge optical emission spectroscopy, J. Anal. At. Spectrom., 2024, 39, 1248–1259 RSC.
- M. R. Webb, F. J. Andrade, G. Gamez, R. McCrindle and G. M. Hieftje, Spectroscopic and electrical studies of a solution-cathode glow discharge, J. Anal. At. Spectrom., 2005, 20, 1218 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ja00330f |
| This journal is © The Royal Society of Chemistry 2025 |