
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegrated technoeconomic and environmental assessment of biogenic polyurethane production†
Alberto
Almena
 *a,
Domenico
Pirone
b,
Susana
Fernández-Prieto
b,
Alberto
Martínez
b and
Mariano
Martin
a
*a,
Domenico
Pirone
b,
Susana
Fernández-Prieto
b,
Alberto
Martínez
b and
Mariano
Martin
a
aDepartment of Chemical Engineering, University of Salamanca, Spain. E-mail: almena@usal.es
bProcter and Gamble. Brussels Innovation Center, Temselaan 100, 1853 Strombeek-Bever, Belgium
First published on 4th April 2025
Abstract
Polyurethane (PU), one of the most consumed materials, is conventionally produced from fossil-based polyols and polyisocyanates. In this case, increasing the PU biogenic carbon content is a strategy followed to enhance its sustainability, e.g., by employing bio-polyols and biogenic polyisocyanates. Bio-based pentamethylene polyisocyanate (PDI-T), with 70% biogenic carbon content, is a prepolymer commercialized for bio-PU production. However, its production route has not yet been widely reported. Thus, herein, a theoretical study was conducted to investigate the manufacturing process of a novel bio-PU gel using castor oil and PDI-T. The biogenic production of PDI-T from molasses was designed. A comprehensive assessment integrating process modelling and simulation, cost estimation, economic analysis, and life-cycle assessment (LCA) methodologies was performed to evaluate the techno-economic and environmental performance of the produced bio-PU. This process showed a molasses-to-PU yield of 0.2 tbio-PU tmolasses−1, with the economic viability reaching a selling price of $15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 per ton, i.e., five-times higher than the price of a conventional PU-gel. Cradle-to-gate LCA showed a global warming potential (GWP) of 22.8 kg CO2e per kg bio-PU, which is four-times higher than the GWP reported in the Ecoinvent database for conventional PU. This methodology allowed the identification of key limitations negatively impacting GWP and production costs, primarily the complexity of bio-PDI-T synthesis, which involves high energy consumption and cumulative emissions from multiple raw materials. This study highlights that substituting fossil-based components with high biogenic carbon alternatives does not always result in climate benefits. Therefore, applying this methodology is pivotal for identifying truly sustainable pathways or suggesting modifications to existing processes, to achieve effective green chemistry transition.
000 per ton, i.e., five-times higher than the price of a conventional PU-gel. Cradle-to-gate LCA showed a global warming potential (GWP) of 22.8 kg CO2e per kg bio-PU, which is four-times higher than the GWP reported in the Ecoinvent database for conventional PU. This methodology allowed the identification of key limitations negatively impacting GWP and production costs, primarily the complexity of bio-PDI-T synthesis, which involves high energy consumption and cumulative emissions from multiple raw materials. This study highlights that substituting fossil-based components with high biogenic carbon alternatives does not always result in climate benefits. Therefore, applying this methodology is pivotal for identifying truly sustainable pathways or suggesting modifications to existing processes, to achieve effective green chemistry transition.
Green foundation1. This study challenges the assumption that maximizing the biogenic carbon content of materials inherently aligns with green chemistry. Rigorous integrated technoeconomic and life-cycle assessments are essential to quantify sustainability benefits and avoid unintended trade-offs.2. Bio-polyurethane gel with a 70% biogenic content, produced from sugarcane molasses and castor oil, requires a fivefold selling price increase for economic viability and exhibits quadruple the global warming impact of fossil-based polyurethane when compared to benchmark LCA available data. 3. Greener pathways for bio-polyurethane production require the electrification of heating, renewable energy sourcing and microbial strain engineering to tolerate higher sugar concentration. The use of waste-derived feedstocks and low-carbon solvents or integrating carbon capture technology may additionally contribute to offset the carbon footprint of the process. However, TEA and LCA must validate whether alternatives align with green chemistry goals. |
1. Introduction
The chemical industry is one of the major contributors to environmental degradation and climate change. This sector is responsible for approximately 2 billion metric tons of CO2 emissions annually, which constitutes more than 5% of global greenhouse gas emissions.1 The chemical sector has been a key focus area to endorse the development and adoption of sustainable practices and technologies. The Environmental Protection Agency (EPA) has defined the concept of green chemistry in 12 main principles with the main objective of reducing and ultimately eliminating the use and generation of hazardous substances.2 Alternatively, policy initiatives implemented for environmental protection such as the European Green Deal include the chemical industry as one of the sectors to undergo green transition and reach net-zero GHG emission performance.3 In the EU, the chemical industry accounts for 155.5 Mt CO2 eq., reporting a similar percentage (5%) of the total emissions in the region. This indicates that this sector achieved a 9% reduction during the period 2012–2021 without experiencing an economic penalty, since the gross value added (GVA) increased by 23%.4 However, environmental protection goals have not been reached to date, and thus, the chemical sector is still pushed to continue its sustainability transition.One of the issues hindering the capability of this sector to reduce its carbon footprint is the production of plastic. Plastics have become bulk materials with the highest growth in production rate. In 2019, the global plastic production reached 368 million metric tons, representing a 20-fold increase in the past 50 years.5 Given the strong reliance on fossil fuels and fossil feedstocks in the manufacturing of plastics, the conventional plastics sector is estimated to generate 1.8 Gt CO2e annually over its entire life cycle, excluding carbon credits from recycling.6 Polyurethanes (PUs) are one of the key players in the plastics industry, constituting materials that possess excellent mechanical, chemical and physical properties, providing great versatility of use within multiple sectors, such as the automotive, construction, furniture, footwear and electronics industries. In 2022, PU production was estimated to be 25.8 million metric tons7 with a market value of above $80 billion.8
Polyurethane (PU) is conventionally synthesized via the addition polymerization of the hydroxyl groups in a polyol and isocyanate functional groups in a polyisocyanate.9 The type of polyol and polyisocyanate selected will determine the properties of the polyurethane and its use as a rigid foam, flexible foam, adhesive, coating, fiber, elastomer, thermoplastic, biogenic material or nanocomposite.10–12 Traditionally, the most widely used polyols (e.g. polyethylene glycol and polypropylene glycol) and polyisocyanates (both aromatic, e.g. toluene diisocyanate, and aliphatic, e.g. hexamethylene diisocyanate) are obtained from petroleum-based resources.13 Extensive research has been conducted with the aim of improving the sustainability of PU materials. The predominant solution involves increasing the content of biogenic carbon in the final material.14 The conventional polyols used in the synthesis of PU are polyether and polyester polyols, which are mainly produced from the reaction between a ‘starter’ polyol and alkylene oxide or dicarboxylic acid, respectively.15 Success has been achieved in substituting them for bio-based polyols obtained from vegetable oils, lignin and carbohydrates.16–18
However, there have been difficulties in achieving an increase in the renewable content of isocyanates.19 The conventional isocyanate production pathway involves the reaction of phosgene and an amine compound. Phosgenation is crucial to form the isocyanate functional group (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO) needed to create the polyurethane linkage. However, despite its efficacy, this route is associated with significant health, safety, and environmental risks due to the highly toxic nature of phosgene. Thus, alternative phosgene-free methods for producing isocyanates have been explored.20 The thermal decomposition of urea derivatives,21 direct amination of carbon monoxide,22 and electrochemical oxidation of amines with CO223 are examples of alternative routes for the production of isocyanates. However, none of them have produced isocyanates with yield and purity values required for the production of PUs on the industrial scale. Furthermore, non-isocyanate polyurethane (NIPU) production methods have also been investigated. These approaches primarily involve synthesizing cyclic carbonates, which are subsequently reacted with amines or polyamine compounds.24 However, the resulting PU materials exhibit low reactivity during the polymerization stage, and thus a lower degree of crosslinking.25 This leads to reduced mechanical strength, elasticity, durability, inferior thermal stability, and challenges in effectively foaming the PU matrix.24–26
CO) needed to create the polyurethane linkage. However, despite its efficacy, this route is associated with significant health, safety, and environmental risks due to the highly toxic nature of phosgene. Thus, alternative phosgene-free methods for producing isocyanates have been explored.20 The thermal decomposition of urea derivatives,21 direct amination of carbon monoxide,22 and electrochemical oxidation of amines with CO223 are examples of alternative routes for the production of isocyanates. However, none of them have produced isocyanates with yield and purity values required for the production of PUs on the industrial scale. Furthermore, non-isocyanate polyurethane (NIPU) production methods have also been investigated. These approaches primarily involve synthesizing cyclic carbonates, which are subsequently reacted with amines or polyamine compounds.24 However, the resulting PU materials exhibit low reactivity during the polymerization stage, and thus a lower degree of crosslinking.25 This leads to reduced mechanical strength, elasticity, durability, inferior thermal stability, and challenges in effectively foaming the PU matrix.24–26
STABiO™, a compound commercialized by Mitsui Chemicals, serves as a prepolymer for the production of bio-polyurethane with diverse applications, including matrices for controlled fragrance release,27 coatings,28 foams,29 adhesives,30 biodegradable tissue material31 and drug delivery.32 This prepolymer is a bio-based aliphatic polyisocyanate, specifically pentamethylene polyisocyanate, designed to achieve at least 70% of its carbon content derived from biomass, in line with the objective of enhancing the content of biogenic carbon in products.33 However, although this product is already commercially available, the complete synthesis route for its production has not yet been investigated or publicly reported in the scientific literature.
In the present work, a theoretical study was conducted to investigate a new process for the production of polyurethane gel using the polyisocyanate mentioned earlier and castor oil as a natural polyol. This approach represents a technically feasible method for the synthesis of bio-polyurethane, achieving the highest biogenic carbon content possible in the final product and contributing to the green transition efforts in the chemical industry. The economic performance and environmental impact of this potentially sustainable route were evaluated. Specifically, the production route for aliphatic pentamethylene polyisocyanate was reconstructed based on Mitsui's patent and a thorough review of the current scientific literature.34 Molasses was selected as the sugar-based biomass feedstock. This complex process involved five reaction stages, encompassing both chemical and biochemical processes, which included biomass pretreatment, sugar fermentation, whole-cell biocatalytic reaction, phosgene synthesis reaction, amine phosgenation and isocyanate oligomerization. The final polymerization of polyisocyanate and castor oil produced a green polyurethane gel. Each of these stages was previously investigated individually.35–37 However, to the best of our knowledge, this is the first work to synthesize and scale up all these subprocesses into a single, complete process producing a polyurethane product with the highest possible biogenic carbon content based on the isocyanate intermediate route. The methodology employed to evaluate this alternative biochemical route combined process modelling and simulation, techno-economic assessment (TEA), and life-cycle assessment (LCA). The findings from this study using the developed methodology will provide insights into the technical and economic feasibility and the environmental implications of producing bio-based polyurethane. The life-cycle perspective aims to explain how producing biogenic products does not necessarily align with reducing the impact of the chemical industry.
2. Methodology description
This study employed a cradle-to-gate approach to conduct the technoeconomic and environmental assessment of the proposed bio-PU production route directly substituting the conventional PU. The production route was designed and flowsheeting was accomplished based on a thorough literature review. The process modelling methodology was employed to estimate the mass and energy balance and size of the equipment involved. The outcome from the process modelling served as reliable secondary data input for the comprehensive life cycle assessment, as well as for the cost estimation involving the economic evaluation. The system boundary encompassed the extraction of raw materials and their transportation to the conversion facility, where all processing stages, from raw material pretreatment to final product purification, take place. Consequently, the assessment did not consider the storage or transportation of the intermediate products. Although the use and disposal of plastics significantly contribute to their environmental impact,38 these stages were excluded from the system boundaries. Bio-based PU produced via amine phosgenation is a ‘drop-in’ plastic showing similar mechanical and chemical properties (e.g. biodegradability and lifespan) as fossil-based PU.39 Therefore, in a direct substitution scenario, the material use and disposal steps in the value chain can be assumed to be identical, with no significant impact on the comparison between bio-PU and fossil-based PU carbon footprints.2.1. Biogenic feedstocks
Molasses is one of the most economical fermentable feedstocks with wide availability across the globe. As a byproduct of the sugarcane and sugar beet refining processes, molasses is produced in large quantities in major sugar-producing regions. Owing to the high fermentable sugar content and low price of molasses, which is recognized as the preferred feedstock for bioethanol production,40 this raw material was selected as the biogenic carbon source for the investigated process. The average molasses composition obtained from sugarcane in the industrial manufacture of sugar is compiled in Table 1.41| Component | Average composition (% WMB) |
|---|---|
| Water | 22.04 |
| Sucrose | 35.70 |
| Glucose | 3.90 |
| Fructose | 5.90 |
| Galactose | 0.02 |
| Raffinose | 0.03 |
| Arabinose | 0.01 |
| Crude protein | 4.90 |
| Polysaccharides | 1.60 |
| Organic acids | 6.20 |
| Ionic salts | 9.50 |
| Ash | 5.20 |
| Solid residue | 5.00 |
Castor oil was selected as the bio-based polyol for this process due to its unique chemical structure, which enables its direct use in the synthesis of polyurethane without extensive preprocessing. Unlike most vegetable oils, castor oil is predominantly composed of ricinolein (87–90%), a triglyceride rich in ricinoleic acid, which naturally contains hydroxyl groups on the 12th carbon of its fatty acid chains. This inherent hydroxyl functionality eliminates the need for chemical modifications such as epoxidation, hydrolysis, and alkoxylation, thus simplifying the production process.42 Its homogeneous hydroxyl group location, long-chain fatty acid structure, low acid value (2–3 mg KOH per g), and high hydroxyl value (164–194 mg KOH per g) facilitate efficient polymerization, leading to the rapid formation of polyurethane. These properties contribute to the production of crosslinked polyurethanes with enhanced elastomeric behaviour, flexibility, and thermal stability.43–45 Due to these advantages, castor oil is already widely used as a feedstock for the production of biogenic crosslinked polyurethane.46
2.2. Process design and modelling of the bio-polyurethane gel production route
The bio-polyurethane gel production route evaluated was divided, as shown in Fig. 1, in seven different subprocesses, as follows: (A) molasses pretreatment, (B) L-lysine HCl synthesis, (C) 1,5-pentanediamine (PDA) synthesis, (D) phosgene synthesis, (E) pentamethylene diisocyanate (PDI) synthesis, (F) pentamethylene polyisocyanate (PDI-T) synthesis and (G) polyurethane gel synthesis. The complete process was modelled using an equation-based mathematical modelling software (GAMS® v.23.5) based on a combination of first principles, dimensionless correlations, heuristic methods and experimental data. A commercial process simulator (CHEMCAD v.8.1.2) was used to model the unit operations involving pure components and complex mixtures when there was a lack of experimental data, such as the separation stages. Surrogate models were generated from the CHEMCAD outcomes and integrated in the GAMS core model. The objective of process modelling was to compute the mass and energy balances and determine the design variables of the equipment involved in the manufacturing process to support both the economic and environmental assessments.47 | ||
| Fig. 1 Diagram representing the sequential set of subprocesses involved in the bio-polyurethane production route. | ||
:1 gH2O gmolasses−1 ratio,48 and then heated to 60 °C to reach a viscosity reduction plateau, minimizing the pumping energy.49 Subsequently, molasses is conditioned to enhance the yield in the subsequent fermentation step, which is the first reaction in the proposed conversion pathway. The molasses is subjected to liquid acid hydrolysis in a continuous stirred tank reactor (Tank1) using 6 M sulfuric acid to achieve a pH value of 2, at 60 °C and residence time of 1 h.50 Under these conditions, sucrose is fully hydrolyzed into fermentable sugars (glucose and fructose).51,52 Additionally, sulfuric acid pretreatment converts the metallic ions present in molasses, which inhibit enzyme activity in biosynthesis mechanisms during fermentation, into precipitated salts.53 Sodium hydroxide solution is later added to neutralize the excess acid in a subsequent neutralization tank (Tank2). The pigments in molasses have been also found to hinder the activity of microorganisms during fermentation. Thus, activated carbon in a 2% w/v ratio is added into the neutralization stirred tank to achieve pigment adsorption and removal without experiencing any sucrose loss.54 All the dispersed solids are later removed in a filter (Filter1). Finally, the fermentable molasses is sterilized in a series of heat exchangers (represented as HX1 and HX2) to prevent the growth of undesirable microorganisms and sent to the first conversion stage. Fig. 2 depicts the flow diagram of the molasses pretreatment process.
 | ||
| Fig. 2 Flow diagram of sugarcane molasses pretreatment. | ||
 | (1) |
 | (2) |
| Parameter | Value | Units |
|---|---|---|
| Initial concentration of fermentable sugars (VS,0) | 120 | kg m−3 |
| Initial threonine concentration (VT,0) | 0.4 | kg m−3 |
| Initial biomass concentration (VX,0) | 0.1 | kg m−3 |
| Oxygen concentration in the medium (Vp,0) | 0.008 | kg m−3 |
| Initial product concentration (Vf) | 0 | kg m−3 |
| Volume of the fermenting media (Vf) | 200 | m3 |
Once the fermentation is completed, the biomass produced is separated from the liquid medium in a rotary filter (Solsep1). Lysine is recovered from the non-converted sugar broth, which is recycled to the fermenter for the next batch, by ultrafiltration.59 The target product is the hydrochloride salt form of L-lysine, i.e.L-lysine-HCl. Its superior chemical stability and solubility in water make the salt more suitable than the raw amino acid for the following reaction step in the proposed biogenic PU production route. Crystallization is the selected purification process producing highly pure L-lysine-HCl.60 Firstly, an evaporator (Evap1) eliminates water and supersaturates the L-lysine solution above its solubility limit, i.e. a concentration superior to 500 kg L-lysine-HCl per m3 solution.61 A supersaturation factor of 1.4 was targeted. With heat, ammonium hydroxide decomposes to ammonia gas and water. Next, the concentrated solution is cooled to ambient temperature and neutralized with excess hydrochloric acid. Then the solution is allowed to crystallize under acidic conditions at 20 °C for 12 h in a stirred tank crystallizer (Crystallizer).62 A particular crystal size distribution is not the aim in the present unit, given that L-lysine-HCl is an intermediate product in the isocyanate production process. Highly pure (>98.5%) L-lysine-HCl crystals are recovered via centrifugation and dried with a continuous flow of warm air. The mother liquor is recycled back and mixed with the feed stream of the crystallizer, i.e. the output from fermentation, for an additional crystallization cycle. A purge stream is considered to prevent mass accumulation within the process. Fig. 3 represents the flow diagram of the proposed process to produce L-lysine HCl from the pretreated sugarcane molasses.
 | ||
| Fig. 3 Flow diagram for the biosynthesis of L-lysine HCl from treated sugarcane molasses. | ||
 | (3) |
An integrated whole-cell biocatalytic reaction process is suggested here, which includes six different unit operations, i.e. bacteria cultivation, cell permeabilization, amine biosynthesis, deprotonation, amine extraction and purification. The flow diagram is displayed in Fig. 4 and operating conditions are compiled in Table 3. A recombinant microorganism is cultivated in Luria Bertani broth that was previously sterilized. Fermentation is performed in a stirred tank (Tank4) at 37 °C for 12 h under controlled aeration to achieve the targeted cell concentration.65 A small quantity of lysine during bacteria growth is added as an inducer. The culture is recovered by centrifugation. Cell permeabilization is required given that bacteria accumulates intracellularly the targeted enzyme. Organic solvents are commonly used to dissolve lipids from cell membranes and make intracellular antigens accessible.66 Here, E. coli cells are suspended in a cold ethanol/water mixture in a stirred tank (Tank5) at 4 °C under pH-controlled conditions with the addition of phosphate buffer. The permeabilized cells are recovered from the liquid medium in a membrane and later fed to the amine biosynthesis reactor (Reactr2) where the L-lysine-HCl is incorporated together with pyridoxal phosphate co-factor. Phosphate buffer is used as the reaction medium to keep the pH value between 5.8 and 7.4 and prevent pH titration during the decarboxylation.67 Under these conditions, PDA is obtained in its hydrochloride salt form (PDA-HCl). The present bioproduction process can achieve a 92% yield.64 The deprotonation of PDA-HCl is achieved in an additional stirred tank (Tank6) with an excess of sodium hydroxide to reach a pH value higher than 10.5, at a reaction temperature of 80 °C, and with continuous agitation for 5 h. Full conversion to PDA is achieved.64
 | ||
| Fig. 4 Flow diagram for the production of 1,5-pentanediamine (cadaverine) hydrochloride via the decarboxylation of L-lysine hydrochloride. | ||
| Unit operation | Parameter | Value | Units |
|---|---|---|---|
| Bacteria cultivation65 | Cultivation temperature (Tcultiv) | 37 | °C |
| Cultivation time (tcultiv) | 12 | h | |
| Initial peptone concentration (VP,0) | 0.456 | kg m−3 | |
| Initial yeast extract concentration (VYE,0) | 0.456 | kg m−3 | |
| L-Lysine inducer concentration (VL,t) | 2 | kg m−3 | |
| E. coli respiration quotient (RQE.coli) | 0.72 | mol CO2/mol O2 | |
| Volume of the fermenting media (Vf) | 24 | m3 | |
| Cell permeabilization64 | Ethanol–water volume ratio | 35 | % (v/v) |
| Permeabilization temperature (Tperm) | 4 | °C | |
| Permeabilization time (tperm) | 0.5 | H | |
| Buffer mass ratio (xb,perm) | 0.2 | m3 kg−1 cells | |
| L-Lysine decarboxylation64 | Decarboxylation temperature (tdecarb) | 37 | °C |
| Decarboxylation time (tdecarb) | 12 | h | |
| Pyridoxal phosphate mass ratio (xpp,decarb) | 0.0001 | kg kg−1L-lysine-HCl | |
| Buffer mass ratio (xb,decarb) | 3.0 | kg kg−1L-lysine-HCl | |
| Permeabilized cells mass ratio (xpc,decarb) | 0.0082 | kg kg−1L-lysine-HCl | |
| Decarboxylation conversion (Xdecarb) | 0.916 | molreacting/molfed | |
| Deprotonation64 | Deprotonation temperature (tdeprot) | 80 | °C |
| Deprotonation time (tdeprot) | 5 | h | |
| NaOH/PDA mass ratio (xNaOH) | 1.6 | kg kg−1 PDA-HCl | |
| Deprotonation conversion (Xdeprot) | 1.0 | molreacting/molfed | |
| PDA extraction67 | Extraction temperature (Textract) | 55 | °C |
| Volume ratio butanol/aq. solution ϕbutanol | 1.0 | mbutanol3 maq. sol−3 | |
| PDA absorption yield (yieldabs) | 0.985 | kgPDAbut kgPDAin−1 | |
| PDA purification (CHEMCAD simulation) | Reflux ratio distillation (R/D) | 0.1 | kg kg−1 |
| Boil up ratio (V/B) | 10.4 | kg kg−1 | |
| Temperature condenser (Tcondenser) | 93 | °C | |
| Temperature boiler (Tboiler) | 176 | °C | |
| PDA recovery yield (yielddis) | 0.952 | kgPDAout kgPDAbut−1 | |
| PDA purity (%purityPDA) | 99.99 | % |
Extraction using an organic solvent is the preferred method for recovering PDA from fermentation broth due to its selectivity, superior energy efficiency and economic performance.68,69n-Butanol has been reported to have high efficiency for recovering PDA from an aqueous medium.67 A packed bed absorption column (Abs_C1) is selected, which facilitates high throughput and improved liquid–liquid contact and diffusion of PDA into the organic phase. Subsequent distillation (Dis_C1) is considered for solvent recovery and PDA purification. The higher boiling point of PDA compared to both water and butanol allows a highly pure stream of cadaverine from the column bottoms to be obtained. The solvent-rich head stream is first decanted to eliminate as much water content as possible and recycled to the absorption column. A purge is considered to prevent material accumulation, while the n-butanol make-up stream compensates for any solvent loss occurring during the purification process. The optimal operating conditions for PDA purification are shown in Table 3, achieving almost 94% PDA recovery.

| (4) |
 | ||
| Fig. 5 Flow diagram of the phosgene synthesis process. | ||
 | (5) |
Subsequently, hot phosgenation is conducted at higher temperatures (120–150 °C) for the amine salts to undergo a substitution reaction with phosgene, forming additional carbamic acid chloride (eqn (6)), which later dechlorinates, producing isocyanate and hydrogen chloride (eqn (7)).76 The high volatility of phosgene requires operating under pressurized conditions (10 bar) to favor phosgene retention in the liquid phase. Chlorinated hydrocarbons, e.g. ortho-dichlorobenzene (o-DCB), are the preferred solvents due to their higher polarity, which allows the dissolution of both amine hydrochlorides, an intermediate of the phosgenation reaction, and phosgene. Excess phosgene is required to hinder urea formation from the isocyanate and amine side reaction.77 Batch processing is recommended to produce aliphatic isocyanates, such as pentamethylene diisocyanate (PDI), given that it has been reported to have superior reaction rates and selectivity for the isocyanate product.78
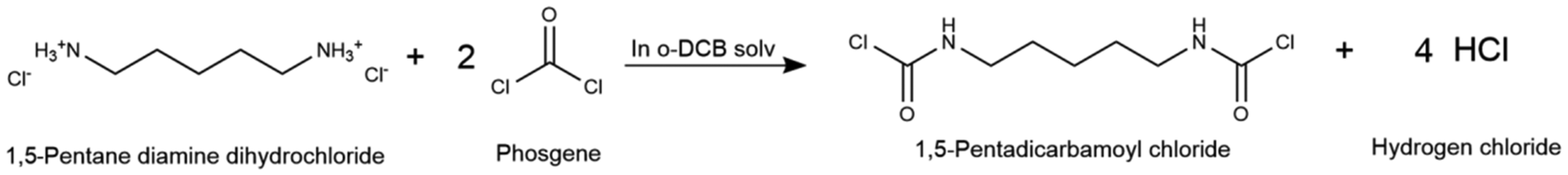 | (6) |
 | (7) |
Fig. 6 displays the proposed process. The liquid-phase phosgenation of PDA is performed in a two-step reaction process using o-DCB as an inert solvent. The liquid phosgene obtained from the phosgene synthesis process is mixed with o-DCB in a 55 wt% ratio in a pressurized stirred tank (Mixer15). Similarly, a 15 wt% PDA solution in o-DCB is prepared in a second stirred tank (Mixer14). Both reactant solutions are pumped to the reactor (Reactor3) at ambient temperature and the required pressure (10 bar). The jacketed stirred tank reactor is first heated to 60 °C for the cold phosgenation to occur. This reaction is exothermic, and thus temperature control is required to keep the reactor close to isothermal conditions and hold the temperature below the boiling point of phosgene.79 The contents of the reactor form a slurry, which is then heated to 150 °C to perform hot phosgenation in the same unit. Substitution and dichlorination reactions are endothermic, and thus heat supply is required to keep the reaction at the required temperature. The total residence time in the reactor can reach 8 h for the process to achieve 96.5% conversion.34 Continuous production is maintained by using multiple equipment units, operating in parallel.
 | ||
| Fig. 6 Flow diagram for PDA phosgenation to produce pentamethylene diisocyanate (PDI). | ||
Excess phosgene and HCl byproduct evaporate under the hot phosgenation temperature conditions. Both gases are purged with a nitrogen flush. A distillation column (Dis_C2) with a partial condenser operating at atmospheric pressure recovers highly pure phosgene at the bottom stream, which is recycled back to the phosgene/o-DCB blend preparation step.80 HCl is recovered as distillate and mixed with water to produce hydrochloric acid as the byproduct, which is suitable to be further converted into chlorine to feed the phosgene synthesis process. Nitrogen is recovered as non-condensable gases and recycled to be compressed and reused as a flushing agent for the next reaction batch.
The PDI product is separated from the liquid stream leaving the phosgenation reactor by distillation (Dis_C3). The o-DCB solvent, together with the unconverted PDA-HCl and dissolved HCl, are recovered in the head stream. PDA deprotonation and HCl neutralization are performed using sodium hydroxide aqueous solution in a packed bed absorption column (Abs_C2) to favor mass transfer between the organic and aqueous phases. Under these mild conditions, aryl halide compounds do not experience nucleophilic substitution reactions, and thus o-DCB remains stable.81 The o-DCB organic stream is recycled back to the PDA/o-DCB blend preparation step, with a purge implemented to prevent accumulation, while the aqueous phase is discarded. Table 4 presents the parameters of the described PDI production process.
| Unit operation | Parameter | Value | Units | |
|---|---|---|---|---|
| Phosgenation34,78 | Molar ratio phosgene:PDA (χphosg,PDA) |
4 | molCCl2O/molPDA | |
| Mass ratio PDA:o-DCB (xPDA,DCB) |
0.15 | kgPDA kgDCB−1 | ||
| Mass ratio phosgene:o-DCB (χphosg,DCB) |
0.55 | kgCCl2O kgDCB−1 | ||
| Pressure (Pphosg) | 10 | Bar | ||
| Temperature cold phosgenation (Tcold phosg) | 60 | °C | ||
| Temperature hot phosgenation (Thot phosg) | 150 | °C | ||
| Reaction time phosgenation (tphosg) | 8 | h | ||
| Phosgenation yield (yieldphosg) | 1.456 | kgPDI kgPDA−1 | ||
| Nitrogen flush34 | Saturation concentration of HCl in DCB (xsatHCl,DCB) | 0.06 (ref. 82) | kgHCl kgDCB−1 | |
| Nitrogen flush ratio (xN2,gases) | 10 | kgN2 kgCCl2O+HCl−1 | ||
| Distillation for phosgene recovery (CHEMCAD simulation) | Reflux ratio distillation (R/D) | 0.2 | kg kg−1 | |
| Boil up ratio (V/B) | 0.5 | kg kg−1 | ||
| Temperature condenser (Tcondenser) | −153 | °C | ||
| Temperature boiler (Tboiler) | 7 | °C | ||
| Non-condensed stream composition | y N2 | 0.998 | molN2/molNC stream | |
| y HCl | 0.002 | molHCl/molNC stream | ||
| y CCl2O | 0 | |||
| Distillate composition | x N2 | 0.038 | kgN2 kgdistillate−1 | |
| x HCl | 0.956 | kgHCl kgdistillate−1 | ||
| x CCl2O | 0.006 | kgCCl2O kgdistillate−1 | ||
| Bottoms composition | x N2 | 0 | kgN2 kgbottoms−1 | |
| x HCl | 0 | kgHCl kgbottoms−1 | ||
| x CCl2O | 1 | kgCCl2O kgbottoms−1 | ||
| Phosgene recovery yield (yielddist,phos) | 0.997 | kgCCl2Obot kgCCl2Oin−1 | ||
| Distillation for PDI recovery (CHEMCAD simulation) | Reflux ratio distillation (R/D) | 1 | kg kg−1 | |
| Boil up ratio (V/B) | 17.2 | kg kg−1 | ||
| Temperature condenser (Tcondenser) | −87 | °C | ||
| Temperature boiler (Tboiler) | 147 | °C | ||
| Distillate composition | x HCl | 0.060 | kgHCl kgdistillate−1 | |
| x PDA | 0.008 | kgPDA kgdistillate−1 | ||
| x DCB | 0.932 | kgDCB kgdistillate−1 | ||
| x PDI | 0 | kgPDI kgdistillate−1 | ||
| Bottoms composition | x HCl | 0 | kgHCl kgbottoms−1 | |
| x PDA | 0.002 | kgPDA kgbottoms−1 | ||
| x DCB | 0.005 | kgDCB kgbottoms−1 | ||
| x PDI | 0.995 | kgPDI kgbottoms−1 | ||
| PDI recovery yield (yielddist,PDI) | 0.996 | kgPDIout kgPDIbut−1 | ||
| Absorption (liq–liq equilibrium) | x aq phaseDCB | 1.6 × 10−4 (ref. 83) | kgDCB kgsolution−1 | |
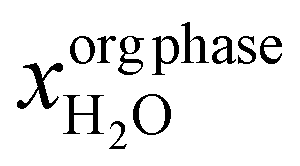
|
2.5 × 10−4 (ref. 84) | kgDCB kgsolution−1 | ||
| Yield HCl in aqueous phase (yieldaq,HCl) | 0.98 | kgHClaq phase kgHClin−1 | ||
 | (8) |
 | ||
| Fig. 7 Flow diagram of the polyisocyanate prepolymer synthesis and purification process, and the subsequent polymerization process to obtain the bio-based polyurethane gel final product. | ||
| Unit operation | Parameter | Value | Units |
|---|---|---|---|
| Oligomerization86 | Reaction temperature (Toligom) | 80 | °C |
| Reaction time (toligom) | 2 | h | |
| Catalyst ratio (toligom) | 0.0004 | kgcat kgPDI−1 | |
| NCO conversion ratio (XNCO) | 0.45 | ||
| Product distribution | |||
| x PDI | 0.25 | kgPDI kgproduct stream−1 | |
| x PDItrimer | 0.47 | kgPDI-T kgproduct stream−1 | |
| x PDIpentamer | 0.20 | kgPDI-P kgproduct stream−1 | |
| x PDIheavy | 0.08 | kgPDI-H kgproduct stream−1 | |
| Thin film distillation (CHEMCAD simulation) | Distillation temperature (TTFdist) | 130 | °C |
| Distillation pressure (PTFdist) | 0.05 | Bar | |
| PDI content in bottom product (xPDA,DCB)86 | 0.03 | kgPDI kgPDI-T−1 | |
| PU polymerization34,92 | Pre-polymerization temperature (Tprepol) | 50 | °C |
| Pre-polymerization reaction time (tprepol) | 0.2 | h | |
| Curing reaction temperature (Tcuring) | 80 | °C | |
| Curing reaction time (tcuring) | 8 | h | |
| Molar ratio NCO:OH (χNCO,OH) |
1.5 | molNCO/molOH | |
| Catalyst (TED) mass ratio (xTED) | 0.03 | kgTED kgcastor oil−1 | |
| Hydroxyl value of castor oil (HNcast oil)43 | 164 | mgKOH gcastor oil−1 |
Together with the desired trimer, higher oligomers (e.g., pentamers, heptamers, and nonamers) are also produced and remain in the prepolymer product. It is crucial to terminate the oligomerization reaction to prevent the predominance of higher oligomers, which would significantly increase the viscosity of the mix. This penalizes the flow properties and hinders the subsequent purification step, which results in a product with poor quality. A 2 h reaction time has been found to achieve desirable product compositions.86 The one-step conversion rate is low, with experimental data reporting a 45% isocyanate (NCO) functional group conversion rate (i.e., 22.5% PDI conversion). Therefore, it is necessary to recover the PDI monomer to increase the product yield and achieve feasible efficiencies in the process. Thin film distillation (DisTF) is an adequate purification step due to the high viscosity of the fluid. Highly pure PDI-T is recovered at the bottom stream from the distillation column, while the evaporated PDI monomer is condensed and recycled back to the reaction stage. Only 3% free monomer content remains in the purified PDI-T.34
 | (9) |
Bulk polymerization following the one-shot method is selected. The flow diagram and the operation parameters for this process are displayed in Fig. 7 and Table 5, respectively. The reactants, i.e., the produced polyisocyanate compound and the selected active hydrogen compound (castor oil), are added to a continuous stirred tank reactor (Prepol_react) operating at 50 °C with a residence time of 10 min to complete the prepolymer reaction.34 Given that the targeted product is polyurethane gel, no blowing agent or surfactant is required in the reaction medium.87 Castor oil is comprised of secondary hydroxyl groups, making it a slow-reacting polyol.46 Thus, a urethanizing catalyst, such as a tertiary amine, is added to enhance polymerization.88 Triethylenediamine in a 3% mass ratio with respect to the bio-polyol present in the reaction medium is considered.89 Heat control with continuous refrigeration is required due to the exothermic nature of the curing reaction.90 Allophanate crosslinking occurs with the degree of crosslinking depending on the NCO:OH functional group molar ratio.91 Castor oil is added to adjust that ratio to 1.5:1.34,45,92 Once the prepolymer reaction is completed, the curing of the polyurethane gel is conducted in the selected mold, inside an oven (Oven) at 80 °C for 8 h to deliver the final product. No purification step is required, and a small fraction of catalyst remains within the polymer matrix without having an impact on the quality of the product.
2.3. Economic evaluation
The economic performance of the polyisocyanate prepolymer and the polyurethane gel was estimated, following the factorial method, as previously described in Almena and Martin (2016).93 Capital expenditure (CAPEX), working capital, and operating expenditure (OPEX) were computed separately. To estimate CAPEX, the process equipment was first conceptually sized to determine the specific variables in its design and materials to conduct the comprising unit operations. The cost of the equipment was obtained from Matches Cost Estimation Tool.94 The Chemical Engineering Plant Cost Index (CEPCI) was used to update equipment cost to 2024 values. The detailed factorial method was selected to estimate the different cost items comprising the CAPEX (see section S1 in the ESI†). Once the fixed capital was estimated, the working capital was assumed to be between 10–20% of CAPEX.95OPEX is the sum of the variable and fixed costs of production. The variable costs of production are proportional to the plant output and comprised of raw materials purchase and utility supply. The results of the process modelling provided the material and utility flow, the cost of which was levelized using market prices for each item. The referenced delivery duty prices (DDP) for raw materials and utilities are provided in section S1 in the ESI.† Conversely, fixed costs of production are not directly dependent on the production rate but on the capacity of the plant. Fixed costs involve different cost allocations such as labor, supervision, and maintenance. Each item was estimated with the corresponding individual factor.95,96
The free on board (FOB) price of the polyurethane gel depends on the market location, ranging between 1800 and 4600 $ per ton.97 Thus, an average FOB of 3000 $ per ton was assumed. Using this value, and considering the entire production, the sold, sales revenue, gross profit and net present value indexes were calculated to measure the economic performance of the plant. The price for the bio-PU gel produced to make a profitable business assuming a plant lifetime of 25 years was estimated from this assessment.
2.4. Life-cycle assessment
The goal of the life-cycle assessment (LCA) was to estimate the global warming potential of the production of the bio-based PU gel material. The LCA complied with the ISO 14040/44 standards and conducted using the SimaPro® 9.5 software. The functional unit was defined as ‘1 kg of polyurethane gel produced by the proposed integrated process’. The carbon intensity score was compared to the conventional fossil-based counterpart to identify potential environmental benefits that may originate from substituting the conventional production process with the alternative proposed.Fig. 8 shows all the stages involved in the production route evaluated. The dashed blue line represents the system boundary of the LCA performed in this work. This study focused on an attributional LCA with a cradle-to-gate approach comprising raw material extraction and pretreatment, amino acid intermediate production, phosgene synthesis, amine intermediate synthesis, diisocyanate and polyisocyanate synthesis, and PU production. The elementary flows crossing the system boundaries include any input material, energy and infrastructure required to produce the bio-PU product, and any airborne, waterborne and solid emissions originating from the process. It was assumed that the entire process is integrated in a single facility, and thus the transportation and storage of intermediate products were not considered. However, the transportation of these raw material supplied to the process was considered. The average environmental burdens of input elementary flows were obtained from updated life-cycle inventory (LCI) databases (i.e. Ecoinvent 3.9.1, Agri-footprint 6.3, USLCI, and Industry data 2.0), which provide comprehensive and well-documented industrial data. The SimaPro module used to represent each input elementary flow is presented in section S2 in the ESI.†
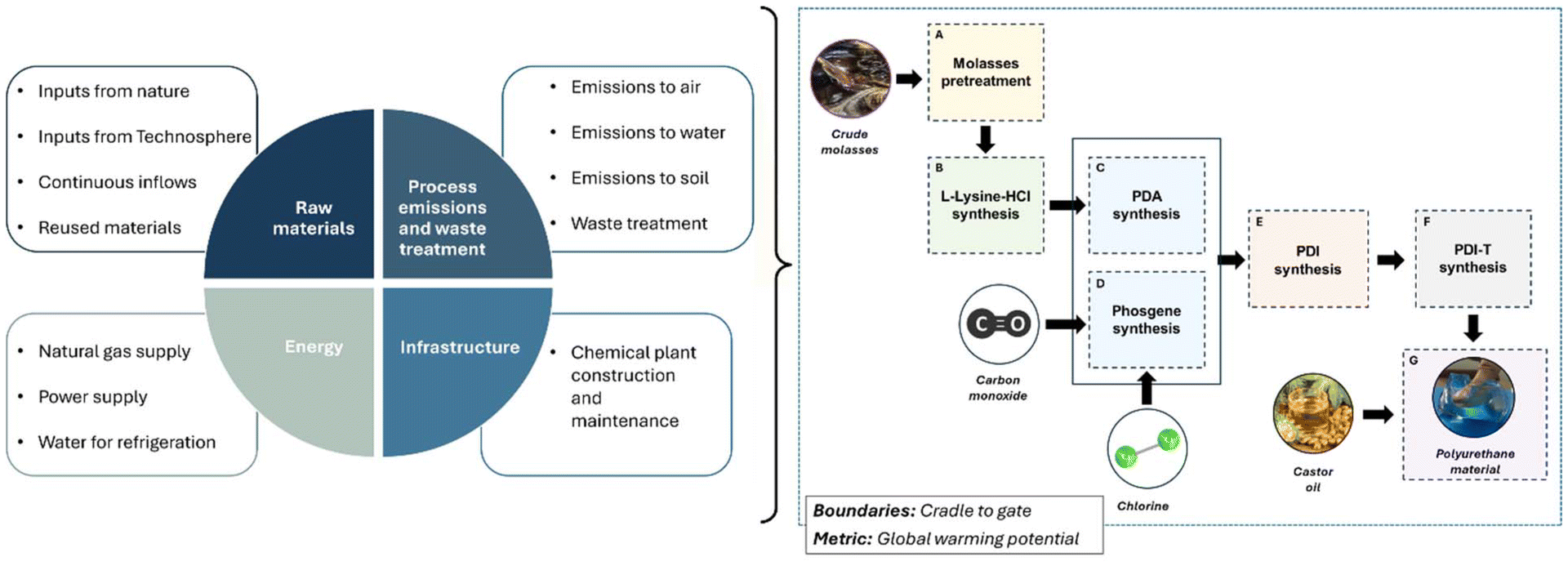 | ||
| Fig. 8 Life-cycle assessment inventory construction and system boundaries of the biogenic polyurethane production supply chain. | ||
No specific module representing castor oil production was found in the SimaPro databases. Thus, to model it, a production and refining process of 1 kg vegetable oil, more specifically palm oil, was used as a template given that its unit operations are similar to castor oil production. Conversely, the castor bean production LCI is reported in the database. This inventory substituted palm fruit bunch in the appropriate yield (300–500 kg crude castor oil per tonne castor seed) found in the literature.98
The life-cycle impact assessment (LCIA) employed was the IPCC 2021 GWP100 (incl. CO2 uptake) V1.02 methodology to classify and characterize climate change impacts.99 This approach estimated the net airborne emissions from the system, separately accounting for biogenic and fossil carbon dynamics. Given that a fraction of biogenic carbon, previously removed from the atmosphere during biomass growth, remains fixed in the PU material, assuming climate neutrality for biogenic carbon emissions could not be applied here. A time horizon of 100 years was considered for the GWP, following the Intergovernmental Panel on Climate Change standards.
3. Results
The results from the techno-economic and environmental assessment are compiled in this section. The largest piece of equipment in the process is the molasses fermenter, and thus the plant size was set to operate a fermenter of 200 m3. Upstream and downstream flows were computed based on the material inflows and production output marked by the correct operation of this equipment. The plant was assumed to operate 7008 h per year, i.e., continuous operation during the whole year considering a capacity factor of 80% to balance maintenance stops and production below maximum capacity. Continuous operation was achieved using multiple units for batch processes.3.1. Process modelling
The whole model is a nonlinear programming (NLP) problem with 21111 variables and 18668 equations. The model was programmed in GAMS® v.23.5 and solved using the CONOPT algorithm. The main results from the process modelling are shown in Table 6, while the complete mass and energy balances are provided in the ESI (section S3†).
| Calculation basis: | Tank size for molasses fermentation equal to 200 m3 | ||
|---|---|---|---|
| Subprocess | |||
| A. Molasses pretreatment | |||
| Raw materials (kg h −1 ) | |||
| Water | 9963.9 | Sulfuric acid | 203.3 |
| Molasses | 2607.4 | Sodium hydroxide | 4.6 |
| Activated carbon (m.u.) | 24.1 | ||
| Utilities (kg h−1) | |||
| Air | 1268.7 | Natural gas | 59.4 |
| Water (refrigeration) | 50503.7 |
Saturated steam (8 bar) | 983.6 |
| Products (kg h−1) | |||
| Purified molasses | 11745.0 |
||
| B. Lysine-HCl synthesis | |||
| Raw materials (kg h−1) | |||
| Water | 535.4 | Hydrogen chloride | 61.9 |
| Threonine | 6.4 | Ammonium hydroxide | 189.7 |
| Utilities (kg h−1) | |||
| Air | 26639.9 |
Natural gas | 815.6 |
| Water (refrigeration) | 8010.2 | Saturated steam (8 bar) | 13514.4 |
| Products (kg h−1) | |||
| L-Lysine HCl | 314.7 | ||
| C. PDA synthesis | |||
| Raw materials (kg h−1) | |||
| Water | 2839.2 | Pyridoxal phosphate | 3.1 × 10−2 |
| Peptone | 9.9 | Monopotassium phosphate | 9.9 |
| Sodium chloride | 9.9 | Butanol (m.u.) | 282.0 |
| Yeast extract | 5.0 | Ethanol (m.u.) | 334.1 |
| Sodium hydroxide | 344.7 | ||
| Utilities (kg h−1) | |||
| Air | 680.0 | Natural gas | 31.2 |
| Water (refrigeration) | 25622.2 |
Saturated steam (8 bar) | 517.8 |
| Products (kg h−1) | |||
| PDA | 154.8 | ||
| D. Phosgene synthesis | |||
| Raw materials (kg h−1) | |||
| Carbon monoxide | 94.4 | Water | 0.4 |
| Chloride | 238.1 | Sodium hydroxide | 0.2 |
| Utilities (kg h −1 ) | |||
| Water (refrigeration) | 4792.8 | ||
| Products (kg h−1) | |||
| Phosgene | 333.0 | ||
| E. PDI synthesis | |||
| Raw materials (kg h−1) | |||
| o-DCB (m.u.) | 116.0 | Sodium hydroxide (aq.) | 176.8 |
| Nitrogen (m.u.) | 499.4 | Water | 858.6 |
| Activated carbon (m.u.) | 24.1 | ||
| Utilities (kg h−1) | |||
| Air | 1071.1 | Natural gas | 50.1 |
| Water (refrigeration) | 11397.8 |
Saturated steam (8 bar) | 830.4 |
| Products (kg h−1) | |||
| PDI | 231.4 | HCl aq | 998.38 |
| F. PDI oligomerization and G. PU polymerization | |||
| Raw materials (kg h−1) | |||
| TMR | 0.1 | TEDA | 9.6 |
| Castor oil | 318.2 | ||
| Utilities (kg h−1) | |||
| Air | 48.3 | Natural gas | 2.3 |
| Water (refrigeration) | 2070.0 | Saturated steam (8 bar) | 23.2 |
| Products (kg h−1) | |||
| PDI-T (intermediate) | 227.1 | PU gel | 554.8 |
Thus, 2600 kg h−1 raw molasses enter the process. Pretreated molasses is fermented to produce L-lysine, which is recovered as hydrochloride salt crystals, obtaining 315 kg h−1L-lysine HCl. The biocatalytic carboxylation process converts the amino acid into an amine intermediate product, producing 155 kg h−1 PDA. The process consumes 333 kg h−1 phosgene, which must be produced without storage. 94 kg h−1 CO and 238 kg h−1 Cl2 are needed to synthesize the phosgene required. The phosgenation process produces 231 kg h−1 PDI, which is further oligomerized to obtain 227 kg h−1 PDI-T prepolymer with 70% biogenic carbon content. As a result, the molasses to PDI-T process yields 87 tPDI-T tmolasses−1.
The final polymerization step requires 318 kg h−1 castor oil to produce 555 kg h−1 PU gel as the final production rate for the whole process. The molasses-to-PU gel yield is 0.2 tbio-PU tmolasses−1. To enhance the energy efficiency and minimize the external heat input in the manufacturing process, heat integration was implemented. Whenever possible, hot process streams were utilized to heat cold streams, achieving an 11% reduction in energy consumption (see Table S28 in the ESI†). Following heat integration, the process required 17.8 kW h per kg of bio-PU produced. The power requirements, which include equipment operation and refrigeration cycles, account for 7.2 kW h kgbio-PU−1.
3.2. Economic evaluation
The process equipment is comprised of a total of 100 units, which includes 18 vessels/tanks, 34 heat exchangers, 4 evaporators, 4 condensers, 8 pumps, 12 compressors, 7 columns, 11 solid separators, 1 air dryer and 1 curing oven. The cost of the equipment increases to 8.8 M$. The detailed factorial method based on this value estimates the CAPEX and working capital to be 51.1 M$ and 7.7 M$, respectively. Considering a lifetime of 25 years for the plant, the annualised capital cost assuming an interest rate of 8.5% is 5 M$ per year.The OPEX estimation gave a value of 44.9 M$ per year. Due to its complexity, the process requires large amount of raw materials, with a total cost of 26.5 M$ per year. Carbon monoxide (22%), molasses (21%) and castor oil (13%) purchases are the biggest contributions. The cost of utilities is important, costing 10.2 M$ per year with an equivalent contribution from natural gas (50%) and electricity (49%). Consumables and packaging costs were assumed to be negligible. Fixed operating costs were estimated to be 8.2 M$ per year. Thus, the total annualized cost including capital and operating cost was 49.9 M$ per year. A breakdown of both the OPEX and CAPEX cost items can be found in section S1 in the ESI.†
The bio-PU produced using the proposed process is not competitive with the conventional PU gel. As shown in Fig. 9a, the economic activity breaks even at a selling price of $11550 per ton, which is nearly four times the average selling price considered ($3000 per ton). However, achieving feasible economic performance requires an even higher selling price. The optimal selling price for the bio-PU was estimated to be five times ($15000 per ton) the assumed average. In this case, the process would generate an annual net profit of $10.3 million, with a payback period of 6 years, meaning a positive cumulative NPV is reached in the 7th year of operation (see Fig. 9b and detailed calculations in section S1 in the ESI†).
 | ||
| Fig. 9 Economic performance of the plant: (a) annual gross profit generated with different selling price ratios for the bio-PU with respect to the current market price for the fossil-based PU gel (ac. 3000 $ per ton); (b) cumulative net present value during the estimated lifetime for the processing plant. | ||
3.3. Life-cycle assessment
The cradle-to-gate LCA performed revealed that the global warming potential of the bio-PU gel is 22.8 kg CO2e per kg of material. Fig. 10a displays that most of the emissions correspond to fossil fuel emissions originating from the whole value chain. The biogenic carbon dynamics are mostly related to the castor oil supply chain. The carbon sequestration results are superior to the biogenic carbon released given that that part of the carbon sequestered from the atmosphere is fixed in the PU gel during the time horizon considered (no product deterioration was assumed). An extra contribution to GWP from land use change caused by castor oil plant cultivation was observed. Three fourths of the carbon footprint of bio-PU are allocated to the polyisocyanate prepolymer supply chain, while the castor oil supply chain is responsible for one fourth of the total score. The polymerization stage has a negligible impact on this value, as shown in Fig. 10b. | ||
| Fig. 10 Global warming potential of bio-based polyurethane production assessed using a cradle-to-gate approach, showing (a) the contributions of different carbon flows—fossil, biogenic, land change use, and CO2 uptake—and (b) the distribution of the carbon footprint across individual reactant supply chains and emissions from the synthesis process. The majority of carbon emissions are fossil-derived, with the production of the bio-polyisocyanate prepolymer representing the largest contributor to the total score. | ||
The score obtained is almost five times the GWP reported in Ecoinvent 3.9.1 for the most similar products, i.e. polyurethane flexible and rigid foams, scoring 4.8 and 4.5 kg CO2e per kgPU, respectively. Similarly, refined castor oil shows a higher impact (9.9 kg CO2e per kgcastor oil) compared to the conventional alternative found in this database. Polyols produced through the alkoxylation of fossil-based alcohols and epoxides report a GWP of 4.0 kg CO2e per kgfossil-polyol. This suggests that despite castor oil being a natural polyol that does not require chemical pre-processing to serve as a crosslinker in PU manufacturing, its GWP is 2.5 times higher than that of the fossil-based alternative according to existing data. The complete life-cycle inventory is provided in the ESI (Tables S7 and S8†), detailing the material and energy inputs, emissions to air and water, and waste outputs. The waste generated was assumed to be treated and disposed of appropriately. Table S7† presents the inventory data from the production of ‘1 kg of polyisocyanate prepolymer’ in the proposed integrated process, while Table S8† provides data from the production of ‘1 kg of polyurethane resin’ using the prepolymer and castor oil.
4. Discussion
The techno-economic and life-cycle assessments generated unfavourable results with respect to the fossil-based polyurethane gel. This outcome is noteworthy, given that green chemistry is generally perceived as a more expensive but environmentally advantageous alternative to conventional chemical processes. However, in this case, the complexity of the process to produce the polyisocyanate prepolymer clearly worsens the economic and environmental performances compared to the traditional process using fossil-based raw materials.PU gel can be produced using castor oil as the polyol compound and an alternative polyisocyanate to that produced in this work. Referring to the Ecoinvent 3.9.1 database, the cradle-to-gate data for the most comparable substances that can be used in synthesizing a similar final product are as follows: toluene diisocyanate (5.9 kg CO2e per kg), methylene diphenyl diisocyanate (4.3 kg CO2e per kg), isophorone diisocyanate (6.3 kg CO2e per kg) and hexamethylene-1,6-diisoncyanate (5.5 kg CO2e per kg). The GWP of the biogenic polyisocyanate assessed here was found to be one order of magnitude larger than that of the reported fossil-based alternatives. These materials only lack the final oligomerization step to produce a prepolymer that can be directly combined with castor oil to produce PU gel. The bio-polyurethane production pathway was thoroughly examined to identify and pinpoint the causes of the significant impact reported, i.e., 41.2 kg CO2e per kgPDI-T.
Similar to the bio-PU gel end product, the fossil-carbon dynamics play a much more significant role than biogenic carbon flow. The motivation of this route was to produce an isocyanate with the greatest content of biogenic carbon (70%). However, the life-cycle assessment showed that the carbon sequestered in the material, i.e., the fraction of CO2 uptake column corresponding to subprocess A in Fig. 11a, is minimal (−0.4 kg CO2e per kgPDI-T) in comparison to the overall GWP score. The primary contributions are illustrated in Fig. 11b. The emissions allocated to the natural gas supply (8% assuming EU market score) and its combustion to meet the process heat requirements (28%) have the biggest impact. The electricity supply chain is the next significant contributor, which is 15% of the total score when the EU mix average impact was assumed. Thus, the energy provision accounts for 50% of the bio-prepolymer emissions.
 | ||
| Fig. 11 Global warming potential of bio-based polyisocyanate prepolymer (PDIT) production assessed using a cradle-to-gate approach, showing (a) the contribution of each subprocess to the different carbon flows—fossil, biogenic, land use change, and CO2 uptake—and (b) the ten largest contributors to the carbon footprint associated with the material. The majority of emissions are fossil-based, with energy supply, solvent use, the molasses supply chain, and direct process emissions identified as the main contributors to the overall score. | ||
The impact of solvent consumption to compensate for losses in the purging streams, which were estimated to be between 10–20 wt% of the recirculating flows to avoid saturation and degradation, is significant. The supply chains for butanol (11%), ethanol (4%), and o-DCB (3%) contributed to the environmental impact of PDI-T. Reducing purge streams through experimental process optimization could decrease the solvent consumption, and consequently the associated impact. The direct CO2 gas emissions generated throughout the process, i.e., from bacterial respiration during the fermentation stages and the decarboxylation of L-lysine, and vented to the atmosphere, contributed 8% to the GWP. Although carbon capture methods could be employed to mitigate these direct emissions, they would incur an extra energy cost. The associated emissions would depend on the chosen energy vector used for these unit operations, which could result in a greater overall environmental impact. Thus, an additional study is required to evaluate the trade-offs between the direct CO2 emissions avoided and the indirect CO2 emissions generated by the implementation of carbon capture technology.
Alternatively, sugarcane molasses cannot be classified as a waste product from sugar production. It currently has various commercial applications and a well-established market, which supports its assignment of an environmental impact through mass allocation in the sugar production process. The sugarcane molasses supply chain accounted for 8% of the total environmental impact of the bio-PDI-T. Substituting molasses with underutilized sugar-based residues, such as bagasse and sugar beet pulp, could eliminate this upstream emission contribution. However, these residues require additional pretreatment to release fermentable sugars, often involving the addition of water that must later be removed. This introduces process complexity and necessitates a detailed assessment to evaluate its overall impact. Sodium hydroxide, which accounts for 6% of the total impact, is a key raw material widely used throughout the process to neutralize acidic conditions. Identifying an alternative neutralizing agent with a lower carbon footprint can help reduce the global warming potential (GWP) of bio-PDI-T production. However, the alternatives reviewed in the Ecoinvent 3.9.1 database and elsewhere, such as potassium hydroxide (4.74 kg CO2e per kg KOH), sodium bicarbonate (1.96 kg CO2e per kg NaHCO3), soda ash (1.69 kg CO2e per kg Na2CO3), and magnesium hydroxide (2.70 kg CO2e per kgMg(OH)2 (ref. 100)), were found to have higher carbon footprints than sodium hydroxide (0.89 kg CO2e per kgNaOH). Finally, the remaining contributions comprised of other raw materials supply chains, infrastructure or waste treatment, reported a combined contribution of 10% to the total GWP associated with the bio-polyisocyanate prepolymer material.
The impact of each subprocess comprising the complex production route described in this work was also measured (see Fig. 12).
 | ||
| Fig. 12 Contribution of each subprocess to the total carbon footprint of 1 kg of the bio-PDI-T prepolymer produced with a cradle-to-gate perspective. Values are compiled in the ESI (Tables S9–S14†). | ||
According to this study, the pretreatment of molasses already has a comparable environmental impact (4.5 kg CO2e per kgPDI-T) to the diisocyanate compounds reported in the Ecoinvent 3.9.1 database. The GWP associated with the molasses supply chain, which involves the cultivation and harvesting of sugarcane, sugar production, impact allocated to molasses subproduct, and molasses transportation, is approximately 3.0 kg CO2e per kgPDI-T. Heat requirements not supplied by the PDI-T production process must be met through natural gas combustion, resulting in emissions of 0.9 kg CO2e per kgPDI-T. The activated carbon input to the process, assumed to account for 10% of the total activated carbon requirement (with the remainder being regenerated and recycled), contributed 0.4 kg CO2e per kg of PDI-T produced. This subprocess consumes minimal electricity, with the remaining emissions are attributed to the other materials used in the pretreatment.
The L-lysine HCl intermediate production showed the largest impact among the subprocesses involved in the manufacture of the bio-prepolymer. The combination of fermentation with crystallization is a high-energy-intensive process. Fermentation requires aqueous cultivation media, generally at low concentrations to enable bacteria growth without substrate or product inhibition. Conversely, crystallization requires water removal to supersaturate the mother liquor for crystals to form and precipitate. Water removal in an evaporator with heat regeneration was assumed to save energy and recover steam that is later used to meet the heat requirements whenever possible. However, the heat demand satisfied with natural gas combustion caused 73% (12.6 kg CO2e per kgPDI-T) of the impact of this subprocess. Evaporation under vacuum conditions was explored, but only 10% energy could be saved, while the equipment cost increased to operate under these conditions, and thus it was discarded. Integrating a series of multi-effect evaporators can represent a potential solution to reduce the heat demand of this unit operation.101 The CO2 generated during fermentation was assumed to be vented to the atmosphere, causing 3 kg CO2e per kgPDI-T. The ammonium salt supply used in the fermentation as the nitrogen source was also remarkable, contributing 1 kg CO2e per kg of bio-polyisocyanate produced.
The synthesis of phosgene is the subprocess with the lowest impact, which is mainly associated with the supply of the two gases. Carbon monoxide was assumed to be produced from the partial combustion of heavy heating oil, while chlorine is obtained as a byproduct from the potassium hydroxide production process. The gas supply added 0.8 kg CO2e per kgPDI-T and 0.6 kg CO2e per kgPDI-T, respectively. The electricity consumed by the ammonia refrigeration cycle used to condense the gases and recycle them back to the reactor had a small contribution (3%) to the subprocess GWP.
The whole-cell biocatalytic reaction process to produce PDA generated 9.7 kg CO2e per kgPDI-T. The butanol supply was shown to be the highest contributor. It was assumed in the process that 20% of the butanol-rich stream recycled is purged, given that it has multiple contaminants that can hinder the absorption operation. This parameter set the butanol consumption, and thus the impact of this material scored 4.6 kg CO2e per kgPDI-T. Ethanol, which is assumed to be produced from ethylene, is used for cell permeabilization and is recycled in a 95% factor. However, the impact of ethanol is high with a value of 1.8 kg CO2e per kgPDI-T. Replacing these materials with their biogenic counterparts, which typically have lower carbon footprints,102 could potentially reduce the impact of this stage. The sodium hydroxide used for the deprotonation of PDA-HCl, assumed to be produced by chlor-alkali electrolysis, had the second largest contribution to this subprocess impact (2.0 kg CO2e per kgPDI-T). The emissions from natural gas combustion during heat supply and the CO2 generated during the decarboxylation of lysine have similar impacts on the final score (0.4 kg CO2e per kgPDI-T).
Finally, the phosgenation and oligomerization subprocesses are reported together due to the simplicity of the latter. This stage is the most electricity-intensive process, contributing the highest environmental impact (5.3 kg CO2e per kgPDI-T). The key contributors include nitrogen compression, which is required to flush excess phosgene from the reactor, and cryogenic refrigeration in the distillation columns to recover phosgene and o-DCB, both of which are highly energy demanding. Additionally, upstream emissions from the production and supply of o-DCB (1.1 kg CO2e per kgPDI-T), sodium hydroxide (0.6 kg CO2e per kgPDI-T) and nitrogen (0.5 kg CO2e per kgPDI-T) are significant. These subprocesses also require heat to operate, causing an impact of 0.8 kg CO2e per kgPDI-T.
The energy supply constitutes 51% of the total impact in the bio-PDI-T pathway. Renewable energy sources were chosen for both heat (onsite biomass-fed boilers) and electricity (wind power). Based on this assumption, the GWP of the prepolymer was significantly reduced to 21.2 kg CO2e per kgPDI-T. This reduction also lowered the bio-PU gel impact by 36%, showing a value of 14.5 kg CO2e per kgPU. However, these values remain substantially higher than that of the fossil-based alternatives found in the evaluated databases. Given that the energy consumption showed the largest contribution to the carbon footprint of the bio-PDI-T prepolymer, and by extension bio-PU, while involving high uncertainty in the energy demand and energy mix impact estimation of the process, a sensitivity assessment was performed to provide a confidence interval. The process modelling included heat integration, which resulted in an 11% reduction in heat demand for process operation. The upper bound for energy contribution corresponded to a scenario where natural gas combustion is used for heat supply, with no heat integration applied. Conversely, the lower bound considered conditions where process optimization reduces the total energy demand (both heat and electricity) by 25% from the benchmark calculated in the process modelling,103 with 100% renewable energy sourcing. Based on these assumptions, the carbon footprints for the bio-PDI-T prepolymer and the bio-PU fall are in the range of 43.1–21.1 kg CO2e per kgPDI-T and 23.5–14.4 kg CO2e per kgPU, respectively.
The last process stage producing the bio-polyurethane gel combines the significant environmental impact estimated for biogenic polyisocyanate and the upstream emissions from the castor oil supply chain. Refined castor oil was found to have a greater impact than the polyurethane materials listed in the Ecoinvent database. Castor beans (2.4 kg CO2e per kgcastor bean) have a much greater GWP than other oil-rich plants, such as rapeseed (−0.9 kg CO2e per kgrapeseed) and palm fruit (−0.8 kg CO2e per kgpalm fruit), which report net negative emissions. Even when the carbon uptake associated with 1 kg of plant for the three species is similar, the positive emissions generated during castor bean production are far greater. 50% of emissions is defined as direct CO2 emissions generated during the activity, which can be associated with land use and plant harvesting. The remainder is primarily attributed to the fertilizers used (such as urea and unspecified inorganic nitrogen and phosphorus fertilizers) and tillage practices.
The large impact of castor bean cultivation explains the higher GWP of castor oil compared to other vegetable oils. However, replacing castor oil with other vegetable oils presents significant challenges due to its unique, almost pure ricinolein content, which provides inherent polyol functionality. Alternative vegetable oils would require additional chemical modifications, such as transesterification, epoxidation and subsequent ring-opening, or alkoxylation steps, to introduce hydroxyl groups and make them suitable for polyurethane production.104 Still, the functionality and distribution of –OH groups would be heterogeneous, leading to less predictable reaction kinetics and polyurethane properties.105 Assessing the environmental impact of these alternatives would require further study, considering the added complexity and resource use of the necessary pretreatment and purification steps. Moreover, the net emissions associated with refined castor oil, which include castor bean cultivation, oil extraction, refining, and transportation, were found to be 2.5 times higher than that of synthetic fossil-based polyols. This raises questions about the suitability of castor oil as a preferred raw material, despite its status as a biogenic polyol, unless the emissions along its supply chain can be significantly reduced.
To assess the overall process in terms of conversion efficiency, economics, and GHG emissions, while also quantifying the impact of each subprocess, five key parameters were evaluated including CAPEX, OPEX, mass loss, GWP and mass efficiency. Fig. 13 illustrates the percentage contribution of each subprocess, highlighting the inefficiencies, cost distribution, and environmental impact across the steps involved in maximizing the biogenic content of polyurethane.
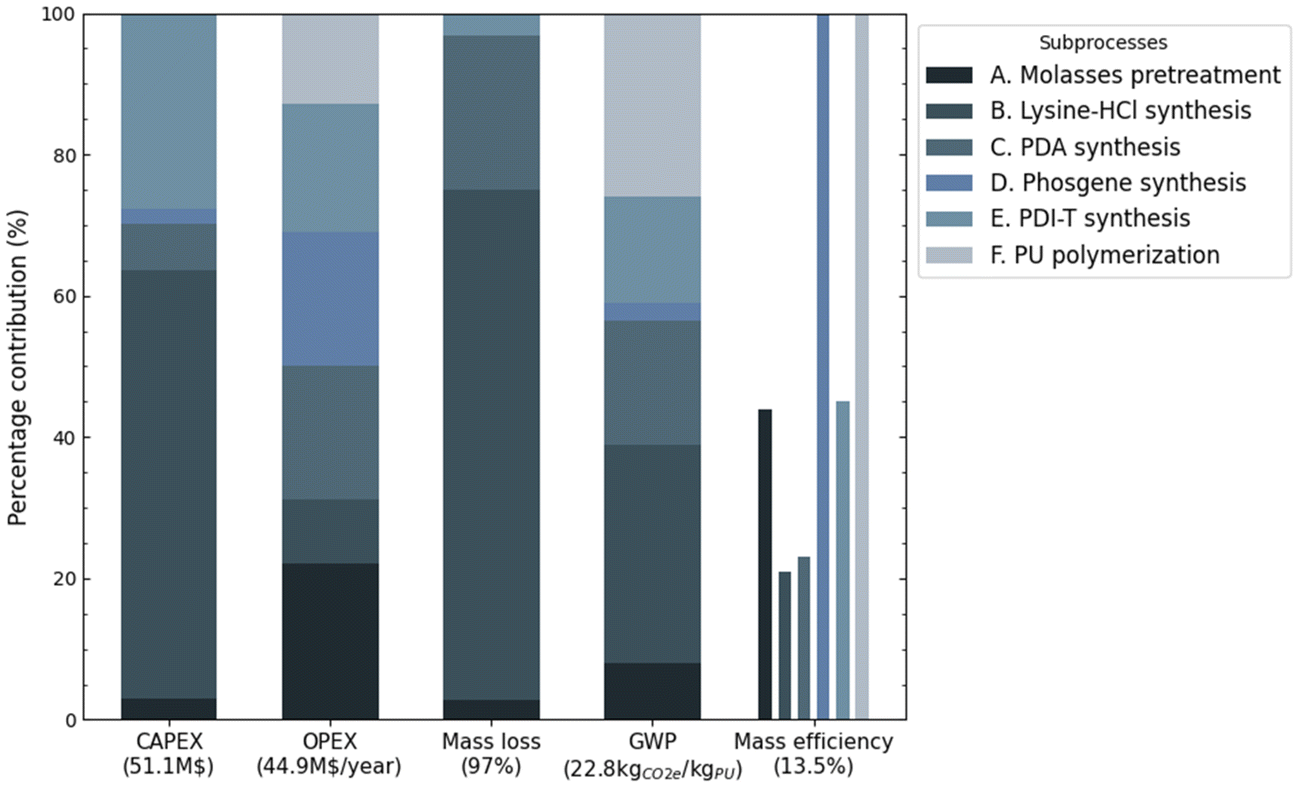 | ||
| Fig. 13 Percentage contribution of each subprocess to the bio-PU production process across five key parameters: CAPEX, OPEX, mass loss, GWP, and mass efficiency. The total values for each parameter are indicated in the x-axis labels. | ||
The overall mass efficiency of the process, which quantifies the proportion of input materials effectively converted into the final product, while excluding non-reactive components such as water or solvents, is 13.5%. This indicates that a substantial part of the input reactants is not converted into the final product but instead lost as by-products or waste streams. The mass loss parameter, which considers all materials entering the process, revealed that only 3% of the total input mass is ultimately retained in the bio-PU polymer. These low efficiency values contribute to the high GWP, given that the large raw material requirements and multiple conversion steps increase the overall carbon footprint of bio-PU.
When assessing the contribution of each subprocess to the overall efficiency, the biological conversion pathways, i.e. lysine-HCl synthesis (subprocess B) and PDA synthesis (subprocess C), exhibited the lowest mass efficiency values of 21% and 23%, respectively. Molasses pretreatment (subprocess A), where fermentable sugars are the intermediate product of interest, and PDI-T synthesis (which includes both amine phosgenation and isocyanate oligomerization steps), demonstrated a higher mass efficiency with nearly 45% of the mass from the reactants retained in the respective subproducts. Notably, phosgene synthesis (subprocess D) and PU polymerization (subprocess F) showed near 100% mass efficiencies, indicating that no significant material losses occur during these steps.
The lysine-HCl synthesis section generates the highest waste, with 72% mass loss primarily due to the removal of large volumes of water, which includes both the water added for the dilution of the reactants and the initial moisture content in molasses, required for amino acid crystallization. This is followed by PDA synthesis, with waste generation driven by solvents purging to maintain their properties during continuous recycling, along with the removal of the aqueous phase after liquid–liquid extraction. In terms of economic evaluation, lysine-HCl synthesis accounts for the highest capital expenditure with 60% of total CAPEX. The need for extensive fermentation before water removal results in large equipment sizes to accommodate the high material flow. The evaporators and crystallizers, essential for subsequent water removal, further increase the capital costs. PDI-T synthesis (Subprocess E) is the second most capital-intensive step, contributing 28% of the total CAPEX, mainly due to its complex separation system for solvent recovery, which includes two cryogenic refrigeration cycles, one absorption and three distillation columns, and a compress gas flushing system. Conversely, the OPEX cost is more equally distributed across subprocesses. Most stages contribute approximately 20% to the total operating cost, except for subprocesses B and F, which show a lower share of around 10%. The GWP column illustrates the contribution of each stage to the overall emissions, as discussed earlier.
5. Limitations of this study
The limitations highlighted above underscore the complexity and inherent uncertainties involved in evaluating the economic and environmental impacts of material production. These factors should be considered when interpreting the results of this study.• Process modelling assumptions: although the process was modelled with high accuracy based on experimental data and established scientific literature, several assumptions were made (detailed in section S4 in the ESI†). These assumptions may not fully align with real-world processes, potentially affecting the reliability of the estimated environmental and economic outcomes.
• Discrepancies in the STABiO™ process: the actual production process for STABiO™ may differ from that described in the patent referenced for this study. These discrepancies can alter the environmental and economic impacts associated with the material.
• Variability in raw material production processes: the processes used for producing the different raw materials may vary considerably, which can lead to differences in the environmental scores reported in this study. These variations are not fully captured in the current model, introducing an additional layer of uncertainty.
• Uncertainty in energy consumption and environmental impact of energy supply: the energy consumption estimates used in this study are subject to significant uncertainty, particularly given that the process can be further optimized when scaled to full operation. Process improvements or optimizations during commercial deployment may lead to a reduced energy demand, which is not fully captured in the current model. Additionally, the environmental impact of energy supply depends heavily on the energy mix (renewable vs. non-renewable sources) used in a specific region or facility. Changes in energy sourcing during future operations can have a substantial impact on the overall environmental footprint of the process.
• Data accuracy uncertainty: there is inherent uncertainty in the accuracy of the data used for this assessment. Variations in the precision and reliability of data sources, such as the LCI data employed from Ecoinvent, may influence the results and conclusions drawn from the analysis.
• Transportation emissions assumptions: transportation was considered using the market average for the European Union (EU). However, emissions allocated to transportation can vary significantly depending on the specific transportation modes and distances, which can impact the final environmental score.
• Economic uncertainty: discounts associated with industrial purchases, alongside uncertainty in equipment costs and estimation factors, present challenges in providing precise economic evaluations. These uncertainties typically introduce a deviation range of 50–100% in cost estimates, which should be considered when interpreting the financial viability of the process.
6. Conclusions
Substituting conventional polyurethane with bio-based compounds of similar formulations presents a significant challenge, particularly regarding the synthesis of isocyanate intermediates. Moving away from traditional amine phosgenation reduces the yields, complicates the industrial scalability, and results in materials with less desirable mechanical and physicochemical properties. Alternatively, increasing the biogenic content enhances the perceived sustainability of the final product, and a comprehensive assessment of the entire value chain revealed significant environmental impacts. Achieving a 70% biogenic content in polyisocyanate prepolymers requires multiple raw materials, and complex and energy-intensive processes, such as crystallization and cryogenic distillation, which increase the carbon footprint. Consequently, the GWP of bio-polyisocyanate was found to exceed fossil-based polyisocyanates by an order of magnitude.Addressing inefficiencies in biological conversion and solvent-intensive steps by optimizing separations and reducing material losses will be pivotal for improving the sustainability without compromising the efficiency. To bridge the carbon footprint gap, the key opportunities include electrification of heating and renewable electricity sourcing, the development of microorganism strains tolerant to higher sugar concentrations, and the use of biogenic solvents with minimal degradation and low-carbon neutralizing agents. Further studies are needed to assess whether using sugar-based residues or incorporating carbon capture technology can effectively reduce the overall environmental impact, given the additional unit operations required.
Similarly, the carbon footprint of castor oil, i.e., the other biogenic reactant in bio-PU production, is double that of conventional polyols. This leads to the bio-polyurethane gel produced having a GWP score four-times higher than the value reported in the Ecoinvent database for this material. Consequently, the economic burden of producing bio-based polyurethane, which requires a selling price five-times higher than the average market price of conventional PU gel to achieve financial viability, is not justified in terms of GWP reduction.
This study revealed that replacing the fossil-derived carbon content with biogenic carbon to formulate a ‘green’ product does not necessarily reduce climate change impacts. A comprehensive life cycle assessment of the entire value chain remains essential for accurately evaluating the environmental performance and identifying strategies for improvement.
Author contributions
Alberto Almena: writing – original draft, writing – review & editing, methodology, investigation, formal analysis, data curation, conceptualization, and funding acquisition. Domenico Pirone: conceptualization, validation, and formal analysis. Susana Fernández-Prieto: conceptualization, validation, supervision, and writing – review & editing. Alberto Martínez: conceptualization, validation, supervision, and writing – review & editing. Mariano Martin: methodology, validation, writing – review & editing, supervision, funding acquisition, and project administration.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this publication.Acknowledgements
This work was funded by the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 101034371.References
- P. Gabrielli, L. Rosa, M. Gazzani, R. Meys, A. Bardow, M. Mazzotti and G. Sansavini, One Earth, 2023, 6, 682–704 CrossRef.
- EPA, The 12 Principles of Green Chemistry, 2023 Search PubMed.
- European Commission, The European Green Deal.
- EEA, Total greenhouse gas emissions in the chemical industry (Indicator), 2024 Search PubMed.
- T. R. Walker and L. Fequet, TrAC, Trends Anal. Chem., 2023, 160, 116984 CrossRef CAS.
- J. Zheng and S. Suh, Nat. Clim. Change, 2019, 9, 374–378 CrossRef.
- Statista, Market volume of polyurethane worldwide from 2015 to 2022, with a forecast for 2023 to 2030, 2023 Search PubMed.
- Precedence Research, Polyurethane Market Size 2022 to 2032.
- M. H. Tran and E. Y. Lee, Environ. Chem. Lett., 2023, 21, 2199–2223 CrossRef CAS.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- S. Wendels and L. Avérous, Bioact. Mater., 2021, 6, 1083–1106 CAS.
- K. S. Chian and L. H. Gan, J. Appl. Polym. Sci., 1998, 68, 509–515 CrossRef CAS.
- R. S. Malani, V. C. Malshe and B. N. Thorat, J. Coat. Technol. Res., 2022, 19, 201–222 Search PubMed.
- M. A. Moyano, M. P. Carbonell-Blasco, F. Arán-Aís and E. Orgilés-Calpena, J. Adhes., 2023, 101, 231–249 CrossRef.
- M. Desroches, M. Escouvois, R. Auvergne, S. Caillol and B. Boutevin, Polym. Rev., 2012, 52, 38–79 CrossRef CAS.
- L. Hojabri, X. Kong and S. S. Narine, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3302–3310 CrossRef CAS.
- Z. Kahlerras, R. Irinislimane, S. Bruzaud and N. Belhaneche-Bensemra, J. Polym. Environ., 2020, 28, 3003–3018 CrossRef CAS.
- E. Głowińska, W. Wolak and J. Datta, J. Polym. Environ., 2021, 29, 2140–2149 Search PubMed.
- J. Niesiobędzka and J. Datta, Green Chem., 2023, 25, 2482–2504 RSC.
- M. V. Zabalov, M. A. Levina and R. P. Tiger, Russ. J. Phys. Chem. B, 2019, 13, 778–788 CrossRef CAS.
- S. Zamani, S. H. E. van der Voort, J.-P. Lange, S. R. A. Kersten and M. P. Ruiz, Polymers, 2023, 15, 2522 Search PubMed.
- F. Ferretti, D. Formenti and F. Ragaini, Rend. Lincei, 2017, 28, 97–115 CrossRef.
- D. Saylik, M. J. Horvath, P. S. Elmes, W. R. Jackson, C. G. Lovel and K. Moody, J. Org. Chem., 1999, 64, 3940–3946 CrossRef CAS.
- M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, RSC Adv., 2013, 3, 4110–4129 RSC.
- A. Gomez-Lopez, F. Elizalde, I. Calvo and H. Sardon, Chem. Commun., 2021, 57, 12254–12265 RSC.
- C. Amezúa-Arranz, M. Santiago-Calvo and M.-Á. Rodríguez-Pérez, Eur. Polym. J., 2023, 197, 112366 CrossRef.
- R. T. Maleeny and J. F. Kinney, U.S. Pat., 6730311, 2004 Search PubMed.
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- R. Jaratrotkamjorn and V. Tanrattanakul, J. Appl. Polym. Sci., 2020, 137, 49310 CrossRef CAS.
- J. Feng, Q. Lu, W. Tan, K. Chen and P. Ouyang, Front. Chem. Sci. Eng., 2019, 13, 80–89 CrossRef CAS.
- P. Szczepańczyk, M. Szlachta, N. Złocista-Szewczyk, J. Chłopek and K. Pielichowska, Polymers, 2021, 13, 946 CrossRef PubMed.
- S. K. Pandey, C. Haldar, D. K. Patel and P. Maiti, in Multifaceted Development and Application of Biopolymers for Biology, Biomedicine and Nanotechnology, ed. P. K. Dutta and J. Dutta, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, pp. 169–202 Search PubMed.
- Mitsui Chemicals, STABiOTM product brochure.
- T. Nakagawa, H. Takeuchi, K. Sato and S. Yamasaki, U.S. Pat., 9376404, 2016 Search PubMed.
- X. Peng, D. Zhang, J. Yuan, H. Yang, M. Li, H. Zhang, H. Niu, C. Zhu, Y. Chen, C. Zhao, T. Guo, Z. Wang, D. Liu and H. Ying, Bioresour. Technol., 2024, 414, 131567 Search PubMed.
- Y. Teng, E. L. Scott, A. N. T. van Zeeland and J. P. M. Sanders, Green Chem., 2011, 13, 624–630 RSC.
- M. Modesti and A. Lorenzetti, Eur. Polym. J., 2001, 37, 949–954 Search PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- S. Sharma, V. Sharma and S. Chatterjee, Sci. Total Environ., 2023, 875, 162627 Search PubMed.
- M. M. Parascanu, N. Sanchez, F. Sandoval-Salas, C. M. Carreto, G. Soreanu and L. Sanchez-Silva, Environ. Sci. Pollut. Res., 2021, 28, 64374–64393 CrossRef CAS PubMed.
- A. Palmonari, D. Cavallini, C. J. Sniffen, L. Fernandes, P. Holder, L. Fagioli, I. Fusaro, G. Biagi, A. Formigoni and L. Mammi, J. Dairy Sci., 2020, 103, 6244–6249 Search PubMed.
- N. B. Trân, J. Vialle and Q. T. Pham, Polymer, 1997, 38, 2467–2473 Search PubMed.
- I. Javni, Z. S. Petrović, A. Guo and R. Fuller, J. Appl. Polym. Sci., 2000, 77, 1723–1734 Search PubMed.
- D. S. Ogunniyi, Bioresour. Technol., 2006, 97, 1086–1091 CrossRef CAS PubMed.
- Y.-C. Chen and W. Tai, Polymers, 2018, 10, 1100 CrossRef PubMed.
- Z. S. Petrović and D. Fajnik, J. Appl. Polym. Sci., 1984, 29, 1031–1040 Search PubMed.
- M. M. Martín, in Industrial Chemical Process Analysis and Design, ed. M. M. Martín, Elsevier, Boston, 2016, pp. 13–60 Search PubMed.
- X. Qiang, J. Luo, S. Guo, W. Cao, X. Hang, J. Liu and Y. Wan, J. Cleaner Prod., 2019, 207, 432–443 Search PubMed.
- N. Mišljenović, R. B. Schüller, E.-O. Rukke and C. S. Bringas, Annu. Trans. – Nord. Rheol. Soc., 2013, 21, 61–68 Search PubMed.
- G. Çalik, M. Berk, F. G. Boyaci, P. Çalik, S. Takaç and T. H. Özdamar, in Engineering and Manufacturing for Biotechnology, ed. M. Hofman and P. Thonart, Springer Netherlands, Dordrecht, 2002, pp. 21–28 Search PubMed.
- E. Tombari, G. Salvetti, C. Ferrari and G. P. Johari, J. Phys. Chem. B, 2007, 111, 496–501 CrossRef CAS PubMed.
- S. Bower, R. Wickramasinghe, N. J. Nagle and D. J. Schell, Bioresour. Technol., 2008, 99, 7354–7362 CrossRef CAS PubMed.
- T. Roukas, Process Biochem., 1998, 33, 805–810 CrossRef CAS.
- X. He, Y. Qi, K. Chen, Y. Li and P. Ouyang, Appl. Biochem. Biotechnol., 2016, 179, 986–996 CrossRef CAS PubMed.
- V. Gopinath and K. M. Nampoothiri, in Encyclopedia of Food Microbiology, ed. C. A. Batt and M. Lou Tortorello, Academic Press, Oxford, 2nd edn, 2014, pp. 504–517 Search PubMed.
- J. Cremer, L. Eggeling and H. Sahm, Appl. Environ. Microbiol., 1991, 57, 1746–1752 Search PubMed.
- B. Stefan, E. Lothar and K. Reinhard, Appl. Environ. Microbiol., 1993, 59, 316–321 CrossRef PubMed.
- O. Anaya-Reza and T. Lopez-Arenas, Bioprocess Biosyst. Eng., 2017, 40, 1033–1048 Search PubMed.
- J. Liu, J.-Z. Xu, Z.-M. Rao and W.-G. Zhang, Microbiol. Res., 2022, 262, 127101 Search PubMed.
- S. An, S. Ahn, A. Letona, J. H. Shin, S. H. Kang, J. Y. Jeong, S. M. Park, J. W. Kim, D. Yu, H. Chung and D. Chung, Food Biosci., 2023, 53, 102845 CrossRef CAS.
- CarlRoth, L-Lysine hydrochloride. Safety data sheet, 2024.
- A.-T. Nguyen, T. Yu and W.-S. Kim, J. Cryst. Growth, 2017, 469, 65–77 CrossRef CAS.
- C.-Y. Huang, W.-W. Ting, Y.-C. Chen, P.-Y. Wu, C.-D. Dong, S.-F. Huang, H.-Y. Lin, S.-F. Li, I.-S. Ng and J.-S. Chang, Biochem. Eng. J., 2020, 156, 107514 Search PubMed.
- Y. Liu, Y. Zheng, H. Wu, W. Zhang, T. Ren, S. You, W. Qi, R. Su and Z. He, J. Chem. Technol. Biotechnol., 2020, 95, 1542–1549 Search PubMed.
- J. Kopp, C. Slouka, O. Spadiut and C. Herwig, Front. Bioeng. Biotechnol., 2019, 7, 328 CrossRef PubMed.
- M. C. Jamur and C. Oliver, Immunocytochemical methods and protocols, 2010, pp. 63–66 Search PubMed.
- Y. Huang, X. Ji, Z. Ma, M. Łężyk, Y. Xue and H. Zhao, RSC Adv., 2021, 11, 23922–23942 Search PubMed.
- H. Li, X. Xu, W. Tan, X. Lu, F. He, S. Xu, W. Tian, K. Chen, G. Li, P. Ouyang, Y. Liu and R. Liang, RSC Adv., 2020, 10, 44728–44735 Search PubMed.
- Y.-G. Hong, H.-J. Kim, J.-M. Jeon, Y.-M. Moon, J.-W. Hong, J.-C. Joo, B.-K. Song, K.-M. Park, S.-H. Lee and Y.-H. Yang, J. Ind. Eng. Chem., 2018, 64, 167–172 CrossRef CAS.
- K. Hollá, M. Polorecká, J. Kubás and M. Ballay, Trans. Res. Procedia, 2021, 55, 1506–1513 Search PubMed.
- A. Bähr, G.-H. Moon, J. Diedenhoven, J. Kiecherer, E. Barth and H. Tüysüz, Chem. Ing. Tech., 2018, 90, 1513–1519 Search PubMed.
- P. Voßnacker, A. Wüst, T. Keilhack, C. Müller, S. Steinhauer, H. Beckers, S. Yogendra, Y. Schiesser, R. Weber, M. Reimann, R. Müller, M. Kaupp and S. Riedel, Sci. Adv., 2024, 7, eabj5186 CrossRef PubMed.
- C. J. Mitchell, W. van der Borden, K. van der Velde, M. Smit, R. Scheringa, K. Ahrika and D. H. Jones, Catal. Sci. Technol., 2012, 2, 2109–2115 RSC.
- G. E. Rossi, J. M. Winfield, C. J. Mitchell, N. Meyer, D. H. Jones, R. H. Carr and D. Lennon, Appl. Catal., A, 2020, 602, 117688 Search PubMed.
- R. Z. Boros, T. Koós, C. Wafaa, K. Nehéz, L. Farkas, B. Viskolcz and M. Szőri, Chem. Phys. Lett., 2018, 706, 568–576 CrossRef CAS.
- F. He, Y. Tang, Z. Lu, Q. Hu, Y. Yang, G. Li, H. Li and K. Chen, RSC Adv., 2023, 13, 31518–31527 RSC.
- S. G. Entelis and O. V. Nesterov, Russ. Chem. Rev., 1966, 35, 917 Search PubMed.
- C. Six and F. Richter, in Ullmann's Encyclopedia of Industrial Chemistry, 2003 Search PubMed.
- Z. Hou, H. Chen, J. Mao, J. Ge and R. Bi, Chem. Eng. Sci., 2023, 280, 119018 CrossRef CAS.
- H. Schelling, T. Mattke, H.-J. Pallasch and K. Thiele, U.S. Pat., 10252912, 2019 Search PubMed.
- K. W. Anderson, T. Ikawa, R. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 10694–10695 Search PubMed.
- Y.-L. Huang, M. Heilig, H. Hasse and J. Vrabec, AIChE J., 2011, 57, 1043–1060 Search PubMed.
- OECD SIDS, SIDS Initial Assessment Report. 1,2-Dichlorobenzene, 2001.
- H. B. Goyal, K. M. Sharan, M. L. Sagu, J. Swarup and K. K. Bhattacharyya, J. Chem. Eng. Data, 1981, 26, 67–70 CrossRef CAS.
- J. Hu, Z. Chen, Y. He, H. Huang and X. Zhang, Res. Chem. Intermed., 2017, 43, 2799–2816 CrossRef CAS.
- J. Zeng, Y. Yang, Y. Tang, X. Xu, X. Chen, G. Li, K. Chen, H. Li, P. Ouyang, W. Tan, J. Ma, Y. Liu and R. Liang, Ind. Eng. Chem. Res., 2022, 61, 2403–2416 Search PubMed.
- D. V. Rosato, D. V. Rosato and M. V. Rosato, in Plastic Product Material and Process Selection Handbook, ed. D. V. Rosato, D. V. Rosato and M. V. Rosato, Elsevier, Oxford, 2004, pp. 333–368 Search PubMed.
- L. Ruiduan, L. Ling, L. Yanjie, W. Ben, Y. J. Jun and Z. Jibo, New Mater. Intell. Manuf., 2018, 1, 54–57 Search PubMed.
- J. Zhang, N. Hori and A. Takemura, Polym. Degrad. Stab., 2020, 179, 109256 CrossRef CAS.
- Y. Zhao and G. J. Suppes, Polym. Eng. Sci., 2015, 55, 1420–1428 CrossRef CAS.
- P. Jutrzenka Trzebiatowska, A. Santamaria Echart, T. Calvo Correas, A. Eceiza and J. Datta, Prog. Org. Coat., 2018, 115, 41–48 CrossRef CAS.
- Y. Guo, J. Kleemann, S. Bokern, A. Kamm, R. P. Sijbesma and Ž. Tomović, Polym. Chem., 2023, 14, 1923–1932 RSC.
- A. Almena and M. Martín, Ind. Eng. Chem. Res., 2016, 55, 3226–3238 CrossRef CAS.
- Matches Engineering, Process equipment cost estimator.
- R. Sinnott and G. Towler, Chemical engineering design: SI Edition, Butterworth-Heinemann, 2019 Search PubMed.
- A. Almena, P. J. Fryer, S. Bakalis and E. Lopez-Quiroga, Sustainable Prod. Consumption, 2019, 19, 181–193 CrossRef.
- Imarc, Polyurethane (PU) Resin Prices, Trend, Chart, Demand, Market Analysis, News, Historical and Forecast Data Report 2024 Edition, 2024.
- V. R. Patel, G. G. Dumancas, L. C. K. Viswanath, R. Maples and B. J. J. Subong, Lipid Insights, 2016, 9, 1–12 CrossRef PubMed.
- V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, C. Péan, S. Berger, N. Caud, Y. Chen, L. Goldfarb, M. I. Gomis, M. Huang, K. Leitzell, E. Lonnoy, J. B. R. Matthews, T. K. Maycock, T. Waterfield, O. Yelekçi, R. Yu and B. Zhou, IPCC, Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2023, DOI:10.1017/9781009157896.
- V.-T. Luong, R. Amal, J. A. Scott, S. Ehrenberger and T. Tran, J. Cleaner Prod., 2018, 202, 1035–1044 CrossRef CAS.
- G. Galán, M. Martín and I. E. Grossmann, Ind. Eng. Chem. Res., 2021, 60, 5558–5573 CrossRef PubMed.
- S. Väisänen, J. Havukainen, V. Uusitalo, M. Havukainen, R. Soukka and M. Luoranen, Renewable Energy, 2016, 89, 401–410 Search PubMed.
- P. Stegmann, V. Daioglou, M. Londo, D. P. van Vuuren and M. Junginger, Nature, 2022, 612, 272–276 CrossRef CAS PubMed.
- Y. Zhang, P. Li and S. Sun, Ind. Crops Prod., 2024, 222, 119546 CrossRef CAS.
- A. Zlatanić, C. Lava, W. Zhang and Z. S. Petrović, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 809–819 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00423c |
| This journal is © The Royal Society of Chemistry 2025 |