
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrocracking of polyolefins over ceria-promoted Ni/BEA catalysts†
Jessie A.
Sun
 ab,
Esun
Selvam
ab,
Alexander
Bregvadze
a,
Weiqing
Zheng
a and
Dionisios G.
Vlachos
*abc
ab,
Esun
Selvam
ab,
Alexander
Bregvadze
a,
Weiqing
Zheng
a and
Dionisios G.
Vlachos
*abc
aDepartment of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy St, Newark, DE 19716, USA. E-mail: vlachos@udel.edu
bCenter for Plastics Innovation, University of Delaware, 221 Academy St, Newark, DE 19716, USA
cDelaware Energy Institute, University of Delaware, 221 Academy St, Newark, DE 19716, USA
First published on 14th March 2025
Abstract
Hydroconversion of polyolefins into primary petrochemical feedstocks, such as naphtha (C5–C12), offers a promising strategy to redirect plastic waste from landfills into the economy. Current hydrocracking strategies predominantly utilize precious metal-based catalysts, as the higher activation energy and stability issues limit the use of Earth-abundant metals (EAM). Herein, we improve the stability of Ni/BEA by incorporating a ceria promoter and demonstrate it in the hydrocracking of low-density polyethylene (LDPE). The Ce-promoted Ni/BEA catalyst achieved over 80% selectivity toward naphtha with the highest-reported naphtha productivity, outperforming current noble metal (Pt and Ru) and EAM (Ni and Co) catalysts. Catalyst evaluation and characterization underscore that ceria directly regulates metal dispersion and redox behavior and mitigates catalyst deactivation by suppressing coke formation. We correlate the promotional role of ceria to its unique hydrogen storage ability, which directly enhances metal hydrogenation ability. This work highlights exciting progress toward optimizing EAM hydrocracking catalysts for polyolefin upcycling.
Green foundation1. The development of polyolefin (PO) deconstruction and upcycling chemistries helps divert plastic waste from landfills, enabling the conservation of the natural environment and non-renewable petrochemical resources. Optimization of Earth-abundant metal (EAM)-based catalysts is crucial for the industrial advancement of sustainable materials and PO waste deconstruction processes, thereby promoting the principles of green chemistry and furthering the field.2. This work presents the use of a ceria-promoted Ni/BEA catalyst for low-density polyethylene (LDPE) hydrocracking. Notably, ceria-promoted Ni/BEA demonstrates high productivity for naphtha generation and the addition of ceria was shown to improve Ni reducibility, resistance to coking, and overall stability. 3. The ceria-promoted Ni/BEA catalyst exhibited enhanced hydrogenation ability even under hydrogen-lean conditions, revealing exciting opportunities for future tuning of reaction pressure requirements for increased energy efficiency. |
Introduction
Plastics have become an irreplaceable commodity in modern society. However, their rapid consumption has led to the mismanagement of over 7000 Mt of end-of-life plastic waste, as only ∼10% is recycled.1–3 This has resulted in their accumulation in landfills, posing significant environmental and human health concerns. Polyolefins (POs) are the most prevalent polymers used today. Due to their structural simplicity and durability, they are widely employed across various societal sectors, including construction, transportation, and telecommunications.4–6 Common POs, including high- and low-density polyethylene (HDPE and LDPE) and polypropylene (PP), constitute nearly 60% of the plastic waste generated annually and are prime candidates for chemical recycling via catalytic hydrocracking.7 Hydrocracking utilizes a bifunctional metal and Brønsted acid catalyst under mild conditions8–10 to deconstruct POs into valuable petrochemical products like naphtha (C5–C12), a primary feedstock for ethylene, propylene, and plastics production. Prior PO hydrocracking work has predominantly used noble metals (Pt, Pd, and Ru) supported on acidic supports (WO3/ZrO2, HY, Beta, and ZSM-5).11–17 Although noble metal-based catalysts are active at milder temperatures, their scarcity and cost limit their industrial applications. Developing efficient Earth-abundant metal (EAM) catalyst alternatives is essential.Bifunctional nickel-based catalysts are appealing EAM alternatives. Previous work utilizing Ni/ZSM-5, Ni/TiO2-A-SG, and Ni/WOx-ZrO2 demonstrated comparable hydrocracking performance to noble metal-based catalysts.11,18,19 However, these catalysts exhibit notably lower naphtha selectivity and often utilize high catalyst/polymer ratios as well as elevated reaction temperatures and pressures.11,18,19 Our group previously demonstrated that nickel supported on BEA zeolite (Ni/BEA) effectively and selectively converts LDPE to naphtha. However, the catalyst is susceptible to coking, which can be mitigated by operating at higher reaction pressures (60 bar).20 Ceria (CeO2) is an inexpensive structural and electronic promoter extensively used in heterogeneous catalysis.21,22 Specifically, doping catalysts with CeO2 can significantly alter metal dispersion, surface acidity, and overall redox behavior.23–28 Recent studies from multiple groups have reported enhanced PO hydrocracking activity using CeO2-promoted Pt/HY catalysts.13,14 These initial insights motivate further exploration of Earth-abundant Ni-based catalysts, particularly the promotional role of ceria and the ceria–metal synergy in PO hydrocracking chemistry, which remain elusive.
Herein, we focus on improving the efficiency and stability of the previously reported Ni/BEA20 catalyst by ceria incorporation. We show the promotional effect of CeO2 doping in enhancing Ni/BEA catalytic activity and stability and demonstrate its superior naphtha selectivity. The impact of Ni–Ce electronic synergy on overall catalyst reducibility, acidity, and morphology is evaluated. Finally, we reveal the critical influence of CeO2 on reaction pressure requirements, metal hydrogenation ability, and enhancing hydrogen storage capacity in polyolefin hydrocracking.
Materials and methods
Catalyst preparation
Nickel on beta zeolite (Ni/BEA) was synthesized using wetness impregnation. Beta zeolite support, BEA(25) (Zeolyst, Zeolite Ammonium Beta Powder, CP814E, Si/Al = 25), was first calcined at 550 °C for 4 h (2 °C min−1 ramp rate) to obtain the H+ form. The zeolite was then impregnated dropwise with an appropriate amount of nickel(II) nitrate hexahydrate (Sigma-Aldrich, Ni(NO3)2·6H2O, 99.999% trace metal) in a DI water solution to achieve 5% Ni loading (5Ni/BEA). Nickel–ceria on BEA zeolite (5Ni%Ce/BEA) was synthesized via co-impregnation using Ni(NO3)2·6H2O and cerium(III) nitrate hexahydrate (Sigma-Aldrich, Ce(NO3)3·6H2O, 99.999% trace metal). A nominal 5% Ni loading was used for all catalysts, with various %Ce metal loadings. Hereafter, the number preceding Ce indicates the weight percent of the added Ce (e.g., 20Ce). For physical mixture experiments, commercial CeO2 (Alfa Aesar by Thermo Scientific, cerium(IV) oxide, 99.5% REO) was purchased and calcined at 550 °C for 4 h in air. 5Ni/CeO2 was synthesized using wetness impregnation.For wetness impregnation synthesis, all catalyst suspensions were continuously stirred using a glass rod on a hot plate set to 70 °C, then dried in air at 110 °C overnight, and calcined in air at 550 °C for 4 h (2 °C min−1 ramp rate). Prior to the reaction, all catalysts were reduced in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (vol%) flow of H2 and He at 350 °C for 2 h with a 10 °C min−1 ramping rate.
:50 (vol%) flow of H2 and He at 350 °C for 2 h with a 10 °C min−1 ramping rate.
Catalyst characterization
:50 (vol%) mixture of H2 and He at 350 °C for 2 h (10 °C min−1 ramping rate) and then transferred to an N2 environment glove box (PO2 < 500 ppm). Furthermore, the samples were pressed onto carbon tape and placed in a transfer vacuum vessel for XPS measurements without air exposure. All data processing was conducted using the Casa XPS software.
 using a molar stoichiometry factor of Ni/CO equal to 1:3.29 Here, nCO represents the moles of CO uptake per mass of catalyst, xNi is the Ni loading (wt%) and MwNi is the molecular weight of Ni.
:50 vol% H2:Ar flow at 300 °C (ramping rate 10 °C min−1) for 2 h. After reduction, the gas flow was switched to pure Ar, the temperature was reduced to 150 °C, and a pre-pyridine saturation spectrum was recorded. Then, 5 μl of liquid pyridine (Sigma-Aldrich, 99.8%) was injected through a septum port using a micro syringe, and the pyridine vapor was allowed to saturate the sample for 30 min. The spectrum of the pyridine-saturated sample was then recorded, and the temperature was increased stepwise from 150 °C to 300 °C in a constant flow of Ar. Finally, spectra were recorded every 15 °C, and peak integration at 1540 cm−1 for pyridinium ions (PyH+) bonded to Brønsted sites and 1450 cm−1 for Py coordinately bonded to Lewis sites (PyL) was performed using the OMNIC 8.2 software. Molar extinction coefficients reported by Tamura et al. were used to calculate acid site densities.30
:50 vol% H2:Ar flow at 300 °C (ramping rate 10 °C min−1) for 2 h. After reduction, the gas flow was switched to pure Ar, and the temperature was reduced to 50 °C under Ar flow. The cell was then purged with D2 flow (30 mL min−1) for 0.5 h. After saturation, the temperature was increased stepwise from 150 °C to 230 °C under a constant flow of D2. Finally, spectra were recorded every 15 °C and analyzed using the OMNIC 8.2 software.
using a molar stoichiometry factor of Ni/CO equal to 1:3.29 Here, nCO represents the moles of CO uptake per mass of catalyst, xNi is the Ni loading (wt%) and MwNi is the molecular weight of Ni.
:50 vol% H2:Ar flow at 300 °C (ramping rate 10 °C min−1) for 2 h. After reduction, the gas flow was switched to pure Ar, the temperature was reduced to 150 °C, and a pre-pyridine saturation spectrum was recorded. Then, 5 μl of liquid pyridine (Sigma-Aldrich, 99.8%) was injected through a septum port using a micro syringe, and the pyridine vapor was allowed to saturate the sample for 30 min. The spectrum of the pyridine-saturated sample was then recorded, and the temperature was increased stepwise from 150 °C to 300 °C in a constant flow of Ar. Finally, spectra were recorded every 15 °C, and peak integration at 1540 cm−1 for pyridinium ions (PyH+) bonded to Brønsted sites and 1450 cm−1 for Py coordinately bonded to Lewis sites (PyL) was performed using the OMNIC 8.2 software. Molar extinction coefficients reported by Tamura et al. were used to calculate acid site densities.30
:50 vol% H2:Ar flow at 300 °C (ramping rate 10 °C min−1) for 2 h. After reduction, the gas flow was switched to pure Ar, and the temperature was reduced to 50 °C under Ar flow. The cell was then purged with D2 flow (30 mL min−1) for 0.5 h. After saturation, the temperature was increased stepwise from 150 °C to 230 °C under a constant flow of D2. Finally, spectra were recorded every 15 °C and analyzed using the OMNIC 8.2 software.
Reactivity tests
Product yields and selectivity to the liquid, solid, and gas products were calculated on a molar carbon basis. Quantification was performed using the following equations:
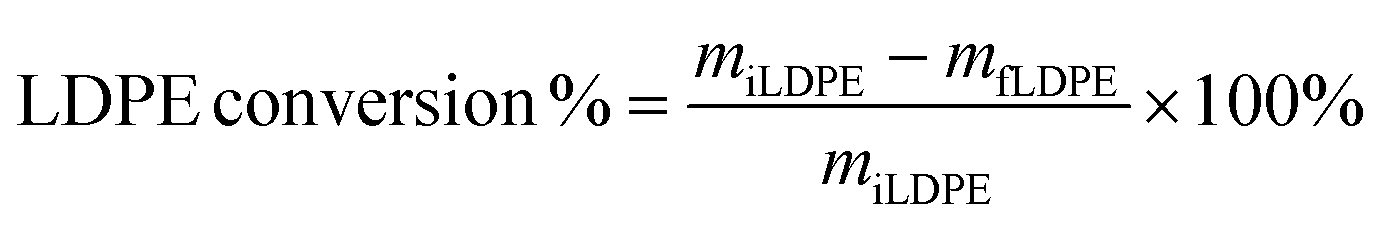
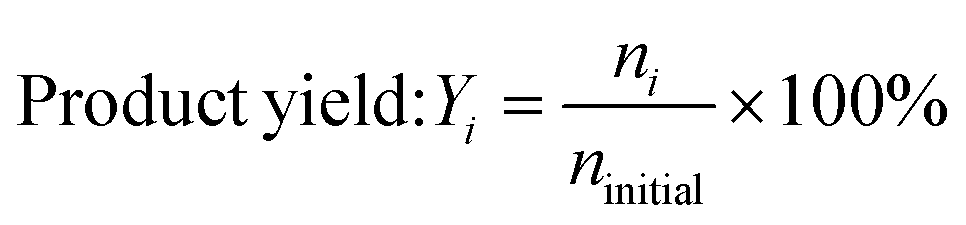
where miLDPE and mfLDPE represent the mass of the initial LDPE polymer and final solid residue, respectively. ni represents the moles of carbon in the product group with i carbons, and ninitial represents the moles of carbon in the initial polymer.

Collection and quantification of gas products extracted from naphthalene hydrogenation probe reactions followed the same procedure. The liquid products were extracted using 0.02 g of n-octacosane as an internal standard and quantified using the same GC-FID and GC-MS methods. Calibration coefficients and retention times for the major liquid products were measured by injecting naphthalene, tetralin (1,2,3,4-tetrahydronaphthalene, Sigma Aldrich), and cis/trans-decalin (decahydronaphthalene, Sigma Aldrich) as standards. Naphthalene conversion, tetralin yield, and selectivity were calculated from the liquid molar quantities measured using GC-FID calibration curves (Fig. S14 and S15†), and the mass of the residual naphthalene solid was obtained gravimetrically.
Results and discussion
Promotional effect of ceria on LDPE hydrocracking
The catalytic performance of 5Ni/BEA (5% Ni by weight) and Ce-promoted 5Ni%Ce/BEA (%Ce = 5, 10, 20, 30, 40 by weight) catalysts for LDPE hydrocracking at 300 °C is shown in Fig. 1. The hydrocracking activity follows a volcano-like trend with increasing Ce incorporation (Fig. 1a). After 1 h reaction time, 5Ni20Ce/BEA (20Ce) exhibited a maximum 72% yield of extractable products (C1–C20), and the LDPE conversion increased from 63% over 5Ni/BEA (5Ni) to 86% over 20Ce. Excess Ce loadings (Ce > 20%) resulted in high solid yields and decreased LDPE conversion (Fig. 1a). N2 physisorption measurements (Table 1) indicate reduced surface area and micropore volume when excess Ce loadings (Ce > 20%) were used, suggesting physical pore blockage and coverage of the Ni and Brønsted acid sites responsible for C–C scission.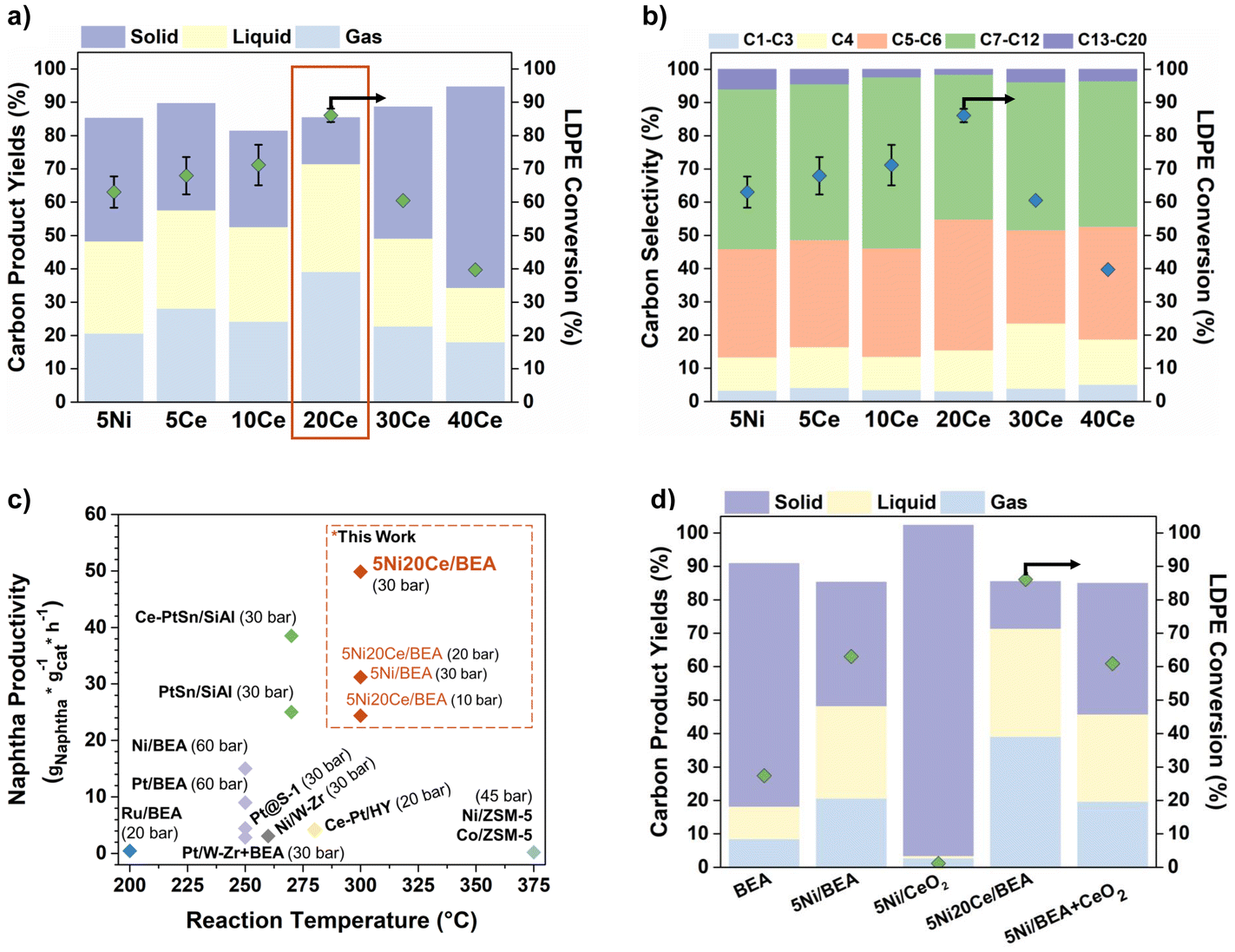 | ||
| Fig. 1 (a) Hydrocracking carbon product yields (solid, liquid, and gas) and conversion for 2 g LDPE using 0.025 g 5Ni%Ce/BEA (% = 0, 5, 10, 20, 30, 40) at 300 °C, 30 bar H2 for 1 h. Error bars represent the conversion standard deviation for 3 repeated experiments. (b) LDPE hydrocracking product selectivity distributions. (c) Naphtha productivity of this work and literature catalysts (Table S1†).11–16,18,20 (d) Physical mixture experiments with total wt% of Ni and Ce at 5% and 20%, respectively, under the same conditions. | ||
| Cat. | TEM dpa (nm) |
CO uptakeb (μmol g−1) |
D
Nib (%) |
BASc (μmol g−1) | LASc (μmol g−1) | BET surface area (m2 g−1) | External surface aread (m2 g−1) |
V
microd (cm3 g−1) |
|---|---|---|---|---|---|---|---|---|
| a Particle diameter (dp) measured from TEM distributions (Fig. 2 and S3†). b Ni dispersion (DNi) measured from CO chemisorption. c Measured from FTIR pyridine adsorption. d Obtained from BET t-plot results. | ||||||||
| BEA | — | — | — | 317 | 230 | 432 | 207 | 0.21 |
| 5Ni | 9.9 ± 6.6 | 75 | 2.9 | 50 | 132 | 432 | 250 | 0.17 |
| 5Ce | 4.5 ± 2.0 | 109 | 4.3 | 78 | 158 | 401 | 229 | 0.16 |
| 10Ce | 3.7 ± 1.6 | 104 | 4.1 | 68 | 182 | 358 | 201 | 0.15 |
| 20Ce | 3.6 ± 1.2 | 105 | 4.1 | 91 | 278 | 303 | 163 | 0.13 |
| 30Ce | — | 76 | 3.0 | — | — | 230 | 132 | 0.09 |
| 40Ce | — | 60 | 2.3 | — | — | 171 | 100 | 0.09 |
The incorporation of ceria does not significantly impact the extractable (C1–C20) product distribution (Fig. 1b, and S1a†); the overall selectivity to liquid products (>C4) remains constant (∼90%). Previous work showed that 5Ni/BEA is highly selective to naphtha (C5–C12) at 60 bar H2 and 250 °C.20 The 20Ce-promoted catalyst achieves a comparable 80% naphtha selectivity at a lower 30 bar H2 pressure and 300 °C. The naphtha productivity, calculated as gnaphtha gcat−1 h−1, underscores that 20Ce outperforms other hydrocracking catalysts (Fig. 1c, and Table S1†) reported thus far. Operating at 300 °C and 30 bar H2, the 20Ce catalyst exhibits a productivity of 50 gnaphtha gcat−1 h−1, which greatly exceeds other EAM catalysts (Ni/BEA, Ni/ZSM-5, Co/ZSM-5, and Ni-WOx-ZrO2) reported in the literature.11,18,20 Furthermore, even under reduced H2 pressures (PH2 < 30 bar), the 20Ce catalyst maintains high activity comparable to that of conventional Pt-based catalysts (Fig. 1c). While noble-metal catalysts may operate at slightly lower temperatures (250–280 °C), the 20Ce catalyst achieves the highest naphtha productivity overall, highlighting its potential as an attractive EAM option for industrial applications.
To obtain insights into the promotional effect of Ce on hydrocracking activity (Fig. 1a, selectivity shown in Fig. S1b†), a physical catalyst mixture of 5Ni/BEA and CeO2 was also investigated (Fig. 1d). The LDPE conversion of 61% was significantly lower than the 86% achieved by 20Ce but comparable to the 63% observed for 5Ni, suggesting that physically adding CeO2 does not impact reactivity. 5Ni/CeO2 was inactive for hydrocracking (<5% LDPE conversion) due to the lack of acid sites, indicating that the 20Ce's improved activity does not stem from Ni/CeO2 particles (Fig. 1d). These results indicate a synergy between Ni and ceria in the 20Ce catalyst that cannot be replicated with a physical mixture.
Structural and electronic impact of Ce doping
The volcano trend in Fig. 1a suggests that an optimal fraction of Ce doping creates synergy with Ni by altering the structural and electronic properties of the catalyst. We first turn to catalyst structural characterization. HRTEM (Fig. 2a & d, Table 1) shows that Ce incorporation reduced the average Ni particle size by ∼2.5×, from an average nanoparticle diameter of dNi ∼ 9.9 nm (5Ni) to 3.6 nm (20Ce). The 5Ni catalyst has a broader distribution of particle sizes (Fig. 2a–c), exhibiting much larger particles dNi ∼ 20 nm and aggregates (dNi > 20 nm, Fig. S2a–c†). TEM d-lattice spacing (ds) measurements31,32 of the 20Ce-promoted catalyst confirmed its compositional heterogeneity, revealing a mixture of Ni nanoparticles on the BEA support (Fig. 2e and S2d†) and Ni particles near CeO2 (Fig. 2f and S2e†). Overall, the 20Ce catalyst exhibited a narrower particle size distribution (Fig. 2d), indicating that ceria prevents Ni agglomeration and improves particle size uniformity (Fig. S2f†). Increased CO uptake from CO chemisorption results (Table 1) further verified that ceria significantly improves Ni metal dispersion. | ||
| Fig. 2 Particle size distributions and HRTEM micrographs of (a–c) 5Ni/BEA (5Ni) and (d–f) 5Ni20Ce/BEA (20Ce). | ||
The Ce promotional effect on metal dispersion and sintering resistance is consistent with prior works.13,14 Metal particle size often plays a vital role in modulating catalytic activity in structure-sensitive reactions, including hydrogen-assisted reactions like PO hydrogenolysis.33–35 However, metal particle size does not significantly affect PO hydrocracking, as cracking is often limited by the strength and density of the zeolite's Brønsted acid sites (BAS) instead.10 Given that the mean Ni particle size and metal dispersion (Table 1, and Fig. S3†) do not change significantly between 5% and 20% Ce loadings, the increased activity shown in Fig. 1a does not strongly correlate with the Ni particle size and dispersion.
The Ni and Ce loadings in each catalyst, verified by XRF (Table S2†) and SEM-EDX elemental mapping (Fig. S4†), were close to the theoretical loadings. Additionally, the formation of segregated bulk CeO2 was not observed (Fig. S4†). BET surface area and pore volume measurements using N2 physisorption are shown in Table 1. A decrease in external surface area and micropore volume is observed as the Ce loading is increased, indicating the presence of CeO2 on both internal and external pore sites. In Fig. S5,† XRD patterns of Ce-promoted catalysts exhibit a noticeable peak at 2Θ = 28.5° corresponding to the CeO2(111) crystal facet.32,36 As the Ce loading increases from 5% to 20%, other prominent diffraction peaks characteristic of the face-centered cubic (fcc) CeO2 fluorite structure [i.e., 2Θ = 33.2° (200), 47.4° (220) and 56.4° (222)] become visible, suggesting the growth of CeO2 particles.32,36 Additionally, the relative peak intensities corresponding to metallic Ni [2Θ = 44.5°(111), 52.1°(200)] and NiO [2Θ = 37.2°(111), 43.5°(200)] decrease significantly.37 Diffraction peaks associated with Ni/NiO are no longer visible in the 20Ce-promoted catalyst, indicating a small Ni crystallite size below the detection limit of XRD and a higher Ni dispersion, consistent with HRTEM (Fig. 2) and CO chemisorption (Table 1) results.
Acid site characterization via pyridine FTIR (Table 1 and Fig. S6†) revealed that the parent BEA zeolite contains high BAS and Lewis acid site (LAS) densities. As expected, the deposition of Ni on the zeolite support significantly reduced both BAS and LAS densities due to acid site coverage. 5Ni exhibited a LAS density of ∼132 μmol gcat−1 and a BAS density of ∼50 μmol gcat−1. Due to the strong Lewis acidity of cerium cations, Ce addition drastically increased the LAS density (Table 1).38 At 20Ce loading, the LAS density increased to ∼278 μmol gcat−1 and the BAS density to ∼91 μmol gcat−1. Increasing Ce loading increases the LAS density but only marginally affects the BAS density. CeO2 is a reducible oxide primarily possessing LAS. Due to oxygen vacancies in the CeO2 crystal lattice, surface hydroxyls can form on Ce3+ sites and create Brønsted acidity, rationalizing the slight increase in BAS.38,39 Yet, we hypothesize that these additional sites do not significantly impact the overall cracking rate since the stronger Brønsted acid sites of the zeolite are responsible for skeletal rearrangement and beta scission.
To understand the redox and electronic properties imparted by Ce incorporation, the catalysts were characterized using TPR and XPS (Fig. 3). TPR profiles for the 5Ni and Ce-promoted Ni catalysts are shown in Fig. 3a. The reduction of 5Ni exhibited a broad peak with multiple local maxima, indicating a heterogeneous mixture of Ni species (Fig. 3a) with a broad particle size distribution. The lower temperature maximum at ∼340 °C has been ascribed to the direct reduction of bulk NiO species located on the external pore sites of the zeolite.23,24 Although the exact attribution of the peaks is difficult to ascertain, the higher temperature peaks at ∼392 °C and 425 °C are associated with the reduction of dispersed NiO species confined within the internal pores and interacting strongly with the support.24,26,28 Upon adding Ce, the main reduction peak becomes unimodal, with different NiO species becoming indistinguishable, reflecting a narrower particle size distribution consistent with the TEM analysis (Fig. 2). Furthermore, the main Ni reduction peak gradually shifts to lower temperatures and includes the partial reduction of Ce4+ to Ce3+. Higher temperature peaks (>400 °C), corresponding to the surface and bulk reduction of pure CeO2, were not observed for Ce-promoted Ni catalysts (Fig. 3a), demonstrating that the strong redox interaction between Ni and CeO2 promotes the co-reduction of both species under milder conditions.
 | ||
| Fig. 3 Characterization of 5Ni%Ce/BEA (%Ce = 0, 5, 10, 20) catalysts: 5Ni (orange), 5Ce (purple), 10Ce (blue), and 20Ce (green). (a) H2-TPR profiles. (b) Air-free XPS spectra of the Ni 3p3/2 region normalized to C 1s at 284.8 eV. | ||
XPS spectra of the Ni 3p3/2 region for the reduced (350 °C) catalysts are shown in Fig. 3b. The 5Ni catalyst exhibits a prominent Ni 3p3/2 peak centered at a binding energy (BE) of 853.7 eV assigned to NiO (Ni2+) and a second peak at 858.6 eV corresponding to the broad satellite.40–42 With the incorporation of 5–10% Ce, the Ni 3p3/2 peak remains centered around 853.7 eV but begins to broaden, implying a more complex electronic environment of Ni oxidation states (Fig. 3b and Table S3†). Upon 20Ce addition, a broad feature at 855.2 eV associated with Ni(OH)2 species (ascribed to BE ∼ 856.2 eV)40–42 becomes more apparent, likely due to the partial reduction of NiO, implying the presence of Niδ+ species. Furthermore, the Ni 3p3/2 peak shifts to a lower BE around 852.7 eV, close to the Ni0 state at 852.6 eV,40–42 indicating contributions from metallic Ni species. The presence of ceria facilitates the reduction of Ni2+ species, as higher fractions of Ni0 (Table S3†) are observed, which further corroborates the improved Ni reducibility observed in TPR (Fig. 3a). Additionally, XPS spectra of the Ce 3d region (Fig. S7†) reveal the co-existence of both Ce4+ and Ce3+ electronic states, indicating the co-reduction of Ni and Ce species (Fig. 3a) at lower temperatures. Clearly, the electronic synergy of Ni and Ce species alters the overall redox properties and enhances the in situ catalyst reduction under reaction conditions.
To investigate the stability of the Ce-promoted catalysts, the coke content on spent catalysts was analyzed using TGA under an air atmosphere. Spent catalysts were collected after a 3.5 h reaction to ensure maximum conversion (Fig. S8†). Fig. 4a shows that the quantity of coke decreased with increasing Ce loading. The 20Ce-promoted catalyst exhibited 6% less total mass loss compared to 5Ni. Furthermore, the composition of the coke was also affected (Fig. 4a); the ratio of light coke (mass loss 100–350 °C) to heavy polyaromatic coke (mass loss >350 °C) is shown in Fig. 4b. Light coke has been attributed to lighter organic residues that can easily be removed under reaction conditions (300 °C, 30 bar H2). Meanwhile, heavy coke deposits form by olefin intermediate oligomerization during hydrocracking, blocking pore openings and active sites, which hinder catalytic activity. Upon adding Ce, an overall reduction in coke is observed (Fig. 4b). We rationalize that the reduction in coke, specifically heavy coke, on the Ce-promoted Ni catalysts implies that ceria promotes the hydrogenation and desorption of adsorbed intermediates (coke precursors).
 | ||
| Fig. 4 TGA thermograms of (a) weight loss (%) and derivative weight loss (% min−1) and (b) light/heavy coke content comparison for the spent 5Ni/BEA (5Ni) and Ce-promoted 5Ni%Ce/BEA (%Ce = 0, 5, 10, 20) catalysts after 3.5 h hydrocracking reaction (2 g LDPE, 0.025 g catalyst, 300 °C, 30 bar). | ||
Direct reuse of the spent 5Ni and 20Ce catalysts in 3 successive reaction cycles revealed significant deactivation (Fig. S9a & b†) and coking, but 20Ce still exhibited higher resistance to coking (Fig. S10a & b†). Ceria likely promotes the desorption of adsorbed species on the Ni site but coking of the Brønsted acid sites, as well as the oligomerization and migration of heavy coke during the reduction process, may contribute to additional deactivation and overall loss of activity after 3 uses. Both catalysts were fully regenerable (via calcination and reduction) and demonstrated comparable activity and naphtha selectivity to the fresh catalysts (Fig. S9c & d and S10c & d†). Additional details can be found in the ESI, Fig. S9–S11.†
Role of H2 and hydrogenation rates
It was previously demonstrated that over 5Ni/BEA, higher hydrogen pressures (PH2) of 60 bar were necessary to limit catalyst coking and achieve adequate LDPE deconstruction and high naphtha yields.20PH2 variation strongly affects PO hydrocracking and correlates strongly with the Ni hydrogenation ability, a critical step in the overall mechanism.20 As such, the hydrocracking of LDPE was conducted under varying PH2 for the 5Ni and 20Ce catalysts, as shown in Fig. 5a. As the PH2 decreased from 30 to 10 bar, the LDPE conversion over 5Ni decreased from 60 to 45% (Fig. 5a, selectivity in Fig. S12a†). In this pressure range, the liquid products had a yellow hue, indicative of olefinic, aromatic, and cyclic alkane precursors to coke (Fig. 5a and S13†). The liquid products remained clear for PH2 ≥ 30 bar, implying less coke precursors. Without ceria, the 5Ni activity increases significantly (from 60% to 88% LDPE conversion) when PH2 increases from 30 to 40 bar. This suggests that the excess hydrogen saturates the Ni surface, stimulating the hydrogenation of adsorbed intermediates and inhibiting coke formation even without Ce incorporation. The LDPE conversion over the 20Ce catalyst also decreased at lower pressures, confirming a positive reaction order in PH2 (Fig. 5a). For PH2 ≤ 20 bar, the liquid products exhibited a yellow hue, and the LDPE conversion reduced from 85% (30 bar) to 54% (10 bar), indicating catalyst deactivation. Nonetheless, 20Ce demonstrates higher activity and naphtha selectivity (Fig. 1c and S12b†) than 5Ni, even under hydrogen-deficient conditions (PH2 < 30 bar). | ||
| Fig. 5 (a) LDPE hydrocracking conversion and images of liquid reaction products at varying H2 pressures for the 5Ni/BEA (5Ni) and 5Ni20Ce/BEA (20Ce) catalysts. Reaction conditions: 2 g LDPE, 0.025 g catalyst, 300 °C, 1 h. (b) Naphthalene hydrogenation reaction conversion and tetralin selectivity over the 5Ni and 20Ce catalysts. Reaction conditions: 1 g naphthalene, 0.025 g catalyst, 300 °C, 1 h. FTIR spectra of (c) O–D and (d) O–H vibrational range for H–D exchange in D2 flow for 5Ni and 20Ce. | ||
Hydrogenation of naphthalene, used as a coke surrogate for polycyclic aromatics, was conducted to probe the promotional effect of Ce on Ni hydrogenation ability (Fig. 5b and S14†). Fig. 5b shows that the 5Ni and 20Ce catalysts displayed similar naphthalene conversions (33% and 39%, respectively), but 20Ce exhibited 1.5× higher selectivity to the primary hydrogenated product, tetralin. The increased selectivity implies that Ce promotes naphthalene hydrogenation, corroborating our findings from TGA (Fig. 4) and pressure variation experiments (Fig. 5a). Our prior work reported that reducible oxide supports, including CeO2, can store and donate H to the metal via spillover, aiding unsaturated intermediates’ hydrogenation during hydrogenolysis.43,44 Likewise, we hypothesize that ceria promotes the hydrogenation of olefin intermediates and reaction products on the nearby Ni sites. This leads to their desorption, thereby prolonging catalyst performance.
Finally, in situ FTIR spectroscopy was used to measure H–D exchange over the 5Ni and 20Ce-promoted Ni/BEA catalysts (Fig. 5c & d). The catalysts were pre-reduced in H2, then a constant flow of D2 was introduced, and the temperature was increased from 50 to 230 °C. At temperatures <100 °C, a peak at 2752 cm−1 appears, attributed to the H–D exchange of non-acidic silanol groups, which can undergo exchange at room temperature (Fig. 5c).45,46 As the temperature increases, prominent O-D free stretching vibrations in the broad frequency range of 2670–2740 cm−1 become more pronounced, indicating the formation of different OD surface species on Ni and Ce.47–50 During H–D exchange, a complementary decrease in the O–H vibrations in the 1600–1700 cm−1 range (Fig. 5d), attributed to hydrogen-bonded OH groups on the zeolite, is also observed.49–51 For the 5Ni catalyst, H–D exchange on the non-acidic silanol groups is noticeable at <100 °C, but primary O–D stretching vibrations do not form until ≥125 °C. In contrast, over the 20Ce catalyst, the Si–O–D exchange is extremely prominent even at 50 °C, and the O–D stretching becomes apparent at temperatures as low as 95 °C and intensifies by ≥110 °C (Fig. 5c). These results imply that Ni dissociates D2 at temperatures as low as 50 °C, and the presence of ceria increases the rate of H–D exchange, especially at lower temperatures. The faster rates of H-D exchange suggest that during PO hydrocracking, ceria may store H that spillovers from the Ni sites, thereby increasing Ni surface availability for further H2 activation and leading to additional active H species available for hydrogenation. Lastly, H2 chemisorption provided additional evidence supporting the role of CeO2 as a potential hydrogen reservoir (Table S4†). The 20Ce catalyst consumed (5.14 μmol H2 gcat−1) ∼ 1.6× more H2 per gcat than the 5Ni catalyst (2.99 μmol H2 gcat−1). This increase in H2 adsorption further suggests that CeO2 likely aids in the storage of activated H from Ni, which promotes additional H2 dissociation and supports the results observed from FTIR H–D exchange.
Discussion
Recent works on Ce-promoted Pt/HY catalysts reported that Ce promotes Brønsted acidity, metal dispersion, reducibility, resistance to coking, and hydrocracking activity.13,14 CeO2 can alter the catalyst morphology (Fig. 2) by potentially increasing active site exposure and improving Ni dispersion. Furthermore, it can store hydrogen, a key factor in enhancing Ni reducibility and hydrogenation. Ni is a widely recognized metal whose hydrogenation ability is sensitive to H2 availability (Fig. 5a). CeO2 can form surface hydroxyls or hydrides (CeOH/CeH) due to the oxygen vacancies carrying excess negative charge near reduced Ce3+ cations.21,52,53 The electron-rich Ce sites adjacent to oxygen vacancies store excess hydrogen as CeOH/CeH species. The formation of oxygen vacancies depends on the local degree of ceria reduction (formation of Ce3+ cations).21,22,54 Under reducing conditions, ceria forms non-stochiometric CeO2−x oxides (0 < x ≤ 0.5), which contain oxygen vacancies.21,22,54 TPR/XPS analyses show (Fig. 3 and S7†) that the synergy between Ni and CeO2 controls the catalyst redox behavior and simultaneously reduces both species under reaction conditions. The proximity of the Ni and ceria interfaces (Fig. 2) enables active H species to readily diffuse from Ni sites to partially reduced ceria (CeO2−x) via spillover.21,55 CeO2−x stores active H (Table S4†), increasing the availability of free sites on the Ni surface, which may promote additional H2 dissociation (Fig. 5c & d). Ceria can donate H to Ni by reverse spillover, facilitating the hydrogenation of intermediates and reaction product desorption (Fig. 5a & b). This prevents Ni deactivation from coking (Fig. 4), enhancing hydrogenation and the apparent hydrocracking rate (Fig. 1). Scheme 1 depicts the hypothesized role of ceria in the reaction. | ||
| Scheme 1 Proposed role of CeO2 in the hydrocracking reaction. | ||
Conclusions
In this work, we reveal the mechanistic role of CeO2 promoters in 5Ni/BEA catalysts for polyolefin hydrocracking. We demonstrated that 20 wt% Ce-promoted 5Ni/BEA is an effective EAM alternative to noble metal-based catalysts with superior LDPE hydrocracking activity and the highest reported naphtha productivity. Ceria stabilized smaller Ni particles (improved metal dispersion) and promoted Ni reducibility while suppressing catalyst coking and deactivation, even under H2-deficient conditions. Probe reactions over Ce-promoted 5Ni/BEA exhibited enhanced H2 activation/dissociation and hydrogenation ability compared to 5Ni/BEA. We hypothesize that a unique property of ceria is its ability to store hydrogen that spillovers, promoting the hydrogenation of adsorbed surface species and facilitating the desorption of reaction products for improved catalytic performance. The significant insights gained from this work open exciting opportunities for tuning the efficiency and stability of EAM catalysts in polyolefin waste deconstruction strategies.Author contributions
J.A.S. and A.B. performed catalyst synthesis and reactivity tests. J.A.S. performed product and catalyst characterization and analysis. E.S. performed SEM imaging. D.G.V., J.A.S. and E.S. conceptualized the project. W.Z. and D.G.V. guided the experimental methodology and investigation. The manuscript was written by J. A. S. and revised by J.A.S., E.S. and D.G.V. with input from all authors. D.G.V. was responsible for project administration and funding acquisition.Data availability
All data needed to evaluate the conclusions in this paper have been included in the main text or the ESI.† Additional data regarding this paper may be requested from the authors.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported as part of the Center for Plastics Innovation (CPI), an Energy Frontier Research Center (EFRC) supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences grant number DE-SC0021166. The authors acknowledge the use of instruments in the Advanced Materials Characterization Lab (AMCL), Keck Center for Microscopy at the University of Delaware. The XPS instrumentation at the University of Delaware was supported by the National Science Foundation under grant number CHE-1428149.References
- O. A. Alabi, K. I. Ologbonjaye, O. Awosolu and O. E. Alalade, J. Toxicol. Risk Assess., 2019, 5, 1–13 Search PubMed.
- P. N. T. Pilapitiya and A. S. Ratnayake, Cleaner Mater., 2024, 11, 100220 CrossRef.
- R. Geyer, in Plastic waste and recycling, Elsevier, Academic Press, 2020, ch. 2, pp. 13–32, DOI:10.1016/B978-0-12-817880-5.00002-5.
- B. Manjula, A. B. Reddy, E. R. Sadiku, V. Sivanjineyulu, G. F. Molelekwa, J. Jayaramudu and K. R. Kumar, in Polyolefin Fibres, Elsevier, Woodhead Publishing, 2 edn, 2017, ch. 18, pp. 539–560, DOI:10.1016/B978-0-08-101132-4.00018-7.
- Y. K. Kim, in Polyolefin Fibres, Elsevier, Woodhead Publishing, 2 edn, 2017, ch. 5, pp. 135–155, DOI:10.1016/B978-0-08-101132-4.00005-9.
- A. D. Patel, Z. O. Schyns, T. W. Franklin and M. P. Shaver, Nat. Commun., 2024, 15, 8733 CrossRef CAS.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Chem, 2021, 7, 1487–1533 Search PubMed.
- J. Sun, J. Dong, L. Gao, Y.-Q. Zhao, H. Moon and S. L. Scott, Chem. Rev., 2024, 124, 9457–9579 CrossRef CAS PubMed.
- H. Wang and S. C. E. Tsang, Cell Rep. Phys. Sci., 2024, 5, 102075 CrossRef CAS.
- Z. Dong, W. Chen, K. Xu, Y. Liu, J. Wu and F. Zhang, ACS Catal., 2022, 12, 14882–14901 CrossRef CAS.
- W.-T. Lee, A. van Muyden, F. D. Bobbink, M. D. Mensi, J. R. Carullo and P. J. Dyson, Nat. Commun., 2022, 13, 4850 CrossRef CAS.
- J. E. Rorrer, A. M. Ebrahim, Y. Questell-Santiago, J. Zhu, C. Troyano-Valls, A. S. Asundi, A. E. Brenner, S. R. Bare, C. J. Tassone and G. T. Beckham, ACS Catal., 2022, 12, 13969–13979 CrossRef CAS.
- H. Wang, T. Yoskamtorn, J. Zheng, P.-L. Ho, B. Ng and S. C. E. Tsang, ACS Catal., 2023, 13, 15886–15898 CAS.
- P. Zhao, W. Guo, Z. Gui, J. Jiang, Z. Zhu, J.-J. Li, L. Zhao, J. Zhou and Z. Xi, ACS Sustainable Chem. Eng., 2024, 12, 5738–5752 CAS.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2021, 7, eabf8283 CAS.
- L. Li, H. Luo, Z. Shao, H. Zhou, J. Lu, J. Chen, C. Huang, S. Zhang, X. Liu and L. Xia, J. Am. Chem. Soc., 2023, 145, 1847–1854 CAS.
- A. Bin Jumah, V. Anbumuthu, A. A. Tedstone and A. A. Garforth, Ind. Eng. Chem. Res., 2019, 58, 20601–20609 CAS.
- J. Shang, Y. Li, Y. Hu, T. Zhang, T. Wang, J. Zhang, H. Yan, Y. Liu, X. Chen and X. Feng, J. Catal., 2024, 430, 115302 CAS.
- L. Chen, J. B. Moreira, L. C. Meyer and J. Szanyi, Appl. Catal., B, 2023, 335, 122897 CAS.
- B. C. Vance, Z. Yuliu, S. Najmi, E. Selvam, J. E. Granite, K. Yu, M. G. Ierapetritou and D. G. Vlachos, Chem. Eng. J., 2024, 487, 150468 CAS.
- A. Trovarelli, Catal. Rev., 1996, 38, 439–520 CrossRef CAS.
- M. Capdevila-Cortada, G. Vilé, D. Teschner, J. Pérez-Ramírez and N. López, Appl. Catal., B, 2016, 197, 299–312 CrossRef CAS.
- D. Li, L. Zeng, X. Li, X. Wang, H. Ma, S. Assabumrungrat and J. Gong, Appl. Catal., B, 2015, 176, 532–541 CrossRef.
- N. Battumur, N. Sergelenbaatar, T. Bold and E. Byambajav, J. CO2 Util., 2023, 68, 102380 CrossRef CAS.
- W. Gac, W. Zawadzki, M. Greluk, G. Słowik, M. Rotko and M. Kuśmierz, Catalysts, 2021, 12, 13 CrossRef.
- W. Gac, W. Zawadzki, G. Słowik, W. Grudziński and S. Dzwigaj, Catal. Today, 2024, 114728, DOI:10.1016/j.cattod.2024.114728.
- W. Gac, W. Zawadzki, M. Rotko, G. Słowik and M. Greluk, Top. Catal., 2019, 62, 524–534 CrossRef CAS.
- X. Wang, L. Zhu, Y. Liu and S. Wang, Sci. Total Environ., 2018, 625, 686–695 CrossRef CAS PubMed.
- C. Bartholomew and R. Pannell, J. Catal., 1980, 65, 390–401 CrossRef CAS.
- M. Tamura, K.-i. Shimizu and A. Satsuma, Appl. Catal., A, 2012, 433, 135–145 CrossRef.
- C. W. Kim, Y. S. Son, A. U. Pawar, M. J. Kang, J. Y. Zheng, V. Sharma, P. Mohanty and Y. S. Kang, J. Mater. Chem. A, 2014, 2, 19867–19872 RSC.
- S. Phoka, P. Laokul, E. Swatsitang, V. Promarak, S. Seraphin and S. Maensiri, Mater. Chem. Phys., 2009, 115, 423–428 CrossRef CAS.
- L. Chen, L. C. Meyer, L. Kovarik, D. Meira, X. I. Pereira-Hernandez, H. Shi, K. Khivantsev, O. Y. Gutiérrez and J. Szanyi, ACS Catal., 2022, 12, 4618–4627 CrossRef CAS.
- M. Tamura, S. Miyaoka, Y. Nakaji, M. Tanji, S. Kumagai, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., B, 2022, 318, 121870 CrossRef CAS.
- J. A. Sun, P. A. Kots, Z. R. Hinton, N. S. Marinkovic, L. Ma, S. N. Ehrlich, W. Zheng, T. H. Epps III, L. T. Korley and D. G. Vlachos, ACS Catal., 2024, 14, 3228–3240 CAS.
- T. S. Sreeremya, A. Krishnan, K. C. Remani, K. R. Patil, D. F. Brougham and S. Ghosh, ACS Appl. Mater. Interfaces, 2015, 7, 8545–8555 CrossRef CAS PubMed.
- J. T. Richardson, R. Scates and M. V. Twigg, Appl. Catal., A, 2003, 246, 137–150 CAS.
- M. I. Zaki, M. A. Hasan, F. A. Al-Sagheer and L. Pasupulety, Colloids Surf., A, 2001, 190, 261–274 CAS.
- M. K. Gnanamani, R. Garcia, G. Jacobs, K. Góra-Marek, D. C. Cronauer, A. J. Kropf and C. L. Marshall, Appl. Catal., A, 2020, 602, 117722 CAS.
- H. Nesbitt, D. Legrand and G. Bancroft, Phys. Chem. Miner., 2000, 27, 357–366 CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, L. W. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- C. Wang, T. Xie, P. A. Kots, B. C. Vance, K. Yu, P. Kumar, J. Fu, S. Liu, G. Tsilomelekis, E. A. Stach, W. Zheng and D. G. Vlachos, JACS Au, 2021, 1, 1422–1434 CrossRef CAS PubMed.
- C. Wang, K. Yu, B. Sheludko, T. Xie, P. A. Kots, B. C. Vance, P. Kumar, E. A. Stach, W. Zheng and D. G. Vlachos, Appl. Catal., B, 2022, 319, 121899 CrossRef CAS.
- R. Salzer, J. Dressler, K.-H. Steinberg, U. Roland, H. Winkler and P. Klaeboe, Vib. Spectrosc., 1991, 1, 363–369 CrossRef CAS.
- U. Roland, R. Salzer and S. Stolle, Investigation of Hydrogen and Deuterium Spillover on Y Zeolites by FT-IR Microscopy–Rate Determining Steps, Elsevier, 1994 Search PubMed.
- A. Badri, C. Binet and J.-C. Lavalley, J. Chem. Soc., Faraday Trans., 1996, 92, 4669–4673 CAS.
- F. Gennari, T. Montini, N. Hickey, P. Fornasiero and M. Graziani, Appl. Surf. Sci., 2006, 252, 8456–8465 CAS.
- H. Zhou, L. Chen, Y. Guo, X. Liu, X.-P. Wu, X.-Q. Gong and Y. Wang, ACS Catal., 2022, 12, 4806–4812 CAS.
- Z. Zhao, G. Gao, Y. Xi, J. Wang, P. Sun, Q. Liu, C. Li, Z. Huang and F. Li, Nat. Commun., 2024, 15, 8444 CAS.
- H. Yamazaki, T. Yokoi, T. Tatsumi and J. N. Kondo, J. Phys. Chem. Lett., 2014, 5, 3528–3531 CAS.
- Z. Wu, Y. Cheng, F. Tao, L. Daemen, G. S. Foo, L. Nguyen, X. Zhang, A. Beste and A. J. Ramirez-Cuesta, J. Am. Chem. Soc., 2017, 139, 9721–9727 CAS.
- K. Werner, X. Weng, F. Calaza, M. Sterrer, T. Kropp, J. Paier, J. Sauer, M. Wilde, K. Fukutani and S. Shaikhutdinov, J. Am. Chem. Soc., 2017, 139, 17608–17616 CAS.
- G. Varvoutis, M. Lykaki, G. E. Marnellos and M. Konsolakis, Catalysts, 2023, 13, 275 CAS.
- W. C. J. Conner and J. L. Falconer, Chem. Rev., 1995, 95, 30 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details regarding catalyst reactivity, reusability, and characterization. See DOI: https://doi.org/10.1039/d5gc00345h |
| This journal is © The Royal Society of Chemistry 2025 |