
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into photoelectrocatalytic lignin oxidation to value-added products using a niobium-doped titanium dioxide photoanode†
Daniela F. S.
Morais
ab,
Luiza M. G.
Sena
ab,
Joana M.
Ribeiro
c,
Telmo
da Silva Lopes
db,
Paula
Dias
 db,
Adélio
Mendes
db,
Carina A. E.
Costa
ab,
Alírio E.
Rodrigues
ab,
Susana R. S.
Pereira
e,
Paula C.
Pinto
e,
Rui A. R.
Boaventura
ab,
Carlos J.
Tavares
c,
Vítor J. P.
Vilar
*ab and
Francisca C.
Moreira
*ab
db,
Adélio
Mendes
db,
Carina A. E.
Costa
ab,
Alírio E.
Rodrigues
ab,
Susana R. S.
Pereira
e,
Paula C.
Pinto
e,
Rui A. R.
Boaventura
ab,
Carlos J.
Tavares
c,
Vítor J. P.
Vilar
*ab and
Francisca C.
Moreira
*ab
aLSRE-LCM – Laboratory of Separation and Reaction Engineering – Laboratory of Catalysis and Materials, Faculty of Engineering, University of Porto, Rua Dr Roberto Frias, 4200-465 Porto, Portugal. E-mail: vilar@fe.up.pt; franciscam@fe.up.pt; Tel: +351918257824 Tel: +351 914332022
bALiCE – Associate Laboratory in Chemical Engineering, Faculty of Engineering, University of Porto, Rua Dr Roberto Frias, 4200-465 Porto, Portugal
cCF-UM-UP – Centre of Physics of Minho and Porto Universities, University of Minho, 4804-533 Guimarães, Portugal
dLEPABE – Laboratory Process Engineering, Environment, Biotechnology and Energy, Faculty of Engineering, University of Porto, Rua Dr Roberto Frias, 4200-465 Porto, Portugal
eRAIZ – Forest and Paper Research Institute, Quinta de São Francisco, 3800-783 Aveiro, Portugal
First published on 1st May 2025
Abstract
The pulp and paper industry generates large quantities of black liquor (BL), a lignin-rich waste stream with the potential to be converted into value-added products, such as low-molecular-weight phenolic products (LMPPs). This study investigates the use of photoelectrocatalysis (PEC) as a promising, yet unexplored, process for lignin oxidation to produce LMPPs. For the first time, a photoanode based on niobium-doped titanium dioxide (TiO2:Nb) and a photoelectrochemical flow cell were applied. Lignin was isolated from Kraft BL (KBL) and used to prepare alkaline Kraft lignin (KL) solutions. The lignin and TiO2:Nb photoanode were thoroughly characterized before and after the PEC process. Six LMPPs, including aldehydes, aromatic acids, and ketones, were identified and quantified during the lignin photoelectrocatalytic oxidation. Applying a constant cell potential of ∼0.8 V transformed the lignin structure, yielding a maximum of 26 ± 1 mg kg−1 of LMPPs after 7 h of reaction and a consumed charge of 5 C gLignin−1. The photoelectrocatalytic oxidation of KL predominantly yielded aromatic acids.
Green foundation1. This work advances green chemistry by introducing a sustainable photoelectrocatalytic approach to valorize lignin, a major waste from the pulp and paper industry. Using a niobium-doped titanium dioxide photoanode and a custom-designed flow cell, lignin was converted into low-molecular-weight phenolic products under mild conditions, promoting waste reduction, resource efficiency, and renewable chemical pathways.2. Quantitatively, this process yielded up to 26 mg kg−1 of phenolic products, primarily aromatic acids, after 7 hours at ∼0.8 V and 5 C gLignin−1. Qualitatively, it represents a sustainable, innovative process for valorizing lignin-rich waste, promoting renewable chemical production and reducing environmental impact. 3. This work can be made greener and elevated by further research by optimizing the photoanode material, improving the reactor design, integrating renewable energy, scaling up the process, and enhancing downstream separation and purification of products. |
1. Introduction
Black liquor (BL) is the major by-product of the pulp and paper industry, where the Kraft process holds 90% of the total production capacity. It contains large amounts of dissolved lignin, together with other organic and inorganic compounds. BL has been concentrated in multi-stage evaporation units and then burned as a low-value fuel to generate electricity, produce heat, and recover cooking chemicals, neglecting the potential of its high lignin content.1,2Lignin is composed of three monomeric structures (p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol) randomly linked through ether (C–O–C) and carbon–carbon (C–C) bonds.3,4 These monomers are the precursors of the primary moieties in the lignin backbone: guaiacyl (G), syringyl (S), and p-hydroxyphenyl (H) units. The mixture of S and G units gives rise to different types of lignin: softwoods (S units), hardwoods (S and G units), and grasses (all three units, with higher production of H units). Lignin also contains various functional groups, such as methoxy, carbonyl, and hydroxyl along with a high C/O ratio.5 All these properties make lignin a great potential feedstock for producing fuel and value-added compounds, such as low-molecular-weight phenolic products (LMPPs).6,7
LMPPs are often reported as major lignin depolymerization products. The most important LMPPs derived from lignin are vanillin (V) and syringaldehyde (Sy),8–10 commonly used as flavoring and fragrance ingredients with important applications in the food and cosmetics industries. They are also widely used in organic synthesis and pharmaceutical applications.9 Their respective acids (vanillic acid – VA, and syringic acid – SA, respectively) and ketones (acetovanillone –VO, and acetosyringone – SO, respectively) are also of interest. They have also been widely used in the chemical and pharmaceutical businesses as intermediary compounds to produce high-value-added compounds.11,12 Moreover, VA and SA have been used as therapeutic agents while possessing antioxidant, antimicrobial, and anti-inflammatory effects.12,13
Several processes have been investigated for the production of value-added compounds from lignin, including alkaline oxidation,10,14,15 hydrogenolysis,16,17 and photocatalysis (PC).18 Alkaline oxidation of lignin with oxygen (O2) as an oxidant is one of the most extensively studied processes for the effective generation of aldehyde products with high yields. However, this process resorts to harsh reaction conditions, namely high temperatures and pressures. The photoelectrocatalysis (PEC) process is a very promising alternative approach because (i) it requires much milder reaction conditions than traditional chemical processes, allowing for operation at ambient pressure and room temperature, (ii) the generation of oxidants (electrons) does not require external high-value reagents, (iii) it is typically low energy consuming, and (iv) it prevents the recombination of the electron/hole (e−/h+) pairs generated by PC since PEC refers to the combination of the PC and electrocatalysis (EC) processes in a standalone device.3,19,20 Photoelectrocatalytic lignin depolymerization is, however, an understudied process. The current research in this field faces several limitations, including: (i) a very limited range of photoanode materials tested, (ii) exclusive reliance on batch electrochemical cells, (iii) the predominant use of lignin model compounds rather than raw lignin, (iv) a lack of quantification for LMPPs, and (iv) a primary focus on ketone production. The effect of charge (constant current density, j, or applied cell potential, Ecell) is also not usually researched for lignin depolymerization by the PEC process.
This work aims to assess the feasibility of applying an innovative photoelectrocatalytic technology for lignin oxidation to produce value-added LMPPs based on a novel niobium-doped titanium dioxide (TiO2:Nb) photoanode and a customized commercial photoelectrochemical flow cell back-side illuminated (BSI) by an ultraviolet A-light-emitting diode (UVA-LED) system. Lignin was isolated from Kraft BL (KBL) obtained from a Portuguese pulp mill and used to prepare alkaline Kraft lignin (KL) solutions, which were then fed into the photoelectrocatalytic cell. Seven phenolic compounds (p-hydroxybenzaldehyde – pHy, syringaldehyde – Sy, vanillin – V, syringic acid – SA, vanillic acid – VA, acetosyringone – SO, and acetovanillone – VO) were identified and quantified during the PEC experiments. The PEC process was carried out at distinct Ecell and compared to the PC and EC processes. The lignin and the TiO2:Nb photoanode were thoroughly characterized both before and after photoelectrocatalytic oxidation. To the best of our knowledge, this is the first time that lignin oxidation by a PEC process has been reported using a TiO2:Nb photoanode and a photoelectrochemical flow cell. The selection of TiO2:Nb as the photoanode stemmed from preliminary tests in our research group involving various photoanode materials (e.g., hematite – α-Fe2O3, aluminum doped zinc oxide – ZnO:Al, gallium doped zinc oxide – ZnO:Ga, antimony doped zinc oxide – ZnO:Sb, tantalum nitride – TaN, strontium titanite doped with ruthenium – SrTiO3:Ru), with TiO2:Nb consistently demonstrating superior stability, activity, and/or selectivity (data not reported).
2. Materials and methods
2.1. Chemicals
Sulfuric acid (H2SO4) with 96% (w/w) purity from Panreac was used to prepare diluted solutions for acidic lignin precipitation. Nitrobenzene (C6H5NO2) with ≥99% (w/w) purity supplied by Merck was used for nitrobenzene oxidation (NO) of lignin samples. Sodium hydroxide (NaOH) with ≥99% (w/w) purity from Merck was used for lignin dissolution. Chloroform (ChCl3) with 99.0–99.4% (w/w) purity supplied by VWR Chemicals was used for liquid–liquid extraction (LLE). Oxalic acid dihydrate (C2H2O4·2H2O) with 99.5–102.5% (w/w) purity and methanol (MeOH, CH4O) with ≥99.8% (w/w) purity (HiPerSolv CHROMANORM®) from PanReac and VWR Chemicals, respectively, were used as eluent solutions in high-performance liquid chromatography (HPLC) analyses. MeOH was also used as a solvent for LLE and solid-phase extraction (SPE). The standards p-hydroxybenzaldehyde (pHy, C7H6O2) with ≥95% (w/w) purity, vanillic acid (VA, C8H8O4) with ≥98% (w/w) purity, syringic acid (SA, C9H10O5) with 98% (w/w) purity, vanillin (V, C8H8O3) with 99% (w/w) purity, syringaldehyde (Sy, C9H10O4) with 98% (w/w) purity, acetovanillone (VO, C9H10O3) with 98% (w/w) purity, and acetosyringone (SO, C10H12O4) with 97% (w/w) purity were supplied by either Thermo Scientific, Merck or Acros Organics. Demineralized water was obtained from a reverse osmosis system (Panice). Ultrapure water was obtained from a Millipore® Direct-Q system (18.2 MΩ cm−1 resistivity at 25 °C).Fluorine-doped tin oxide (FTO) glass substrates (soda-lime type, TEC-7–7 Ω sq−1, 2.2 mm thickness) were supplied by Greatcell Solar Materials Pty Ltd. The glass composition was ∼70% silicon dioxide (SiO2), ∼9% calcium oxide (CaO), and ∼4% magnesium oxide (MgO), among other minor constituents. Derquim LM 01 alkaline detergent from Panreac and ethanol (EtOH, C2H6O) with 70% (v/v) purity from VWR were used for glass substrate cleaning. A conductive silver paste (Ferro GmbH GSSP SP 1963) containing 50–80% iron (Fe) was used to allow the electrical conduction from the FTO thin film to the non-conductive glass-side. A titanium![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :niobium (Ti:Nb) target (96:4 wt%) with 99.9% (w/w) purity from FHR Anlagenbau GmbH was used for the magnetron sputtering deposition of a TiO2:Nb thin film onto FTO glass substrates. Research grade argon (Ar) with 99.999% (w/w) purity and oxygen (O2) with 99.999% (w/w) purity, both from Air Liquid, were used as working and reactive gas, respectively, for the deposition of TiO2:Nb thin films.
:niobium (Ti:Nb) target (96:4 wt%) with 99.9% (w/w) purity from FHR Anlagenbau GmbH was used for the magnetron sputtering deposition of a TiO2:Nb thin film onto FTO glass substrates. Research grade argon (Ar) with 99.999% (w/w) purity and oxygen (O2) with 99.999% (w/w) purity, both from Air Liquid, were used as working and reactive gas, respectively, for the deposition of TiO2:Nb thin films.
2.2. Lignin: source, isolation, and characterization
Industrial Eucalyptus globulus (hardwood) KBL was collected from a Portuguese pulp mill at the outlet of the Kraft digester. Lignin was isolated from KBL by slow acidification with H2SO4:H2O 1:1 (v/v) up to pH 4–5 under agitation and at low temperature (10 °C). The precipitated KL was recovered by centrifugation (10 min at 3500 rpm), washed two times with demineralized water, and freeze-dried for at least 48 h.
2.2.2.1. Gravimetric analysis for inorganic content determination. The inorganic content of KL was determined by gravimetric quantification after incineration at 600 °C for 6 h of 100 mg of KL.21 This determination was performed in duplicate. The relative error was lower than 2%.
2.2.2.2. Nitrobenzene oxidation for low-molecular-weight phenolic product determination. KL was subjected to NO to determine the LMPPs as already described elsewhere.22 Briefly, 30 mg of isolated freeze-dried lignin was dissolved in 7 mL of 2 M NaOH aqueous solution in a Teflon vessel. A volume of 0.45 mL of nitrobenzene was added to the reaction mixture and heated up to 170 °C for 4 h in a stainless-steel reactor. The oxidized cooled-down lignin was subjected to LLE with chloroform, followed by acidification with H2SO4
:H2O 1:1 (v/v) and a second LLE to recover the LMPPs. The resulting organic phase was evaporated under reduced pressure (Bibby rotary vacuum evaporator, model Re 100), and the dried sample was dissolved in 10 mL of MeOH. The solution was then analyzed by HPLC following the procedure provided in the ESI.† NO and HPLC analyses were performed in duplicate.
2.2.2.3. Fourier-transform infrared spectroscopy for lignin functional group determination. KL (before and after oxidation) was characterized by universal attenuated total reflectance Fourier-transform infrared spectroscopy (UATR-FT-IR) analysis to detect the functional groups of lignin. A PerkinElmer Spectrum Two™ FT-IR Spectrometer equipped with a deuterated triglycine sulfate (DTGS) detector was used, and a diamond plate accessory was employed during measurements. The FT-IR spectrum was recorded between 4000 cm−1 to 500 cm−1 wavenumbers at 4 cm−1 resolution and 16 scans per sample.
2.3. Fabrication and characterization of photoanodes
:Nb with 10 cm diameter was used in a planar configuration. The chamber was evacuated prior to the depositions with a primary rotary pump and a turbo molecular pump to achieve a base pressure of around 10−4 Pa. The target–substrate distance was kept at 6.5 cm, and the substrate holder was heated up to 430 K. Ar was used as working gas to enable plasma formation, and O2 was used as reactive gas at a flow rate of 7 sccm. Before the deposition, 2 min of etching at 500 V was performed under an Ar atmosphere at a pressure of 1.8 Pa in continuous rotation. This process removes oxides and impurities possibly accumulated and retained on the surface of the substrates and creates atomic defects in the substrate, enabling better nucleation of the film during deposition. This was followed by a dummy deposition of 2 min for target cleaning (burn). The films were deposited for 5 min at a pressure of around 0.22 Pa, with a target current density fixed at 12.7 mA cm−2, a bias voltage of −60 V, and an Ar flow of 40 sccm. These deposition conditions enable an effective ∼2 at% Nb substitutional doping in TiO2. The substrate holders were mounted onto a rotating 6-position support controlled by a motor. Four samples were deposited in a single batch.
2.3.3.1. Scanning electron microscopy combined with energy-dispersive X-ray spectroscopy. An FEI Nova SEM 200 scanning electron microscope (SEM) in combination with an energy-dispersive X-ray spectrometer (EDX) was used at the Materials Characterization Services (CEMAT), University of Minho, Portugal, to evaluate the morphology and elemental composition of the TiO2:Nb thin films. EDX data were obtained using an accelerating voltage of 15 kV with a scanning resolution of 129 eV for mapping and local analysis using the standardless ZAF quantification method. The obtained data were analyzed using EDAX Genesis APEX 2i software.
2.3.3.2. X-ray diffraction. X-ray diffraction (XRD) analysis was performed to investigate the crystallographic structure of the TiO2:Nb thin films using a Bruker AXS D8 advanced Discovery diffractometer at the CEMAT, University of Minho, Portugal. The diffraction patterns were acquired using grazing incidence X-ray diffraction (GIXRD), with an incident angle of 1°, a step of 0.02°, and an integration time of 1 s. CuKα radiation was used with a wavelength (λ) of 1.5406 Å.
2.3.3.3. X-ray photoelectron spectroscopy. X-ray photoelectron spectroscopy (XPS) was used to evaluate the surface composition of the film at the Materials Research Center (CENIMAT-i3N), NOVA University of Lisbon, Portugal. The experiments were performed using a Kratos Axis-Supra instrument equipped with monochromatic Al-Kα radiation (1486.6 eV). A co-axial electron neutralizer was used to minimize surface charging. The photoelectron spectra were collected from a take-off angle of 90° relative to the sample surface. The measurement was done in a constant analyzer energy lens mode (CAE) with a pass energy of 160 eV for the survey spectra and 40 eV for the high-resolution spectra. Emission currents of 15 mA and 20 mA were used for the survey spectra and high-resolution spectra, respectively. Charge referencing was done by setting the lower binding energy of the C 1s hydrocarbon peak (the most intense component of the C 1s spectrum) at 284.8 eV. Unifit software was used to fit the recorded experimental XPS spectra.
2.3.3.4. Optical properties. Measurements of optical transmittance and reflectance were carried out using a Shimadzu UV-2501 PC spectrophotometer in the 200 nm to 900 nm λ range. From these measurements, the absorbance coefficient (α, in m−1) was calculated as follows:26
 | (1) |
The indirect optical band-gap energy (Eg) was obtained from the Tauc plot by extrapolating to zero the dependence of eqn (2):27,28
| (αE)1/pT = B(E − Eg) | (2) |
| E = hv | (3) |
2.4. Experimental setup
2.4.1.1. Lignin. Cyclic voltammetry (CV) analysis was carried out in a three-electrode glass cell to infer about the lignin susceptibility for oxidation. The cell was composed of a 3 mm diameter freshly polished glassy carbon electrode (Chemical Instruments) as the working electrode, a 99.9% pure platinum (Pt) wire (Alfa Aesar®) as the counter-electrode, and a silver/silver chloride electrode in saturated potassium chloride solution (Ag/AgCl/sat. KCl) (Metrohm) as the reference electrode. The measured potential vs. Ag/AgCl/sat. KCl was converted into the reversible hydrogen electrode (RHE) potential scale following the Nernst equation (eqn (4)).31 Tests were performed at room temperature in the dark using a ZENNIUM workstation (Zahner Elektrik) controlled by Thales software package (Thales Z 2.0). CVs were recorded at a scan rate of 100 mV s−1 using 1 M NaOH (pH = 13.2) and aqueous lignin solutions containing 1, 10, 30, and 60 g L−1 lignin (in 1 M NaOH, pH = 13.2).
| ERHE = EAg/AgCl/sat.KCI + 0.059pH + 0.197. | (4) |
2.4.1.2. TiO2:Nb photoanodes. Linear-sweep voltammetry (LSV) analysis was conducted on a photoelectrocatalytic cell device, known as the “Cappuccino” cell,32 to characterize the fabricated TiO2:Nb photoanodes. A standard three-electrode configuration was employed comprising the prepared TiO2:Nb photoanodes (1.2 × 3.0 cm2) as the working electrode, a 99.9% pure Pt wire (Alfa Aesar®) as the counter electrode, and an Ag/AgCl/sat. KCl (Metrohm) as the reference electrode. The measured potential vs. Ag/AgCl/sat. KCl was converted into the RHE potential scale following the Nernst equation (eqn (4)). The active photoanode surface area was 0.283 cm2, defined by an internal mask. (Photo)current density–voltage (j–V) characteristic curves were obtained by applying an external potential bias to the cell and measuring the generated (photo)current. Data were recorded in an AUTOLAB electrochemical station (Methrom, PGSTAT302N) at a scan rate of 10 mV s−1 using 1 M NaOH (pH = 13.2), and aqueous lignin solutions containing 1, 10, 30, and 60 g L−1 lignin (in 1 M NaOH, pH = 13.2). The measurements were performed at room temperature in the dark and under illumination using a UVA-LED illumination system that provides an intense emission band centered at 365 nm (L14310-100 LED-head unit – Lightningcure® LC-L1, Hamamatsu Photonics; standard irradiance of 1000 mW cm−2 at 1 cm from the lens tip). The photoanode was BSI, i.e., the light passes first through the FTO glass and then through the TiO2:Nb thin film, avoiding absorption losses from the electrolyte in front-side illuminated (FSI) configurations. The distance between the “Cappuccino” cell and the UVA-LED system was adjusted to produce the same photocurrent as in the two-electrode PEC system employed for lignin oxidation (Section 2.4.2.) (11.8 cm vs. 8.1 cm, respectively).
 | ||
| Fig. 1 (a) Stainless-steel current collector of e−Cell, and (b) scheme of the experimental setup for the PEC process. | ||
The e−Cell reactor was incorporated into a system (Fig. 1b) containing: (i) a recirculation glass vessel thermostatically controlled and magnetically stirred, (ii) a refrigerated heating circulating bath (Grant, model ecocool 150R) for temperature control of solution in the recirculation glass vessel, (iii) a gear pump (model BVP-Z, pump head Ismatec Z-142), (iv) a Velleman® power supply (model LABPS3005DN, 0–5 A, 0–30 V), (v) a digital multimeter (Range®, model RE64), and (iv) the UVA-LED illumination system (L14310-100 LED-head unit – Lightningcure® LC-L1, Hamamatsu Photonics; standard irradiance of 1000 mW cm−2 at 1 cm from the lens tip). The cell-to-LED distance was 8.1 cm, enabling full photoanode illumination with an irradiance of 120 ± 10 mW cm−2 (radiant power of 1.2 ± 0.1 W), determined by ferrioxalate actinometry35 with Fe3+ = 10 mM. All the system components were connected using PTFE tubing.
KL was dissolved in 1 M NaOH aqueous solution to a final concentration of 60 g L−1 (pH = 13.2). A volume of 200 mL of this KL solution was employed, resulting in a photoanode active surface area-to-solution volume ratio of 5.0 m2 m−3. The system operated at a Reynolds number (Re) of 1750 (flow rate = 75.5 L h−1) to overcome mass transfer limitations.33,34 Photoelectrocatalytic lignin oxidation was performed at constant Ecell and room temperature (controlled). No pH control was performed. A control sample was taken after placing the KL solution in the recirculation glass vessel and allowing it to reach 25 ± 1 °C. The beginning of the reactions was marked by the switching on of the power supply and/or the illumination system. Samples were taken at predetermined time intervals up to 7 h. Experiments were carried out in duplicate.
Two additional trials were performed: a PC experiment, which only involves the illumination of the TiO2:Nb photoanode (here functioning as the photocatalyst; no application of Ecell); and an EC experiment, which only entails the application of a constant Ecell (no illumination). Furthermore, a blank trial without the application of Ecell or illumination was also conducted.
2.5. Analytical determinations
HPLC, pH, temperature, and viscosity determinations are described in the ESI.†2.6. Calculations
The efficiency of the photoelectrocatalytic oxidation of lignin was evaluated in terms of yield of LMPPs and selectivity to V and Sy. The yield of a given LMPP was determined on a mass basis (mg kg−1) viaeqn (5).14 Selectivity to V and Sy was expressed as the mass ratio of aldehyde/aromatic acid (eqn (6)).10,14 | (5) |
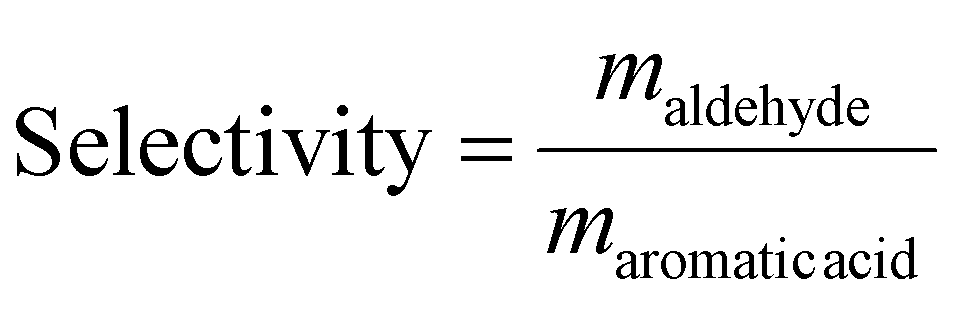 | (6) |
The consumed specific charge was calculated according to eqn (7) (in C gLignin−1) and eqn (8) (in Ah L−1):36
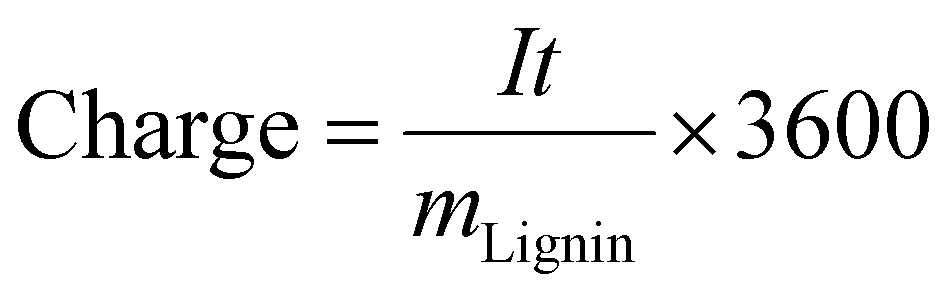 | (7) |
 | (8) |
The energy consumption for electrical current supply (ECo., in kW h m−3) for the e−Cell reactor operation was calculated from eqn (9):37
 | (9) |
The apparent quantum yield (AQY, in %) was calculated according to eqn (10),38 considering the generated LMPPs since the stoichiometry of lignin oxidation to the different LMPPs is unclear.
 | (10) |
3. Results and discussion
3.1. Lignin characterization
:H:S provides the basic information about the nature of the lignin. The yields and types of the LMPPs obtained by NO are presented in Table 2.
| LMPPsa | ||||||||
|---|---|---|---|---|---|---|---|---|
| pHy | VA | SA | V | Sy | VO | SO | Total yield | |
| a Reported on dry weight and corrected to nonvolatile solids weight after deducing inorganics content; pHy – p-hydroxybenzaldehyde; VA – vanillic acid; SA – syringic acid; V – vanillin; Sy – syringaldehyde; VO – acetovanillone; SO – acetosyringone. | ||||||||
| mg kg−1 | 10 × 101 | 1.9 × 103 | 1.3 × 104 | 1.7 × 104 | 6.9 × 104 | 9.0 × 102 | 2.4 × 103 | 1.0 × 105 |
The obtained results are in agreement with the expected ones for hardwood KL, which yields mainly S-type units along with some G-type ones.10,14 The low NO yields of LMPPs obtained are indicative of a lignin with a highly condensed structure, and a large number of C–C bonds,43 which are more difficult to break than C–O bonds.4,44–46 This might be an indication that the KL used in this work was difficult to depolymerize. Nevertheless, the obtained NO yields for LMPPs can be very variable (Table SI-1†) due to the different feedstocks, pulping processes, lignin heterogeneities, and isolation procedures, as also reported by Villar et al.47 The obtained NO yields for KL are however within the expected range for most LMPPs.
 | ||
| Fig. 2 FT-IR spectra of Kraft lignin before (black line) and after (pink line) photoelectrocatalytic oxidation at ∼0.8 V using TiO2:Nb photoanodes for 7 h. Each colored band represents a different region for the occurrence of specific chemical bonds.48–50 | ||
The characteristic peaks of lignin are related to aromatic- and ether-containing structures: aliphatic and phenolic OH-groups (3385 cm−1),51 C–H stretching in methyl groups of the side chain (2939 cm−1),9,11,51–54 and aromatic ring vibrations of the phenyl-propane skeleton (1598 cm−1, 1517 cm−1, and 1424 cm−1).9,49,52,53,55–57 No bands were found in the range of 1740–1655 cm−1, which originate from the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching in unconjugated ketones, carbonyls and ester groups, and conjugated aldehydes and carboxylic acids.49,53,55,57,58
O stretching in unconjugated ketones, carbonyls and ester groups, and conjugated aldehydes and carboxylic acids.49,53,55,57,58
Characteristic vibrations of S-type units were found at 1329 cm−1 (ref. 53–55, 57 and 58) and 1111 cm−1 (ref. 53–55 and 58) G-type units were found alongside S-type units at 1216 cm−1 (ref. 9, 55 and 57) and 828 cm−1 (ref. 9, 11, 53, 55, 56 and 58). This is consistent with the predominance of S-type units observed for NO in section 3.1.2. There was also contamination detection, revealed by the residual content of sugars at 1043 cm−1, which is assigned to C–O stretching in cellulose, hemicellulose, and lignin or C–O–C stretching in cellulose and hemicellulose.56,57
From the recorded spectra, it can be concluded that the KL is mainly composed of S-type units, along with some G-type units. The main functional groups identified in the lignin structure were hydroxyl and methyl.
After lignin photoelectrocatalytic oxidation, the analysis of the FT-IR spectrum suggests that the lignin structure underwent complex chemical transformations that involved the modification and/or removal of –OH and C–O bonds, and structural rearrangement of lignin units (S-type and G-type), especially S-type ones. These are perceptible by analyzing the bands: (i) disappearance at 3385 cm−1 and 1216 cm−1, (ii) appearance at 996 cm−1, and (iii) intensity increase at 1111 cm−1. Furthermore, the absence of a peak at ∼1700 cm−1 suggests that lignin was not transformed into CO or carbonyl functional groups,59,60 or that only low contents were produced.
3.2. TiO2:Nb photoanode characterization
3.2.1.1. Morphological and textural characteristics (SEM-EDX). Fig. 3a.1, 2, 3b.1 and 2 present the SEM micrographs of TiO2:Nb thin films on FTO glass substrates, with the corresponding elemental mapping from EDX presented in Fig. 3a.3, 3b.3, and Table 3.
 | ||
| Fig. 3 SEM-EDX analysis of the (a) unused and (b) used TiO2:Nb thin films. | ||
| Sample | O (at%) | Sn (at%) | Ti (at%) | Si (at%) | Nb (at%) | C (at%) | Mg (at%) | Al (at%) |
|---|---|---|---|---|---|---|---|---|
| TiO2:Nb unused | 58.5 | 26.7 | 12.2 | 1.9 | 0.7 | 0 | 0 | 0 |
| TiO2:Nb used | 51.2 | 8.5 | 3.6 | 0 | 0.4 | 23.9 | 10.4 | 2.0 |
Before the utilization of TiO2:Nb photoanodes for lignin photoelectrocatalytic oxidation, the SEM micrographs show a well-adhered and dense semiconductor film with a rather smooth texture and relatively uniform distribution with particle sizes below 1 μm. The EDX results confirm that the primary elemental constituents are oxygen (O), tin (Sn), and titanium (Ti). Silicon (Si) and Nb are also found. The presence of Si may originate from the FTO glass substrate, possibly due to the beam spot incident on the sample edge of the FTO glass substrate during the EDX analysis.25 The presence of Sn is likely attributed to the FTO coating itself. From the EDX analysis, 12.2 at% Ti and 0.7 at% Nb are detected, which is in agreement with other similar depositions,25 and within the measurement error of Ti(96):Nb(4) wt% (2.1 at% of Nb).
The used TiO2:Nb thin film has a different morphology. This is probably the result of the deposition of an organic film that mainly contains carbon (C) and magnesium (Mg) elements (Table 3), which probably resulted from lignin deposition or its LMPPs. Carbon comes from the aromatic ring structure,61 while inorganic elements such as Mg and aluminum (Al) are present in the wood chips and dissolved into the KL.62 This also explains the decrease in the quantification of elements with film usage: the deposition of an organic film hinders the quantification of the underneath film. The deposition of lignin and/or lignin derivatives on the TiO2:Nb surface led to the formation of aggregated particles (average particle size >2 μm), as seen on the SEM micrograph. Oxygen remained stable because an oxidation process took place, which also led to the deposition of this element. The absence of silver (Ag) indicates that the painted conductive silver paste (section 2.3.1.) did not suffer any abrasion and remained stable.
3.2.1.2. Crystallographic structure (XRD). XRD data presented in Fig. 4 indicate no apparent differences between unused and used TiO2:Nb thin films on the FTO glass substrates, confirming that both materials are composed of FTO and TiO2 in rutile and anatase phases. The highest intensity diffraction peaks are all related to FTO: (211) at 2θ = 51.6°, (200) at 2θ = 37.8°, (101) at 2θ = 33.6°, and (110) at 2θ = 26.5°. The presence of reflections from both (110) rutile and (101) anatase phases is expected, as previously reported by Ribeiro et al.25 for reactive DC magnetron sputtering of Ti
:Nb (96:4 wt%) at a 6.5–7.5 sccm of reactive oxygen flow rate.
 | ||
| Fig. 4 XRD for the unused and used TiO2:Nb thin films. The XRD patterns (upper graph) were compared with those of TiO2 rutile (ICDD PDF card 00-021-1276), TiO2 anatase (ICDD PDF card 00-064-0863), and SnO2 (FTO, ICDD PDF cards 00-046-1088 and 01-070-6995). Dotted values indicate experimental data and lines indicate Rietveld refined data. | ||
Furthermore, XRD patterns reveal a preferred orientation, which agrees with that obtained from the Rietveld refinement (Table SI-3†) for the unused TiO2:Nb thin film. The volume and the lattice parameters of the TiO2:Nb unit cell parameters did not change significantly for the unused and used film. However, the Rietveld refinement shows less crystalline rutile peaks, and no anatase phase is discerned. The average crystallite size increased 3-fold after using the semiconductor film (50 nm vs. 150 nm). All these results corroborate that the deposition of an organic layer may be occurring.
3.2.1.3. Surface composition (XPS). To further investigate potential surface modifications, XPS analysis was conducted for the unused (Fig. 5) and used (Fig. 6) TiO2:Nb thin films on the FTO glass substrates. It allows the determination of the chemical and structural information of the TiO2:Nb film composition. Elemental composition quantification (Table 4) was derived from the XPS fits for the O 1s and C 1s singlets, and Nb 3d and Ti 2p doublet core levels with the respective fitting parameters listed in Tables SI-4, SI-5, SI-6, and SI-7,† respectively. For the used TiO2:Nb thin film, XPS fits were not applicable to the doublets Ti 2p and Nb 3d, which did not allow for its quantification. The Lorentzian–Gaussian (L–S) relative profile was considered for the XPS fits in doublet peaks. Si traces were detected in the survey spectra due to the incidence of the beam spot on the sample border, as previously highlighted.
 | ||
| Fig. 5 XPS spectra with the respective survey scan and XPS fits for the C 1s and O 1s core lines for the unused TiO2:Nb thin film. | ||
 | ||
| Fig. 6 XPS spectra with the respective survey scan and XPS fits for the C 1s and O 1s core lines for the used TiO2:Nb thin film. | ||
| Sample | C (at%) | O (at%) | Ti (at%) | Nb (at%) | Si (at%) |
|---|---|---|---|---|---|
| TiO2:Nb unused | 59 | 30 | 5.0 | 0.3 | 4.9 |
| TiO2:Nb used | 63 | 37 | 0 | 0 | 0.1 |
For the unused TiO2:Nb thin film, the quantification of the elemental composition derived from the XPS fits (not considering C) for the Ti 2p, O 1s and Nb 3d core levels were as follows: 73 at% for O, 12% for Ti, and 0.7% for Nb. An over-stoichiometry in oxygen occurred for the TiO2:Nb thin film (1:6), explaining the higher quantification of oxygen than expected (73% vs. 66%).25
On analyzing the O 1s core level, the fit of the TiO2:Nb thin films included three components – C1, C2, and C3 (Table SI-4,† and Fig. 5 and 6). For the unused film, these are associated with Ti–O bonds (C1), Nb–O bonds and defective oxygen (vacancies) (C2), and surface adsorbed oxygen and hydroxide impurities (C3).25 For the used film, the contribution C1 (531.6 eV) is attributed to organic CO bonds, C2 (532.5 eV) is ascribed to aliphatic CO bonds or esters (C–O–C), and C3 (533.6 eV) is associated with aromatic and aliphatic CO–O* bonds.63 These organic bonds indicate surface-adsorbed organic compounds, probably lignin along with LMPPs. Contributions C2 and C3 (532–534 eV) can also be related to surface-adsorbed oxygen and hydroxide, and metal carbonates. Surface-adsorbed species such as Ti–OH might have been adsorbed from water, and metal carbonates from CO2 reaction with air.64 The area ratio between C1 and C2 is approximately 60:40% for the unused TiO2:Nb thin film, and it increases to almost 70:30% for the used film.
A detectable quantity of C 1s arises from adventitious carbon contamination, which results from sample exposition to the atmosphere and is a commonly used charge reference for XPS spectra. Although carbon can also come from polymer compounds, core binding energies for adventitious carbon contamination (284.8–288.5 eV) and carbon polymers (284.8–290 eV) are very close.65 Hence, a definitive identification of the carbon species cannot be determined.
For the unused TiO2:Nb thin film, a spin–orbital (L–S) splitting with 2.8 eV of width is observed for the Nb 3d core level (Table SI-6†), which is in agreement with the literature.25,66 The peak position of Ti 2p3/2 in Fig. 5 and Table SI-7† corresponds to that of the Ti4+ oxidation state.66,67 The shape of the Ti 2p excludes the presence of traceable amounts of Ti3+, Ti2+, and Ti0,68 as no other peaks are perceptible. The binding energies associated with Ti 2p (458–462 eV; Table SI-7†) and Nb 3d (207–213 eV; Table SI-6†) are indicative of Ti–O and Nb–O bonds, respectively. No Ti–Ti or Nb–Nb metal bonds are present, as no binding energies at 454.1 eV and 202.4 eV, respectively, occur (Fig. 5 and 6, spectra).65 This indicates that the TiO2 films were effectively doped with Nb. After the use of TiO2:Nb in photoelectrocatalytic lignin oxidation, the metal elements (Ti, Nb, and Si) were not possible to be quantified, which indicates the deposition of a layer, probably lignin and/or lignin LMPPs. Moreover, Mg and calcium (Ca) traces were noticed (Fig. 6, survey), which could have arisen from lignin itself. As the wood chips can contain inorganic species, these might not be completely removed upon lignin precipitation and washing.62
3.2.1.4. Optical properties. Fig. 7 presents the UV-Vis spectrometry, and the respective Tauc plot for the TiO2:Nb thin films on the FTO glass substrates before and after usage for photoelectrocatalytic oxidation of lignin. The film thickness was determined to be around 300 nm.25
 | ||
| Fig. 7 Representations of the (a) optical transmittance (solid lines) and reflectance (dashed lines) for unused (light green) and used (dark green) TiO2:Nb thin film photoanodes over the wavelength (λ), and (b) Tauc plots for the optical band-gap energy (Eg) determination for (b.1) unused and (b.2) used TiO2:Nb thin film photoanodes, respectively, with 300 nm thickness. | ||
The semiconductor film not subjected to lignin photoelectrocatalytic oxidation presents higher values of both transmittance and reflectance when compared to the used material by 1.3- and 1.5-fold, respectively. The observed reflectance signal oscillations arise from the multiple internal light reflections in the film, which is an effect of the film thickness.25
For λ = 365 nm, transmittance and reflectance values of 17% and 12% are obtained for the unused TiO2:Nb thin film, respectively, and of 9% and 12% for the used one, respectively. Overall, the higher absorption capability (and lower transmittance) of the used TiO2:Nb thin film agrees with the results obtained in the previous sections, where there has been an indication of film adsorption on the TiO2:Nb surface. These differences lead to the consideration that that there are changes in the α value and, as a result, in the Eg value (derived from Tauc plots – Fig. 7b). As expected, a higher Eg value is obtained for the unused TiO2:Nb thin film (2.95 ± 0.08 eV) when compared to the used one (2.56 ± 0.01 eV). However, the attained Eg value is far from the expected value of ∼3.24 eV reported by Ribeiro et al.25 for the as-deposited TiO2:Nb thin film with a 7.0 sccm oxygen flow rate for 5 min. As the Eg values for the TiO2 phases are different (3.2 eV for anatase and 3.0 eV for rutile),69 this observed difference could be explained by the diffraction peaks identified in section 3.2.1.2. As ∼41 wt% of rutile is observed vs. only ∼5 wt% of anatase, an Eg value closer to that of rutile is expected. Ribeiro et al.25 observed that increasing reactive oxygen flow rates improved the Eg value due to enhancement of the anatase phase crystallization, which was not observed here.
Upon the use of the TiO2:Nb thin film, the Eg value significantly decreased by 0.39 eV (2.56 ± 0.01 eV – Fig. 7b.2). This band-gap narrowing could be due to carbon adsorption on the surface, and incorporation into the titania lattice, shifting the conduction band (CB) of TiO2 and introducing surface states near the valence band (VB) of the semiconductor, leading to a reduced band-gap.70–72
3.2.2.1. Lignin. Fig. 8 shows the CV analysis conducted in a three-electrode glass cell equipped with a glassy carbon working electrode to study the electrochemical behavior of lignin aqueous solutions (0 g L−1 to 60 g L−1 in 1 M NaOH).
 | ||
| Fig. 8 Cyclic voltammograms acquired in a three-electrode glass cell with a glassy carbon electrode acting as the working electrode at a scan rate of 100 mV s−1 for (a) 1 M NaOH (pH = 13.2) and 1 g L−1 lignin (pH = 13.2) aqueous solutions, (b) lignin aqueous solutions at different concentrations (pH = 13.2), and (c) 60 g L−1 lignin (pH = 13.2) aqueous solution over 10 consecutive cycles. | ||
The wide CV scan displayed in Fig. 8a allows the identification of the onset potentials for the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER): ∼1.60 VRHE and ∼0.60 VRHE, respectively. This agrees with the reported bibliography on glassy carbon electrodes tested in highly-alkaline environments.73 An anodic peak (b1) can be observed in the absence of lignin at ∼1.40 VRHE (with onset at ∼1.30 VRHE). This signal can be associated with carbon oxidation,74 but given its small peak j, it did not significantly interfere in the attempts to identify other oxidation reactions. After adding 1 g L−1 lignin, an oxidation peak appeared at ∼1.20 VRHE (a1), the oxygen evolution potential (OEP) slightly shifted to more cathodic potentials (∼1.50 VRHE), and the OER kinetics decreased. In the potential range of 1.10–1.60 VRHE (Fig. 8a, inset), a higher current response was obtained when compared with the scenario in the absence of lignin. This indicates that lignin electro-oxidation is thermodynamically more favorable than the OER.75 This anodic peak at ∼1.20 VRHE can likely be attributed to the electro-oxidation of lignin on the glassy carbon electrode. Previous studies have reported that lignin electro-oxidation occurs at potentials below that for the OER.76 The CV curves show no traces of a corresponding reduction peak, demonstrating that lignin electro-oxidation is an irreversible reaction.1,77,78
Fig. 8b shows a linear correlation (R2 = 0.995) between the lignin concentration and j up to 10 g L−1 of lignin, together with growing current responses up to 30 g L−1 of lignin. From 30 g L−1 of lignin, a current density plateau was observed. These results suggest that lignin underwent direct charge transfer oxidation at the glassy carbon electrode surface, and also indicate that (i) electrode surface poisoning may have occurred due to the adsorption of lignin and LMPPs, as supported by SEM and XPS analyses, (ii) the electrode surface may have become saturated with lignin, and/or (iii) the reaction rate may have become limited by mass transport of lignin to the electrode surface. The application of 10 consecutive CV cycles (Fig. 8c) at a constant lignin concentration resulted in a gradual decrease in j, which can be attributed to the deactivation of the surface of the glassy carbon electrode due to lignin and LMPP adsorption. Thus, the electrode surface poisoning may have played a major role. Note that despite the occurrence of electrode surface poisoning, the electrode activity was fully restored after a simple physical cleaning procedure involving rinsing with water.
3.2.2.2. TiO2:Nb photoanodes. Fig. 9 shows the LSV analysis carried out to study the (photo)electrochemical behavior of the fabricated TiO2:Nb photoanodes in the absence and presence of lignin. j–V curves under dark conditions showed a current increase for a potential higher than ∼1.60 VRHE for 1 M NaOH (0 g L−1 lignin), and ∼1.50 VRHE for 60 g L−1 lignin. These onset potentials of dark current indicate the occurrence of the OER. Under light conditions, the onset potential of the j–V curves was ∼0.5 VRHE for 1 M NaOH and ∼0.20 VRHE for 60 g L−1 lignin (Fig. 9a). An anodic oxidation peak/wave (c1) seems to be occurring at ∼0.2–0.5 VRHE in the presence of lignin (Fig. 9a), which can be ascribed to its oxidation. Nevertheless, the shape of the j–V curves in Fig. 9b suggests an overlap between these two mechanisms: lignin oxidation and the OER. Upon increasing the lignin concentration (from 0 g L−1 to 10 g L−1), the j value also increased at ∼0.2–0.5 VRHE, although not linearly. From 10 g L−1 onwards, no perceptible differences in j occurred. This trend is consistent with that observed for the glassy carbon electrode. It indicates that lignin likely underwent direct charge transfer oxidation at the TiO2:Nb photoanode, and that more evident limiting factors in the electrochemical process may have emerged at higher lignin concentrations, including electrode surface poisoning due to lignin and lignin-derived product adsorption, electrode surface saturation, and mass transport limitations. Once again, the photoelectrode activity was completely restored following a simple physical cleaning procedure that involved rinsing with water.
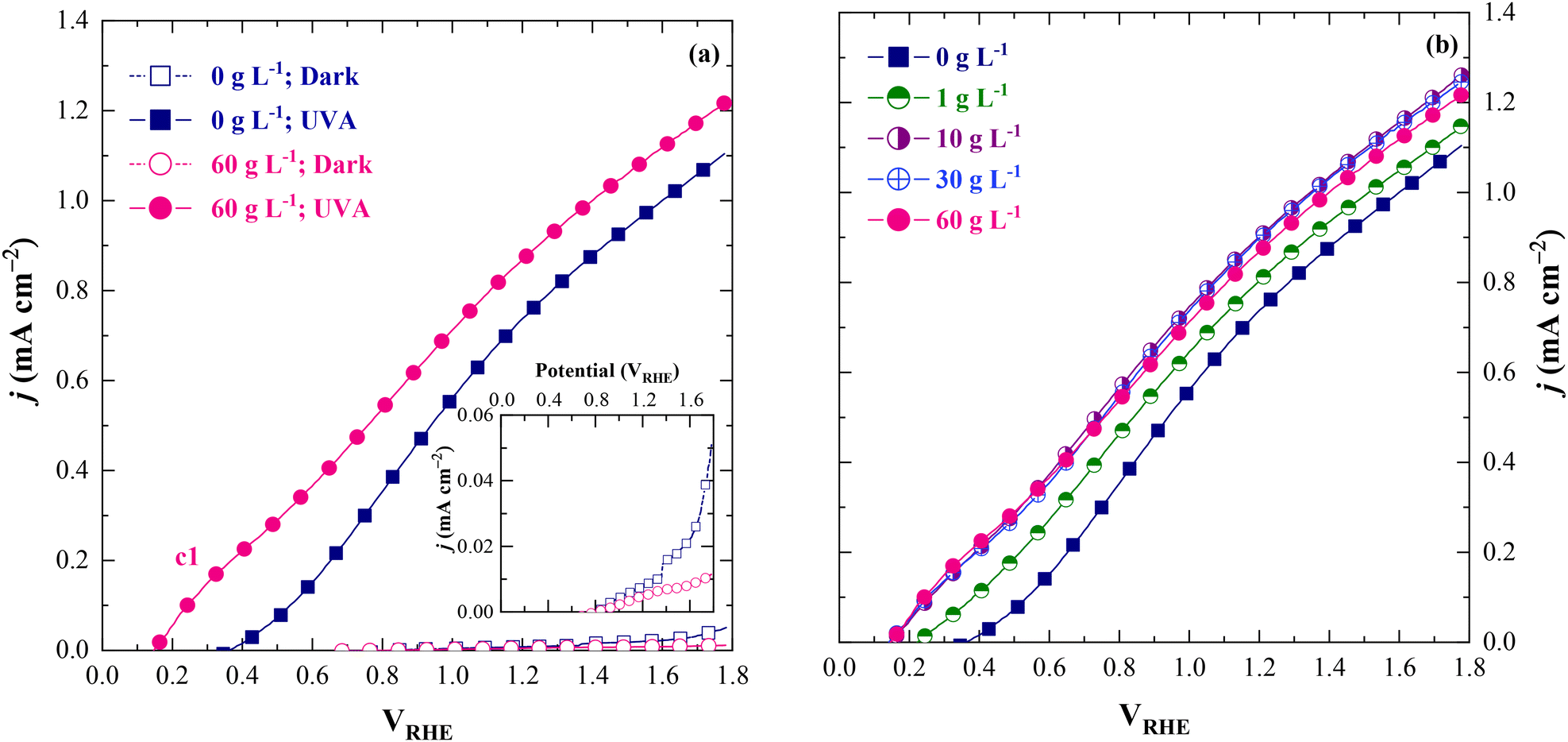 | ||
| Fig. 9 J–V characteristic curves acquired in a three-electrode “Cappuccino” cell with the fabricated TiO2:Nb photoanodes acting as a working electrode at a scan rate of 10 mV s−1 for (a) 1 M NaOH (pH = 13.2) and 60 g L−1 lignin (pH = 13.2) aqueous solutions under dark and light conditions, and (b) lignin aqueous solutions at different concentrations (pH = 13.2) under light conditions. | ||
3.3. Photoelectrocatalytic oxidation of lignin to value-added products
Lignin oxidation trials were conducted with an initial lignin content of 60 g L−1 to minimize errors in the HPLC analysis when low yields of LMPPs were generated, while minimizing mass transfer limitations of lignin to the TiO2:Nb photoanode surface. The lignin oxidation by PEC (using a two-electrode setup) was assessed for different applied constant Ecell values between ∼0 V and ∼2.5 V. The generation of LMPPs was not detected in the blank trial (without the application of Ecell or illumination).The content of each LMPP was assessed over time for PEC at different applied Ecell values. The results are available in Fig. SI-2.† The content of most LMPPs increased over time regardless of the Ecell value, reaching a maximum content after 7 h of reaction. Based on these kinetic profiles, yields of LMPPs were assessed in detail after 7 h of reaction and are presented in Fig. 10.
 | ||
| Fig. 10 Yields of combined LMPPs after 7 h of reaction for lignin oxidation using the photoelectrocatalysis (PEC) process at different applied cell potentials, and photocatalysis (PC) and electrocatalysis (EC) processes at ∼0.8 V. Conditions: TiO2:Nb (photo)anode/photocatalyst; [NaOH] = 1.0 M; [lignin]0 = 60 g L−1; T = 25 ± 1 °C; Re = 1750; a UVA-LED illumination system employed in PEC and PC processes (λ = 365 nm, irradiance = 120 ± 10 mW cm−2) under BSI. All tests were performed in duplicate. Error bars represent mean errors. Legend: V – vanillin, Sy – syringaldehyde, VA – vanillic acid, SA – syringic acid, VO – acetovanillone, and SO – acetosyringone. p-Hydroxybenzaldehyde (pHy) was not detected. | ||
For the PEC process, the increase of the applied Ecell from ∼0 V to ∼0.8 V resulted in increased yields of LMPPs. This positive effect can be attributed to enhanced direct oxidation of lignin on the surface of the TiO2:Nb photoanode at a higher applied Ecell value. It was previously reported that lignin serves as an electron donor through O–H bond breaking, producing protons (H+) and consuming photogenerated h+.3 For applied Ecell > 0.8 V, the yields of LMPPs decreased, most likely due to the generation of strong oxidative reactive oxygen species (ROS) able to overoxidize lignin. ROS such as the hydroxyl radicals (HO˙) can adsorb on the TiO2:Nb surface (M), forming the so-called superoxide MO viaeqn (11) and (12).79 The redox couple MO/M can act as a mediator in the EC process, unselectively oxidizing lignin and the formed LMPPs. Other ROS such as the superoxide radical (O2˙−), hydroperoxyl radical (HO2˙), and hydrogen peroxide (H2O2) can also be formed.79
| H2O + M → M(HO˙) + H+ + e− | (11) |
| M(HO˙) → MO + H+ + e− | (12) |
Regarding the nature of the LMPPs formed, S-type units were predominant, being produced in a higher amount than G-type ones, with no H-type units being detected. This agrees with the expected generation of LMPPs from hardwood Kraft lignin oxidation. Even though the FT-IR analysis does not show CO bond stretching (section 3.1.3.), the modification/elimination of C–O bonds and structural rearrangement of lignin units corroborate the obtained results. The absence of bond stretching might be the result of the low obtained yields. LMPPs can be arranged in the following order with respect to the yield of their generation upon the application of PEC at a constant Ecell of ∼0.8 V: SA > SO > Sy > VA > V > VO. Regardless of the unit type, aromatic acids were always generated in higher amounts compared to aldehydes and ketones. This can be attributed to the continuous oxidation of lignin to the aromatic acid forms (SA and VA). The lower yields of aromatic aldehydes and ketones may be due to their overoxidation, as other authors have previously reported.80,81 To further evaluate the aldehyde overoxidation hypothesis, the analyses of process selectivity to aldehyde generation were conducted. This was performed by examining the V/VA and Sy/SA ratios. The higher the ratios, the higher the selectivity towards aldehydes. Ratios of 2.7 ± 0.2 for V/VA and 5.1 ± 0.2 for Sy/SA were achieved at the beginning (t = 0 h) of all the experiments. Apart from EC at ∼0.8 V, all other experiments resulted in a decrease of selectivity of ∼2-fold for V/VA and of ∼5-fold for Sy/SA after 7 h of reaction time. This indicates that the generated aromatic acids (VA and SA) are likely products of further oxidation of the corresponding aldehydes, as previously stated. For the EC process, the decrease in selectivity was only 1-fold (V/VA) and 2.5-fold (Sy/SA), indicating a higher effect of h+ on C–O bond oxidation to CO. A higher decrease is expected for S-type units, as they are more reactive.
For an applied Ecell of ∼0.8 V, the obtained results demonstrate a cumulative effect of the PC and EC processes on the overall PEC process (Fig. 10). Total yields of 19 ± 1 mg kg−1 and 7 ± 2 mg kg−1 were obtained for PC and EC, respectively, while the PEC process afforded a total yield of 26 ± 1 mg kg−1. The absence of synergistic effects of EC and PC on PEC might indicate that (i) charge carrier separation was not optimized, highlighting the complexity of achieving synergistic effects for PEC processes, and/or (ii) lignin might have a high propensity to react quickly with generated h+ and/or ROS. The higher efficiency of the PC process compared to the EC process can be related to a higher ability of h+ to oxidize lignin compared to electro-oxidation mechanisms.
The generation of LMPPs from lignin (photo)electro-oxidation was then analyzed as a function of the total consumed charge and ECo. (Table SI-8†). The highest yield was obtained after a consumed charge of 5 C gLignin−1 and an ECo. of 0.06 kW h m−3 for the PEC process at a constant Ecell of ∼0.8 V. For PEC at Ecell > 0.8 V, the increase in charge and electrical consumption did not translate into a higher yield of LMPPs. This might be explained by the higher competition with the OER along with an increasing LMPP overoxidation degree. The AQY for the total LMPPs obtained under the PEC process at ∼0.8 V was 0.0016%.
4. Conclusions
The fabricated TiO2:Nb photoanode was able to photoelectrooxidize lignin, with direct charge transfer playing a crucial role. However, the adsorption of lignin and lignin-derived products on the photoanode surface occurred immediately after the beginning of the reaction, affecting its activity. The PEC process was responsible for the modification/elimination of C–O bonds, and the structural rearrangement of lignin units. Applying the PEC process at ∼0.8 V provided the highest yield of LMPPs – 26 ± 1 mg kg−1 after 7 h of reaction with a consumed charge of 5 C gLignin−1. The yield of the PEC process resulted from the cumulative effects of the yields from the PC and EC processes. S-type units were predominant, with no H-type units detected, consistent with the expected formation of LMPPs from the oxidation of hardwood KL. Aromatic acids were the predominant LMPPs generated, and their formation can be attributed to the overoxidation of aldehydes. The maximum yield of LMPPs was low, highlighting the need for further developments in the PEC technology applied to lignin oxidation.Author contributions
Daniela F. S. Morais: data curation, investigation, methodology, visualization, and writing – original draft; Luiza M. G. Sena: investigation; Joana M. Ribeiro: investigation; Telmo da Silva Lopes: conceptualization, investigation, and writing – review & editing; Paula Dias: conceptualization, supervision, and writing – review & editing; Adélio Mendes: supervision; Carina A. E. Costa: investigation, methodology, and writing – review & editing; Alírio E. Rodrigues: supervision; Susana R. S. Pereira: resources; Paula C. Pinto: resources; Rui A. R. Boaventura: writing – review & editing; Carlos J. Tavares: supervision and writing – review & editing; Vítor J. P. Vilar: conceptualization, funding acquisition, supervision, and writing – review & editing; Francisca C. Moreira: conceptualization, funding acquisition, methodology, project administration, supervision, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by Fundação para a Ciência e a Tecnologia, I.P./MCTES through national funds under project LigTech – EXPL/EAM-AMB/0216/2021 (https://doi.org/10.54499/EXPL/EAM-AMB/0216/2021). This research was also supported by UID/50020 of LSRE-LCM – Laboratory of Separation and Reaction Processes – Laboratory of Catalysis and Materials – funded by Fundação para a Ciência e a Tecnologia, I.P./MCTES through national funds; and ALiCE – LA/P/0045/2020 (https://doi.org/10.54499/LA/P/0045/2020). Daniela F. S. Morais, Luiza M. G. Sena, Joana M. Ribeiro, and Telmo da Silva Lopes acknowledge FCT for their PhD scholarship (references: SFRH/BD/146476/2019 – https://doi.org/10.54499/SFRH/BD/146476/2019; 2022.11876.BD; SFRH/BD/147221/2019 – https://doi.org/10.54499/SFRH/BD/147221/2019 and SFRH/BD/147426/2019 – https://doi.org/10.54499/SFRH/BD/147426/2019, respectively). Paula Dias, Vítor J. P. Vilar, and Francisca C. Moreira acknowledge the FCT Individual Call to Scientific Employment Stimulus (CEECIND/02862/2018, CEECIND/01317/2017 (https://doi.org/10.54499/CEECIND/01317/2017/CP1399/CT0010), and 2023.07337.CEECIND (https://doi.org/10.54499/2023.07337.CEECIND/CP2834/CT0009, respectively). Carlos J. Tavares acknowledges the funding from FCT through the Strategic Funds project reference UIDB/04650/2020-2023.References
- R. C. P. Oliveira, M. Mateus and D. M. F. Santos, Chronoamperometric and chronopotentiometric investigation of Kraft black liquor, Int. J. Hydrogen Energy, 2018, 43(35), 16817–16823, DOI:10.1016/j.ijhydene.2018.01.046.
- D. D. S. Argyropoulos, C. Crestini, C. Dahlstrand, E. Furusjö, C. Gioia, K. Jedvert, G. Henriksson, C. Hulteberg, M. Lawoko, C. Pierrou, J. S. M. Samec, E. Subbotina, H. Wallmo and M. Wimby, Kraft lignin: A Valuable, sustainable resource, opportunities and challenges, ChemSusChem, 2023, 16(23), e202300492, DOI:10.1002/cssc.202300492.
- J. King and S. S. C. Chuang, Photoelectrochemical conversion of lignin to hydrogen: Lignin as an electron donor, Catal. Commun., 2021, 149, 106219, DOI:10.1016/j.catcom.2020.106219.
- X. Li, Y. Ding, X. Pan, Y. Xing, B. Zhang, X. Liu, Y. Tan, H. Wang and C. Li, Scission of C–O and C–C linkages in lignin over RuRe alloy catalyst, J. Energy Chem., 2022, 67, 492–499, DOI:10.1016/j.jechem.2021.10.040.
- C. Yang, H. Chen, T. Peng, B. Liang, Y. Zhang and W. Zhao, Lignin valorization toward value-added chemicals and fuels via electrocatalysis: A perspective, Chin. J. Catal., 2021, 42(11), 1831–1842, DOI:10.1016/s1872-2067(21)63839-1.
- M. Zirbes and S. R. Waldvogel, Electro-conversion as sustainable method for the fine chemical production from the biopolymer lignin, Curr. Opin. Green Sustainable Chem., 2018, 14, 19–25, DOI:10.1016/j.cogsc.2018.05.001.
- F. M. Casimiro, C. A. E. Costa, C. M. Botelho, M. F. Barreiro and A. E. Rodrigues, Kinetics of oxidative degradation of lignin-based phenolic compounds in batch reactor, Ind. Eng. Chem. Res., 2019, 58(36), 16442–16449, DOI:10.1021/acs.iecr.9b02818.
- P. C. R. Pinto, E. Silva and A. E. Rodrigues, Lignin as Source of Fine Chemicals: Vanillin and Syringaldehyde, in Biomass Conversion: The Interface of Biotechnology, Chemistry and Materials Science, ed. C. Baskar, S. Baskar and R. S. Dhillon, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 381–420 Search PubMed.
- D. Gao, D. Ouyang and X. Zhao, Controllable oxidative depolymerization of lignin to produce aromatic aldehydes and generate electricity under mild conditions with direct biomass fuel cells as flexible reactors, Chem. Eng. J., 2024, 479, 147874, DOI:10.1016/j.cej.2023.147874.
- P. C. R. Pinto, E. A. Borges da Silva and A. E. Rodrigues, Insights into oxidative conversion of lignin to high-added-value phenolic aldehydes, Ind. Eng. Chem. Res., 2011, 50(2), 741–748, DOI:10.1021/ie102132a.
- X. Xiao, S. Sun, Y. Qi, S. Hao, W. Zhang and X. Qiu, Selective electrocatalytic oxidation of C(OH)–C bond in lignin with Pt@CeO2 toward the synthesis of benzoic acid, Electrochim. Acta, 2023, 470, 143377, DOI:10.1016/j.electacta.2023.143377.
- C. Srinivasulu, M. Ramgopal, G. Ramanjaneyulu, C. M. Anuradha and C. Suresh Kumar, Syringic acid (SA) - A review of its occurrence, biosynthesis, pharmacological and industrial importance, Biomed. Pharmacother., 2018, 108, 547–557, DOI:10.1016/j.biopha.2018.09.069.
- J. Kaur, M. Gulati, S. K. Singh, G. Kuppusamy, B. Kapoor, V. Mishra, S. Gupta, M. F. Arshad, O. Porwal, N. K. Jha, M. V. N. L. Chaitanya, D. K. Chellappan, G. Gupta, P. K. Gupta, K. Dua, R. Khursheed, A. Awasthi and L. Corrie, Discovering multifaceted role of vanillic acid beyond flavours: Nutraceutical and therapeutic potential, Trends Food Sci. Technol., 2022, 122, 187–200, DOI:10.1016/j.tifs.2022.02.023.
- P. C. R. Pinto, C. E. Costa and A. E. Rodrigues, Oxidation of lignin from Eucalyptus globulus pulping liquors to produce syringaldehyde and vanillin, Ind. Eng. Chem. Res., 2013, 52(12), 4421–4428, DOI:10.1021/ie303349j.
- F. M. Casimiro, C. A. E. Costa, C. Vega-Aguilar and A. E. Rodrigues, Hardwood and softwood lignins from sulfite liquors: Structural characterization and valorization through depolymerization, Int. J. Biol. Macromol., 2022, 215, 272–279, DOI:10.1016/j.ijbiomac.2022.06.067.
- B. Yan, W. Ding, X. Lin, Q. Cai and S. Zhang, Selective hydrogenolysis of lignin for phenolic monomers with a focus on β−O−4 cleavage and C=O hydrodeoxygenation, Fuel, 2022, 320, 123732, DOI:10.1016/j.fuel.2022.123732.
- W. Jiang, J.-P. Cao, C. Zhu, M. Zhao, Z.-H. Ni, X.-Y. Zhao, J.-X. Xie, L. Zhao, Y.-P. Zhao and H.-C. Bai, Catalytic hydrogenation of aromatic ring over ruthenium nanoparticles supported on α-Al2O3 at room temperature, Appl. Catal., B, 2022, 307, 121137, DOI:10.1016/j.apcatb.2022.121137.
- M. Cui, C. Liang, W. Zhao, X. Liu, L. Dong, D. Wang, S. Fu, Z. Jiang, F. Wang and X. Wei, Fabrication of Z-scheme CdS/H5PMo10V2O40/g-C3N4 for the photocatalytic depolymerization of lignin into aromatic monomers, Fuel Process. Technol., 2022, 238, 107481, DOI:10.1016/j.fuproc.2022.107481.
- K. Wu, M. Cao, Q. Zeng and X. Li, Radical and (photo)electron transfer induced mechanisms for lignin photo- and electro-catalytic depolymerization, Green Energy Environ., 2023, 8(2), 383–405, DOI:10.1016/j.gee.2022.02.011.
- R. Ayub and A. Raheel, High-value chemicals from electrocatalytic depolymerization of lignin: Challenges and opportunities, Int. J. Mol. Sci., 2022, 23(7), 3767, DOI:10.3390/ijms23073767.
- C. A. E. Costa, P. C. R. Pinto and A. E. Rodrigues, Lignin fractionation from E. globulus Kraft liquor by ultrafiltration in a three stage membrane sequence, Sep. Purif. Technol., 2018, 192, 140–151, DOI:10.1016/j.seppur.2017.09.066.
- P. C. R. Pinto, E. A. B. da Silva and A. E. Rodrigues, Comparative study of solid-phase extraction and liquid-liquid extraction for the reliable quantification of high value added compounds from oxidation processes of wood-derived lignin, Ind. Eng. Chem. Res., 2010, 49(23), 12311–12318, DOI:10.1021/ie101680s.
- F. Francisco, P. Dias, D. Ivanou, F. Santos, J. Azevedo and A. Mendes, Synthesis of host–guest hematite photoelectrodes for solar water splitting, ChemNanoMat, 2019, 5(7), 911–920, DOI:10.1002/cnma.201900141.
- T. Lopes, P. Dias, R. Monteiro, A. Vilanova, D. Ivanou and A. Mendes, A 25 cm2 solar redox flow cell: Facing the engineering challenges of upscaling, Adv. Energy Mater., 2022, 12(5), 2102893, DOI:10.1002/aenm.202102893.
- J. M. Ribeiro, F. C. Correia, F. J. Rodrigues, J. S. Reparaz, A. R. Goñi and C. J. Tavares, Transparent niobium-doped titanium dioxide thin films with high Seebeck coefficient for thermoelectric applications, Surf. Coat. Technol., 2021, 425, 127724, DOI:10.1016/j.surfcoat.2021.127724.
- M. Fox, Optical Properties of Solids, Oxford University Press, Oxford, 2nd edn, 2010 Search PubMed.
- S. Landi, I. R. Segundo, E. Freitas, M. Vasilevskiy, J. Carneiro and C. J. Tavares, Use and misuse of the Kubelka-Munk function to obtain the band gap energy from diffuse reflectance measurements, Solid State Commun., 2022, 341, 114573, DOI:10.1016/j.ssc.2021.114573.
- J. Tauc, R. Grigorovici and A. Vancu, Optical properties and electronic structure of amorphous germanium, Phys. Status Solidi B, 2006, 15(2), 627–637, DOI:10.1002/pssb.19660150224.
- J. Klein, L. Kampermann, B. Mockenhaupt, M. Behrens, J. Strunk and G. Bacher, Limitations of the Tauc plot method, Adv. Funct. Mater., 2023, 33(47), 2304523, DOI:10.1002/adfm.202304523.
- B. D. Viezbicke, S. Patel, B. E. Davis and D. P. Birnie, Evaluation of the Tauc method for optical absorption edge determination: ZnO thin films as a model system, Phys. Status Solidi B, 2015, 252(8), 1700–1710, DOI:10.1002/pssb.201552007.
- R. Krol, Photoelectrochemical Measurements, in Photoelectrochemical Hydrogen Production. Electronic Materials: Science & Technology, ed. R. Krol and M. Grätzel, Springer New York, New York, 2012, pp. 69–117 Search PubMed.
- T. Lopes, L. Andrade, H. A. Ribeiro and A. Mendes, Characterization of photoelectrochemical cells for water splitting by electrochemical impedance spectroscopy, Int. J. Hydrogen Energy, 2010, 35(20), 11601–11608, DOI:10.1016/j.ijhydene.2010.04.001.
- D. F. S. Morais, J. C. B. Lopes, M. M. Dias and V. J. P. Vilar, Moreira FC, e−NETmix: A pioneering electrochemical flow reactor with enhanced mass transfer, Chem. Eng. J., 2024, 481, 148244, DOI:10.1016/j.cej.2023.148244.
- C. H. Rosa, D. F. S. Morais, G. R. Rosa, J. H. Z. dos Santos, J. C. B. Lopes, M. M. Dias, R. Montes, R. Rodil, J. B. Quintana, G. A. Cerrón-Calle, S. Garcia-Segura, C. J. Tavares, V. J. P. Vilar and F. C. Moreira, Selective electrochemical oxidation of organic compounds in a mass transfer-enhanced electrochemical flow reactor (e−NETmix), J. Environ. Chem. Eng., 2024, 12(5), 113424, DOI:10.1016/j.jece.2024.113424.
- M. Rios-Enriquez, N. Shahin, C. Durán-de-Bazúa, J. Lang, E. Oliveros, S. H. Bossmann and A. M. Braun, Optimization of the heterogeneous Fenton-oxidation of the model pollutant 2,4-xylidine using the optimal experimental design methodology, Sol. Energy, 2004, 77(5), 491–501, DOI:10.1016/j.solener.2004.03.027.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, Inc., New York, 2nd edn, 2000 Search PubMed.
- N. Chu, D. Wang, H. Wang, Q. Liang, J. Chang, Y. Gao, Y. Jiang and R. J. Zeng, Flow-electrode microbial electrosynthesis for increasing production rates and lowering energy consumption, Engineering, 2023, 25, 157–167, DOI:10.1016/j.eng.2021.09.015.
- R. Ricka, M. Přibyl and K. Kočí, Apparent quantum yield – Key role of spatial distribution of irradiation, Appl. Catal., A, 2023, 658, 119166, DOI:10.1016/j.apcata.2023.119166.
- P. C. R. Pinto, C. Oliveira, C. A. Costa, A. Gaspar, T. Faria, J. Ataíde and A. E. Rodrigues, Kraft delignification of energy crops in view of pulp production and lignin valorization, Ind. Crops Prod., 2015, 71, 153–162, DOI:10.1016/j.indcrop.2015.03.069.
- H. Wang, Y. Pu, A. Ragauskas and B. Yang, From lignin to valuable products – strategies, challenges, and prospects, Bioresour. Technol., 2019, 271, 449–461, DOI:10.1016/j.biortech.2018.09.072.
- C. A. E. Costa, F. M. Casimiro, C. Vega-Aguilar and A. E. Rodrigues, Lignin valorization for added-value chemicals: Kraft lignin versus lignin fractions, ChemEngineering, 2023, 7(3), 42, DOI:10.3390/chemengineering7030042.
- M. Yamamura, T. Hattori, S. Suzuki, D. Shibata and T. Umezawa, Microscale alkaline nitrobenzene oxidation method for high-throughput determination of lignin aromatic components, Plant Biotechnol., 2010, 27(4), 305–310, DOI:10.5511/plantbiotechnology.27.305.
- J.-M. Ha, K.-R. Hwang, Y.-M. Kim, J. Jae, K. H. Kim, H. W. Lee, J.-Y. Kim and Y.-K. Park, Recent progress in the thermal and catalytic conversion of lignin, Renewable Sustainable Energy Rev., 2019, 111, 422–441, DOI:10.1016/j.rser.2019.05.034.
- R. S. Weber and K. K. Ramasamy, Electrochemical oxidation of lignin and waste plastic, ACS Omega, 2020, 5(43), 27735–27740, DOI:10.1021/acsomega.0c03989.
- D. Gao, D. Ouyang and X. Zhao, Electro-oxidative depolymerization of lignin for production of value-added chemicals, Green Chem., 2022, 24(22), 8585–8605, 10.1039/d2gc02660k.
- S. Guadix-Montero and M. Sankar, Review on catalytic cleavage of C–C inter-unit linkages in lignin model compounds: Towards lignin depolymerisation, Top. Catal., 2018, 61(3–4), 183–198, DOI:10.1007/s11244-018-0909-2.
- J. C. Villar, A. Caperos and F. García-Ochoa, Oxidation of hardwood Kraft-lignin to phenolic derivatives. Nitrobenzene and copper oxide as oxidants, J. Wood Chem. Technol., 1997, 17(3), 259–285, DOI:10.1080/02773819708003131.
- R. Md Salim, J. Asik and M. S. Sarjadi, Chemical functional groups of extractives, cellulose and lignin extracted from native Leucaena leucocephala bark, Wood Sci. Technol., 2021, 55(2), 295–313, DOI:10.1007/s00226-020-01258-2.
- O. Y. Abdelaziz and C. P. Hulteberg, Physicochemical characterisation of technical lignins for their potential valorisation, Waste Biomass Valorization, 2016, 8(3), 859–869, DOI:10.1007/s12649-016-9643-9.
- A. B. D. Nandiyanto, R. Oktiani and R. Ragadhita, How to read and interpret FTIR spectroscope of organic material, IJoST, 2019, 4(1), 97, DOI:10.17509/ijost.v4i1.15806.
- P. Yan, Z. Xu, C. Zhang, X. Liu, W. Xu and Z. C. Zhang, Fractionation of lignin from eucalyptus bark using amine-sulfonate functionalized ionic liquids, Green Chem., 2015, 17(11), 4913–4920, 10.1039/c5gc01035g.
- D. Shao, J. Liang, X. Cui, H. Xu and W. Yan, Electrochemical oxidation of lignin by two typical electrodes: Ti/Sb SnO2 and Ti/PbO2, Chem. Eng. J., 2014, 244, 288–295, DOI:10.1016/j.cej.2014.01.074.
- A. Tejado, C. Peña, J. Labidi, J. M. Echeverria and I. Mondragon, Physico-chemical characterization of lignins from different sources for use in phenol-formaldehyde resin synthesis, Bioresour. Technol., 2007, 98(8), 1655–1663, DOI:10.1016/j.biortech.2006.05.042.
- C.-M. Popescu, M.-C. Popescu, G. Singurel, C. Vasile, D. S. Argyropoulos and S. Willfor, Spectral characterization of eucalyptus wood, Appl. Spectrosc., 2016, 61(11), 1168–1177, DOI:10.1366/000370207782597076.
- M. N. Mohamad Ibrahim, N. Zakaria, C. S. Sipaut, O. Sulaiman and R. Hashim, Chemical and thermal properties of lignins from oil palm biomass as a substitute for phenol in a phenol formaldehyde resin production, Carbohydr. Polym., 2011, 86(1), 112–119, DOI:10.1016/j.carbpol.2011.04.018.
- J.-L. Wen, B.-L. Xue, F. Xu and R.-C. Sun, Unveiling the structural heterogeneity of bamboo lignin by in situ HSQC NMR technique, BioEnergy Res., 2012, 5(4), 886–903, DOI:10.1007/s12155-012-9203-5.
- C. A. Esteves Costa, W. Coleman, M. Dube, A. E. Rodrigues and P. C. Rodrigues Pinto, Assessment of key features of lignin from lignocellulosic crops: Stalks and roots of corn, cotton, sugarcane, and tobacco, Ind. Crops Prod., 2016, 92, 136–148, DOI:10.1016/j.indcrop.2016.07.032.
- M. Fodil Cherif, D. Trache, N. Brosse, F. Benaliouche and A. F. Tarchoun, Comparison of the physicochemical properties and thermal stability of organosolv and Kraft lignins from hardwood and softwood biomass for their potential valorization, Waste Biomass Valorization, 2020, 11(12), 6541–6553, DOI:10.1007/s12649-020-00955-0.
- J. Kim, Y. Um, S. Han, T. Hilberath, Y. H. Kim, F. Hollmann and C. B. Park, Unbiased photoelectrode interfaces for solar coupling of lignin oxidation with biocatalytic CC bond hydrogenation, ACS Appl. Mater. Interfaces, 2022, 14(9), 11465–11473, DOI:10.1021/acsami.1c24342.
- E. Melro, A. Filipe, D. Sousa, B. Medronho and A. Romano, Revisiting lignin: A tour through its structural features, characterization methods and applications, New J. Chem., 2021, 45(16), 6986–7013, 10.1039/d0nj06234k.
- Z. Dai, X. Shi, H. Liu, H. Li, Y. Han and J. Zhou, High-strength lignin-based carbon fibers via a low-energy method, RSC Adv., 2018, 8(3), 1218–1224, 10.1039/c7ra10821d.
- A. Lenong, A. J. Ragauskas and J. S. Hsieh, Metal profiling of southeastern U.S. softwood and hardwood furnish, Tappi J., 2000, 83(8), 97 Search PubMed.
- Z. Tian, L. Zong, R. Niu, X. Wang, Y. Li and S. Ai, Recovery and characterization of lignin from alkaline straw pulping black liquor: As feedstock for bio–oil research, J. Appl. Polym. Sci., 2015, 132(25), 42057, DOI:10.1002/app.42057.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci., 2011, 257(7), 2717–2730, DOI:10.1016/j.apsusc.2010.10.051.
- T. F. Scientific , Carbon X-ray photoelectron spectra, carbon electron configuration, and other elemental information 2024 [https://www.thermofisher.com/pt/en/home/materials-science/learning-center/periodic-table/non-metal/carbon.html].
- M. Z. Atashbar, H. T. Sun, B. Gong, W. Wlodarski and R. Lamb, XPS study of Nb-doped oxygen sensing TiO2 thin films prepared by sol-gel method, Thin Solid Films, 1998, 326(1–2), 238–244, DOI:10.1016/s0040-6090(98)00534-3.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V, Cu and Zn, Appl. Surf. Sci., 2010, 257(3), 887–898, DOI:10.1016/j.apsusc.2010.07.086.
- R. Zanoni, G. Righini, A. Montenero, G. Gnappi, G. Montesperelli, E. Traversa and G. Gusmano, XPS analysis of sol–gel processed doped and undoped TiO2 films for sensors, Surf. Interface Anal., 2004, 22(1–12), 376–379, DOI:10.1002/sia.740220182.
- D. Reyes-Coronado, G. Rodríguez-Gattorno, M. E. Espinosa-Pesqueira, C. Cab, R. de Coss and G. Oskam, Phase-pure TiO2 nanoparticles: Anatase, brookite and rutile, Nanotechnology, 2008, 19(14), 145605, DOI:10.1088/0957-4484/19/14/145605.
- M.-S. Wong, S.-W. Hsu, K. K. Rao and C. P. Kumar, Influence of crystallinity and carbon content on visible light photocatalysis of carbon doped titania thin films, J. Mol. Catal. A:Chem., 2008, 279(1), 20–26, DOI:10.1016/j.molcata.2007.09.024.
- S. Rehman, R. Ullah, A. M. Butt and N. D. Gohar, Strategies of making TiO2 and ZnO visible light active, J. Hazard. Mater., 2009, 170(2–3), 560–569, DOI:10.1016/j.jhazmat.2009.05.064.
- H. Abdullah, M. M. R. Khan, H. R. Ong and Z. Yaakob, Modified TiO2 photocatalyst for CO2 photocatalytic reduction: An overview, J. CO2 Util., 2017, 22, 15–32, DOI:10.1016/j.jcou.2017.08.004.
- V. Bansal, J. D. Benck, B. A. Pinaud, Y. Gorlin and T. F. Jaramillo, Substrate selection for fundamental studies of electrocatalysts and photoelectrodes: Inert potential windows in acidic, neutral, and basic electrolyte, PLoS One, 2014, 9(10), e107942, DOI:10.1371/journal.pone.0107942.
- Y. Yi, G. Weinberg, M. Prenzel, M. Greiner, S. Heumann, S. Becker and R. Schlögl, Electrochemical corrosion of a glassy carbon electrode, Catal. Today, 2017, 295, 32–40, DOI:10.1016/j.cattod.2017.07.013.
- J. Li, W. Zhou, Y. Huang and J. Gao, Lignin-assisted water electrolysis for energy-saving hydrogen production with Ti/PbO2 as the anode, Front. Energy Res., 2021, 9, 762346, DOI:10.3389/fenrg.2021.762346.
- K. Beliaeva, N. Grimaldos-Osorio, E. Ruiz-López, L. Burel, P. Vernoux and A. Caravaca, New insights into lignin electrolysis on nickel-based electrocatalysts: Electrochemical performances before and after oxygen evolution, Int. J. Hydrogen Energy, 2021, 46(72), 35752–35764, DOI:10.1016/j.ijhydene.2021.01.224.
- N. Di Fidio, J. W. Timmermans, C. Antonetti, A. M. Raspolli Galletti, R. J. A. Gosselink, R. J. M. Bisselink and T. M. Slaghek, Electro-oxidative depolymerisation of technical lignin in water using platinum, nickel oxide hydroxide and graphite electrodes, New J. Chem., 2021, 45(21), 9647–9657, 10.1039/d1nj01037a.
- F. Bateni, R. Ghahremani and J. A. Staser, Electrochemical oxidative valorization of lignin by the nanostructured PbO2/MWNTs electrocatalyst in a low-energy depolymerization process, J. Appl. Electrochem., 2020, 51(1), 65–78, DOI:10.1007/s10800-020-01451-y.
- M. Panizza and G. Cerisola, Direct and mediated anodic oxidation of organic pollutants, Chem. Rev., 2009, 109(12), 6541–6569, DOI:10.1021/cr9001319.
- O. Movil-Cabrera, A. Rodriguez-Silva, C. Arroyo-Torres and J. A. Staser, Electrochemical conversion of lignin to useful chemicals, Biomass Bioenergy, 2016, 88, 89–96, DOI:10.1016/j.biombioe.2016.03.014.
- P. Parpot, A. P. Bettencourt, A. M. Carvalho and E. M. Belgsir, Biomass conversion: attempted electrooxidation of lignin for vanillin production, J. Appl. Electrochem., 2000, 30(6), 727–731, DOI:10.1023/A:1004003613883.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00038f |
| This journal is © The Royal Society of Chemistry 2025 |