
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The importance of ester cleavage in the butylamine pretreatment of hybrid poplar†
Joseph M.
Palasz
 a,
Anagha
Krishnamoorthy
a,
Raynald A.
Giovine
b,
Xueli
Chen
a,
Venkataramana
Pidatala
ac,
Emine A.
Turumtay
ac,
Tyrell S. A.
Lewis
d,
Edward E. K.
Baidoo
ac,
Chang
Dou
d,
Hemant
Choudhary
ce,
Ning
Sun
d and
Blake A.
Simmons
*ac
a,
Anagha
Krishnamoorthy
a,
Raynald A.
Giovine
b,
Xueli
Chen
a,
Venkataramana
Pidatala
ac,
Emine A.
Turumtay
ac,
Tyrell S. A.
Lewis
d,
Edward E. K.
Baidoo
ac,
Chang
Dou
d,
Hemant
Choudhary
ce,
Ning
Sun
d and
Blake A.
Simmons
*ac
aBiological Systems and Engineering Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. E-mail: basimmons@lbl.gov
bPines Magnetic Resonance Center (PMRC) – Core Facility, College of Chemistry, University of California, Berkeley, CA 94720, USA
cJoint BioEnergy Institute, Emeryville, CA 94608, USA
dAdvanced Biofuels and Bioproducts Process Development Unit, Lawrence Berkeley National Laboratory, Emeryville, CA 94608, USA
eDepartment of Bioresource and Environmental Security, Sandia National Laboratories, Livermore, CA 94550, USA
First published on 27th June 2025
Abstract
This work explores the “in-and-out” pretreatment of hybrid poplar with butylamine as a distillable protic solvent and reagent. The butylamine solvent can be removed by vacuum distillation with >95% solvent removal in all cases, providing a valuable scheme for efficient solvent recovery and recycling. Running the reaction with neat butylamine at 140 °C for 3 hours results in high yields of monosaccharides (90% glucose and 71% xylose) after enzymatic digestion, and a good tolerance to water content with no significant reduction in glucose yield up to an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water:butylamine ratio. We investigate the mechanisms of this pretreatment using powder X-ray diffraction, thermogravimetric analysis, fluorescence microscopy, elemental analysis, solid state and solution state nuclear magnetic spectroscopy to observe chemical and material markers of the pretreatment chemistry. The results suggest that the butylamine leaves the macro and microstructural properties of the lignocellulose relatively unaltered, but conducts targeted ester cleavage chemistry to remove cross-links between the various biopolymers and partially solubilize the lignin component of the biomass. These findings should act to guide future development of pretreatment chemistry for the development of biorefinery processes, and assist in the utilization of biomass as a starting point for chemical syntheses.
:1 water:butylamine ratio. We investigate the mechanisms of this pretreatment using powder X-ray diffraction, thermogravimetric analysis, fluorescence microscopy, elemental analysis, solid state and solution state nuclear magnetic spectroscopy to observe chemical and material markers of the pretreatment chemistry. The results suggest that the butylamine leaves the macro and microstructural properties of the lignocellulose relatively unaltered, but conducts targeted ester cleavage chemistry to remove cross-links between the various biopolymers and partially solubilize the lignin component of the biomass. These findings should act to guide future development of pretreatment chemistry for the development of biorefinery processes, and assist in the utilization of biomass as a starting point for chemical syntheses.
Green foundation1. This work makes significant contributions to advancing sustainable biofuel production through the demonstration of protic amine solvents that can be efficiently recovered after biomass pretreatment.2. Running the reaction with neat butylamine results in high yields of monosaccharides (90% glucose and 71% xylose) after enzymatic digestion, and a good tolerance to water content. 3. It is recommended that future work be focused on developing closed-loop solvent systems with near-100% recovery efficiency. This would include exploring alternative solvent recovery methods, such as membrane separation or adsorption techniques. |
Introduction
As we pursue modernizing our chemical and energy production to efficiently utilize agricultural and forestry waste biomass, it is important to develop efficient conversion technologies for unlocking the potential of these biomass based resources. It has been estimated that there can be more than 1 billion tons of non-food lignocellulosic biomass produced annually in the United States alone.1,2 Non-food waste biomass is of particular interest because most is discarded or burned for energy. Unlocking that biomass for the production of fuels, materials and chemicals will allow us to supply industries from wherever plants grow.The vast majority of waste biomass occurs in the form of lignocellulose, which is an assembly of three biopolymers; cellulose, hemicellulose and lignin. As polysaccharides, the cellulose and hemicellulose fractions can be utilized to generate sugars for microbial fermentations. These bioprocesses can produce a variety of valuable products, including certain high-interest aviation fuel precursors.3–6 While the enzymatic depolymerization of the cellulose and hemicellulose components is well developed.7 The lignocellulose still requires chemical or physical pretreatment for the enzymes to gain access to the polysaccharide components of the biomass.8,9 While fermentation derived bioprocesses based on lignocellulose conversion include several biology focused technologies, the pretreatment of lignocellulose to enable those downstream operations is fundamentally a materials and chemistry problem.
Many biomass pretreatment technologies have been explored over several decades, and many such approaches have resulted in efficient sugar release from a wide variety of potential bio-energy feedstocks. The scalability necessary for fuel and platform chemical production has prevented many of the contemporary pretreatment technologies from being cost-effective. The energy, hardware and reagent costs of pretreatment are of paramount importance in the viability of any lignocellulose derived process at scale.10,11 To substantially reduce these costs, as well as the potential safety impacts of large-scale biomass pretreatment, it is important to develop pretreatment reagents and solvents which can be recycled in a closed-loop system where a small volume of solvent can be continuously recycled to treat a much larger quantity of biomass over time.
Alkaline pretreatments of lignocellulosic biomass are widely studied as they can enable selective lignin and hemicellulose dissolution.12,13 These alkaline pretreatments have been found to operate with a variety of mechanisms including the disruption of crystalline cellulose, the dissolution and removal of lignin and the deacylation of the various ester functionalities. However, the use of mineral bases, while common, requires expensive recovery steps.14 Ammonia gas, aqueous ammonia, and alkylamines are examples of alkaline reagents that can greatly simplify the pretreatment solvent recovery and recycle process and enable an “in-and-out” approach to lignocellulose deconstruction yielding dry pretreated biomass with no carbon losses.12,15–19 Alkylamines in particular have started to appear as effective pretreatment reagents which combine some of the effects of organosolv and alkaline processes.21,22 Alkanolamines have recently been explored as reactive solvents with very promising results.23,24 Butylamine was originally explored by Tanaka et al. for the pretreatment of corn stover.25 As a simple alkylamine (pKa ∼10.7) with intermediate polarity and boiling point (78 °C), it is a good starting point in terms of molecular identity to explore the underlying mechanisms of alkylamine based pretreatments following an in-and-out pretreatment scheme.
Hybrid poplar has been identified as a potential high-yielding bioenergy crop due to its high productivity and ability to regrow from trunks after harvest.26 Hardwoods (and more generally eudicots) represent a promising but difficult class of lignocellulose to unlock due to their tightly interwoven structure and high lignin content when compared to more common grassy feedstocks (e.g. corn stover, sugarcane begasse).19,27 As a hardwood, hybrid-poplar possesses syringyl rich lignin with monolignol conjugates of p-hydroxybenzoate which play a significant, but unidentified, structural role in their cell-walls. The work herein studies samples of a hybrid poplar clone which is a cross between Populus deltoides and Populus nigra as a potentially valuable feedstock for bioenergy and bioproduct processes.
This work explores a pretreatment protocol on hybrid poplar using butylamine as an alkaline reagent and protic solvent that can be removed by distillation, thereby eliminating the losses of soluble sugars and small molecules (Scheme 1). We also explore the reaction space of butylamine pretreatment to understand how conditions such as water content and atmosphere influence the efficacy of the pretreatment process. We investigate the mechanisms of this pretreatment with powder X-ray diffraction (pXRD), thermal gravimetric analysis (TGA), fluorescence microscopy, solution and solid state nuclear magnetic resonance (NMR) spectroscopy to identify the material and chemical changes that occur during pretreatment in the structure of poplar. We maintain a specific focus on unravelling those cross-linking moieties within biomass that are chemically attacked by the butylamine pretreatment, with the aims of informing lignocellulose chemistry towards more selective, facile and scalable processes.
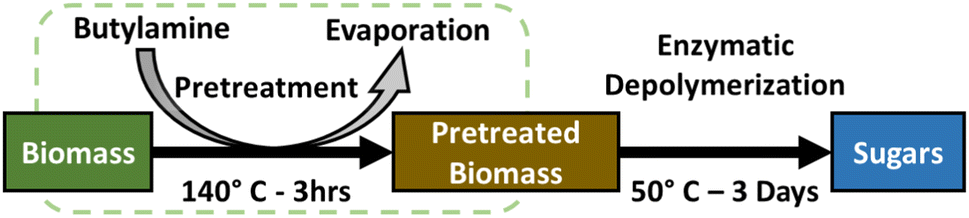 | ||
| Scheme 1 Schematic of the in-and-out pretreatment process explored within this work. The process involves an initial pretreatment step with butylamine at elevated temperature, followed by evaporative drying to yield a dry solid which is efficiently digested by a cellulase/hemicellulase enzyme cocktail to monomeric sugars. | ||
Results and discussion
The in-and-out pretreatment of hybrid poplar with butylamine
To begin exploring the activity of butylamine as an in-and-out pretreatment solvent for hybrid poplar, we first assessed our hybrid poplar biomass samples following the NREL compositional analysis protocol.28 We chose to use the pretreatment conditions of 140 °C for three hours with a 15% solid biomass loading as an intermediate baseline condition. The results of both compositional analysis of hybrid poplar and the pretreatment runs are shown in Fig. 1. The sample has a glucan content of 45 ± 0.4%, with a total lignin content of 29.0 ± 0.1% and xylan content of 16.8 ± 0.1% demonstrating this sample of hybrid poplar may be a compelling source of glucose and lignin fractions. | ||
| Fig. 1 The NREL compositional analysis of the hybrid poplar sample used in this study reported in percentages of the total dry-weight of the biomass (top) and the recovered sugar yields achieved following pretreatment and enzymatic hydrolysis of hybrid poplar with varying pretreatment conditions (bottom). | ||
We first tested the influence of varying solvent composition during pretreatment in glass pressure tubes. Under the tested conditions the butylamine pretreatment performed well both as a neat solvent and as an additive in an aqueous solution, yielding >84% glucose release in all test cases with neat butylamine yielding the most glucose at 90.76 ± 1.50% yield. Untreated hybrid poplar yielded a modest 26.9 ± 0.13% glucose and 21.27 ± 0.19% xylose. Only in the 1:16 butylamine to water ratio was there a significant loss in glucose compared to neat butylamine with the yield dropping to 84.44 ± 1.70%. This is a promising result as it indicates the butylamine pretreatment is tolerant of relatively substantial quantities of water, which may eliminate the need for multiple costly drying steps in a potential biorefinery scheme. The xylan yields varied more substantially, however yields remained about 60% in all cases.
To test the influence of atmospheric composition on the butylamine pretreatment reaction we ran the same pretreatment conditions with 15% solid loading of hybrid poplar with neat butylamine in 75 mL Parr reactors with N2, O2 and CO2. The glucose yields for N2, O2 and CO2 were 94.51 ± 4.1%, 97.86 ± 0.51% and 96.51 ± 2.26% respectively. Unfortunately transfer losses from the Parr reactors prevented the collection of meaningful solvent removal data, although the samples were still extensively dried. The samples ran under a CO2 atmosphere appeared fully solid after cooling, which was likely due to reactions between CO2 and butylamine. Surprisingly, this did not meaningfully affect the sugar yields. The increase in glucose yield across all three samples is attributed to the superior mixing and heat transfer in the parr reactor compared to the prior runs which were conducted in glass pressure tubes. The minimal difference in sugar release with varying atmospheric conditions suggests that the presence of oxygen does not substantially influence the outcome of the pretreatment event, indicating oxidative lignin depolymerization is not a significant contributor to this process. The tolerance to a CO2 atmosphere allows us to envisage a scheme where the reactivity of amines as pretreatment solvents and alkaline scrubbing reagents may be utilized simultaneously.
Overall these results demonstrated promising sugar yields and established that butylamine is a relatively resilient reagent for pretreatment of hybrid poplar, retaining its activity in a wide variety of concentrations and atmospheric conditions. As butylamine is a simple amine, we felt it would be prudent to investigate how butylamine altered the structure of the lignocellulose to enable this heightened activity of the subsequent enzymatic digestion steps.
The mechanism of butylamine pretreatment
Since this pretreatment scheme seems both effective and simple, we chose to pursue some mechanistic characterization to provide insight into how the butylamine pretreatment alters the structure of the biomass. From this point we chose to focus on pretreatment with 100% butylamine under ambient atmosphere. While the sugar yields of aqueous amine pretreatments are consistently high, we suspected that the inclusion of water in the pretreatment system may complicate the reaction pathways, and make it more difficult for us to identify the important mechanism of this pretreatment.As lignocellulose is first, and foremost, a material, we started with several material characterization techniques to observe the changes during pretreatment. Since the primary structural component of plan cell walls are crystalline fibers of cellulose, pXRD has been a valuable tool for assessing whether the cellulose matrix has been altered.29,30 Many pretreatment approaches, including ionic liquid, ammonia and soda pulping report substantial changes to the cellulose crystallinity and suggest that it is an essential operation in those pretreatment reactions.31–33 Contrasting that precedent, the butylamine pretreated hybrid poplar samples showed a near identical pXRD pattern, suggesting that the morphology of the cellulose remains relatively unaltered (Fig. 2A). Thermogravimetric decomposition measurements of lignocellulose have been used to assess changes to both composition34 and cellulose order.35 We employed TGA and the resulting first derivative DTG curve (Fig. 2B) as a secondary technique to assess the how butylamine pretreatment altered the biomass. The TGA curves of untreated and pretreated hybrid-poplar are again similar, showing only a modest change in the shape and peak temperature of the crystalline cellulose decomposition peak near 350 °C.
 | ||
| Fig. 2 pXRD spectra (A) of untreated (black) and butylamine pretreated (blue) hybrid poplar samples, TGA (B) of untreated (black) and butylamine pretreated (blue) hybrid poplar samples, and fixed-contrast fluorescence microscopy images of untreated (C) and pretreated (D) milled particles of hybrid poplar. | ||
To add further qualitative characterization we utilized the autofluorescence of the lignin under fluorescence microscopy to image the hybrid poplar before (Fig. 2C) and after pretreatment (Fig. 2D). The biomass particles did not noticeably change size, nor lose the microstructure of the cell wall ordering, further validating the assertion that the butylamine pretreatment is not altering the material structure or ordering of the cell walls. There was a clear change in the level of contrast in the samples after pretreatment, suggesting that the fluorescent lignin component is being modified or relocated within the biomass during the pretreatment and evaporation process. These results demonstrated that very modest changes to the material properties of the lignocellulose are sufficient to achieve the goal of increasing enzyme accessibility to the biomass. This specificity implies that only a few chemical operations may be necessary for high-yielding pretreatment, and may be possible to achieve in more facile conditions if those modifications can be identified.
To start exploring the chemical changes which have occurred in the pretreated biomass, we used cross-polarization magic-angle-spinning (CP-MAS) solid state nuclear magnetic resonance (ssNMR) spectroscopy. When sufficiently ball-milled to a fine particle size, lignocellulosic biomass can be packed into a 4 mm ssNMR rotor and yield sufficient signal to obtain an informative 13C CP-MAS spectra. The 13C CP-MAS spectra for untreated hybrid poplar, butylamine pretreated hybrid poplar and the solid residual after the enzymatic hydrolysis step are shown in Fig. 3. In further confirmation of the previous materials characterization, the CP-MAS spectra of both untreated and pretreated hybrid poplar are strikingly similar. While not quantitative, peaks attributable to cellulose dominate the spectra for both untreated and butylamine pretreated biomass, reflecting the principal component in the biomass. A small, but noticeable, peak attributable to acyl carbon functionalities is a present around ∼173 ppm in the untreated biomass, which broadens into the baseline in the pretreated spectra, alluding to, but not confirming the modification of acyl carbon functionality during pretreatment. The most stark difference between the untreated and pretreated biomass is the presence of peaks attributable to the butylamine alkyl chain in the 10–50 ppm region of the pretreated biomass. This indicates some inclusion of butylamine groups in the pretreated solid. We suspect some quantity of butylamine reacts via acid–base reactions to form non-volatile butylammonium salts with the carboxylate and phenolate functional groups present in the biomass, as well as with cleaved acetate functionalities resulting in butylammonium acetate. We also expected some chemical derivatization of the biomass through amidation and amination reactions which have been shown to occur in ammonia and other amine based pretreatments. The residual solid after enzymatic hydrolysis clearly shows a higher intensity of the aromatic lignin peaks in the as well as the aryl methoxy peak at ∼55 ppm indicating an enrichment of lignin within the residue. Peaks in the carbohydrate region remain in residual solid, reflecting incomplete depolymerization of the polysaccharides,36 or some residual lignin–carbohydrate complexes which have prevented complete digestion.37 Relative to the lignin aromatic signals appearing from 130–160 ppm, the butylamine derived signals in the 10–40 ppm region decrease in intensity in the residual solid. We take this to indicate that some of the butylamine functionalized species may be water soluble and removed with the aqueous hydrolysate. This may present an issue for fermentation processes if the sugar-rich hydrolysate stream contains butylamine functionalized small-molecules and oligomers.
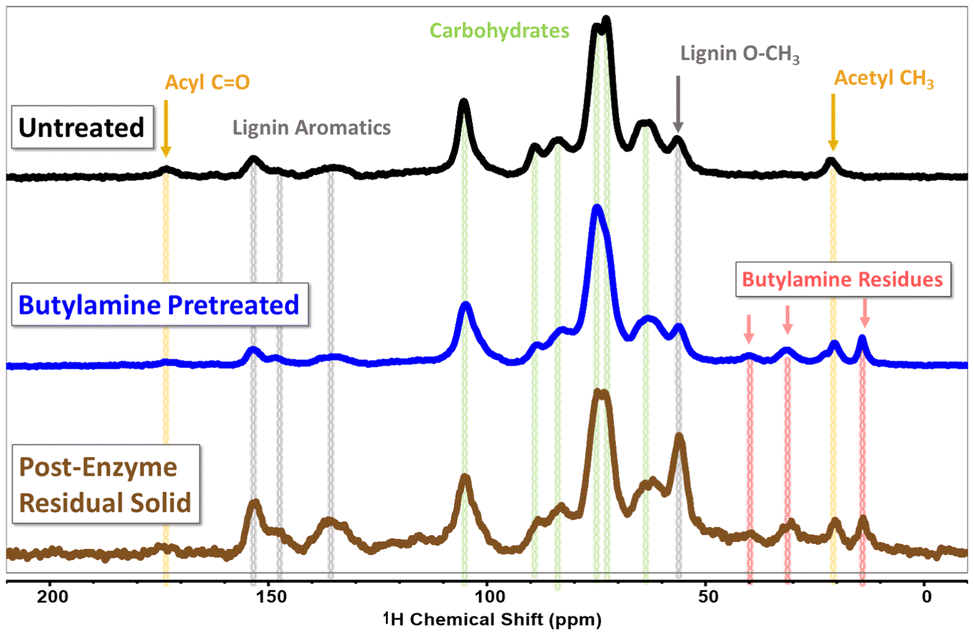 | ||
| Fig. 3 Solid state 13C CP-MAS NMR spectra of untreated (black, top) butylamine pretreated (blue, middle) and the lignin-rich solid residual after enzymatic saccharification (brown, bottom). Peaks assigned to carbohydrate (cellulose and hemicellulose) are marked with green vertical lines. Peaks associated with lignin aromatic moieties are marked with grey lines. Peaks associated with the butyl chain of butylamine are marked with pink lines and peaks associated with acetyl or acyl carbons are marked with yellow. | ||
While we could observe the presence of butylamine residues within the pretreated biomass, we wanted to understand what moieties within the biomass were functionalized. Solution-state NMR can provide information about the chemical linkages present within biomass and has been a valuable tool for the identification of linkages present in the lignin and hemicellulose fractions.38 To start we chose to study the DMSO extracts of untreated and pretreated biomass. DMSO is a strong polar solvent which can dissolve both lignin and some polysaccharide components of biomass, and is widely available in deuterated form.
Unsurprisingly, the extraction of untreated biomass with DMSO-d6 yielded no identifiable soluble products. The pretreated biomass visibly released a dark brown extract. The absence of soluble components in the raw biomass is likely due to the tightly interconnected character of the lignocellulose. This may include various covalent and mechanical crosslinks between the various biopolymer components. In the 13C NMR spectra of the pretreated extract, the major component is N-butylacetamide which we suspect is derived from amidation reactions cleaving acetyl groups in the biomass.
The 1H–13C HSQC spectra of the pretreated extract allows us to identify the major biopolymer components which are solubilized (Fig. 4B and C). The extract contains a lignin with mostly S monomers and minor quantities of guaiacyl (G) and p-hydroxybenzoyl (pHB) moieties. The βO-4 motif is the major assigned linkage observed. This corresponds well with the literature description of most hardwood and poplar lignins.19,39,40 There is a minimal appearance of resinol or phenylcoumaran linkages but both are observable in small quantities. Some free cinnamyl alcohol groups can also be observed in concentrated extract. It has been reported that lignin synthesized from p-hydroxybenzoylated syringyl alcohol subunits produces unique β–β linkages which we suspect may be present in this lignin fraction, however we could not assign their resonances without appropriate reference compounds due to heavy overlap with reported lignin carbohydrate complexes.41–43
 | ||
| Fig. 4 13C NMR spectra of untreated and pretreated hybrid poplar extracted with DMSO-d6 (A) and 1H–13C HSQC of the butylamine pretreated sample shown in the aromatic region (B) and the carbohydrate region. (C) HSQC spectra are color coded when major identifiable linkages and structural motifs when confidently assigned. Syringyl subunits (green), guaiacyl units (blue) and para-hydroxybenzoyl units (magenta) appear in the aromatic region, and B-O4 linkages (purple), xylan (brown) and aryl O-Me groups (teal) appear in the aliphatic region. | ||
The DMSO-D6 extract also contains some xylan, indicating some release of some hemicellulose after pretreatment. It is unclear at this point if this xylan is covalently bound to the released lignin, although some phenolic glycoside linkages could be detected at 4.71 ppm/99.29 ppm. These results indicate that both lignin and hemicellulose portions of the biomass have been made soluble by the pretreatment reaction, demonstrating a marked difference in chemical behavior between the untreated and pretreated biomass.
Additionally, we examined the DMSO-D6 extracts of samples pretreated with 50 wt% aqueous butylamine to observe some of the changes to the reaction space when water was included in the pretreatment (Fig. S16†). The aqueous pretreated sample showed higher relative concentrations of p-hydroxybenzoic acid and acetic acid versus their respective n-butyl amides. This demonstrates a preference for hydrolytic ester cleavage over amidation in the aqueous condition. This suggests that higher water content pretreatments may minimize the direct chemical reactions between butylamine and the biomass, enabling more efficient solvent removal and recovery. Additionally the aqueous sample showed a higher solubilization of hemicellulosic components, suggesting that the inclusion of water aided in the dissolution and release of the polysaccharide components. This is likely due to the increased polarity of the mixed aqueous solvent versus neat butylamine.
Studying the DMSO extracts hints at some chemical changes in the biomass which may explain the dramatic differences in solubility. We also wanted to clarify which species were dissolving into the butylamine during the pretreatment reaction versus simply being released. To do this, we conducted a typical pretreatment with 15% biomass in butylamine at 140 °C for 3 hours. Instead of evaporating the butylamine to give a dry pretreated biomass, the slurry was press filtered through a 0.2 μm PTFE syringe filter to collect a butylamine filtrate. This filtrate was then concentrated in vacuo and NMR samples prepared from the residual solids. The residual solids after press filtration were also washed several times with butylamine to remove any other soluble components. The DMSO-d6 extraction protocol was repeated to identify any “released” components which were not actively dissolved in the butylamine during pretreatment. The results of this are displayed in Fig. 5. The dominant component dissolved by the butylamine pretreatment is lignin, with a small fraction of xylan. It is still unclear if this xylan is part of a covalent lignin–carbohydrate complex (LCC), or simply free hemicellulose. The released material in the residual solid is a clean xylan fraction with little trace of aromatic moieties. In both extraction studies, there was a substantial concentration of components with N-butyl tail groups visible in the alkyl region of the HSQC spectra, providing evidence of butylamine inclusion into the pretreated biomass. Elemental analysis of the biomass before and after pretreatment (Table 1) reveals a significant increase in the nitrogen content of the pretreated biomass, increasing from 0.339 ± 0.100% to 1.972 ± 0.091%. Since these results were obtained after extensive drying of the biomass under elevated temperature and vacuum we hypothesized that this nitrogen was likely in the form of non-volatile derivatives of butylamine, rather than trapped solvent.
 | ||
| Fig. 5 1H–13C HSQC spectra taken in DMSO-d6 of the residual solid resulting from press filtering the wet pretreatment slurry (top, grey) and the contents of the butylamine filtrate (bottom, red). | ||
| Untreated poplar | Butylamine pretreated poplar | ||
|---|---|---|---|
| C | 44.935 ± 0.205% | C | 47.533 ± 0.145% |
| H | 6.590 ± 0.334% | H | 7.529 ± 0.428% |
| N | 0.339 ± 0.100% | N | 1.972 ± 0.091% |
| S | 0.000 ± 0.000% | S | 0.000 ± 0.000% |
To explore the nature of the residual butylamine derivatives, we used LC-MS to investigate the major organic soluble small-molecule products. The results from this are shown in Table 2. The major detected compounds were n-butylamine, N-butylacetamide, N-butyl-4-hydroxybenzamide (pHBAmide) and 4-((butylimino)methyl)phenol. The major detected acid was 4-hydroxybenzoic acid (pHBA), with trace (<1 μM) quantities of 4-hydroxybenzaldehyde, protocatechuic acid and salicylic acid detected.
| Assigned compound | Structure | Concentration (μM) | Formula | Observed m/z | RT |
|---|---|---|---|---|---|
| Amines, amides and imines | |||||
| n-Butylamine |

|
421.148 | C4H11N | 73.09 | 1.82 |
| N-Butylacetamide |

|
1291.408 | C6H13NO | 115.1 | 3.3 |
| N-Butyl-4-hydroxybenzamide |

|
97.100 | C11H15NO2 | 193.111 | 5.26 |
| 4-((Butylimino)methyl)phenol |

|
22.956 | C11H15NO | 177.115 | 7.06 |
| Acids and aldehydes | |||||
| 4-Hydroxybenzoic acid |
|
19.525 | C7H6O3 | 138.031 | 2.93 |
| 4-Hydroxybenzaldehyde |

|
1.234 | C7H6O2 | 122.037 | 3.86 |
| Protocatechuic acid |

|
1.743 | C7H6O4 | 154.024 | 2.48 |
| Salicylic acid |

|
0.517 | C7H6O3 | 138.031 | 5.4 |
N-Butylacetamide occurring in the highest concentration is unsurprising. Hardwood hemicellulose is reported to have a concentration of acetylated monosaccharides44 which we can expect to be cleaved under amidation conditions. While we did not include quantification of acetyl groups in the compositional analysis, the 1291 μM is consistent with a 1–3% acetyl group content in the starting biomass which falls within a typical range for hardwood samples. The high boiling point of the acetamide explains why it remains in the dried biomass in higher concentrations than residual n-butylamine, which we suspect is a combination of physisorbed and chemisorbed in the biomass. The detection of pHBA and pHBAmide in relative concentrations of 19.525 μM and 97.100 μM respectively demonstrates both the successful cleavage of the lignin-esters, as well as the dominance of the amidation chemistry over hydrolysis under these conditions. 4-Hydroxybenzaldehyde is a known oxidative decomposition product of poplar lignin,45 and the high reactivity of aldehydes towards primary amines provides a convincing explanation of the detection of the 4-((butylimino)methyl)phenol.
To verify the observation of these compounds, N-butylacetamide, N-butyl-4-hydroxybenzamide and 4-((butylimino)methyl)phenol were prepared synthetically to use as standards for LC-MS, 1D and 2D NMR experiments and were used to validate their presence in the HSQC and HMBC spectra of the pretreated extracts discussed earlier.
Infrared spectroscopy further reveals the extent of this amidation chemistry (Fig. 6). Untreated hybrid poplar samples show a strong absorption feature at 1733 cm−1 which can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of ester moieties throughout the biomass. Following butylamine pretreatment we observe a complete loss of this feature. Two prominent new features appear in the spectra of pretreated biomass. Centered around 1638 cm−1 and 1559 cm−1 we assign them as the CO stretch and N–H bend of the n-butyl amides. This supports the previous observation of small-molecule amides within the extracts of pretreated biomass, but also demonstrates that most, if not all, of the ester functionality is cleaved during the pretreatment reaction.
O stretch of ester moieties throughout the biomass. Following butylamine pretreatment we observe a complete loss of this feature. Two prominent new features appear in the spectra of pretreated biomass. Centered around 1638 cm−1 and 1559 cm−1 we assign them as the CO stretch and N–H bend of the n-butyl amides. This supports the previous observation of small-molecule amides within the extracts of pretreated biomass, but also demonstrates that most, if not all, of the ester functionality is cleaved during the pretreatment reaction.
 | ||
| Fig. 6 ATR-FTIR of untreated (black) and butylamine pretreated (blue) hybrid poplar samples showcasing the loss of the 1733 cm−1 ester stretching peak in the untreated biomass, and the appearance modes at 1638 cm−1 and 1559 cm−1 which are assigned to amide CO stretching and N–H bending respectively. | ||
This pervasive ester cleavage mirrors the behavior of ammonia based pretreatments, however previous protocols showed much more extensive cellulose disruption than what we have observed with this butylamine pretreatment.15,18–20 The high sugar release we were able to obtain with this pretreatment strategy allows us to postulate that the disruption of cellulose crystallinity may be an unnecessary step in biomass pretreatment. Although some reports have suggested deacetylation of biomass is a vital pretreatment step,46 we find it difficult to conclude that the simple removal of clip-off components from the lignin and hemicellulose would result in such dramatic differences in solubility of the lignin and hemicellulose before and after pretreatment. An alternative hypothesis is that deacetylation is diagnostic of concomitant structural ester cleavage which is the true transformation necessary for effective pretreatment. Monolignol conjugates have been demonstrated to play a vital role in the synthesis of lignin within the cell wall,47 although the specific role of p-hydroxybenzoyl (pHB) conjugates has remained elusive. γ-Esters of uronic acids in hemicellulose have been repeatedly observed in hardwood lignin–carbohydrate complexes and are a known cross-linkage motif within biomass.36,37,48–52 There is evidence that pHB conjugates play an important structural role in poplar lignocellulose,53 suggesting that some ester cross-linking between pHB conjugates may be an additional motif in poplar cell walls. In monocots, monolignol conjugates of coumaric and ferulic acid are known cross-linkage groups between lignin and the polysaccharide fractions47,54 so we believe it is a reasonable suspicion that pHB plays a similar role in hardwoods. However, truly conclusive identification of this linkage chemistry has been difficult to obtain.
To attempt to obtain evidence of cleaved lignin–lignin and lignin–hemicellulose linkages, we conducted a fractionation scheme to isolate the soluble biopolymer components from the pretreated biomass. First, pretreated poplar was washed extensively with methanol to remove soluble lignin and organic small molecules from the biomass. This methanol filtrate was then concentrated, followed by the addition of diethyl ether to induce precipitation of the lignin fraction. This precipitate was washed extensively with additional ether to remove any residual small-molecule species which previously dominated the 2D NMR spectra of the extracts. A schematic of the various sample isolation methods is shown in Fig. S17.† The resulting fraction appeared to be an isolated lignin fraction which was completely soluble in DMSO-d6. In addition to HSQC spectra, which show the directly adjacent carbon and proton resonances in the molecule, we also employed a TOCSY:HSQC which helps to reveal connectivity between nearby protons within a bonding network.55 This allows for the identification of linkages between two distinct moieties within biomass, as evidenced by the appearance of cross-peaks between signals in the traditional HSQC spectra. The aromatic region of the HSQC and TOCSY:HSQC of this fraction are shown in Fig. 7A. The HSQC of this fraction reveals that there is a substantial amount of pHB moieties remaining in the biomass. Because this broad pHB feature is distinct from both pHBA and pHBAmide reference compounds, and it remains with the precipitated lignin fraction even after extensive washing, we can only conclude that these pHB moieties are still directly bound to the lignin. This is further evidenced by the broadness of this feature in the 1H dimension. The TOCSY:HSQC reveals cross peaks with the pHB proton signal at 7.80 ppm with carbons at 38.58 and 31.65 which assign to the C1 and C2 carbons of the N-butyl amide of lignin bound pHB. We take this as strong evidence that butylamine has cleaved the acyl functionality of these groups in the native biomass.
 | ||
| Fig. 7 Overlaid 1H–13C HSQC (colored) and TOCSY:HSQC (black) spectra (A) of the ether precipitated lignin fraction dissolved in DMSO-d6. Syringyl subunits (green), guaiacyl subunits (blue), p-hydroxybenzoyl subunits (magenta) and residual cinnamyl alcohol moieties (orange) are labelled by color. 1H–13C HSQC of the hemicellulose rich aqueous filtrate shown in the aromatic region (B) and the aliphatic region.(C) Xn labels mark the resonances associated with linear xylan chains, (brown) and Un labels mark peaks associated with xylan bound 4-methyl-glucuronic acid (teal). | ||
From the previous FTIR results it appears that most ester linkages have been cleaved during pretreatment, which suggests that these residual pHB groups are likely etherified with the lignin. We propose that the likely linkage sites are either in the α or β ether position of the lignin subunits. This will need to be studied more in depth with 13C enriched biomass to make the identification of the exact attachment chemistries conclusive. Discussion of ether linked pHB has been quite limited, as it is typically only referenced as a “clip-off” compound, but this result suggests that pHB may play a larger role as a branch point for lignin polymerization. This theory may help explain the dramatic differences in lignin solubility before and after pretreatment, as cleavage of the branching groups leads to linear lignin which may be more easily solubilized. To study the water soluble biopolymers, the pretreated and methanol washed biomass was then extracted extensively with warm water (60 °C) over several days to generate an aqueous filtrate which is rich in water soluble hemicelluloses. The combined aqueous extract was then dried and redissolved in D2O. The HSQC of this fraction is shown in Fig. 7B and C. The resulting fraction is expectedly rich in water soluble polysaccharides, with the major identifiable component being deacetylated glucuronoxylan.56,57 Ester linkages between glucuronic acid and lignin have been established previously.48,49,52 We suspect that the cleavage of this ester linkage by butylamine is important for the release of hemicellulose during pretreatment. Between 3.2–2.7 ppm/39 ppm in the 1H/13C dimensions, there is a strong signal which we assign as the alpha carbon of various n-butylated moieties. Unfortunately the solubility of this fraction was minimal enough that direct amidation of any suspected uronic acids was also not observable. The unexpected inclusion of some pHB moieties in this hemicellulose fraction may suggest that pHB may also serve as a lignin–hemicellulose cross-linkage motif in a similar fashion to ferulic acid in other angiosperms,54,58 however this will certainly need to be studied further to give the claim more substance. If true, it may imply a multifaceted structural role of pHB in the cross-linkages between the various components of hardwood lignocellulose.
These results allow us to propose a mechanistic picture of the butylamine pretreatment of hybrid poplar. As the butylamine permeates into the lignocellulose matrix, cleavage of ester cross-linkages between the interconnected biopolymers facilitates the release and physical separation of the lignin and polysaccharide components of the biomass. This release and dissolution allows the cellulase and hemicellulase enzymes access to begin digestion of glycosidic linkages. A visualization of this proposed mechanism is shown in Scheme 2.
 | ||
| Scheme 2 Diagram of proposed mechanism of butylamine pretreatment, showing pHB and glucuronate ester cross-linkages (left) and the cleaved amidation products (right). Below shows the proposed structural changes to lignocellulose after the lignin (grey) has been released from the interconnected structure of cellulose (green) and hemicellulose (orange). | ||
Conclusions
In this work we demonstrate the high efficacy of butylamine as a pretreatment solvent for hybrid poplar. The butylamine can be effectively removed from the biomass by evaporation, providing a viable route for future solvent recycling efforts. We explore the material and chemical effects the butylamine pretreatment has on the properties of lignocellulose. We find that butylamine serves as a targeted pretreatment solvent, showing little signs of cellulose dissolution, decrystallization and minimal alteration of the macrostructure of the biomass samples.The main evidence of pretreatment appears in the increased solubility of lignin, and the cleavage of ester linkages within the biomass. These results suggest a mechanism which involves butylamine permeation into the lignocellulose, followed by ester cleavage, and the release of lignin from the matrix. The concomitant release of clip-off phenolics and acetate groups may provide a route for isolation of fine-chemical fractions directly from a pretreatment process. The detailed study of the structures of hybrid poplar biomass before and after pretreatment suggests that 4-hydroxybenzoate esters in biomass may act as branch points or cross-linkers within lignocellulose, and the breakage of those ester linkages provides a tractable mechanism to unravel the lignocellulose network. This importance of ester cleavage on the pretreatment process will allow us to pursue more facile, and scalable lignocellulose deconstruction chemistries which can target lignocellulose cross-linkages at lower temperatures, concentrations and with non-toxic reagents.
While a fundamental understanding of pretreatment chemistry is important, there are many additional steps necessary to establish butylamine, or other amine, pretreatments as viable processes for large-scale utilization. Lab and pilot scale demonstrations of solvent recycling will be essential for establishing the limits of solvent reuse, nitrogen loss to side-reactions, and obtaining realistic technoeconomic analysis of the energetics and costs of the process. While the high reactivity of butylamine enables efficient pretreatment, its reactivity and toxicity are very important considerations. The high volatility of butylamine provides the benefit of lowering the energy cost of the solvent recovery process,59 however with higher volatility comes a higher flammability risk. Because this pretreatment showed promising results under high aqueous loadings, this may assist with mitigating those risks, and additionally improve downstream toxicity issues by favoring hydrolysis reactions over amidation chemistry.60 Full optimization and exploration of the aqueous amine space will be an important area of study. Dilution of the pretreatment solvent with residual moisture from the feedstock biomass, will require clever process design to maintain pretreatment efficacy over many solvent reuse cycles.
While this work shows butylamine to be very effective for pretreatment of hybrid poplar, many of the downstream process considerations may favor the selection of other alkylamines. We expect the mechanistic results from this work will be applicable to the whole family of alkylamine pretreatment reagents. We also believe this ester cleavage mechanism can be extrapolated to other angiosperm feedstocks (bamboo, corn, rice, switchgrass, sugarcane, etc.) which also feature prominent ester cross-linkages within the cell wall structure. This understanding lets us approach a pretreatment chemistry which can unlock a wide variety of plant biomass for chemical and biological biorefinery processes.
Experimental methods
Hybrid poplar sourcing and pre-processing
Hybrid poplar samples were obtained from Idaho National Laboratory (sample ID 4e9a41fd-c55a-a65a-fd7b36cc0aaa). The samples were sourced from the Boardman Tree Farm in Morrow County, Oregon managed by Greenwood Resources and handled and inventoried by Idaho National Laboratory. The hybrid is a cross between Populus deltoides and Populus nigra with clone OP 367 (433). The samples were ground using a knife mill with a round slit 2 mm outlet screen. The milled sample was allowed to air dry until the total moisture content of the sawdust was repeatedly measured under 10%. The sample was stored in a cool dark environment and used without any further processing.Butylamine pretreatment of hybrid poplar (tube scale)
Following previously established protocols,25,59 hybrid poplar (300 mg) was placed in a tared tube flask. Pretreatment solvent (1.7 g butylamine or butylamine:water solutions) was then added and the vessel was sealed with a Teflon stopper. The tube was placed in an oil-bath preheated to 140 °C for 3 hours. After the allotted time the tube flasks were removed from the oil bath, allowed to cool for 30 minutes, before the stopper was removed and the flasks transferred to a vacuum oven. The pretreated samples were then dried at 80 degrees celsius under house vacuum for at least 12 hours. After drying the tube + dried biomass was weighed again, and the residual dry biomass weight was used to determine the amount of solvent removed by evaporation.
Butylamine pretreatment of hybrid poplar (75 mL Parr scale)
Reactions under controlled atmosphere were run in a Parr instrument company series 5000 multiple reactor system. The reactor vessels used had a total volume of 75 mL and were fitted with a thermostat, pressure sensor and rupture disk with a pressure rating of 200 psi. Reactions were set up by loading 1.5 g of untreated biomass, 8.5 g of butylamine and a Teflon stir bar into the reactor. The atmosphere was exchanged 5 times with 1 bar N2, O2 or CO2 before the reactors were closed. The stirring speed was set to 200 rpm. The reactors were brought to temperature (140 °C) before being allowed to stir at temperature for 3 hours before being allowed to cool to room temperature. The reactors were then opened, covered with a kimwipe, and dried in a vacuum oven at 80 °C overnight.Butylamine pretreatment of hybrid poplar (1 L Parr scale)
A 1 L 4520 Parr bench-top reactor (Parr Instrument Company, Moline, IL) was used to prepare larger quantities of butylamine pretreated biomass. 30 g of poplar was mixed with 170 g of butylamine. The reactors were stirred at 75 rpm, and heated to 140 °C for 3 hours. The mixtures were vacuum-dried at 80 °C following pretreatment, and the weights were recorded at each stage. The dried pretreated biomass was then used for the characterization of pretreated biomass.Fractionation protocol to isolate poplar lignin and hemicellulose
12 g of pretreated hybrid poplar biomass was suspended in 250 mL of methanol in a round bottom flask and stirred for 24 hours. The solid was filtered, and returned to the flask for two more identical washing steps. After the final washing with methanol the solid was dried under vacuum to remove any residual methanol, before being resuspended in 250 mL of warm water (60 °C) for 24 hours. The slurry was again sequentially filtered and washed with water a total of three times and the aqueous filtrates were combined and dried in vacuo to result in the hemicellulose rich fraction shown in Fig. 7B and C.The methanol filtrates from the first washing steps were combined and concentrated in vacuo. The resulting concentrate was a dark brown oil, which was resuspended in methanol until homogeneous. The mixture was then diluted with diethyl ether, resulting in a beige precipitate which was left to settle for 1 hour. The solid was then filtered and dried further under vacuum at 80 °C. This solid was then used as the isolated lignin fraction shown in Fig. 7A.
Enzymatic hydrolysis protocol
Once the biomass was sufficiently dried to remove major traces of butylamine, the biomass was rehydrated with 1.4 mL deionized water for every 300 mg of biomass. This slurry was agitated and allowed to rest for at least 30 minutes to ensure full hydration. The pH of the resulting slurry was then adjusted to within 4.75–5.25 using a 5% solution of H2SO4. After the pH had stabilized within the desired region, 1 M citric acid buffer (pH 5, 100 μL/300 mg biomass), 2% sodium azide (100 μL/300 mg biomass), and enzyme solution (novozymes HTEC3:CTEC3 9:1 ratio diluted with distilled water to 100 μL/300 mg biomass) were added. The dilution of the enzyme cocktail (with a CTec 3:HTec 3 ratio of 9:1 by volume from Novozymes) was adjusted to ensure a 15% dry solids loading of the slurry with 30 mg of enzymes per gram of biomass. The samples were incubated under agitation for 3 days at 50 °C.
Preparation of reference compounds for NMR and HP-LC analysis
N-Butylacetamide was prepared by refluxing butylamine in acetic acid overnight with a catalytic amount of Zn(OAc)2 as reported by Brahmachari and coworkers.61N-Butyl-4-hydroxybenzamide was prepared from methyl-4-hydroxybenzoate as reported by Aitken et al.62 4-((Butylimino)methyl)phenol was prepared from 4-hydroxybenzaldehyde with an equimolar amount of butylamine as reported by Marchand et al.63 All compounds matched their reported syntheses and characterization.Characterization methods
:HSQC) were collected on a Bruker AVANCE NEO 500 MHz instrument equipped with a 5 mm BBO Prodigy CryoProbe utilizing previously reported pulse programs.51 Other 1D experiments were run on a 400 MHz JEOL instrument with a ECZL400S console and a 5 mm HFX probe. 2D spectra were processed with 20 Hz exponential and a 90° sin squared apodization factors in both dimensions and baselined with a full automatic Whittaker Smoother protocol in both dimensions using MestReNova version 15.0.1. 2D spectra were referenced using the solvent residual signal of DMSO-d6 appearing at 39.52 ppm in the 13C dimension and 2.50 ppm in the 1H dimension.
The moisture content of the raw biomass was evaluated by drying 0.5 to 2 grams of the sample in porcelain crucibles using a conventional oven (Binder GmbH, Germany) for 18–24 hours. The dried biomass was subjected to further heating in a muffle furnace at 575 °C for at least 6 hours to determine the ash content. Crucibles were permitted to cool in desiccators, and their weights were recorded between each heating phase.
Moisture content % of biomass:
 | (1) |
Ash content % of biomass:
 | (2) |
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI.† Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.Conflicts of interest
BAS has a financial interest in Illium Technologies, Caribou Biofuels, and Erg Bio. XC, AK, JMP, VRP, and BAS are named inventors on at least one related patent application. All other authors have no financial interest to disclose.Acknowledgements
This work was supported by the DOD Tri-Service Biotechnology for a Resilient Supply Chain (T-BRSC) program through award AWD00007196 to Lawrence Berkeley National Laboratory. The portion of the work conducted at the Joint BioEnergy Institute was supported by the U.S. Department of Energy, Office of Science, Biological and Environmental Research Program, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the U.S. Department of Energy. The portion of the work conducted at the Advanced Biofuels and Bioproducts Development Unit was supported by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Bioenergy Technologies Office. We thank Dr Hasan Celik and Pines Magnetic Resonance Center's Core NMR Facility (PMRC Core) for spectroscopic assistance. The instruments used in this work were supported by the PMRC Core. We thank Nick Setterini for assistance collecting the powder X-ray diffraction spectra. Sandia National Laboratories, a multimission laboratory managed and operated by National Technology and Engineering Solutions of Sandia LLC, a wholly owned subsidiary of Honeywell International Inc. for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA0003525. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. Any subjective views or opinions that might be expressed in this paper do not necessarily represent the views of the U.S. Department of Energy, U.S. Department of Defense, or the United States Government.References
- C. Somerville, Science, 2006, 312, 1277–1277 CrossRef CAS PubMed.
- M. Langholtz , Oak Ridge National Laboratory (ORNL), Oak Ridge, TN (United States), Bioenergy Knowledge Discovery Framework (BEKDF); Oak Ridge National Laboratory (ORNL), Oak Ridge, TN (United States), Bioenergy Knowledge Discovery Framework (BEKDF), DOI: 10.23720/bt2023/2316165.
- D. M. Morris, R. L. Quintana and B. G. Harvey, ChemSusChem, 2019, 12, 1646–1652 CrossRef CAS PubMed.
- K. E. Rosenkoetter, C. R. Kennedy, P. J. Chirik and B. G. Harvey, Green Chem., 2019, 21, 5616–5623 RSC.
- N. R. Baral, M. Yang, B. G. Harvey, B. A. Simmons, A. Mukhopadhyay, T. S. Lee and C. D. Scown, ACS Sustainable Chem. Eng., 2021, 9, 11872–11882 CrossRef CAS.
- C.-L. Liu, T. Tian, J. Alonso-Gutierrez, B. Garabedian, S. Wang, E. E. K. Baidoo, V. Benites, Y. Chen, C. J. Petzold, P. D. Adams, J. D. Keasling, T. Tan and T. S. Lee, Biotechnol. Biofuels, 2018, 11, 285 CrossRef CAS PubMed.
- J. S. Van Dyk and B. I. Pletschke, Biotechnol. Adv., 2012, 30, 1458–1480 CrossRef CAS PubMed.
- G. Brodeur, E. Yau, K. Badal, J. Collier, K. B. Ramachandran and S. Ramakrishnan, Enzyme Res., 2011, 2011, 787532 Search PubMed.
- A. R. Mankar, A. Pandey, A. Modak and K. K. Pant, Bioresour. Technol., 2021, 334, 125235 CrossRef CAS PubMed.
- A. K. Chandel, V. K. Garlapati, A. K. Singh, F. A. F. Antunes and S. S. da Silva, Bioresour. Technol., 2018, 264, 370–381 CrossRef CAS PubMed.
- V. Balan, ISRN Biotechnol., 2014, 2014, 463074 Search PubMed.
- J. S. Kim, Y. Y. Lee and T. H. Kim, Bioresour. Technol., 2016, 199, 42–48 CrossRef CAS PubMed.
- J. Yang, M. Sun, L. Jiao and H. Dai, Polymers, 2021, 13(23), 4166 CrossRef CAS PubMed.
- P. Bajpai, in Green chemistry and sustainability in pulp and paper industry, Springer International Publishing, Cham, 2015, pp. 11–39 Search PubMed.
- S. P. S. Chundawat, R. Vismeh, L. N. Sharma, J. F. Humpula, L. da Costa Sousa, C. K. Chambliss, A. D. Jones, V. Balan and B. E. Dale, Bioresour. Technol., 2010, 101, 8429–8438 CrossRef CAS PubMed.
- A. Mittal, R. Katahira, B. S. Donohoe, S. Pattathil, S. Kandemkavil, M. L. Reed, M. J. Biddy and G. T. Beckham, ACS Sustainable Chem. Eng., 2017, 5, 2544–2561 CrossRef CAS.
- J. R. Meyer, S. B. Waghmode, J. He, Y. Gao, D. Hoole, L. da Costa Sousa, V. Balan and M. B. Foston, Biomass Bioenergy, 2018, 119, 446–455 CrossRef CAS.
- F. Teymouri, L. Laureano-Perez, H. Alizadeh and B. E. Dale, Bioresour. Technol., 2005, 96, 2014–2018 CrossRef CAS PubMed.
- V. Balan, L. da C. Sousa, S. P. S. Chundawat, D. Marshall, L. N. Sharma, C. K. Chambliss and B. E. Dale, Biotechnol. Prog., 2009, 25, 365–375 CrossRef CAS PubMed.
- S. P. S. Chundawat, R. K. Pal, C. Zhao, T. Campbell, F. Teymouri, J. Videto, C. Nielson, B. Wieferich, L. Sousa, B. E. Dale, V. Balan, S. Chipkar, J. Aguado, E. Burke and R. G. Ong, J. Visualized Exp., 2020, 158, e57488 Search PubMed.
- L. Xu, M. Cao, J. Zhou, Y. Pang, Z. Li, D. Yang, S.-Y. Leu, H. Lou, X. Pan and X. Qiu, Nat. Commun., 2024, 15, 734 CrossRef CAS PubMed.
- S. Ntakirutimana, T. Xu, H. Liu, J.-Q. Cui, Q.-J. Zong, Z.-H. Liu, B.-Z. Li and Y.-J. Yuan, Green Chem., 2022, 24, 5460–5478 RSC.
- E. C. Achinivu, S. Frank, N. R. Baral, L. Das, M. Mohan, P. Otoupal, E. Shabir, S. Utan, C. D. Scown, B. A. Simmons and J. Gladden, Green Chem., 2021, 23, 8611–8631 RSC.
- E. C. Achinivu, M. Mohan, H. Choudhary, L. Das, K. Huang, H. D. Magurudeniya, V. R. Pidatala, A. George, B. A. Simmons and J. M. Gladden, Green Chem., 2021, 23, 7269–7289 RSC.
- M. Tanaka, C. W. Robinson and M. Moo-Young, Biotechnol. Bioeng., 1985, 27, 362–368 CrossRef CAS PubMed.
- P. Sannigrahi, A. J. Ragauskas and G. A. Tuskan, Biofuels, Bioprod. Biorefin., 2010, 4, 209–226 CrossRef CAS.
- T. Treasure, R. Gonzalez, H. Jameel, R. B. Phillips, S. Park and S. Kelley, Biofuels, Bioprod. Biorefin., 2014, 8, 755–769 CrossRef CAS.
- J. Sluiter, A. Sluiter, C. Scarlata, D. Templeton, D. Crocker and B. Hames, Laboratory Analytical Procedure (LAP), 2012, https://www.nrel.gov/docs/gen/fy13/42618.pdf Search PubMed.
- P. Ahvenainen, I. Kontro and K. Svedström, Cellulose, 2016, 23, 1073–1086 CrossRef CAS.
- S. Singh, G. Cheng, N. Sathitsuksanoh, D. Wu, P. Varanasi, A. George, V. Balan, X. Gao, R. Kumar, B. E. Dale, C. E. Wyman and B. A. Simmons, Front. Energy Res., 2014, 2 DOI:10.3389/fenrg.2014.00062.
- G. Cheng, P. Varanasi, R. Arora, V. Stavila, B. A. Simmons, M. S. Kent and S. Singh, J. Phys. Chem. B, 2012, 116, 10049–10054 CrossRef CAS PubMed.
- L. da C. Sousa, J. Humpula, V. Balan, B. E. Dale and S. P. S. Chundawat, ACS Sustainable Chem. Eng., 2019, 7, 14411–14424 CrossRef CAS.
- C. Driemeier, M. T. B. Pimenta, G. J. M. Rocha, M. M. Oliveira, D. B. Mello, P. Maziero and A. R. Gonçalves, Cellulose, 2011, 18, 1509–1519 CrossRef CAS.
- M. Carrier, A. Loppinet-Serani, D. Denux, J.-M. Lasnier, F. Ham-Pichavant, F. Cansell and C. Aymonier, Biomass Bioenergy, 2011, 35, 298–307 CrossRef CAS.
- T. P. Schultz, G. D. McGinnis and M. S. Bertran, J. Wood Chem. Technol., 1985, 5, 543–557 CrossRef CAS.
- D. Min, C. Yang, V. Chiang, H. Jameel and H. Chang, Fuel, 2014, 116, 56–62 CrossRef CAS.
- D. Min, Q. Li, V. Chiang, H. Jameel, H. Chang and L. Lucia, Fuel, 2014, 119, 207–213 CrossRef CAS.
- J. Ralph and L. Landucci, in Lignin and lignans: advances in chemistry, ed. C. Heitner, D. Dimmel and J. Schmidt, CRC Press, 2010, pp. 137–243 Search PubMed.
- R. Martín-Sampedro, J. I. Santos, Ú. Fillat, B. Wicklein, M. E. Eugenio and D. Ibarra, Int. J. Biol. Macromol., 2019, 126, 18–29 CrossRef PubMed.
- Y. Mottiar, S. D. Karlen, R. E. Goacher, J. Ralph and S. D. Mansfield, Plant Biotechnol. J., 2023, 21, 176–188 CrossRef CAS PubMed.
- F. Lu, J. Ralph, K. Morreel, E. Messens and W. Boerjan, Org. Biomol. Chem., 2004, 2, 2888–2890 RSC.
- F. Lu, S. D. Karlen, M. Regner, H. Kim, S. A. Ralph, R.-C. Sun, K. Kuroda, M. A. Augustin, R. Mawson, H. Sabarez, T. Singh, G. Jimenez-Monteon, S. Zakaria, S. Hill, P. J. Harris, W. Boerjan, C. G. Wilkerson, S. D. Mansfield and J. Ralph, Bioenerg. Res., 2015, 8, 934–952 CrossRef CAS.
- T.-Q. Yuan, S.-N. Sun, F. Xu and R.-C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS PubMed.
- S. Gille and M. Pauly, Front. Plant Sci., 2012, 3, 12 CAS.
- W. Schutyser, J. S. Kruger, A. M. Robinson, R. Katahira, D. G. Brandner, N. S. Cleveland, A. Mittal, D. J. Peterson, R. Meilan, Y. Román-Leshkov and G. T. Beckham, Green Chem., 2018, 20, 3828–3844 RSC.
- P. Wen, Z. Chen, Z. Lian and J. Zhang, Bioresour. Technol., 2023, 385, 129459 CrossRef CAS PubMed.
- S. D. Karlen, C. Zhang, M. L. Peck, R. A. Smith, D. Padmakshan, K. E. Helmich, H. C. A. Free, S. Lee, B. G. Smith, F. Lu, J. C. Sedbrook, R. Sibout, J. H. Grabber, T. M. Runge, K. S. Mysore, P. J. Harris, L. E. Bartley and J. Ralph, Sci. Adv., 2016, 2, e1600393 CrossRef PubMed.
- J. Larsbrink and L. Lo Leggio, Essays Biochem., 2023, 67, 493–503 CrossRef PubMed.
- M. L. Gandla, M. Derba-Maceluch, X. Liu, L. Gerber, E. R. Master, E. J. Mellerowicz and L. J. Jönsson, Phytochemistry, 2015, 112, 210–220 CrossRef PubMed.
- S.-Y. Jeong, E.-J. Lee, S.-E. Ban and J.-W. Lee, Carbohydr. Polym., 2021, 270, 118375 CrossRef CAS PubMed.
- H. Nishimura, A. Kamiya, T. Nagata, M. Katahira and T. Watanabe, Sci. Rep., 2018, 8, 6538 CrossRef PubMed.
- T. Watanabe and T. Koshijima, Agric. Biol. Chem., 1988, 52, 2953–2955 CAS.
- Y. Zhao, X.-H. Yu and C.-J. Liu, Front. Plant Sci., 2021, 12, 755576 CrossRef PubMed.
- D. M. de Oliveira, A. Finger-Teixeira, T. R. Mota, V. H. Salvador, F. C. Moreira-Vilar, H. B. C. Molinari, R. A. C. Mitchell, R. Marchiosi, O. Ferrarese-Filho and W. D. dos Santos, Plant Biotechnol. J., 2015, 13, 1224–1232 CrossRef CAS PubMed.
- J. Ralph, J. M. Marita, S. A. Ralph, R. D. Hatfield, F. Lu, R. M. Ede, J. Peng and L. L. Landucci, Advances in lignocellulosics characterization, TAPPI Press, Atlanta, Ga, 1999, pp. 55–108 Search PubMed.
- P. Vincent, F. Ham-Pichavant, C. Michaud, G. Mignani, S. Mastroianni, H. Cramail and S. Grelier, Polymers, 2021, 13(13), 2044 CrossRef CAS PubMed.
- J. Zhou, X. Du, S. Zhou and S. Wu, Cellulose, 2023, 30, 4855–4871 CrossRef CAS.
- R. Samuel, M. Foston, N. Jiang, L. Allison and A. J. Ragauskas, Polym. Degrad. Stab., 2011, 96, 2002–2009 CrossRef CAS.
- E. C. Achinivu, B. W. Blankenship, N. R. Baral, H. Choudhary, R. Kakumanu, M. Mohan, E. E. K. Baidoo, C. D. Scown, A. George, B. A. Simmons and J. Gladden, Chem. Eng. J., 2023, 147824 Search PubMed.
- C. Zhao, Q. Shao and S. P. S. Chundawat, Bioresour. Technol., 2020, 298, 122446 CrossRef CAS PubMed.
- G. Brahmachari, S. Laskar and S. Sarkar, J. Chem. Res., Synop., 2010, 34, 288–295 CrossRef CAS.
- R. A. Aitken, A. D. Harper, R. A. Inwood and A. M. Z. Slawin, J. Org. Chem., 2022, 87, 4692–4701 CrossRef CAS PubMed.
- P. Marchand, L. Griffe, M. Poupot, C.-O. Turrin, G. Bacquet, J.-J. Fournié, J.-P. Majoral, R. Poupot and A.-M. Caminade, Bioorg. Med. Chem. Lett., 2009, 19, 3963–3966 CrossRef CAS PubMed.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- B. Amer, R. Kakumanu, Y. Tian, A. Eudes and E. E. K. Baidoo, Targeted analysis of phenolic compounds by LC-MS, 2021 protocols.io DOI:10.17504/protocols.io.byhcpt2w.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc01795e |
| This journal is © The Royal Society of Chemistry 2025 |