
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible-light-mediated late-stage N-functionalization of unprotected peptides: introducing the aza-Zimmerman–O'Connell–Griffin reaction†
Sara
Bacaicoa
 ,
Mario
Martos
,
Wilma Yhlen
Graf
,
August
Runemark
,
Ganesh H.
Shinde
,
Ganesh S.
Ghotekar
and
Henrik
Sundén
*
,
Mario
Martos
,
Wilma Yhlen
Graf
,
August
Runemark
,
Ganesh H.
Shinde
,
Ganesh S.
Ghotekar
and
Henrik
Sundén
*
Department of Chemistry and Molecular Biology, University of Gothenburg, Medicinaregatan 17 Natrium, 41390 Gothenburg, Sweden. E-mail: henrik.sunden@chem.gu.se
First published on 20th June 2025
Abstract
Amides represent a crucial class of compounds in organic chemistry, forming key structural components of biologically significant molecules such as amino acids and peptides. There is a growing need for sustainable methods to synthesize amides that do not rely on coupling reagents or extensive derivatization of carbonyl compounds. In this context, we present an alternative approach for amide synthesis and α-amino acid and peptide functionalization, utilizing the ketene intermediate generated through the photomediated Zimmerman–O'Connell–Griffin (ZOG) rearrangement. This method offers a more sustainable route for late-stage functionalization of peptides, with potential applications in medicinal chemistry, providing a greener alternative to conventional synthetic strategies.
Green foundation1. The present reaction is one of the few examples of amide coupling that proceeds through photogenerated ketenes. We hope this chemistry will inspire chemists to develop new photochemical transformations that utilize photonic energy rather than chemical energy to generate activated carbonyl reagents for amide couplings and related reactions.2. In our developed reaction, we can convert a broad plethora of amines into amides and N-functionalized peptides. The reaction generally proceeds in high yields. Purification typically requires no column chromatography, which saves solvent. The reaction does not require any reagents—only light is needed. 3. The reaction could become even greener if we manage to make it solvent-free and reduce the reaction time. We are currently working on expanding the scope of the reaction, which could naturally lead to new transformations and contribute to the growing repertoire of green chemical processes. |
1 Introduction
Amides are a cornerstone in organic chemistry, playing pivotal roles in biochemistry, pharmaceuticals, and materials science. Their widespread presence in both natural products and synthetic molecules highlights their importance, with amide bond formation recognized as the most frequently employed reaction in medicinal chemistry.1 The formation of amides typically involves the reaction of a carboxylic acid with an amine, a process often facilitated by coupling reagents (Scheme 1a).2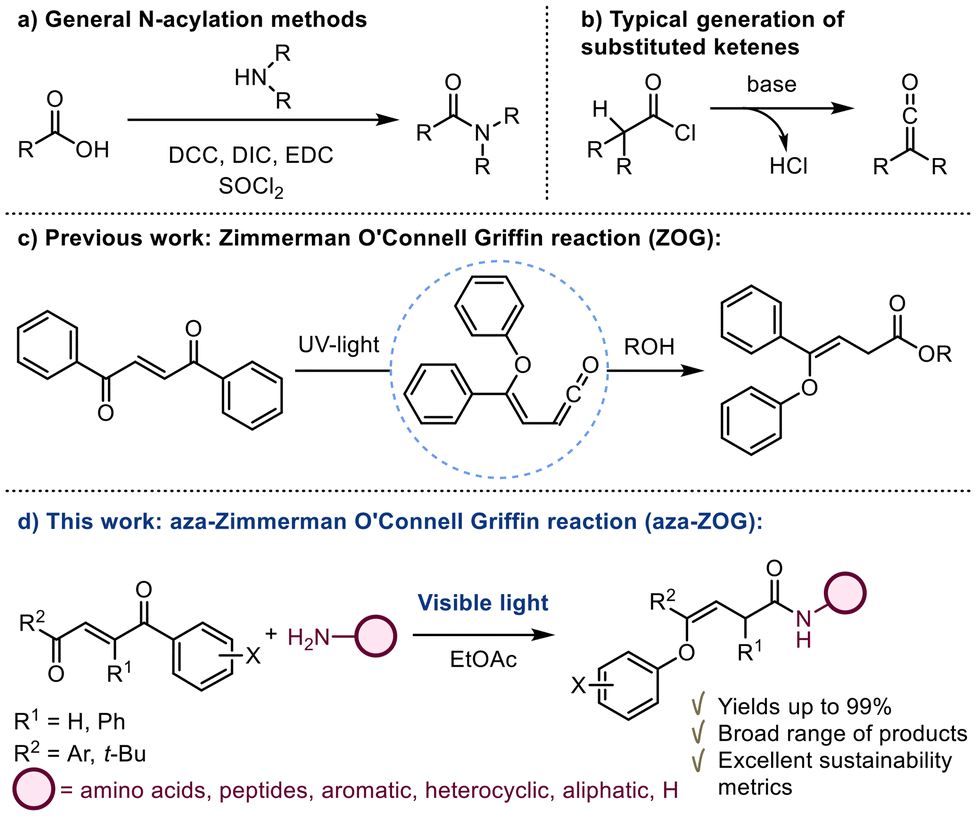 | ||
| Scheme 1 (a) N-Acylation by using coupling reagents on carboxylic acids. (b) Typical generation strategy of substituted ketenes. (c) Previous work: ZOG reaction generating ketenes solely by UV irradiation. (d) This work: development of the visible-light-driven aza-ZOG reaction. | ||
Alternatively, amides can also be synthesized through the reaction of amines with ketenes, a method that offers distinct advantages in terms of reactivity and simplicity. Both approaches are widely recognized for their efficiency and versatility, catering to various synthetic needs.3
However, amide coupling reactions are not without drawbacks. A major limitation is their poor sustainability, as many coupling reagents generate stoichiometric amounts of waste. Additionally, some reactions produce corrosive byproducts, such as HCl or HBr, which can degrade sensitive reagents or products or complicate purification and require specialized handling (Scheme 1b). These factors highlight the need for more sustainable and efficient methods in amide synthesis.
From the perspective of green chemistry, the Zimmerman–O'Connell–Griffin (ZOG) reaction represents an underdeveloped yet promising methodology.4–6 This reaction facilitates the photogeneration of ketenes from 1,2-dibenzoylethylenes (DBE) under mild reaction conditions, without the need for additional reagents (Scheme 1c). Originally, the reaction was conducted under UV light, but it suffered from low yields, limiting its synthetic utility. In 2023, our group reported a significant advancement in this area, demonstrating a visible light promoted synthesis of substituted β-lactams and β-lactones via a regioselective, version of the ZOG reaction, wherein the ketene intermediate was intercepted in a [2 + 2] cycloaddition by imines or aldehydes, respectively.7 Building on this work, we hypothesized that the ketene generated through the ZOG rearrangement could be intercepted by amine nucleophiles, offering a sustainable and efficient strategy for amide synthesis (Scheme 1d). Herein, we present a visible-light-driven, reagent-free, and chemoselective amidation reaction, termed the aza-Zimmerman–O'Connell–Griffin (aza-ZOG) reaction, which exhibits a broad substrate scope, including unprotected amino acids and peptides. This methodology highlights the potential of the ZOG reaction as a green and versatile tool for organic synthesis, particularly in the context of late-stage functionalization and peptide chemistry.
2 Results and discussion
2.1 Optimization
Our study of the aza-ZOG reaction commenced by performing an optimization by reacting (E)-1,4-diphenylbut-2-ene-1,4-dione (E-DBE) 1a with 1H-1,2,4-triazole 4a.Initial optimisation established our standard conditions with ethyl acetate as an excellent reaction solvent giving 5aa in 95% yield. Other solvents were also employed to evaluate the influence of the solvent in the reaction. Firstly, less polar aprotic solvents were investigated, namely toluene and hexane (Table 1, entries 2 and 3). The reaction proceeded efficiently in dry toluene, affording 5aa in a 97% yield (Table 1, entry 2). However, when n-hexane was used as the reaction medium, no formation of product 5aa was observed, and all Z-DBE remained unreacted, likely due to the low solubility of the starting materials in this solvent (Table 1, entry 3).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield % of 5aaa |
| a Yields measured by NMR quantification against standard dimethylsulfone. b Isolated yield. | ||
| 1 | None | 95b |
| 2 | Toluene as solvent | 97 |
| 3 | n-Hexane as solvent | n.r. |
| 4 | Acetonitrile as solvent | 67 |
| 5 | Dichloromethane as solvent | 85 |
| 6 | Tetrahydrofuran as solvent | 98 |
| 7 | Diethyl ether as solvent | 91 |

Dry polar aprotic solvents were well tolerated, yielding moderate to excellent conversions of 5aa, with yields reaching up to 98% with THF as the reaction solvent (Table 1, entry 6). Despite these results, ethyl acetate was selected as reaction solvent for further investigations due to its low toxicity, biodegradability, low environmental impact, pharmaceutical compatibility and favorable performance under standard conditions (Table 1, entry 1). Consequently, these conditions were employed in the exploration of the substrate scope of the aza-ZOG.
2.2 Substrate scope of amino acids
After establishing the optimal conditions for the aza-ZOG reaction, the investigation was extended to explore the reactivity of various N-nucleophiles with E-DBE, including amino acids (Scheme 2). | ||
| Scheme 2 Substrate scope of amino acids run under optimal conditions of Table 1. aYield from the upscaling experiment. bReaction performed at 0.125 mmol scale. c1.05 equivalents of nucleophile. d2 equivalents of nucleophile. e4 equivalents of nucleophile. | ||
Initially, 1a was reacted with the methyl ester of L-phenylalanine, yielding product 3aa in 63% after purification via column chromatography. Subsequently, the reaction was performed using unprotected L-phenylalanine as a nucleophile, where a simple filtration through a syringe filter was sufficient to obtain the pure product 3ab in quantitative yield.
Similarly, L-proline underwent the reaction under the same conditions, affording 3ac in quantitative yield. Remarkably, we were able to scale this reaction up to 3 mmol without detriment in performance while avoiding chromatographic purification, rendering 97% yield of 3ac (see ESI for scaling up protocols†). The reaction of 1a with pipecolic acid (2d) rendered the targeted product 3ad as a mixture of rotamers in 58% yield. However, when unprotected alanine was employed as a nucleophile, the yield decreased significantly to a 15%, necessitating purification via column chromatography (not included in Table 1). A potential explanation could be that alanine forms an electron-donor–acceptor (EDA) complex with E-DBE (1a). This observation is in line with previous studies from our research group on EDA chemistry that have demonstrated that DBE can act as an acceptor in EDA mediated reactions, forming EDA complexes with electron-rich, sterically unhindered amines.8–10 To mitigate this effect, the bulkier L-tert-butyl leucine was tested as a nucleophile for the aza-ZOG, resulting in a clean and quantitative reaction, yielding 99% of 3ae.
Following this trend, sterically hindered amino acids containing a tert-butyl ester were evaluated to suppress side reactivity. tert-Butyl L-alaninate afforded product 3af in quantitative yield, a trend that was consistently observed for 3ag, 3ah, 3ai, 3aq, and 3ar. To functionalize L-tyrosine via the aza-ZOG reaction, the substrate O-tert-buty-L-tyrosine was employed to prevent undesired reactivity from the phenolic –OH group, affording the desired product 3aj in 99% yield.
Reducing the equivalents of di-tert-butyl esters of L-aspartic acid and L-glutamic acid from 1.2 to 1.05 equivalents was necessary to obtain clean products 3ak and 3al after filtration in 95% and 94% yields respectively. The reason for this reduction of equivalents was that the di-tert-butyl esters exhibited higher solubility in ethyl acetate than other tert-butyl esters of amino acids. In the case of product 3am, in which an amide group is also present, increasing the equivalents of tert-butyl L-glutaminate from 1.2 to 2 equivalents resulted in complete selectivity towards the amine. A similar scenario was observed for 3an, where 4 equivalents of tert-butyl L-asparaginate were used to suppress the formation of byproducts. As no chromatography was required, the excess material could be recovered.
It was anticipated that employing a tert-butyl protecting group on the secondary alcohol of O-(tert-butyl)-L-threonine would prevent the side reaction in which the hydroxyl group acts as a nucleophile, successfully resulting in a quantitative yield of 3ao. Interestingly, when E-DBE was reacted with tert-butyl L-serinate, containing an unprotected primary alcohol, complete selectivity for amine functionalization was observed, yielding 3ap in 99%.
2.3 Substrate scope of various N-nucleophiles
Furthermore, the reactivity of this rearrangement was investigated using a variety of amines (Scheme 3). Unsubstituted heterocyclic amines, including 1H-1,2,4-triazole, 1H-benzo[d][1,2,3]triazole, and 1H-pyrazole, successfully participated in the reaction, affording the corresponding products 5aa, 5ab, and 5ac in 95%, 76%, and 84% yield, respectively. Substituted heterocyclic amines containing electron-donating groups, such as 3,5-dimethy-L-1H-pyrazole, 3-phenyl-1H-pyrazole, and 3-(p-tolyl)-1H-pyrazole, demonstrated good nucleophilicity, yielding products 5ad, 5ae, and 5af in 69%, 89%, and 76% yield, respectively. Additionally, the heterocyclic amine 3-(4-fluorophenyl)-1H-pyrazole, bearing an electron-withdrawing p-fluorophenyl group, exhibited excellent reactivity, producing 5ag in 92% yield. Electron-deficient anilines methyl 4-aminobenzoate and 4-aminobenzonitrile were effective nucleophiles, providing the corresponding products 5ah and 5ai in good yields of 80% and 73%, respectively. tert-Butyl phenylglycinate also worked well in the transformation delivering the 5aj in 56% yield. Of note, is that the N-phenyl glycine is a competent donor in EDA reactions together with unsaturated carbonyl compounds.11 Further strengthening the hypothesis that bulky amines divert the reactivity from EDA chemistry to the ZOG reactivity. In line with this observation, bulky amines, such as triphenylmethanamine, also proved to be effective nucleophiles in this transformation, affording the desired product 5ak in 92% yield. Similarly, the aliphatic amine tert-butyl amine demonstrated good reactivity, yielding 5el in 78% after isolation. Finally, the reaction of E-DBE with ammonium acetate was explored, wherein ammonium acetate functioned as a nucleophile, leading to the formation of the primary amide 5am in 85% yield. | ||
| Scheme 3 Substrate scope with amines as nucleophiles run under the optimal conditions of Table 1. aThe reaction was performed using 0.2 mmol of 1a and 1.07 equivalents of 1H-1,2,4-triazole. bThe reaction was performed using 0.2 mmol of 1a and 4.2 equivalents of 1H-benzo[d][1,2,3]triazole. cThe reaction was performed using 0.11 mmol of 1a and 6.6 equivalents of 1H-pyrazole. d1.4 equivalents of nucleophile. | ||
2.4 Substrate scope of E-DBE
The reactivity of various E-DBE derivatives possessing different steric and electronic properties was also investigated (Scheme 4). The di-p-methoxy-substituted DBE (1b) exhibited good compatibility with the ZOG rearrangement when reacted with L-proline, affording product 3bc in 98% yield. Similarly, the di-p-chloro-substituted E-DBE underwent the reaction efficiently, yielding 3cc in high yield when coupled with L-proline. | ||
| Scheme 4 Substrate scope of E-DBE run under the optimal conditions of Table 1. aIrradiated for 48 hours. | ||
For more sterically hindered E-DBE derivatives, such as the substrate bearing two naphthyl groups (1d), the reaction proceeded with a yield of 61% for 3de when employing the tert-butyl ester of L-alanine as the nucleophile. When E-DBE 1e, incorporating an additional phenyl group, was subjected to the reaction with L-proline, the formation of a new stereogenic centre was observed post-rearrangement, resulting in 3ec with a 99% yield and a diastereomeric ratio of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
Additionally, a E-DBE derivative in which one of the phenyl substituents was replaced by a tert-butyl group was evaluated, demonstrating that the rearrangement still proceeded successfully, yielding 3fc in 67%. However, a prolonged irradiation time of 48 hours was required for complete consumption of the starting material.
The mono-p-methoxy-substituted DBE (1g) displayed total selectivity for the migration of the unsubstituted phenyl group during the rearrangement, presumably due to stabilization of the radical intermediate IV (Scheme 5) by the phenyl substituent, yielding 3gc in 99% with a diastereomeric ratio of 5:1. This selective migration of the unsubstituted phenyl group was also observed in the formation of 3he and 3ie, obtained in 63% and 65% yield, respectively, with diastereomeric ratios of 7:5.
 | ||
| Scheme 5 Proposed mechanism for aza-ZOG rearrangement. | ||
In contrast, when a mono-p-bromo-substituted DBE (3j) lacking a stabilizing phenyl ring was tested, a near-equimolar (1:1) mixture of products was obtained for 3je, suggesting that both phenyl groups migrated with comparable probability.
2.5 Proposed mechanism for the aza-ZOG reaction
Scheme 5 illustrates the proposed mechanism of the aza-ZOG rearrangement. Upon irradiation, the E-DBE undergoes rapid isomerization to the Z isomer I within 30 minutes. Subsequently, continued irradiation excites the n–π* electrons of one ketene which subsequently perform an ipso attack on the phenyl group located on the opposite side of the molecule, forming a new oxygen-carbon bond, resulting in the formation of the intermediate II. Following this step, electronic reorganization occurs in which the aryl ring migrates, yielding the ketene intermediate III. The highly reactive ketene is then attacked by a nitrogen-containing nucleophile, leading to the formation of the final product (5aa).2.6 Functionalization of peptides with the aza-ZOG reaction
The applicability of the aza-ZOG rearrangement was extended to peptide functionalization (Scheme 6). The initial attempt involved the reaction of L-phenylalanyl-L-phenylalanine (6a) under the optimized conditions outlined in Table 1, successfully yielding the functionalized product (7aa) in 73% yield. | ||
| Scheme 6 Scope of peptides functionalized with the aza-ZOG rearrangement. aThe reaction solvent was dry ethyl acetate, 1.2 equivalents of diphenylalanine were used respect 0.1 mmol of E-DBE and the product was cleaned by cooling down to −20 °C and syringe filtration. bWas used 1 equivalent of E-DBE and the reaction was irradiated for 48 hours. cThe reaction was irradiated for 48 hours and was purified by wash of the solids with diethyl ether and centrifugation. dYield from the upscaling experiment. | ||
Subsequently, E-DBE was reacted with L-alanyl-L-phenylalanine (6b) using ethyl acetate as the solvent; however, no reaction was observed, and both the dipeptide and Z-DBE were recovered unreacted. It was suspected that this result was due to a solubility issue, and consequently the solvent was changed to dry DMF, a medium in which peptides exhibit higher solubility. Under these modified conditions, the reaction proceeded successfully, affording the desired product (7ab) in 57% yield. Furthermore, L-leucyl-L-proline (6c) and glycylglycine (6e) were successfully functionalized with E-DBE, yielding 7ac in 71% and 7ae in 61% under the established peptide functionalization conditions.
However, a slight modification of the reaction conditions was necessary to obtain 7ad in 73% yield, employing 1 equivalent of E-DBE to prevent functionalization at the primary amide of the L-glutamine residue. Additionally, an extended reaction time of 48 hours was necessary to ensure complete consumption Z-DBE for this same reaction. For the synthesis of the functionalized tripeptide 7af (92% yield), the reaction was irradiated for 48 hours to maximize the conversion of Z-DBE. Due to the insolubility of 7af in organic solvents, purification was achieved through a simple washing step with diethyl ether and centrifugation to remove any residual unreacted Z-DBE. In addition, we were able to scale this reaction up to 2.5 mmol without detriment in performance while avoiding chromatographic purification rendering 70% yield of 7af (see ESI for scaling up protocols†).
2.7 Application of the protocol installing a linker on peptides
We hypothesized that the (Z)-4-phenoxy-4-phenylbut-3-en fragment remaining after N-functionalizing peptides with E-DBE under visible light could serve as a linker for late-stage functionalization of unprotected peptides. Our investigation revealed that hydrolysis of the O-phenyl enolate could be achieved using trifluoroacetic acid (TFA), yielding the structure 8 in 80% yield as shown in Scheme 7. The structure 8 is a N-functionalized unprotected peptide with a linker that contains a ketene functional group, susceptible of undergoing several chemical transformations. These transformations include the formation of imines, hydrazones or oximes to create bifunctional molecules,12 keto–enol reactions or reduction to hydroxyl group. | ||
| Scheme 7 Application of the aza-ZOG reaction for functionalizing peptides with a ketyl linker by hydrolysis with trifluoroacetic acid and reactivity on the linker. | ||
The N-functionalized peptide 8 was then reacted with a stoichiometric amount of biotin hydrazide, resulting in the formation of a hydrazone bond, yielding bifunctional molecule 9 in 93% crude yield with a Z/E stereoisomer ratio of 1:6. Considering the critical role of biotin in chemical biology13 and its relevance in the development of novel therapeutics,14 including antibody–drug conjugates, this methodology for late-stage functionalization of unprotected peptides presents promising opportunities for the discovery of new bioactive molecules.
2.8 Sustainability assessment
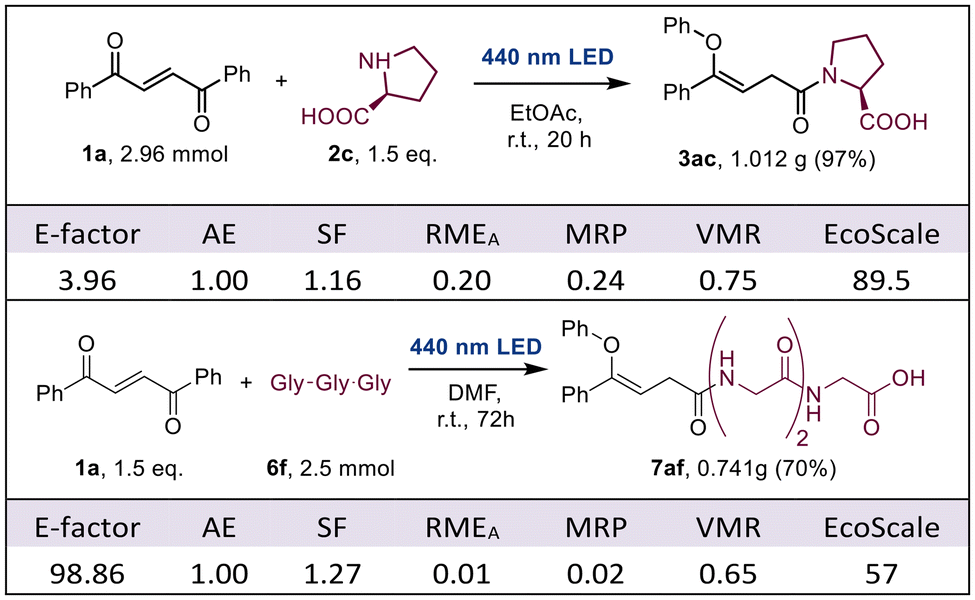
Encouraged by the excellent results observed, we decided to carry out an unbiased assessment of the environmental impact of the aza-ZOG reaction under the conditions described herein. We thus selected the gram scale preparation of 3ac and 7af as representative examples of our methodology. Regarding quantitative metrics, we calculated the E-factor,15 stoichiometric factor (SF),16 atom economy (AE),17 Andraos’ reaction mass efficiency (RMEA)18 and materials recovery parameter (MRP).19,20 The four metrics were combined along with yield into a vector magnitude ratio (VMR) to provide a quick view of the overall material efficiency of the protocol. To address qualitative aspects, we determined the EcoScale scores for both synthetic protocols (Table 2).21|
|
||||||
|---|---|---|---|---|---|---|
| E-Factor | AE | SF | RMEA | MRP | VMR | EcoScale |
| 3.96 | 1.00 | 1.16 | 0.20 | 0.24 | 0.75 | 89.5 |
|
|
||||||
|---|---|---|---|---|---|---|
| E-Factor | AE | SF | RMEA | MRP | VMR | EcoScale |
| 98.86 | 1.00 | 1.27 | 0.01 | 0.02 | 0.65 | 57 |

Beginning with the synthesis of 3ac, the obtained metrics highlight the excellent material efficiency of the aza-ZOG reaction. The E-factor is remarkably low, firmly within the desired range for the elaboration of bulk chemicals (1 to 5), greatly benefitting from avoiding chromatography. In addition to near-quantitative yield, the intrinsic mechanism of this transformation implies a perfect 1.00 (i.e., 100%) AE value. The SF deviates from ideality slightly due to the excess 2c required, although this is mostly negated by the possibility of completely recovering the unreacted material. Indeed, almost complete recovery of both solvent and excess 2c was achieved, which shows in the outstanding values of RMEA and MRP and plays a huge role in the excellent E-factor observed. Overall, the material efficiency of this reaction is excellent, with a VMR of 0.75 (of 1). Regarding the EcoScale, this protocol is only held back slightly due to the flammability of ethyl acetate and the requirement for dry conditions but is otherwise firmly classified as excellent (above 75 points).
The numbers for the preparation of 7af are significantly lower although they are still very good considering the significantly higher difficulty of the transformation in hand. The E-factor is within the desired range for the manufacture of pharmaceuticals (25 to 100). AE is still 1, being the same type of reaction and SF is a touch higher than before, due to the lower yield, but still good, nonetheless. RMEA and MRP are both lower due to the lower relative recovery of materials, which is limited to the diethyl ether used to precipitate and wash the material. Still, the material efficiency is very good, with a VMR of 0.65. The EcoScale highlights the effects of the lower yield and the solvents used, although remains an acceptable protocol (50 to 75 points). At this point, it is important to note that the lower numbers observed mostly come from the necessity of using DMF to solubilize 6f, which will be present in one way or another on most protocols using it as a substrate. Remarkably, we still completely avoid chromatographic purification, which would add 1500 to 2500 points to the E-factor and 10 penalty points to the EcoScale. We also dealt with logistic constraints which forced us to rely on azeotropic distillation with toluene to remove the DMF, rather than using a high vacuum apparatus. Avoiding toluene and recovering the solvent would imply an E-factor of around 17 and an EcoScale of 62, besides much increased RMEA and MRP scores. In general, the numbers obtained for both protocols highlight the exceptional sustainability of the aza-ZOG reaction.
3. Conclusions
In summary, we have developed a novel transformation by adapting the ZOG rearrangement to accommodate nitrogen nucleophiles, resulting in the aza-ZOG reaction. This methodology demonstrates broad substrate scope, tolerating a wide range of N-nucleophiles including N-heterocycles, aromatic and aliphatic amines, sterically hindered amines, and ammonium salts. The reaction is effective for the direct functionalization of amino acids (up to 99% yield) and unprotected peptides (up to 92% yield), with scalability and minimal purification requirements, eliminating the need for chromatographic separation in many cases. This compares exceptionally well with recent advances in photochemical peptide bond formation.22,23 Sustainability metrics further highlight the environmental compatibility of this protocol. Importantly, the installation of ketene linkers on peptides enables subsequent derivatization, offering valuable opportunities for late-stage peptide modification and the development of bifunctional biomolecules.Author contributions
The conceptualization of the project was developed by the authors H. Sundén and S. Bacaicoa. The experimental and analytical work was carried out by S. Bacaicoa, W. Yhlen Graf, M. Martos, A. Runemark, G. Shinde, G. Ghotekar. The sustainability assessment was performed by M. Martos. The manuscript and ESI were written by S. Bacaicoa, H. Sundén and M. Martos.†Conflicts of interest
There are no conflicts to declare.Data availability
The data underlying this study are available in the published article and its ESI.†Acknowledgements
S. B. Acknowledges the Wilhelm och Martina Lundgrens Vetenskapsfond for providing funding to this project (application 2024-GU-4684). M. M. acknowledges the European Commission for a Marie Skłodowska-Curie Actions postdoctoral fellowship under project OTHOx (HORIZON, grant agreement num. 101152118).HRMS analysis was performed at the Proteomics Core Facility, Sahlgrenska Academy, Gothenburg University, with financial support from SciLifeLab and BioMS.
References
- D. G. Brown and J. Boström, Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone?, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed
.
- V. R. Pattabiraman and J. W. Bode, Rethinking amide bond synthesis, Nature, 2011, 480, 471–479 CrossRef CAS PubMed
- L. S. Hegedus, J. Montgomery, Y. Narukawa and D. C. Snustad, A contribution to the confusion surrounding the reaction of ketenes with imines to produce .beta.-lactams. A comparison of stereoselectivity dependence on the method of ketene generation: acid chloride/triethylamine vs photolysis of chromium carbene complexes, J. Am. Chem. Soc., 1991, 113, 5784–5791 CrossRef CAS
- H. E. Zimmerman, H. G. C. Durr, R. G. Lewis and S. Bram, Mechanistic Organic Photochemistry. V.1 Phenyl Migration in a New Photochemical Reaction, J. Am. Chem. Soc., 1962, 84, 4149–4150 CrossRef CAS
- H. E. Zimmerman, H. G. Durr, R. S. Givens and R. G. Lewis, The Photochemistry of Dibenzoylethylenes. Mechanistic and Exploratory Organic Photochemistry. XXII, J. Am. Chem. Soc., 1967, 89, 1863–1874 CrossRef CAS PubMed
- G. W. Griffin and E. J. O'Connell, Photochemistry of Dibenzoylethylene; A Novel Photochemical Rearrangement and Reduction, J. Am. Chem. Soc., 1962, 84, 4148–4149 CrossRef CAS
- A. Runemark, M. Martos, M. Nigríni, F. M. Amombo Noa, L. Öhrström and H. Sundén, Photochemical Access to Substituted β-Lactams and β-Lactones via the Zimmerman–O'Connell–Griffin Rearrangement, Org. Lett., 2023, 25, 5520–5524 CrossRef CAS PubMed
- A. Runemark, S. C. Zacharias and H. Sundén, Visible-Light-Driven Stereoselective Annulation of Alkyl Anilines and Dibenzoylethylenes via Electron Donor–Acceptor Complexes, J. Org. Chem., 2021, 86, 1901–1910 CrossRef CAS PubMed
- A. Runemark and H. Sundén, Aerobic Oxidative EDA Catalysis: Synthesis of Tetrahydroquinolines Using an Organocatalytic EDA Active Acceptor, J. Org. Chem., 2022, 87, 1457–1469 CrossRef CAS PubMed
- C.-W. Hsu and H. Sundén, α-Aminoalkyl Radical Addition to Maleimides via Electron Donor–Acceptor Complexes, Org. Lett., 2018, 20, 2051–2054 CrossRef CAS PubMed
- A. Runemark and H. Sundén, Overcoming Back Electron Transfer in the Electron Donor–Acceptor Complex-Mediated Visible Light-Driven Generation of α-Aminoalkyl Radicals from Secondary Anilines, J. Org. Chem., 2023, 88, 462–474 CrossRef CAS PubMed
- A. F. M. Noisier, M. J. Johansson, L. Knerr, M. A. Hayes, W. J. Drury III, E. Valeur, L. R. Malins and R. Gopalakrishnan, Late-Stage Functionalization of Histidine in Unprotected Peptides, Angew. Chem., Int. Ed., 2019, 58, 19096–19102 CrossRef CAS PubMed
- H. P. Lesch, M. U. Kaikkonen, J. T. Pikkarainen and S. Ylä-Herttuala, Avidin-biotin technology in targeted therapy, Expert Opin. Drug Delivery, 2010, 7, 551–564 CrossRef CAS PubMed
- C. M. Dundas, D. Demonte and S. Park, Streptavidin–biotin technology: improvements and innovations in chemical and biological applications, Appl. Microbiol. Biotechnol., 2013, 97, 9343–9353 CrossRef CAS PubMed
- R. A. Sheldon, The E Factor: fifteen years on, Green Chem., 2007, 9, 1273–1283 RSC
- R. A. Sheldon, Atom efficiency and catalysis in organic synthesis, Pure Appl. Chem., 2000, 72, 1233–1246 CrossRef CAS
- B. Trost, The Atom Economy—A Search for Synthetic Efficiency, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed
- J. Andraos, Unification of Reaction Metrics for Green Chemistry: Applications to Reaction Analysis, Org. Process Res. Dev., 2005, 9, 149–163 CrossRef CAS
-
J. Andraos, Reaction green metrics: problems, exercises, and solutions, CRC Press, 2018 Search PubMed
- J. Andraos, Useful Tools for the Next Quarter Century of Green Chemistry Practice: A Dictionary of Terms and a Data Set of Parameters for High Value Industrial Commodity Chemicals, ACS Sustainable Chem. Eng., 2018, 6, 3206–3214 CrossRef CAS
- K. Van Aken, L. Strekowski and L. Patiny, EcoScale, a semi-quantitative tool to select an organic preparation based on economical and ecological parameters, Beilstein J. Org. Chem., 2006, 2, 3 Search PubMed
- A.
K. Mishra, G. Parvari, S. K. Santra, A. Bazylevich, O. Dorfman, J. Rahamim, Y. Eichen and A. M. Szpilman, Solar and Visible Light Assisted Peptide Coupling, Angew. Chem., Int. Ed., 2021, 60, 12406 CrossRef CAS PubMed
- O. G. Mountanea, D. Psathopoulou, C. Mantzourani, M. G. Kokotou, E. A. Routsi, D. Tzeli, C. G. Kokotos and G. Kokotos, An Efficient Light-Mediated Protocol for the Direct Amide Bond Formation via a Novel Carboxylic Acid Photoactivation Mode by Pyridine-CBr4, Chem. – Eur. J., 2023, 29, e20230055 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, upscaling experiments, green metrics, general synthetic procedure for N-functionalized α-amino acids 3aa–3ar, general synthetic procedure for amides 5aa–5an, general synthetic procedure for substrate scope of DBE 3bc–3je, synthetic procedure for the synthesis of N-functionalized peptides 7aa–7af, synthesis of 8, synthesis bifunctional molecule 9, and characterization by 1H, 13C{1H} NMR, HRMS, IR, melting point and physical description of all molecules synthesized. See DOI: https://doi.org/10.1039/d5gc02108a |
| This journal is © The Royal Society of Chemistry 2025 |