
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-promoted catalyst-free aminolytic degradation for chemical recycling of single and mixed plastic wastes†
Kai
Cheng
 ,
Yu-I.
Hsu
* and
Hiroshi
Uyama
*
,
Yu-I.
Hsu
* and
Hiroshi
Uyama
*
Department of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka, 565-0871, Japan. E-mail: yuihsu@chem.eng.osaka-u.ac.jp; uyama@chem.eng.osaka-u.ac.jp
First published on 16th May 2025
Abstract
The large-scale production of commodity plastics with poor degradability presents significant environmental and human health challenges. Instead of relying on landfilling or mechanical recycling, chemical recycling of plastic waste provides an effective strategy to alleviate environmental pollution while transforming “trash” into “treasure”. Pioneering studies have demonstrated the effective depolymerization of polyester- and polycarbonate-based materials using nucleophiles. However, this process often requires catalysts, elevated temperatures, or high nucleophile-to-polymer ratios, posing obstacles to the development of chemical recycling for industrialized applications. To address these limitations, we propose a solvent-promoted catalyst-free strategy for the aminolysis of post-consumer polyester- and polycarbonate-based waste. Certain polar aprotic solvents stand out among commonly used organic solvents due to their ability to effectively promote aminolytic degradation under catalyst-free conditions. By leveraging these selected solvents, the aminolysis process can be carried out efficiently under mild conditions. This work provides a simple and versatile method for recycling single and mixed post-consumer plastic wastes into the corresponding monomers.
Green foundation1. Developing catalyst-free reactions is a critical focus as one of the principles of green chemistry. This work proposes a solvent-promoted strategy to convert diverse end-of-life plastic wastes into the corresponding monomers. By employing the selected solvent, the reaction time is reduced, and the aminolysis processes are efficiently conducted under mild and catalyst-free conditions.2. Compared to no DMAc-involved reactions (e.g. 21 h of PLA conversion time with 17% monomer yield), the polymer conversion time is reduced, while the monomer yield increases under the addition of optimized amounts of DMAc solvent (e.g. 3.2 h of PLA conversion time with >99% monomer yield). 3. Further research should be dedicated to recycling the solvent after the reaction is finished and using it in the next recycling cycle, making the chemical recycling process more sustainable. |
Introduction
Plastics are widely utilized across various fields, significantly contributing to social development and improving the quality of human life due to their advantages such as light weight, durability, and formability.1–3 However, commodity plastics are primarily designed for performance and functionality, often neglecting considerations of degradability and recyclability.4–6 With increasing demand and expanding production capacity, the generation of plastic waste has grown at an alarming rate, and it has been projected that by 2060, between 155 and 265 million tons of plastic waste will be produced annually worldwide.7 The accumulation of massive amounts of plastic waste poses severe threats to environmental sustainability and human health.8–10Compared to petroleum-based plastics, biodegradable plastics derived from renewable resources have gained more attention recently.11,12 Polylactic acid (PLA), known for its biomass-derived origin and biodegradability, is the first biobased thermoplastic polyester material, with global production increasing annually.13,14 Despite its biodegradability, the degradation of PLA wastes typically requires specific microbes, favorable temperature, and humidity to produce its final degradation products, CO2 and H2O—conditions that are usually met only in industrial composting conditions.15–17 When PLA waste is discarded in natural or marine environments, degradation is significantly prolonged,18–20 leading to pollution comparable to that caused by non-degradable petroleum-based plastics.
In stark contrast, chemical recycling to monomers (CRM) or value-added chemicals provides diverse opportunities to valorize plastic wastes, enabling the transformation of “trash” into “treasure” in a straightforward and time-efficient way.21–23 Common CRM methods for PLA include hydrolysis,24–27 methanolysis,28–31 alcoholysis,32–34 and aminolysis,35 often in combination with an organo-catalyst36,37 or metal-based catalyst,38,39 predominantly Zn-based complexes.40–44 For instance, Liu et al.42 demonstrated the effectiveness of guanidinium zinc complexes as catalysts for the alcoholysis and aminolysis of PLA, finding that aminolysis was significantly more efficient than alcoholysis. Beyond single PLA recycling, Spicer et al.45 explored the effects of various Lewis acid/base catalysts on the glycolysis of mixed plastics containing PLA. Their study selectively depolymerized PLA based on the different depolymerization kinetics of each polymer under specific catalysts. These advancements contribute to the discovery of novel catalysts for more efficient PLA depolymerization. However, adhering to the principles of green chemistry, catalyst-free processes remain a key priority.46,47 In industrial applications, the removal or recycling of catalysts from crude products is necessary to ensure monomer purity, increasing the purification costs.48 Furthermore, catalyst-involved reactions may be more challenging to scale up compared to catalyst-free methods.49 Additionally, achieving high monomer yields often requires a high molar ratio of nucleophiles (methanol, ethylene glycol, etc.) to PLA and elevated temperatures, further limiting industrial application. Consequently, there is a strong demand for the development of efficient and mild methods for the chemical recycling of PLA materials—an objective that remains challenging to achieve.
Polar aprotic solvents are of significant interest due to their large dipole moments, high dielectric constants, and excellent solubility for a wide range of materials.50 Typical polar aprotic solvents, such as N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), and acetonitrile, are widely used in SN2 reactions, where they greatly enhance the reactivity of nucleophiles.51–53 Previous works42 have demonstrated that certain polar aprotic solvents play a crucial role in facilitating polymer degradation processes without the use of catalysts. For instance, Zhang et al.54 investigated the effect of various solvents on the methanolysis of PLA and identified DMF as particularly effective, achieving a 96% depolymerization rate under catalyst-free conditions. Similarly, Zhou et al.49 selected DMSO as the optimal solvent for the aminolysis of bisphenol A-based polycarbonate (BPA-PC) plastics, achieving a 99% yield of bisphenol A monomer at room temperature and under catalyst-free conditions.
Based on these considerations, we hypothesized that appropriate solvents could enhance the solvation effect and accelerate the degradation process of PLA under mild and catalyst-free conditions. In this study, we investigated the effect of various organic solvents, ranging from polar and non-polar to biobased solvents, on the aminolysis of commercial PLA pellets using ethanolamine under catalyst-free conditions. A systematic investigation was further performed to explore the interactions in the solvent-promoted PLA aminolysis reaction and proposed possible mechanisms behind this facilitating effect. In addition to PLA, this strategy was generalized to other common thermoplastic resins including BPA-PC and polyethylene terephthalate (PET), indicating the universality of this strategy. In the end, recycling of single post-consumer PLA, BPA-PC, and PET wastes, composite wastes, and mixed wastes was conducted to highlight the applicability of this strategy under various recycling situations. To our knowledge, we demonstrated the first solvent-promoted catalyst-free strategy for the aminolysis recycling of various post-consumer plastic wastes by selecting effective solvents. This work provided new ideas for addressing the issue of plastic waste accumulation and advancing the sustainable development of waste plastics (Scheme 1).
 | ||
| Scheme 1 Schematic illustration of this work: aminolytic degradation for chemical recycling of plastic wastes under mild and catalyst-free conditions. | ||
Results and discussion
Solvent selection for the catalyst-free aminolysis of PLA pellets
The PLA aminolysis reactions were all conducted at 60 °C for 5 h under catalyst-free conditions (Fig. S1†). Initially, various common solvents were selected as candidates for the aminolysis of PLA pellets. As displayed in Table 1, in most cases, the addition of solvent greatly promoted the aminolysis of PLA compared with merely an ethanolamine-involved reaction (entry 1). Specifically, N,N-dimethylacetamide (DMAc), acetonitrile, tetrahydrofuran (THF), chloroform, and dimethyl sulfoxide (DMSO) exhibited a prominent facilitating effect on PLA aminolysis, with over 90% yield of N-LEA monomer (entries 2–6). Meanwhile other organic solvents demonstrated a less favorable promoting effect (entries 7–12) or minimal ability (entries 13–16) to enhance the aminolytic degradation pace of PLA.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Conversionb (%) | Yieldc (%) | Greennessd |
| a Reaction conditions: PLA 2003D pellets (288 mg, 4 mM relative to the repeating unit, NatureWorks® 2003D, Mw = 190.0 kg mol−1, Đ = 1.51), EA (0.61 mL, 10 mM, 2.5 equiv.), solvent 2 mL, NMP (N-methyl-2-pyrrolidone as internal standard, 39.7 mg, 0.4 mM, 0.1 equiv.), 60 °C, 5 h. b Conversion (%) = (minitial − mfinal)/minitial × 100%, minitial and mfinal denote PLA mass before and after the reaction, respectively. c N-LEA yield was determined by 1H NMR spectroscopy (400 MHz, DMSO-d6, 298 K). Yield = (integralN-LEA × 3 × molarNMP)/(3 × molarPLA) × 100%. The integral of N-LEA was calculated from the peak integration of 1.18–1.22 ppm, d, 3H. The details of 1H NMR results are shown in Fig. S38–53.† d The greenness of each solvent is classified as recommended, problematic, hazardous, or highly hazardous based on the references.55,56 | ||||
| 1 | None | 21 | 15 | — |
| 2 | DMAc | >99 | >99 | Hazardous |
| 3 | Acetonitrile | >99 | 99 | Problematic |
| 4 | THF | >99 | 97 | Problematic |
| 5 | Chloroform | >99 | 96 | Highly hazardous |
| 6 | DMSO | 97 | 91 | Problematic |
| 7 | Toluene | 90 | 86 | Problematic |
| 8 | Dimethyl isosorbide | 89 | 84 | Recommended |
| 9 | DMF | >99 | 80 | Hazardous |
| 10 | Ethanol | 69 | 71 | Recommended |
| 11 | Isopropanol | 41 | 49 | Recommended |
| 12 | Hexane | 35 | 38 | Hazardous |
| 13 | γ-Valerolactone | 9 | 5 | Problematic |
| 14 | Ethyl acetate | 98 | Trace | Recommended |
| 15 | Acetone | 88 | Trace | Recommended |
| 16 | Cyrene | Trace | Trace | Problematic |
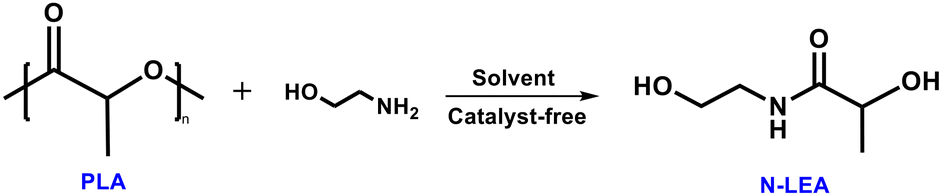
The findings in Table 1 shed light on the effect of different solvents on PLA aminolysis. Intrigued by these results, further analysis of the solvent parameters shown in Table S1† was utilized to elucidate the solvent selection criteria. Hansen solubility parameters including δD (dispersion forces), δp (dipolar intermolecular forces), and δH (hydrogen bonds) are generally utilized to anticipate the solubility of a polymer in a specific solvent theoretically.57 A lower difference Δδ between polymers and solvents indicates better solubility.58 The Δδ between PLA and ethanolamine is so high (Δδ = 9.24) that it takes 21 h for PLA pellets to fully convert into the ethanolamine nucleophile (Fig. 4). A lower conversion time (3.2 h) could be observed after introducing the DMAc solvent into the PLA aminolysis reaction system due to the smaller difference (Δδ = 0.86) between PLA and DMAc. The Kamlet–Taft parameters include three factors: π* (polarity), α (hydrogen bond donor or acidity), and β (hydrogen bond acceptor or basicity), determining the physiochemical properties of solvents.59,60 As shown in Table 1, the solvents with over 90% yield (entries 2–6) of N-LEA monomer after PLA aminolysis exhibited either high polarity (π*) or high relative basicity (β–α), especially for aprotic solvents with high polarity like DMAc and DMSO. The π* values of DMAc and DMSO are 0.88 and 1.00, respectively, and the β–α values are both 0.76. However, the conversion of PLA and the yield of N-LEA using DMSO were lower than those using DMAc. This might be attributed to the higher Δδ difference between PLA and DMSO (Δδ = 2.62) than between PLA and DMAc (Δδ = 0.86), delaying the PLA conversion and affecting the monomer yield. In contrast, dimethylformamide (DMF), as another high-polarity aprotic solvent, demonstrated a satisfactory PLA conversion but had a less favorable effect on PLA aminolysis (entry 9), corresponding to a higher difference Δδ between PLA and DMF and a lower β–α value compared with DMAc and DMSO.
To better understand the reasons for the different effects of solvents on PLA aminolysis, the interactions between various solvents (Fig. 1a, marked with red dots) and PLA pellets were further investigated by 1H NMR spectroscopy. As shown in Fig. 1b and S2,† the chemical shifts of the proton peak connected to tertiary carbon and the proton peak of the methyl group of PLA were 5.16 and 1.57 ppm in CHCl3, respectively. These peak signals exhibited obvious shifts in different solvents, following a general trend of lower chemical shifts with higher polarity or relative basicity of the solvents. Especially for DMAc, it shifted to 4.28 and 0.68 ppm, respectively, indicating a stronger interaction with the carbonyl group of PLA than other solvents. Additionally, interaction between different solvents and ethanolamine was also detected; specifically, both ethanolamine and DMAc showed evident chemical shifts in mixed solvents, implying their strong interaction (Fig. 1c and d).
 | ||
Fig. 1 (a) Relationship of polarity (π*), relative basicity (β–α) of different solvents, and the corresponding N-LEA yield in PLA aminolysis shown in Table 1. The solvents highlighted with red dots were further investigated for their interactions with PLA. Detailed polarity and relative basicity parameters of different solvents are listed in Table S1.† (b) 1H NMR chemical shifts of the proton connected with the tertiary carbon in PLA with chloroform, acetonitrile, THF, DMF, DMSO, or DMAc solvent. (c) 1H NMR chemical shifts of the methylene proton in ethanolamine with chloroform, DMF, acetonitrile, THF, DMAc, or DMSO solvent and (d) 1H NMR chemical shifts of the DMAc, EA, and mixed DMAc and EA (volume ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvents (400 MHz, CDCl3-d1, 298 K). :1) solvents (400 MHz, CDCl3-d1, 298 K). | ||
DMAc-promoted catalyst-free aminolysis for PLA-based materials
Encouraged by these results, the DMAc solvent was selected for further experiments due to its optimal promoting effect in catalyst-free PLA aminolysis. After elucidating the interactions between different solvents and PLA pellets, the depolymerization conditions of PLA pellets were optimized considering different molar ratios of ethanolamine to PLA, and DMAc to PLA. The PLA conversion and N-LEA yield achieved optimal performances when the molar ratios of ethanolamine to PLA and DMAc to PLA were 2 and 5, respectively (Fig. S3†). Based on the optimal conditions, the N-LEA yield changes in 5 h were monitored, with >99% of the final yield by 1H NMR and 90% of the isolated yield (Fig. 2 and S4†). | ||
| Fig. 2 Aminolysis processes of commercial PLA, BPA-PC, and PET pellets promoted by DMAc solvent under catalyst-free conditions. Yield denotes the isolated monomer yield, which was calculated based on eqn (S2).† | ||
The aminolysis of PLA promoted by DMAc was further investigated by gel permeation chromatography (GPC) to monitor the changes in molecular weight (Mn) and polydispersity (Đ) within 5 h (Fig. S5†). A steady decrease in Mn could be observed in the initial depolymerization stage, while Đ remained slightly changed. After 1 h, the Mn sharply decreased from 125.9 kg mol−1 to 66.3 kg mol−1, and simultaneously Đ increased from the original 1.51 to 1.78, indicating more multiform PLA chain lengths. After that, both Mn and Đ displayed an obvious decline, suggesting that the longer PLA chains were depolymerized into shorter chains, resulting in a more uniform chain length. The GPC test for aminolysis of PLA without DMAc was also conducted (Fig. S6†), distinguishing the promotion effect of DMAc.
After observing the Mn changes during PLA aminolysis, 1H NMR tests were conducted to examine the relative concentration variations of each component in this process, aiming to uncover its mechanism. The 1H NMR results in Fig. 3a indicate that DMAc-promoted catalyst-free PLA aminolysis followed a two-step depolymerization mechanism based on random chain scission (Fig. S7†), which was concluded from the peak presence of oligomers and agreed with the previous studies.42,54 In the presence of ethanolamine and DMAc, the PLA chain (5.16 and 1.58 ppm) initially cleaves into α-oligomers (4.36 and 1.48 ppm) and Ω-oligomers (5.26 and 1.52 ppm), resulting in the formation of oligomers, followed by a subsequent decrease (Fig. 3b). After 3 h, the proton peaks of oligomers and PLA were all diminished and converted to N-LEA monomers (4.18 and 1.38 ppm). Extending the reaction time increased the N-LEA yield (Fig. S3c†).
 | ||
| Fig. 3 (a) 1H NMR spectra of PLA aminolysis promoted by DMAc under catalyst-free conditions (400 MHz, CDCl3-d1, 298 K). Reaction conditions: PLA (1 eq.), EA (2 eq.), and DMAc (5 eq.) at 60 °C for 5 h. For protons of the internal methine group, the different circles are assigned to PLA (blue), α-oligomers (red), Ω-oligomers (yellow), and N-LEA monomers (green). For protons of the methyl group, the different triangles are assigned to PLA (blue), α-oligomers (red), Ω-oligomers (yellow), and N-LEA monomers (green). (b) The PLA, PLA oligomers, and N-LEA monomer percentage changes within 5 h, and (c) the proposed mechanism for DMAc-promoted PLA aminolysis under catalyst-free conditions. | ||
In light of the above results and previous studies,31,42,54 a depolymerization mechanism for PLA aminolysis without catalysts is proposed as illustrated in Fig. 3c. Due to the high polarity and uneven electronic density distribution, the DMAc solvent is depicted with a “δ+δ−” electron pair. The “δ−” region interacts with the proton in ethanolamine, while the “δ+” region interacts with the oxygen in the carbonyl group of PLA (I). The amine group in ethanolamine functions as a nucleophile, targeting the carbon in the carbonyl group of PLA and forming an intermediate product (II). After that, the ester bond in the PLA chain is cleaved, generating the PLA oligomers (III). With sufficient ethanolamine and DMAc, the PLA oligomers gradually depolymerize into the final N-LEA monomers (IV).
In addition to the PLA 2003D pellets, the DMAc-promoted catalyst-free strategy was applied to other PLA-based post-consumer wastes. For instance, PLA-based cups and fibrous towels could be recycled to N-LEA monomer under no catalyst-involved conditions promoted by DMAc even at R.T. in 5 h, achieving 93% and 87% of the N-LEA isolated yield, respectively (Table 2, entries 1 and 2 and Fig. S23 and S27†), implying a minimal effect of additives or fillers in post-consumer plastics on applying this strategy. The morphological changes in post-consumer PLA wastes during aminolysis at R.T. and 60 °C were observed by scanning electron microscopy. As revealed in Fig. S8,† for PLA fibrous towels at 60 °C, the continuous PLA fibers with uniformly distributed diameters progressively converted into discontinuous fibers at 3 min and short fibers at 7 min. After that, the PLA fibers were twisted at 10 min, eventually turning into a sticky paste at 20 min. However, it was not until the reaction had run for 30 min that discontinuous PLA fibers could be observed at R.T. Subsequently, more short fibers were seen at 1 h, and twisted fibers appeared at 2 h. For the PLA cup at 60 °C, the smooth and flat PLA flakes gradually displayed plenty of grooves on the surface at the reaction for 1 min, resulting from the etching effect of ethanolamine promoted by DMAc. After 3 min, a great number of cracks appeared on its surface. Meanwhile at R.T., the surface morphology of PLA flakes went from grooves to intricate cracks, with a relatively slower aminolysis rate than the reaction at 60 °C. These morphological changes confirmed the effectiveness of DMAc-promoted aminolytic degradation for post-consumer PLA-based wastes and demonstrated the temperature sensitivity of this strategy.
| Entry | Plastic wastea | Time (h) | Temperature (°C) | After the reaction | Recycled monomer (yieldb) |
|---|---|---|---|---|---|
| a Plastic wastes were pre-treated before the aminolysis reaction, as shown in Fig. S19.† b Yield corresponds to isolated monomer yields, which were calculated based on eqn (S2).† | |||||
| 1 |

|
5 | R.T. |

|

|
| 2 |
|
5 | R.T. |

|

|
| 3 |

|
5 | R.T. |

|

|
| 4 |
|
5 | R.T. |

|

|
| 5 |

|
8 | 100 °C |

|

|
| 6 |

|
8 | 100 °C |

|

|
DMAc-promoted catalyst-free aminolysis for BPA-PC- and PET-based materials
The facilitating effect of catalyst-free PLA aminolysis promoted by DMAc solvent motivated us to extend this depolymerization strategy to other materials. Considering that the majority of commercially available plastics are non-biodegradable petroleum-based polymers, it is highly desirable to implement this strategy for the sustainable management of post-consumer plastic wastes. Besides PLA-based materials, BPA-PC and PET are petroleum-based polymers that rank among the most widely produced plastics, with annual production capacities surpassing 5 and 30 million tons, respectively.61,62 BPA-PC-based materials are commonly used in products such as disks, goggles, and containers,62 whereas PET-based materials are used in applications such as packaging and textile materials.63For the aminolytic degradation of BPA-PC-based materials, the optimization of DMAc-promoted aminolysis of commercial BPA-PC pellets was initially performed (Fig. S9 and 10†). Based on the optimal conditions, the reaction proceeded smoothly even at room temperature, yielding 77% bisphenol-A (BPA) and 63% 2-oxazolidone (2-Oxa.) in the isolated products (Fig. 2). In contrast, a significantly prolonged BPA-PC conversion time (87 h vs. 0.6 h) was observed under DMAc-free conditions, meanwhile leading to a sharp decrease of BPA monomer yield by 1H NMR (11% vs. 97%) after 5 h of reaction (Fig. 4). Additionally, the GPC traces during BPA-PC aminolysis with or without DMAc solvent were also analyzed to highlight the crucial role of the DMAc solvent in this reaction (Fig. S11 and 12†). To figure out the chain cleavage mechanism of DMAc-promoted BPA-PC aminolysis, 1H NMR spectra were monitored and proton peaks corresponding to bi-carbonated BPA, mono-carbonated BPA, and BPA monomer were revealed in the early depolymerization stage (Fig. S13†), suggesting that the chain cleavage mechanism followed a random chain scission pathway (Fig. S14†). Inspired by these results, we generalized this strategy to recycle end-of-life BPA-PC disks and goggles. Interestingly, favorable isolated yields of BPA (72% and 79%) and 2-Oxa. (69% and 68%) monomers were successfully recycled from BPA-PC disks and goggles (Table 2 (entries 3 and 4) and Fig. S24, 25, S28 and 29†). The recycled BPA monomer can be reused for BPA-PC polymerization, while the recycled 2-Oxa. monomer holds potential as a valuable building block for synthetic intermediates in the fields of medicine and pesticides.49,64
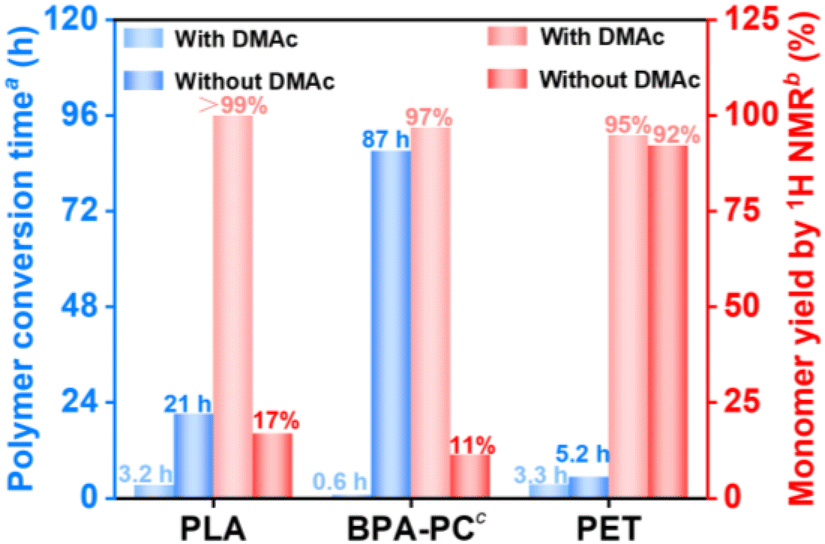 | ||
| Fig. 4 The polymer conversion time and monomer yield of commercial PLA, BPA-PC, and PET pellets for the aminolysis reaction with or without the DMAc solvent. aThe conversion time was defined as the time of polymer conversion over 99%, which was determined by using eqn (S1).†bThe monomer yield was calculated by introducing NMP solvent as an internal standard. cThe monomer yield by 1H NMR of BPA-PC was calculated by using a bisphenol A monomer. The reaction conditions of each polymer were based on optimized conditions and 1H NMR details referred to in Fig. S18.† | ||
For the aminolysis of PET-based materials, a higher reaction temperature (100 °C) was chosen compared with post-consumer PLA or BPA-PC wastes (R.T.), as PET pellets remained nearly intact at the R.T. of DMAc-promoted aminolysis. The optimal reaction conditions for PET pellets were initially explored (Fig. S15†), revealing that ethanolamine played a more significant role than the DMAc solvent in enhancing the monomer yield. This was supported by the observation that a higher ratio of ethanolamine to PET resulted in a higher BHETA monomer yield, while the BHETA yield changed slightly as the DMAc-to-PET ratio increased. It was also verified by the results from Fig. 4 that the DMAc addition in the PET aminolysis reaction shortened the PET conversion time (3.3 h vs. 5.2 h), concurrently with a slight increase of BHETA monomer yield by 1H NMR (95% vs. 92%). The BHETA isolated yield recycled from PET pellets was 86% by using the DMAc-promoted aminolysis strategy (Fig. 2) and its mechanism was proposed as shown in Fig. S17.† Following the depolymerization of PET pellets, this strategy was applied to PET-based end-of-life materials, producing satisfactory BHETA isolated yields of 90% and 94% from PET bottles and packages, respectively (Table 2 (entries 5 and 6) and Fig. S26 and S30†). The above results showed the facileness and versatility of this strategy, indicating that the addition of DMAc solvent enhanced the polymer conversion rate and monomer yield simultaneously, especially for PLA- and BPA-PC-based materials.
DMAc-promoted catalyst-free aminolysis for composite and mixed plastic wastes
Although favorable yields were achieved from recycling various single plastic wastes, plastic waste streams in reality generally contain other polymers, which can notably affect the depolymerization rate of the targeted polymer. Furthermore, mixed polyester-based wastes tend to undergo depolymerization simultaneously under similar conditions due to their similar backbone carbonyl structures, rendering it challenging to separate each monomeric component.65 Therefore, it is essential to selectively depolymerize the targeted plastic waste from other plastic wastes and facilitate easy separation of the monomers.Given that commodity plastics are typically composed of multiple types of plastics, we first depolymerized composite plastic wastes using the DMAc-promoted aminolysis strategy. As displayed in Fig. 5a and b, high yields of BHETA (91%) and N-LEA (90%) monomers could be recycled from the composite PE/PET package and PS/PLA fork wastes, respectively. Meanwhile, polyethylene (PE) and polystyrene (PS) components in composite wastes remained intact supported by the ATR-FTIR results (Fig. S32 and 33†). This suggested the advantageous effect of selective aminolysis, which selectively depolymerized the targeted plastic into monomers while leaving the unreacted plastic intact. For mixed plastic wastes, as illustrated in Fig. 5c, varieties of monomers with favorable yields were recycled step-wise (Fig. S35†). Intriguingly, the yields of BPA and 2-oxazolidone recycled from mixed BPA-PC and PET wastes were comparable to those from single post-consumer BPA-PC wastes, suggesting that PET had minimal impact on this reaction and thus enabling beneficial selective aminolysis in step 1. SEM was utilized to observe the morphological changes of PET flakes in this aminolysis process (Fig. S34†). In the step 1 reaction, the surface of PET flakes remained smooth and intact during 5 h at R.T. until BPA-PC was completely depolymerized into the corresponding monomers. However, when the temperature was elevated to 100 °C in the step 2 reaction, plenty of cracks appeared on the surface of PET flakes at the initial time. With the prolonged reaction time, the roughness increased, and a great number of grooves were displayed on the PET surfaces, resulting from the synergistic etching effect of ethanolamine and DMAc. The resulting high specific surface area of PET could effectively enhance the aminolysis process at the interfaces between PET and ethanolamine. The distinct morphological differences of PET flakes indicated that the aminolysis process of PET was highly temperature-dependent, which could be applied to separate PET wastes from other plastic wastes. Eventually, the BHETA monomer with 90% yield was recycled in step 2. For mixed PLA and PET wastes shown in Fig. 5d, 89% yield of the N-LEA monomer and 87% yield of the BHETA monomer could be recycled from PLA and PET wastes, respectively, in a sequential manner (Fig. S36†), further confirming the effectiveness of selective aminolysis in this strategy.
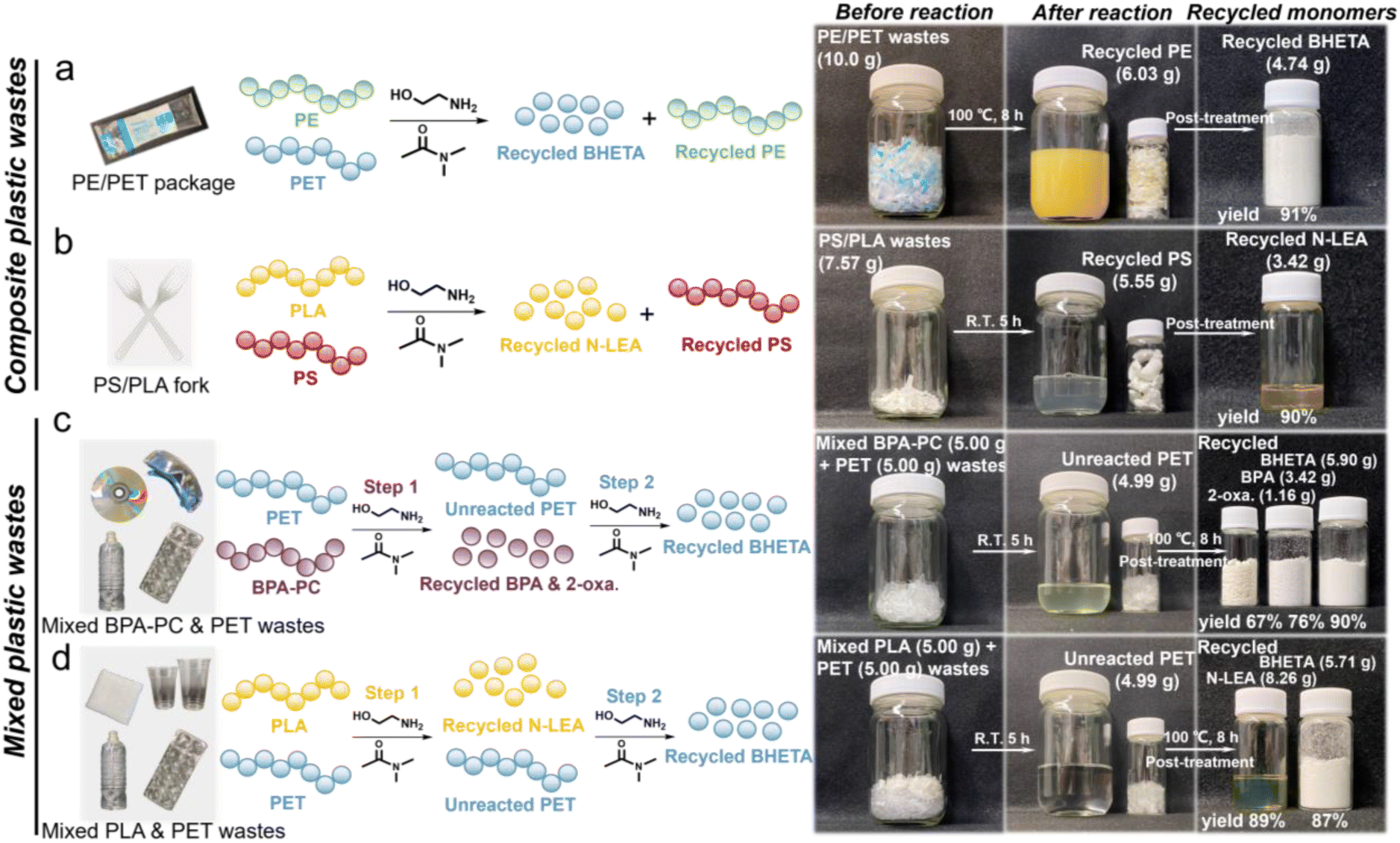 | ||
| Fig. 5 Selective aminolysis processes for composite plastic wastes: (a) PE/PET syringe package and (b) PS/PLA fork. Selective aminolysis processes for mixed plastic wastes in a step-wise manner: (c) mixed BPA-PC (goggles and disk) and PET (bottle and package), and (d) PLA (cup and towel) and PET (bottle and package). | ||
Evaluations of reaction mildness in this work
To distinguish the significance of this work, the reaction mildness was evaluated from five factors: reaction time, temperature, molar ratio of nucleophile to polymer, catalyst loading, and solvent. As presented in Table 3 and Fig. S37,† reaction conditions using different depolymerization methods including aminolysis, glycolysis, and methanolysis were collected for better comparisons. According to these results, the addition of the solvent facilitates the reaction at a lower temperature and with a reduced nucleophile dosage, rendering the reaction significantly milder, which is beneficial for the industrialized application. This could be attributed to the fact that the carefully selected solvent could accelerate the swelling rate of the polymer, which is as important as other kinetic factors such as temperature, catalyst, etc.66 Along with the nucleophile, the polymer surfaces are more likely to swell, dissolve, and depolymerize simultaneously. Once the reaction system becomes homogeneous, the depolymerization rate is accelerated. The role of the solvent is to shorten the transition time from a heterogeneous to a homogeneous reaction system, as further verified by the results shown in Fig. 4. Furthermore, aminolysis is observed to be much more efficient than glycolysis or methanolysis, which can be attributed to the stronger nucleophile (amine group), in line with a previous study.42 In addition, as shown in Table S3,† the evaluation of reaction mildness for recycling composite/mixed plastics in a step-wise manner was compared with other works as well, distinguishing the broad applicability of mild reaction conditions in this strategy for overcoming different recycling obstacles.| Entry | Method | Time (h) | Temperature (°C) | Nucleophile–polymer ratioa (mol/mol) | Catalystb (mol/mol) | Solventc (mol/mol) | References |
|---|---|---|---|---|---|---|---|
| a The nucleophile–polymer ratio refers to the molar ratio of the nucleophile to the polymer (relative to the repeating unit). b Catalyst loading normally denotes the molar ratio of the catalyst to the polymer, otherwise mass ratios of the catalyst to the polymer are marked with *. c Solvent stands for the molar ratio of the solvent to the polymer (relative to the repeating unit). | |||||||
| PLA | |||||||
| 1 | Aminolysis | 5 | 60 | 2 | 0 | 5 | This work (Natureworks 2003D) |
| 2 | Aminolysis | 5 | R.T. | 2 | 0 | 5 | This work (PLA wastes) |
| 3 | Aminolysis | 1 | 100 | 4 | 0 | 0 | Shao et al.67 |
| 4 | Aminolysis | 1 | 60 | 3.5 | 0.01 | 10.4 | Liu et al.42 |
| 5 | Glycolysis | 2 | 60 | 17.3 | 0.01 | 0 | Liu et al.42 |
| 6 | Glycolysis | 2 | 150 | 20 | 0.15 | 0 | Spicer et al.45 |
| 7 | Methanolysis | 8 | 140 | 25 | 0 | 9.7 | Zhang et al.54 |
| 8 | Methanolysis | 3 | R.T. | 5.9 | 0.05 | 3.0 | Yang et al.5 |
| 9 | Methanolysis | 4 | 120 | 4.5 | 0.03* | 0 | Manal et al.31 |
| 10 | Methanolysis | 4 | 140 | 35.6 | 0.05* | 0 | Wang et al.30 |
| BPA-PC | |||||||
| 1 | Aminolysis | 5 | R.T. | 2 | 0 | 10 | This work |
| 2 | Aminolysis | 5 | R.T. | 2 | 0 | 14.1 | Zhou et al.49 |
| 3 | Glycolysis | 2 | 150 | 20 | 0.15 | 0 | Spicer et al.45 |
| 4 | Glycolysis | 2 | 130 | 10 | 0.05 | 0 | Arifuzzaman et al.68 |
| 5 | Methanolysis | 3 | 70 | 20.9 | 0.05 | 8.4 | Yang et al.5 |
| 6 | Methanolysis | 1 | 120 | 11.7 | 0.03* | 0 | Manal et al.31 |
| 7 | Methanolysis | 0.5 | 125 | 9 | 0.05* | 0 | Parida et al.69 |
| 8 | Methanolysis | 2 | 140 | 50 | 0.10 | 18.5 | Wang et al.70 |
| PET | |||||||
| 1 | Aminolysis | 8 | 100 | 8 | 0 | 2.5 | This work |
| 2 | Aminolysis | 24 | 80 | 7.9 | 0 | 0 | Zhou et al.49 |
| 3 | Aminolysis | 0.5 | 140 | 20 | 0.1 | 0 | Demarteau et al.71 |
| 4 | Glycolysis | 2 | 180 | 20 | 0.15 | 0 | Spicer et al.45 |
| 5 | Glycolysis | 10 | 180 | 11.5 | 0.05 | 0 | Yang et al.5 |
| 6 | Glycolysis | 2 | 180 | 10 | 0.05 | 0 | Arifuzzaman et al.68 |
| 7 | Glycolysis | 0.5 | 196 | 34.5 | 0.025* | 0 | Wang et al.30 |
| 8 | Methanolysis | 20 | 100 | 15.8 | 0.05 | 6.03 | Yang et al.5 |
Considering the economic benefits of recycling, we further compared the prices of reagents used for depolymerization and recycled monomers in this work based on information from the official Sigma-Aldrich website (Table S4†). Interestingly, the prices of recycled monomers were prominently higher than the cost-effective ethanolamine and DMAc solvent, implying that DMAc-promoted aminolysis under catalyst-free conditions holds promising potential for large-scale industrial production from recycled plastic wastes.
Conclusions
In summary, we developed a solvent-promoted strategy for the aminolysis of post-consumer polyester and polycarbonate wastes (PLA, BPA-PC, and PET-based materials) under mild, catalyst-free conditions. Among the various organic solvents tested, polar solvents with high polarity (π*) and relative basicity (β–α), such as DMAc, exhibited the most effective facilitation of PLA aminolysis. The interactions within the DMAc-promoted PLA aminolysis reaction system under catalyst-free conditions were thoroughly investigated using NMR spectroscopy, proposing a possible reaction mechanism. Inspired by the favorable effect of DMAc in promoting PLA depolymerization, this strategy was extended to other commercially available PET and BPA-PC resins, as well as mass-produced post-consumer waste materials. A variety of monomers derived from PET and BPA-PC materials were successfully recycled with high yields, highlighting the simplicity and versatility of this approach for effective depolymerization. Notably, selective aminolysis was efficiently performed on post-consumer composite and mixed wastes for converting the targeted polymers into monomers. Finally, the mildness of the reaction conditions was evaluated from five key aspects, emphasizing the significance of this work. This study offers a novel approach for addressing plastic accumulation issues by transforming “trash” into “treasure” using cost-friendly ethanolamine nucleophiles and DMAc solvent.Author contributions
Kai Cheng: writing – original draft, methodology, investigation and conceptualization. Yu-I Hsu: writing – review & editing, visualization, methodology, funding acquisition and conceptualization. Hiroshi Uyama: supervision and funding acquisition.Data availability
The data supporting this article are included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Science and Technology Agency (JST) PRESTO Grant Number JPMJPR23N4, the Environment Research and Technology Development Fund JPMEERF21S11900 of the Environmental Restoration and Conservation Agency of Japan, and the Japan Society for the Promotion of Science (JSPS) KAKENHI Grants (22K21348 and 23K26717). K. C. would like to thank the China Scholarship Council (CSC) for supporting the scholarship.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- N. Simon, K. Raubenheimer, N. Urho, S. Unger, D. Azoulay, T. Farrelly, J. Sousa, H. van Asselt, G. Carlini, C. Sekomo, M. L. Schulte, P.-O. Busch, N. Wienrich and L. Weiand, Science, 2021, 373, 43–47 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- X. Tang and E. Y.-X. Chen, Chem, 2019, 5, 284–312 CAS.
- R. Yang, G. Xu, B. Dong, X. Guo and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 9860–9871 CrossRef CAS.
- M. Hong and E. Y.-X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- L. Lebreton and A. Andrady, Palgrave Commun., 2019, 5, 6–16 CrossRef.
- J. M. Millican and S. Agarwal, Macromolecules, 2021, 54, 4455–4469 CrossRef CAS.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- K. D. Nixon, Z. O. G. Schyns, Y. Luo, M. G. Ierapetritou, D. G. Vlachos, L. T. J. Korley and T. H. Epps III, Nat. Chem. Eng., 2024, 1, 615–626 CrossRef.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS PubMed.
- J. Yu, S. Xu, B. Liu, H. Wang, F. Qiao, X. Ren and Q. Wei, Eur. Polym. J., 2023, 193, 112076–112105 CrossRef CAS.
- C. Sun, S. Wei, H. Tan, Y. Huang and Y. Zhang, Chem. Eng. J., 2022, 446, 136881–136902 CrossRef CAS.
- J.-G. Rosenboom, R. Langer and G. Traverso, Nat. Rev. Mater., 2022, 7, 117–137 CrossRef PubMed.
- A. Takagi, Y.-I. Hsu and H. Uyama, ACS Appl. Polym. Mater., 2024, 6, 13307–13318 CrossRef CAS.
- S. Tian, Y. Jiao, Z. Gao, Y. Xu, L. Fu, H. Fu, W. Zhou, C. Hu, G. Liu, M. Wang and D. Ma, J. Am. Chem. Soc., 2021, 143, 16358–16363 CrossRef CAS PubMed.
- S. Kakadellis and G. Rosetto, Science, 2021, 373, 49–50 CrossRef CAS PubMed.
- M. He, Y.-I. Hsu and H. Uyama, J. Hazard. Mater., 2024, 474, 134819–134835 CrossRef CAS PubMed.
- A. R. Bagheri, C. Laforsch, A. Greiner and S. Agarwal, Glob. Chall., 2017, 1, 1700048–1700052 CrossRef PubMed.
- G.-X. Wang, D. Huang, J.-H. Ji, C. Völker and F. R. Wurm, Adv. Sci., 2021, 8, 2001121–2001146 CrossRef CAS PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- H. Q. Li, H. A. Aguirre-Villegas, R. D. Allen, X. L. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. García, I. Hermans, M. S. Kim, J. Z. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. S. Castillo, K. L. Sánchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorst, M. M. Wright, J. Y. Wu, V. M. Zavala, P. Z. Zhou and G. W. Huber, Green Chem., 2022, 24, 8899–9002 RSC.
- C. Shi, E. C. Quinn, W. T. Diment and E. Y.-X. Chen, Chem. Rev., 2024, 124, 4393–4478 CrossRef CAS PubMed.
- M. Yagihashi and T. Funazukuri, Ind. Eng. Chem. Res., 2010, 49, 1247–1251 CrossRef CAS.
- C.-X. Liu, K. Liu, Y. Xu, Z. Wang, Y. Weng, F. Liu and Y. Chen, Angew. Chem., Int. Ed., 2024, 63, e202401255 CrossRef CAS PubMed.
- Y. Ma, X. Guo, M. Du, S. Kang, W. Dong, V. Nicolosi, Z. Cui, Y. Zhang and B. Qiu, Green Chem., 2024, 26, 3995–4004 RSC.
- X. Song, X. Zhang, H. Wang, F. Liu, S. Yu and S. Liu, Polym. Degrad. Stab., 2013, 98, 2760–2764 CrossRef CAS.
- R. Yang, G. Xu, B. Dong, H. Hou and Q. Wang, Macromolecules, 2022, 55, 1726–1735 CrossRef CAS.
- Z. Wang, Y. Wang, S. Xu, Y. Jin, Z. Tang, G. Xiao and H. Su, Polym. Degrad. Stab., 2021, 190, 109638–109647 CrossRef CAS.
- A. K. Manal, G. Saini and R. Srivastava, Green Chem., 2024, 26, 3814–3831 RSC.
- F. A. Leibfarth, N. Moreno, A. P. Hawker and J. D. Shand, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4814–4822 CrossRef CAS.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS.
- T. M. McGuire, A. Buchard and C. Williams, J. Am. Chem. Soc., 2023, 145, 19840–19848 CrossRef CAS PubMed.
- F. Wu, Y. Wang, Y. Zhao, M. Tang, W. Zeng, Y. Wang, X. Chang, J. Xiang, B. Han and Z. Liu, Sci. Adv., 2023, 9, eade7971 CrossRef CAS PubMed.
- O. Coulembier, S. Moins, J.-M. Raquez, F. Meyer, L. Mespouille, E. Duquesne and P. Dubois, Polym. Degrad. Stab., 2011, 96, 739–744 CrossRef CAS.
- D. Hubble, S. Nordahl, T. Y. Zhu, N. Baral, C. D. Scown and G. Liu, ACS Sustainable Chem. Eng., 2023, 11, 8208–8216 CrossRef CAS.
- M. Hofmann, C. Alberti, F. Scheliga, R. R. R. Meißner and S. Enthaler, Polym. Chem., 2020, 11, 2625–2629 RSC.
- K. Z. Wang, Z. H. Sun, W. D. Guo, M. G. Chen, C. L. Zhu, J. C. Fei, Y. M. Liu, H. Y. He, Y. Cao and X. H. Bao, ChemSusChem, 2023, 16, e202301128 CrossRef CAS PubMed.
- P. McKeown, L. A. Román-Ramírez, S. Bates, J. Wood and M. D. Jones, ChemSusChem, 2019, 12, 5233–5238 Search PubMed.
- L. A. Román-Ramírez, P. McKeown, M. D. Jones and J. Wood, ACS Catal., 2019, 9, 409–416 Search PubMed.
- S. Liu, L. Hu, J. Liu, Z. Zhang, H. Suo and Y. Qin, Macromolecules, 2024, 57, 4662–4669 CrossRef CAS.
- X. Zhu, H. Wang, B. Liu, D. Yang, F. Liu and X. Song, Polym. Degrad. Stab., 2024, 219, 110625–110634 CrossRef CAS.
- R. Yang, G. Xu, C. Lv, B. Dong, L. Zhou and Q. Wang, ACS Sustainable Chem. Eng., 2020, 8, 18347–18353 CrossRef CAS.
- A. J. Spicer, A. Brandolese and A. P. Dove, ACS Macro Lett., 2024, 13, 189–194 CrossRef CAS PubMed.
- A. Sarkar, S. Santra, S. K. Kundu, A. Hajra, G. V. Zyryanov, O. N. Chupakhin, V. N. Charushin and A. Majee, Green Chem., 2016, 18, 4475–4525 Search PubMed.
- J. C. Warner, A. S. Cannon and K. M. Dye, Environ. Impact Assess. Rev., 2004, 24, 775–799 CrossRef.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- X. Zhou, M. Chai, G. Xu, R. Yang, H. Sun and Q. Wang, Green Chem., 2023, 25, 952–959 RSC.
- A. Jordan, C. G. J. Hall, L. R. Thorp and H. F. Sneddon, Chem. Rev., 2022, 122, 6749–6794 Search PubMed.
- A. J. Parker, Chem. Rev., 1969, 69, 1–32 CrossRef CAS.
- J. Miller and A. J. Parker, J. Am. Chem. Soc., 1961, 83, 117–123 CrossRef CAS.
- C. P. Ashcroft, P. J. Dunn, J. D. Hayler and A. S. Wells, Org. Process Res. Dev., 2015, 19, 740–747 CrossRef CAS.
- Q. Zhang, C. Hu, P. Y. Li, F. Q. Bai, X. Pang and X. Chen, ACS Macro Lett., 2024, 13, 151–157 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- F. Russo, F. Galiano, F. Pedace, F. Aricò and A. Figoli, ACS Sustainable Chem. Eng., 2020, 8, 659–668 CrossRef CAS.
- L. P. Novo and A. A. S. Curvelo, Ind. Eng. Chem. Res., 2019, 58, 14520–14527 CrossRef CAS.
- H. T. Yu, Y. Wang, L. Chen, C. Y. Wei, T. C. Mu and Z. M. Xue, Green Chem., 2023, 25, 7807–7816 RSC.
- M. J. Kamlet, J. L. Abboud and R. W. Taft, J. Am. Chem. Soc., 1977, 99, 6027–6038 CrossRef.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS.
- E. Barnard, J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 Search PubMed.
- J. G. Kim, Polym. Chem., 2020, 11, 4830–4849 RSC.
- S. Debnath, M. Parit, A. P. Brown, J. Pearson, B. Saha and B. W. Boudouris, Chem. Mater., 2024, 36, 10259–10266 CrossRef CAS.
- D. J. Kerr, J. M. White and B. L. Flynn, J. Org. Chem., 2010, 75, 7073–7084 CrossRef CAS PubMed.
- J. J. R. Arias, E. Barnard and W. Thielemans, ChemSusChem, 2022, 15, e202200625 CrossRef PubMed.
- R. A. Clark and M. P. Shaver, Chem. Rev., 2024, 124, 2617–2650 CrossRef CAS PubMed.
- L. Shao, Y.-C. Chang, C. Hao, M.-e. Fei, B. Zhao, B. J. Bliss and J. Zhang, Green Chem., 2022, 24, 8716–8724 RSC.
- M. Arifuzzaman, B. G. Sumpter, Z. Demchuk, C. Do, M. A. Arnould, M. A. Rahman, P.-F. Cao, I. Popovs, R. J. Davis, S. Dai and T. Saito, Mater. Horiz., 2023, 10, 3360–3368 RSC.
- D. Parida, A. Aerts, L. V. Perez, C. Marquez, S. Vloemans, K. Vanbroekhoven, E. Feghali and K. Elst, Chem. Eng. J., 2024, 497, 154390–154398 CrossRef CAS.
- N. Wang, Q. Zhang, Z. Sun, H. Zhang, C. Hu, H. Sun, X. Pang and X. Chen, ACS Macro Lett., 2025, 14, 377–384 CrossRef CAS PubMed.
- J. Demarteau, I. Olazabal, C. Jehanno and H. Sardon, Polym. Chem., 2020, 11, 4875–4882 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc01068c |
| This journal is © The Royal Society of Chemistry 2025 |