
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe low-temperature selective oxidation of alcohols and a one-pot oxidative esterification using an IBS(III/V)/oxone catalysis†
Ryutaro Kondo,
Muhammet Uyanik * and
Kazuaki Ishihara*
* and
Kazuaki Ishihara*
Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa, Nagoya 464-8603, Japan. E-mail: muha@chembio.nagoya-u.ac.jp; ishihara@cc.nagoya-u.ac.jp
First published on 7th July 2025
Abstract
The IBS/oxone catalyst system has been improved for selective alcohol oxidation at near-room temperature (30 °C), overcoming the limitations of the conventional high-temperature (70 °C) method. An NMR spectroscopy analysis identified the rate-determining step, leading to the development of optimized conditions based on the pre-generation of IBS(III) and a phase-transfer catalyst. These improvements enhanced the functional-group tolerance and substrate scope, including thermally unstable, acid-sensitive, or overoxidation-prone alcohols. Furthermore, the system was successfully applied to one-pot oxidative esterification reactions, demonstrating its versatility and synthetic utility in mild oxidation processes.
Green foundation1. This work introduces a low-temperature (30 °C), transition metal-free IBS(III/V)/oxone catalytic system for the selective oxidation of alcohols and one-pot oxidative esterification, offering a sustainable alternative to conventional oxidation methods that rely on toxic heavy metals, transition metal catalysts, or elevated temperatures.2. This method employs oxone, a green terminal oxidant, and avoids hazardous reagents and energy-intensive conditions, while achieving high chemoselectivity and broad substrate scope—including acid-sensitive and overoxidation-prone alcohols. 3. Future work will focus on further improving sustainability by replacing nitromethane or acetonitrile with greener solvents, and by developing recyclable or immobilized IBS catalysts to enhance operational efficiency and reduce waste. |
Introduction
The selective oxidation of alcohols to carbonyl compounds is a fundamental transformation in organic synthesis that plays a key role in the production of fine chemicals, pharmaceuticals, and functional materials.1 The development of environmentally benign and efficient methods for this transformation remains a significant challenge and a central objective in green and sustainable chemistry.1,2 Traditional oxidation methods often rely on heavy-metal oxidants, such as chromium(VI) and manganese(IV), or transition-metal catalysts, such as ruthenium and copper.1 In recent years, hypervalent iodine compounds3 and nitroxyl radicals4 have emerged as promising metal-free alternatives, offering high efficiency with reduced toxicity. However, many existing methods still suffer from drawbacks, such as the use of stoichiometric hazardous oxidants, harsh reaction conditions, or limited substrate scope, emphasizing the need for more sustainable approaches.1–4 The 2-iodoxybenzenesulfonic acid (IBS)5/oxone catalyst system is an example of a metal-free alternative, developed by our group,6 that has demonstrated significant potential for alcohol oxidation under non-aqueous conditions (Scheme 1a).7,8 Oxone, a commercially available triple inorganic salt (KHSO5·0.5KHSO4·0.5K2SO4),9 serves as a terminal oxidant, facilitating the stepwise oxidation of iodine species from I(I) to I(III) and subsequently to I(V). As illustrated in Scheme 1b,6 pre-IBS 1 is initially oxidized to IBS(III) 2, starting the catalytic process. The oxidation cycle then proceeds via a reversible I(III)/I(V) redox equilibrium, where 2 is further oxidized to IBS(V) 3, which subsequently oxidizes the alcohol substrate and thus regenerates 2. | ||
| Scheme 1 IBS/oxone catalysis for alcohol oxidation. | ||
Despite its advantages, the original IBS/oxone system requires elevated temperatures (typically 70 °C), which limited its functional-group tolerance and substrate scope (Scheme 1a).6a,d,e High-temperature conditions frequently lead to overoxidation, dehydration, and undesired side reactions, particularly for thermally unstable or acid-sensitive substrates and those prone to overoxidation. To address these challenges, we explored two key strategies: (1) enhancing the oxone solubility using a solid–liquid phase-transfer catalyst (PTC) and (2) identifying the rate-determining step of the catalytic cycle and optimizing the reaction conditions accordingly, aiming to develop a more versatile, energy-efficient, and substrate-tolerant oxidation protocol. The pursuit of lower-temperature catalytic oxidations without compromising efficiency, particularly for sensitive and synthetically relevant substrates, remains a central objective in contemporary green chemistry.10
We have previously demonstrated that a PTC improves oxone solubility, thus facilitating efficient oxidation in the IBS/oxone-catalyzed oxidative dearomatization of phenols.11 Here, we extend this approach to alcohol oxidation to enhance the reaction scope and efficiency. More importantly, an NMR analysis identified the rate-determining step under non-aqueous conditions to be the oxidation of 1 to 2 prior to entering the catalytic cycle (Scheme 1b). Based on these findings, we hypothesized that using 2 directly instead of 1 would accelerate the oxidation process. Indeed, this modification significantly improved the reaction efficiency, allowing alcohol oxidation at lower temperatures while maintaining high chemoselectivity (Scheme 1c). Furthermore, we extended the IBS/oxone system to achieve a one-pot oxidative esterification under mild conditions, demonstrating its versatility and practical utility.
Results and discussion
To evaluate the performance of the IBS/oxone system at lower temperatures, we selected the oxidation of 5-nonanol (4a) to 5-nonanone (5a) in the presence of pre-IBS 1a (2 mol%) and powdered oxone6 in acetonitrile as the model reaction (Scheme 2a). Under conventional high-temperature conditions (70 °C),6a the reaction proceeded rapidly. However, at 30 °C, the reaction rate significantly decreased, highlighting the limitations of the existing system. To improve the efficiency of the oxidation at lower temperatures, we introduced tetrabutylammonium hydrogen sulfate as a solid–liquid PTC to enhance the solubility of oxone in the organic solvent. As anticipated, this modification reduced the reaction time from 24 h to 11 h. | ||
| Scheme 2 Initial investigation of the IBS/oxone catalysis at lower temperatures. | ||
To gain deeper insight into the catalytic mechanism, we conducted a series of NMR experiments to monitor the iodine oxidation process (Scheme 2b; I(I): black, I(III): blue, I(V): red).
In CD3CN (in the presence of the PTC), the oxidation of I(I) to I(III) was identified as the rate-determining step, as the conversion of 1a to 2a was slow, whereas the subsequent oxidation of 2a to 3a proceeded rapidly (Scheme 2b, left). The influence of electron-donating substituents was then examined in CD3CN. The 4,5-dimethyl-substituted pre-IBS 1c exhibited significantly faster consumption than 1a, attributed to both enhanced solubility12 and the electronic effects6 of the methyl groups. In contrast, the 4-methyl derivative 1b reacted at a rate comparable to 1a, suggesting that a substantial increase in solubility, as observed for 1c, might be an important factor in achieving marked rate acceleration in this system. Despite these variations in I(I) consumption rates, all corresponding IBS(III) intermediates (2a, 2b, and 2c) remained consistently at very low or undetectable levels (Scheme 2b, left). This uniformly low accumulation–even for the potentially faster-forming 2c–indicates their extremely rapid subsequent oxidation to IBS(V) species, supporting the oxidation of I(I) to I(III) as the overall rate-determining step. In D2O, the oxidation of 1a to 2a proceeded rapidly, but the subsequent oxidation of 2a to 3a was slower (Scheme 2b, right).
Although the mechanism by which 1 is oxidized by oxone is not yet fully understood, these pronounced differences in reaction profiles and identified rate-determining steps between CD3CN and D2O can be explained by solubility effects. Both oxone and 1a are poorly soluble in organic solvents, which slows the initial oxidation to 2a, while 2a and 3a are more soluble, allowing faster oxidation of 2a to 3a. In contrast, in water, all species (1a, 2a, 3a, and oxone) are fully dissolved, resulting in faster oxidation steps than in organic solvents. It is also important to note that different amounts of oxone were used in each solvent: 10 equivalents in CD3CN and 4 equivalents in D2O. These loadings were selected to reflect and accommodate the distinct solubility and reactivity profiles of oxone and the iodine species in each solvent. While absolute oxone concentration may influence reaction rates, the chosen conditions were optimized to clearly reveal the distinct kinetic bottlenecks in each medium. Therefore, these variations in oxone loading do not affect the validity of our mechanistic conclusions.
Building on these mechanistic insights, we developed a new strategy to improve the efficiency of the IBS/oxone system. We hypothesized that using IBS(III) 2 as the starting species could eliminate the slow initiation step, thereby accelerating the oxidation process. IBS(III) 2 can be easily synthesized from pre-IBS 1 using peracetic acid in the presence of sulfuric acid.13
To evaluate this approach, we compared the outcomes of the oxidation of 4a using pre-IBS 1c or IBS(III) 2c (1 mol%) (Scheme 3a, Method B). Although the reactions proceeded smoothly at room temperature (23–26 °C), we used a thermostatic bath set to 30 °C in most experiments to ensure consistent temperature control and reproducibility. To our delight, the use of 2c resulted in a faster reaction (red vs. black in the graph). This improvement was attributed to the rapid formation of 3c from 2c, eliminating the need for the slow initial oxidation step under non-aqueous conditions.
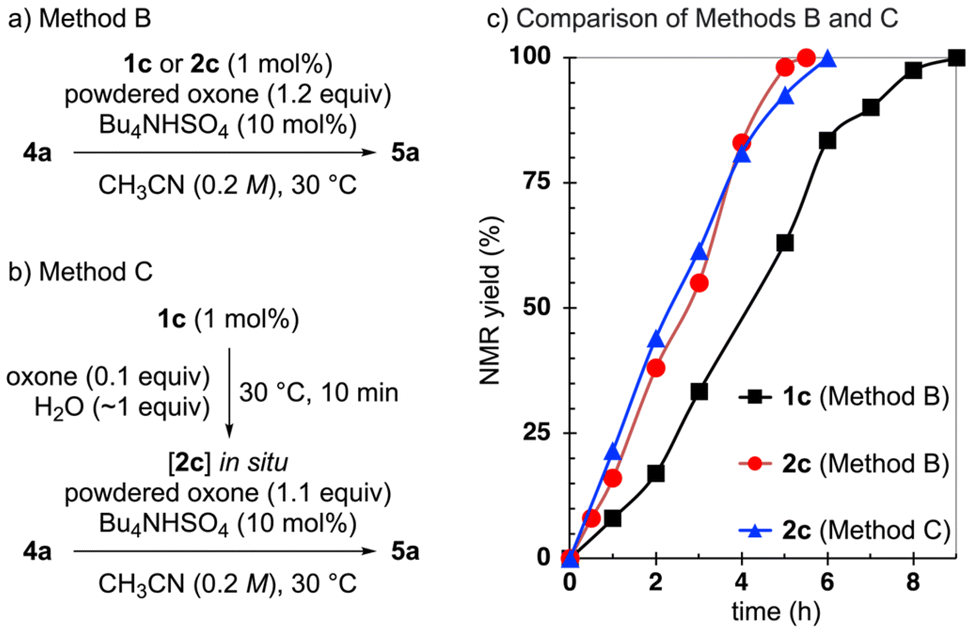 | ||
| Scheme 3 Design of new procedures, Methods B and C. | ||
While pre-formed IBS(III) 2 successfully accelerated the reaction, we also explored an alternative strategy to generate IBS(III) 2 in situ, allowing the use of the more cost-effective and easily handled precatalyst pre-IBS 1. As 1 can be rapidly oxidized to 2 in water, we designed a modified procedure in which a small amount of water (∼1 equiv.) was added to 1 and oxone to pre-generate 2 in situ (Scheme 3b, Method C; blue vs. black in Scheme 3c). Although the catalytic activity of IBS decreases significantly in aqueous conditions, we anticipated that the dehydrating effect of oxone (specifically its K2SO4 component) would mitigate the impact of the small amount of water used. Thus, we found that the reaction proceeded rapidly, at a comparable rate to Method B, when 2c was pre-generated in situ before adding the alcohol substrate (blue vs. red in Scheme 3c). Furthermore, we compared the performance of IBS(III) 2a–2c when using Method C. As the previous NMR experiments had led us to expect (Scheme 2b), 2c exhibited the highest reactivity, further confirming that electron-donating substituents enhance the oxidation efficiency (Scheme S1†).
To demonstrate the generality and applicability of the improved IBS/oxone system using 2c in the presence of a PTC, we explored the oxidation of various primary and secondary alcohols. We selected nitromethane based on our previous findings,6a which showed that it consistently provides superior chemoselectivity for the conversion of primary alcohols to aldehydes, particularly compared to conventional solvents such as acetonitrile. Here, we particularly focused on substrates that have posed challenges under conventional high-temperature conditions that use pre-IBS 1a in the absence of a PTC. Under these conventional conditions, overoxidation and side reactions are frequently observed, particularly for electron-rich benzylic alcohols, acid-sensitive terpenoids, and functionalized aliphatic alcohols.
To further illustrate these issues, we examined representative substrates prone to these side reactions. The oxidation of 4-methoxybenzyl alcohol (4b) under conventional high-temperature conditions yielded a complex mixture containing 4-methoxybenzaldehyde (5b) in low yield, with the complex mixture likely resulting from dehydration and overoxidation pathways (Scheme 4a, Method A). Similarly, 1,2,3,4-tetrahydro-1-naphthol (4c) yielded α-tetralone (5c) in low yield, likely due to overoxidations leading to the formation of quinonoid byproducts (Scheme 4b, Method A). In contrast, the low-temperature oxidation conditions developed here (Method B) effectively suppressed these competing pathways, affording 5b and 5c in significantly improved yields.
 | ||
Scheme 4 Comparison of conventional and current oxidation methods: representative examples. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Anhydrous Na2SO4 was used as a desiccant in both the conventional and current oxidation methods to prevent aldehyde hydration to hemiacetals during the oxidation of the primary alcohols.6a Anhydrous Na2SO4 was used as a desiccant in both the conventional and current oxidation methods to prevent aldehyde hydration to hemiacetals during the oxidation of the primary alcohols.6a | ||
The oxidation of electron-rich benzylic alcohols (4d–f), 6-methoxy-2-naphthalenemethanol (4g), and 9-anthracenemethanol (4h) proceeded efficiently under low-temperature conditions (Method B), affording the corresponding aldehydes in high yield (Scheme 5a). Similarly, both primary and secondary acid-sensitive alcohols were efficiently oxidized under the improved conditions. Primary alcohols, including (−)-myrtenol (4i), (−)-nopol (4j), and the epoxide-containing aliphatic alcohol 4k, could be successfully oxidized using either Method B or C (Scheme 5a), while secondary alcohols such as (−)-isopulegol (4l) and (−)-cis-verbenol (4m) were effectively converted to ketones using Method C (Scheme 5b). In contrast, oxidation under high-temperature conditions (Method A) led to side reactions and the formation of complex mixtures of products. Additionally, the oxidation of Boc-protected amino alcohol 4n and the highly lipophilic steroid-derived alcohol 4o afforded the corresponding ketones in high yield, demonstrating the broad functional-group tolerance and applicability of the new system (Scheme 5b).
 | ||
| Scheme 5 Chemoselective oxidation of alcohols. aUnless otherwise noted, the reactions were performed with 1.0 mmol of 4 under the optimized conditions using Method B. Yields from conventional Method A (see Scheme 4) are shown in brackets. b1a or 2c (5 mol%). cIn EtOAc (0.1 M). dPowdered oxone (1.6 equiv.). e1a or 2c (10 mol%). fA complex mixture containing unidentified side products was obtained. gDibenzyl ether (a dehydrative dimerization byproduct) was obtained in 70% yield. hMethod C. For details, see the ESI.† | ||
The IBS/oxone catalyst system naturally operates under acidic conditions due to the KHSO4 component of oxone. Moreover, as the oxidation progresses, KHSO4 is gradually generated as a byproduct, further increasing the acidity of the reaction medium. Lowering the reaction temperature to 30 °C not only moderates the oxidative strength but also reduces the acidity, thereby enabling a milder yet effective oxidation process. This adjustment expands the substrate scope to include acid-sensitive and overoxidation-prone alcohols, which are challenging substrates for the high-temperature conditions. An alternative approach to controlling acidity involves using K2CO3-buffered oxone, which we previously demonstrated to be effective in the IBS/oxone-catalyzed oxidative dearomatization of phenols.11b However, during alcohol oxidation, this approach substantially reduces the oxidative power of the system, making it unsuitable for the present reaction (Scheme S2†).
To further demonstrate the utility of the improved IBS/oxone system, we explored its applicability to the formation of lactones8e and oxidative esterification reactions. The oxidation of 1,4-butanediol (6) under low-temperature conditions using in situ pre-generated 2c (Method C) led to the selective oxidation of one hydroxyl group, followed by an intramolecular cyclization and further oxidation, affording γ-butyrolactone (7) in high yield (Scheme 6a). In contrast, the conventional high-temperature conditions using 1a (Method A) resulted in a lower yield, likely due to the dehydration of the lactol intermediate under the harsh conditions applied.
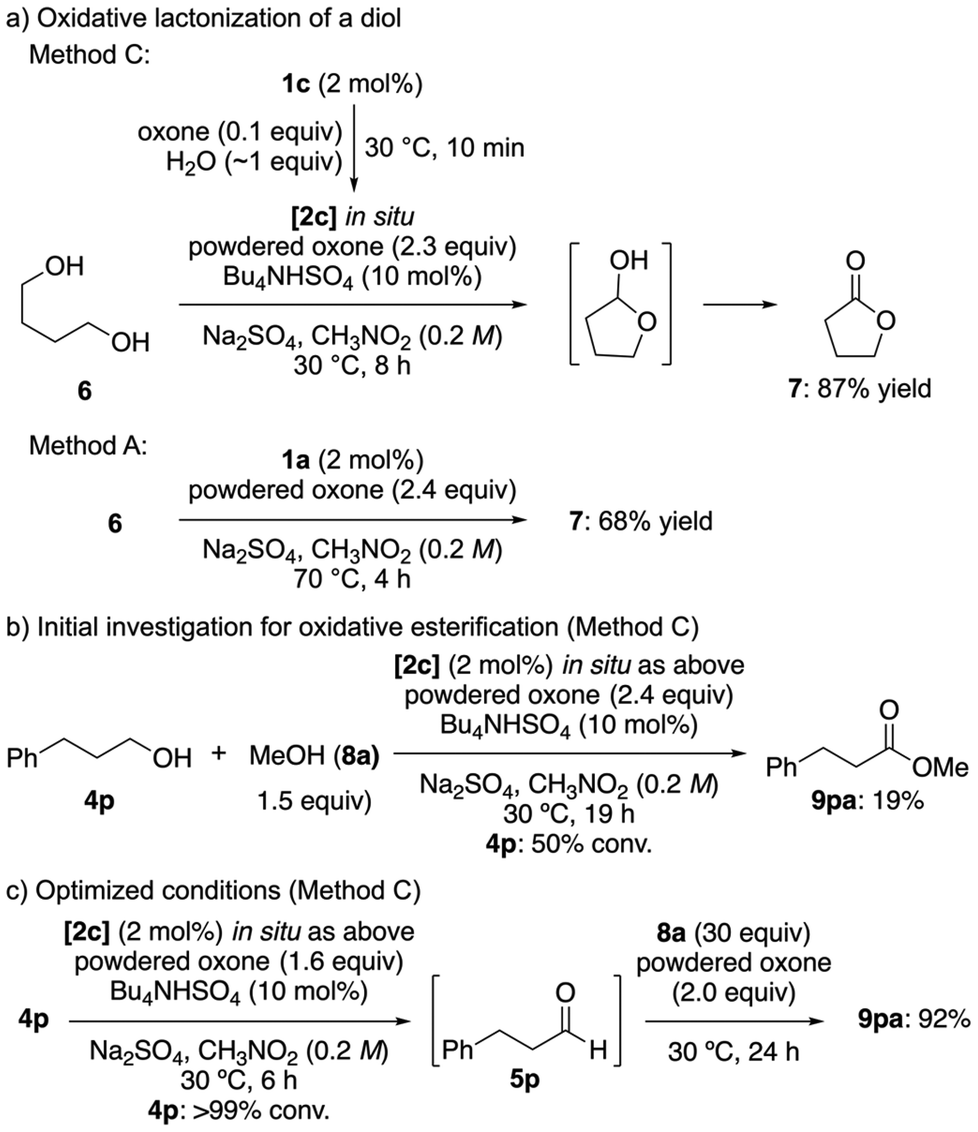 | ||
| Scheme 6 Oxidative lactonization and esterification of alcohols. | ||
Encouraged by this result, we extended the method to encompass intermolecular oxidative esterification reactions. Esters are widely used in pharmaceuticals, cosmetics, and fine chemicals, and they also serve as protecting groups in organic synthesis. Conventional esterification reactions from alcohols typically require multiple steps,14 whereas the present method enables a direct esterification via a selective oxidation to aldehydes.
We initially examined the oxidation of 3-phenyl-1-propanol (4p) in the presence of methanol (8a) to yield methyl 3-phenylpropionate (9pa) (Scheme 6b). However, the presence of 8a likely inhibited ligand exchange7k between IBS(V) and the substrate alcohol 4p, suggesting that oxidative esterification in the presence of two different alcohols is challenging. To overcome this issue, we first allowed the complete oxidation of 4p to the aldehyde 5p prior to the addition of 8a (Scheme 6c). This modification prevented interference between the two alcohols during the initial oxidation step, thereby facilitating an efficient oxidative esterification. Optimization studies revealed that using 1.6 equivalents of oxone for the oxidation of 4p to 5p, followed by the addition of 2.0 equivalents of oxone and an excess of 8a, afforded 9pa in 92% yield (Scheme 6c; for details, see Table S1†). This stepwise oxone addition is feasible because overoxidation is less problematic at room temperature, allowing the controlled oxidation of the aldehyde intermediate without the occurrence of significant side reactions.
With the optimized conditions in hand, we explored the substrate scope for the oxidative esterification (Scheme 7). Replacing methanol (8a) with ethanol (8b) or 2-propanol (8c) yielded the corresponding esters, even though the bulkier 8c required a higher temperature for the final hemiacetalization/oxidation step. The oxidation of a propargyl alcohol (4q), benzyl alcohol (4r), and dibenzyl alcohols (4s, 4t) also proceeded efficiently, affording ethyl ester 9qb and methyl esters 9ra–9ta at elevated temperatures.
 | ||
| Scheme 7 Oxidative esterification of alcohols. Unless otherwise noted, the reactions were performed with 1.0 mmol of 4. Conditions for the hemiacetalization/oxidation step are shown in parentheses. aROH 8 (40 equiv.). bMeOH 8a (60 equiv.). c10 mmol of 4 were used. | ||
To demonstrate the synthetic utility of this method, we applied it to the total synthesis of phlomic acid, a naturally occurring axially chiral allene.15 In the two-step method reported by Ma et al., the esterification requires the use of toxic trimethylsilyldiazomethane.16 In contrast, our one-pot oxidative esterification approach afforded ester 9ua in 96% yield (Scheme 7), surpassing the efficiency of the literature method while eliminating the need for hazardous reagents.
To quantitatively validate the green credentials of our approach, we performed an EcoScale analysis.17 For the oxidation of primary alcohols (4b to 5b), the present low-temperature method achieved an excellent EcoScale score of 76, significantly higher than the score of 45 obtained with our previous high-temperature protocol.6a This improvement is primarily attributed to the substantially improved yield and milder reaction conditions. A similarly notable enhancement was observed for the oxidation of secondary alcohols (4c to 5c), with the EcoScale score increasing from 19.5 to 68 (see ESI† for detailed calculations).
To further contextualize the advantages of our method, it is valuable to consider its position relative to state-of-the-art biocatalytic approaches. Biocatalysis, typically employing enzymes such as alcohol dehydrogenases (ADHs), oxidases (AOXs), or galactose oxidase variants (GOase), represents a premier strategy in green chemistry, capable of delivering unparalleled selectivity under exceptionally mild, aqueous conditions, often utilizing oxygen as a benign oxidant.18 Complementing these highly specialized enzymatic systems, chemocatalytic methods like the one developed herein offer distinct and complementary advantages, primarily centered on broader substrate scope, operational simplicity, and robustness. Our system utilizes standard laboratory reagents and common synthetic techniques, bypassing the complexities associated with enzyme stability, cofactor recycling, and enzyme-specific optimization. Therefore, it provides a readily accessible and practical alternative for a wide variety of functionalized alcohols, especially in cases where suitable enzymes may not be readily available or applicable.
Ultimately, the choice between biocatalytic and chemocatalytic approaches depends strategically on the target substrate, required reaction conditions, and practical constraints. The present IBS/oxone catalysis expands the green oxidation toolbox by offering a highly practical, efficient, and easily implementable chemocatalytic option.
Conclusions
In conclusion, we have developed a significantly improved IBS/oxone catalyst system that enables efficient and selective oxidation of alcohols at 30 °C with broad substrate applicability. This advance was achieved through a mechanistically guided strategy involving the pre-generation (or efficient in situ formation) of the active IBS(III) species, along with enhanced solubility of oxone via a phase-transfer catalyst (PTC). These modifications not only accelerated the oxidation process but also effectively suppressed side reactions, thereby expanding the scope to include thermally unstable, acid-sensitive, and oxidation-prone substrates. Lowering the operational temperature from 70 °C to 30 °C represents a significant step toward more energy-efficient and sustainable synthetic methodologies. The versatility and practicality of this system were further demonstrated by its successful application to one-pot oxidative lactonization and, notably, highly efficient oxidative esterification. These examples highlight its potential as an atom-economical alternative to conventional multi-step processes. Overall, this work not only expands the utility of hypervalent iodine catalysis but also demonstrates its promise as a practical and sustainable platform for key oxidative transformations, addressing the growing demand for greener and more streamlined processes in the synthesis of pharmaceuticals and fine chemicals.Author contributions
M. U. and K. I. developed the concept and conceived the experiments. R. K. performed the experiments. R. K. and M. U. analyzed the data. M. U. prepared the manuscript with assistance from K. I.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
Financial support for this project was partially provided by JSPS KAKENHI grants 23H05467 (to K. I.) and 21H01932 (to M. U.). We extend our gratitude to Dr K. Oyama (Nagoya University) for the elemental analyses.References
- (a) S. V. Ley, Comprehensive Organic Synthesis, Pergamon, Oxford, 3rd edn, 1999, ch. 2, vol. 7, pp. 251–327 Search PubMed; (b) J. E. Backvall, Modern Oxidation Methods, Wiley-VCH, New York, 2004 CrossRef; (c) G. Tojo and M. Fernandez, Oxidation of Alcohols to Aldehydes and Ketones, Springer, Berlin, 2006 Search PubMed; (d) RSC Green Chemistry Series No. 28, in Transition Metal Catalysis in Aerobic Alcohol Oxidation, ed. F. Cardona and C. Parmeggiani, Royal Society of Chemistry, London, 2015 Search PubMed; (e) M. Uyanik and K. Ishihara, in Patai's Chemistry of Functional Groups, ed. I. Marek, B. Olofsson and Z. Rappoport, John Wiley & Sons, Chichester, 2019, pp. 261–306 Search PubMed.
- (a) T. F. S. Silva and L. M. D. R. S. Martins, Molecules, 2020, 25, 748 CrossRef CAS PubMed; (b) Y. Zhang, C. Cao and G. Li, Biomass, 2022, 2, 103–115 CrossRef CAS; (c) C. Corberán, M. E. González-Pérez, S. Martínez-González and A. Gómez-Avilés, Appl. Catal., A, 2024, 474, 211–223 CrossRef; (d) N. Ayoub, Z. Hamie, J. Toufaily, E. Guénin and G. Enderlin, ACS Sustainable Chem. Eng., 2024, 12, 9568–9590 CrossRef CAS.
- (a) M. Ochiai and K. Miyamoto, Eur. J. Org. Chem., 2008, 4229–4239 CrossRef CAS; (b) T. Dohi and Y. Kita, Chem. Commun., 2010, 46, 2073–2085 RSC; (c) M. Uyanik and K. Ishihara, Chem. Commun., 2009, 46, 2086–2099 RSC; (d) Topics in Current Chemistry, in Hypervalent Iodine Chemistry, ed. T. Wirth, Springer, Berlin, 2016, vol. 373 Search PubMed; (e) Iodine Catalysis in Organic Synthesis, ed. K. Ishihara and K. Muñiz, Wiley-VCH, Weinheim, 2022 Search PubMed; (f) F. V. Singh, S. E. Shetgaonkar, K. Manjula and T. Wirth, Chem. Soc. Rev., 2022, 51, 8102–8139 RSC; (g) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2024, 124, 11108–11860 CrossRef CAS PubMed.
- (a) A. E. J. de Nooy, A. C. Besemer and H. van Bekkum, Synthesis, 1996, 1153–1176 CrossRef CAS; (b) R. A. Sheldon and I. W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS; (c) D. Leifert and A. Studer, Chem. Rev., 2023, 123, 10302–10380 CrossRef CAS PubMed.
- (a) A. Y. Koposov, D. N. Litvinov, V. V. Zhdankin, M. J. Ferguson, R. McDonald and R. R. Tykwinski, Eur. J. Org. Chem., 2006, 4791–4795 CrossRef CAS; (b) I. A. Mironova, P. S. Postnikov, R. Y. Yusubova, A. Yoshimura, T. Wirth, V. V. Zhdankin, V. N. Nemykin and M. S. Yusubov, Beilstein J. Org. Chem., 2018, 14, 1854–1858 CrossRef CAS PubMed.
- (a) M. Uyanik, M. Akakura and K. Ishihara, J. Am. Chem. Soc., 2009, 131, 251–262 CrossRef CAS PubMed; (b) M. Uyanik, R. Fukatsu and K. Ishihara, Org. Lett., 2009, 11, 3470–3473 CrossRef CAS PubMed; (c) M. Uyanik and K. Ishihara, Aldrichimica Acta, 2010, 43, 83–91 CAS; (d) M. Uyanik and K. Ishihara, Org. Synth., 2012, 89, 105–114 CrossRef CAS; (e) K. Ishihara, M. Uyanik, Y. Isogai and S. Ohara, JP Patent Reg. No. JP-4590514(B2), 2010 Search PubMed.
- (a) L.-Q. Cui, K. Liu and C. Zhang, Org. Biomol. Chem., 2011, 9, 2258–2265 RSC; (b) Y. Tanaka, T. Ishihara and T. Konno, J. Fluor. Chem., 2012, 137, 99–104 CrossRef CAS; (c) F. Drouet, G. Masson and J. Zhu, Org. Lett., 2013, 15, 2854–2857 CrossRef CAS PubMed; (d) V. C. Purohit, S. P. Allwein and R. P. Bakale, Org. Lett., 2013, 15, 1650–1653 CrossRef CAS PubMed; (e) C. Si, K. R. Fales, A. Torrado, K. Frimpong, T. Kaoudi, H. G. Vandeveer and F. G. Njoroge, J. Org. Chem., 2016, 81, 4359–4363 CrossRef CAS PubMed; (f) E. Nisigaki, K. Sugamoto, M. Nishida and Y. Matsushita, Chem. Lett., 2016, 45, 746–748 CrossRef CAS; (g) J. Zhang, Y.-F. Ling, G.-X. Wang, L. Zhang and J. Luo, Org. Biomol. Chem., 2018, 16, 4784–4788 RSC; (h) N. Ayoub, C. Bergère, J. Toufaily, E. Guénin and G. Enderlin, New J. Chem., 2020, 44, 11577–11583 RSC; (i) Z. Tan, X. Ju, H. Wu, W. Dong, J. C. Leung, X. Hou, H. Lee, A. Granger, J. M. Paolillo, S. V. Dimeo, C. Chen, L. Wu, J. C. Lorenz, M. Sarvestani, F. Buono, R. Frutos, T. G. Tampone, X. Huang, G. Zhang, Y. Wang, E. Spinelli, Z. Lei and J. J. Song, Org. Process Res. Dev., 2024, 28, 78–91 CrossRef CAS; (j) K. Bensberg, A. Savvidis, F. Ballaschk, A. Gómez-Suárez and S. F. Kirsch, Chem. – Eur. J., 2024, 30, e202304011 CrossRef CAS PubMed; (k) H. Jiang, T.-Y. Sun, X. Wang, Y. Xie, X. Zhang and H. F. Schaefer, Org. Lett., 2017, 19, 6502–6205 CrossRef CAS PubMed.
- (a) A. P. Thottumkara, M. S. Bowsher and T. K. Vinod, Org. Lett., 2005, 7, 2933–2936 CrossRef CAS PubMed; (b) A. Schulze and A. Giannis, Synthesis, 2006, 257–260 CAS; (c) P. C. B. Page, L. F. Appleby, B. R. Buckeley, S. M. Allin and M. J. McKenzie, Synlett, 2007, 1565–1568 CrossRef CAS; (d) T. Miura, K. Nakashima, N. Tada and A. Ito, Chem. Commun., 2011, 47, 1875–1877 RSC; (e) S. Seth, S. Jhulki and J. N. Moorthy, Eur. J. Org. Chem., 2013, 2445–2452 CrossRef CAS; (f) T. Yakura, A. Yamada, N. Noda, T. Fujiwara and H. Nambu, Asian J. Org. Chem., 2014, 3, 421–424 CrossRef CAS; (g) A. Maity, S.-M. Hyun, A. K. Wortman and D. C. Powers, Angew. Chem., Int. Ed., 2018, 57, 7205–7209 CrossRef CAS PubMed.
- (a) H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3371 CrossRef CAS PubMed; (b) S. Alvi, V. Jayant and R. Ali, ChemistrySelect, 2022, 7, e202200704 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- (a) M. Uyanik, T. Mutsuga and K. Ishihara, Molecules, 2012, 17, 8604–8616 CrossRef CAS PubMed; (b) M. Uyanik, T. Mutsuga and K. Ishihara, Angew. Chem., Int. Ed., 2017, 56, 3956–3960 CrossRef CAS PubMed.
- J. N. Moorthy, K. Senapati, K. N. Parida, S. Jhulki, K. Sooraj and N. N. Nair, J. Org. Chem., 2011, 76, 9593–9601 CrossRef CAS PubMed.
- (a) G. F. Koser, G. Sun, C. W. Porter and W. J. Youngs, J. Org. Chem., 1993, 58, 7310–7312 CrossRef CAS; (b) M. W. Justik, Tetrahedron Lett., 2007, 48, 3003–3007 CrossRef CAS; (c) Y. Ishiwata and H. Togo, Synlett, 2008, 2637–2641 CAS.
- (a) S. Tang, J. Yuan, C. Liu and A. Lei, Dalton Trans., 2014, 43, 13460–13470 RSC; (b) L. Ding, P. Su, Y. Shi, L. Zhu, H. Zhang and B. Wang, Ind. Eng. Chem. Res., 2024, 63, 18667–18698 CrossRef CAS.
- K. Aitzetmüller, N. Tsevegsüren and K. Vosmann, Lipid/Fett, 1997, 99, 74–78 CrossRef.
- X. Jiang, J. Zhang and S. Ma, J. Am. Chem. Soc., 2016, 138, 8344–8347 CrossRef CAS PubMed.
- F. Van Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2, 3 Search PubMed.
- (a) F. Hollmann, I. W. C. E. Arends, K. Bühler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226–265 RSC; (b) J. Dong, E. Fernández-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS PubMed; (c) H. Puetz, E. Puchl'ová, K. Vranková and F. Hollmann, Catalysts, 2020, 10, 952 CrossRef CAS; (d) C. D. F. Milagre and H. M. S. Milagre, Alcohol dehydrogenase-catalyzed oxidation, Curr. Opin. Green Sustainable Chem., 2022, 38, 100694 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, additional information, experimental protocols, nuclear magnetic resonance spectra. See DOI: https://doi.org/10.1039/d5gc01737h |
| This journal is © The Royal Society of Chemistry 2025 |